
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly concepts for multicompartment nanostructures
André H.
Gröschel
 *a and
Axel H. E.
Müller
*b
*a and
Axel H. E.
Müller
*b
aMolecular Materials, Department of Applied Physics, Aalto University School of Science, FIN-00076 Aalto, Espoo, Finland. E-mail: andre.groschel@aalto.fi
bInstitute of Organic Chemistry, Johannes Gutenberg-University, D-55099 Mainz, Germany. E-mail: axel.mueller@uni-mainz.de
First published on 17th June 2015
Abstract
Compartmentalization is ubiquitous to many biological and artificial systems, be it for the separate storage of incompatible matter or to isolate transport processes. Advancements in the synthesis of sequential block copolymers offer a variety of tools to replicate natural design principles with tailor-made soft matter for the precise spatial separation of functionalities on multiple length scales. Here, we review recent trends in the self-assembly of amphiphilic block copolymers to multicompartment nanostructures (MCNs) under (semi-)dilute conditions, with special emphasis on ABC triblock terpolymers. The intrinsic immiscibility of connected blocks induces short-range repulsion into discrete nano-domains stabilized by a third, soluble block or molecular additive. Polymer blocks can be synthesized from an arsenal of functional monomers directing self-assembly through packing frustration or response to various fields. The mobility in solution further allows the manipulation of self-assembly processes into specific directions by clever choice of environmental conditions. This review focuses on practical concepts that direct self-assembly into predictable nanostructures, while narrowing particle dispersity with respect to size, shape and internal morphology. The growing understanding of underlying self-assembly mechanisms expands the number of experimental concepts providing the means to target and manipulate progressively complex superstructures.
 André H. Gröschel | André H. Gröschel obtained his PhD in 2012 from University of Bayreuth in Germany focusing on controlled polymerization of diblock co- and triblock terpolymers as well as their self-assembly behavior to multicompartment nanostructures. He further studied the hierarchical self-assembly of polymer-based patchy nanoparticles their application potential as dispersants and compatibilizer. During a postdoc stay at the department of Applied Physics in Aalto University (Helsinki, Finland), he entered the field of biomimetic and responsive nanocomposites, photonic materials and dynamic systems. |
 Axel H. E. Müller | Axel H. E. Müller obtained his PhD in 1977 from the Johannes Gutenberg University in Mainz, working with G. V. Schulz. From 1999–2012, he was professor and chair of Macromolecular Chemistry at the University of Bayreuth. In October 2012, he retired from Bayreuth and moved back to Mainz University to continue research as a Fellow of the Gutenberg Research College. His research interest focuses on the design of complex polymer and hybrid structures by controlled/living polymerization techniques and on self-organized nanostructures obtained from them in bulk and solution. Structures of interest include multicompartment micelles, soft Janus particles, and hybrids of polymers and inorganic or biological materials. He has published over 400 research papers and edited various books. He is also Senior Editor of the journal Polymer. Awards include the IUPAC MACRO Distinguished Polymer Scientist Award (2004) and the Hermann Staudinger Prize of the German Chemical Society (2012). In 2011, he became the first Fellow of the Polymer Chemistry Division of the ACS not living in the United States. |
1. Introduction
Self-assembly describes the spontaneous arrangement of building blocks into larger structures with well-defined symmetry, complex architecture and (preferably) long-range order. As a versatile tool for the energy-efficient bottom-up structuring of bulk materials, surface patterns or nanoobjects in solution,1–5 self-assembly fuels the innovative design of nanomaterials that are “smart”, self-healing, programmed for target-oriented motion, and serve as miniaturized components for nanoelectronics or nanooptics.6–14 The interaction potential between building blocks has diverse origin, but the universal driving force is to reach thermodynamic equilibrium by minimization of the free energy. To facilitate equilibration, mobility is most important as otherwise building blocks cannot rearrange upon the applied stimulus and are instead kinetically trapped in local energy minima (although there are also concepts deliberately targeting these minima). In that regard, block copolymers, i.e., two or more covalently linked segments with intrinsic incompatibility, are very attractive building blocks appreciated for several advantageous and relevant features:• Versatile synthetic protocols for diblock, triblock or multiblock copolymers from a large selection of functional monomers; each block may respond selectively to a specific physical field;
• Control over short-range attraction by block-block linkage (covalent, coordinating, electrostatic, hydrogen bonding); long-range repulsion by nature of the monomer and block incompatibility;
• Variable domain sizes and particle geometry through block lengths and additives (co-solvent, supramolecular interaction, loading with cargo);
• Reasonably fast diffusion kinetics in solvents (surfaces/interfaces) and stability of aggregates;
Block copolymer self-assembly is a rapidly evolving field of high diversity with great progress in structural control and functionalization. Besides synthetic encoding of material properties and aggregation behavior into individual blocks, the self-assembly process itself can be altered in specific directions as well. From the vast collection of directing agents (stimuli or additives) virtually any change of physical field strength is conceivable (e.g., solvent polarity, ionic strength, electric or magnetic field, etc.). Sequences of orthogonal stimuli can be utilized to address blocks in varying order with the aim to bridge hierarchies through their sequential collapse. Directed and hierarchical self-assembly of block copolymers and knowledge about driving forces aid in creating near-monodisperse (hybrid) materials with narrow size distribution and well-defined internal symmetry. This review does not to encompass all aspects of self-assembly, but tries to summarize recent developments in spontaneous and directed solution self-assembly of ABC triblock terpolymers and its structural analogues (e.g. blends of diblock copolymers) into core- and corona-compartmentalized nanostructures. The first section introduces tools to tune block–block and block–solvent interactions and manipulate self-assembly through external stimuli. The second part examines these tools in more detail on concrete examples of multicompartment nanostructure (MCN) formation: first, on diblock copolymers, followed by blending of two diblock copolymers through non-covalent interactions and finally, covalently linked ABC triblock terpolymers and capabilities for hierarchical self-assembly. The review concludes with applications of MCNs and discusses obstacles that need to be tackled with to unfold their full potential for future technologies.
2 Controlled and directed self-assembly
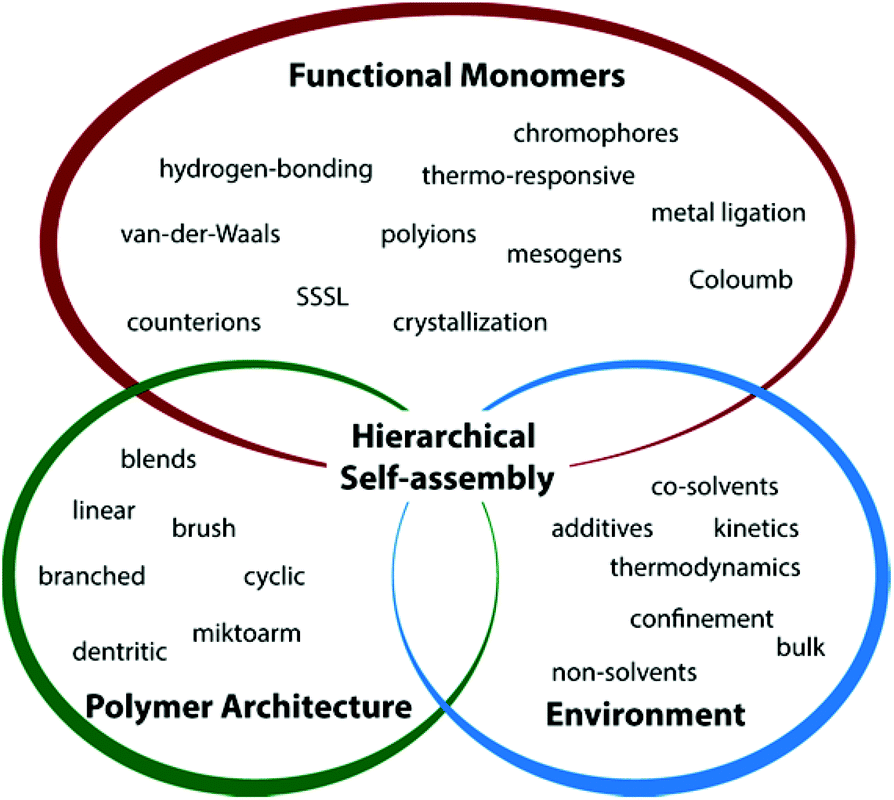
The formation of MCNs with multiple defined nanophases requires connected polymer blocks (architecture) with distinct differences in physical properties (monomer functionality) that spontaneously self-assemble in bulk or solution (environmental parameters). In solution, block–solvent interactions dominate and polymer chains are physically bound within individual nanostructures of defined geometry (also topology or simply morphology). Throughout this review we will use “geometry” for the shape of nanostructures and “morphology” when addressing their internal structure. Lipids – naturally occurring amphiphiles that form membranes in water (liposomes, cells) – are the design inspiration for amphiphilic block copolymers, i.e., hydrophilic and hydrophobic segments covalently linked to one molecule (more general, solvophilic/solvophobic).15–17 The spontaneous orientation of the “philic” segment towards the solvent and the “phobic” segment to form the particle core results in defined geometries of well-controlled size. Much effort has been invested to pinpoint parameters that govern diblock copolymer self-assembly in selective solvents into spherical and cylindrical micelles, bicontinuous networks and vesicles.18–26 The block length, NA, the molar volume, Vm,A, and the density, ρA, of the solvophobic block together give the core volume, Vcore, via the aggregation number, Z.27–30 The packing parameter, p = v/aelc, as defined by Israelachvili for surfactants, is applicable to block copolymers and a measure for chain packing at the core–solvent interface. Thereby, ae is the equilibrium interfacial area of the hydrophilic head group, lc the critical length of the hydrophobic tail and v the segment volume.15,31 Note that this approximation does not account for a corona block with variable volume, which requires extension to more sophisticated theoretical descriptions.32 Values for p mark pronounced morphological transitions from spheres at p ≤ 1/3 to cylinders 1/3 < p ≤ 1/2, and bilayer sheets/vesicles at p ≥ 1. Today, the formation of these geometries is well-understood for diblock copolymers and structural transition involve: fusion of spheres into cylinders, cylinder entanglement to sheets (resembling “octopus”-shape) bending of sheets to “jellyfish”-like aggregates and roll-up to closed shells or vesicles (also polymersomes).18,33,34 A comparable level of understanding would be also desirable for ABC triblock terpolymers, but the increased number of interaction parameters makes the reliable prediction of particle shape and block arrangement difficult. It is however, imperative to develop a concise theory for triblock terpolymer self-assembly in order to open up a whole new playground of MCNs with unprecedented structural complexity.A multitude of parameters allow manipulating the self-assembly process into various directions. The most relevant setscrews may be divided into three major categories (Fig. 1):
• polymer architecture,
• intrinsic block functionality, and
• external stimulation.
Polymer architecture. The polymer architecture reflects the number of polymer blocks, the block sequence (more than two blocks) and how blocks are connected to each other. Polymers made from two monomers are commonly termed AB diblock copolymers and from three different monomers ABC triblock terpolymers. The addition of a third block complicates the prediction of the self-assembly outcome, because a total of eight independent parameters has to be considered in solution:35 three polymer–polymer interactions, χAB, χAC, χBC, three polymer–solvent interactions χAS, χBS, χCS (S = solvent) and two block volume fractions ϕA, ϕB (ϕC = 1 − (ϕA + ϕB)). Linear block sequences thus often require agents to guide blocks into the desired position, but they also display more complex self-assembly behavior with access to hierarchies under proper self-assembly conditions as will be examined in section 3.3.36 When blocks are not connected in sequence, but in a single junction point, they are designated as miktoarm star terpolymers.37 This architecture demonstrates particular straightforward self-assembly behavior, because the star-like arrangement of the polymer arms leads to packing frustration and intricate phase behavior. Molecular bottlebrushes are giant single molecules with AB, ABA or ABC sequence of brush segments along (or concentric to) the polymeric backbone prepared by either “grafting to”, “grafting through” or “grafting from” strategies.38–41 The dimension of these molecules reaches the micrometer scale and superstructures in solution or in bulk display unusual large domain sizes.42,43Cyclic, (hyper-)branched and multiarm star architectures with three or more segments have received less attention as building blocks for MCN formation so far, but individual studies give a glimpse of their structuring potential.44–48
Monomer functionality. The strength of microphase separation depends on the segment–segment incompatibility, χ, which is an intrinsic property of the employed monomers and higher values correspond to stronger enthalpic repulsion. For any given self-assembly scenario, first the desired physical property of the blocks will be chosen from a vast selection of available monomers. Thereby, liquid crystalline, (semi-)crystalline and mesogen-modified blocks form frustrated self-assemblies with unusual (rigid) shape where nano-domains are further sub-compartmentalized by the mesogen dimension.49 Light-responsive chromophores (e.g., azobenzene dyes)50 often adapt a rod-like conformation and light-stimulated changes to this conformation likewise alter polarity, solubility and chain packing. Related are fluorocarbon or hydrocarbon blocks with extraordinary high χ-parameter close to or above the super strong segregation limit (SSSL) with preference for dense chain packing. In contrast to mere microphase separation, other concepts rely on electrostatic attraction between polyions or multivalent counterions.51 Depending on the nature of the counterion, charges on the polymer chains are compensated when approaching charge neutralization increasing χ and solvophobicity. And even monovalent counterions are used to finetune the degree of swelling or de-swelling of microphases. Ions may further act as binding units with exceptional selectivity when combined with specific coordination sites; metal–ligand complexes are reversible binding sites that allow the construction of multiblock copolymers by linking of end-tethered blocks.52 Hydrogen bonds are similarly selective and reversible with binding strength controlled through the supramolecular motifs (number of acceptors and donors).53 Besides the possibility to physically attach functional moieties during post-polymerization modification, complementary binding sites may chemically stitch block copolymers or even link entire compartments.
Environmental triggers comprise any change in physical field strength that directly affects polymer solubility, e.g. non-solvents, ionic strength or temperature, light, oxidative state, and several others.9,54,55 In order to work properly, some stimuli require the complementary monomer functionality. Other external fields such as electric, magnetic, acoustic or flow fields are feasible to direct the self-assembly of polymeric particles or hard colloids, but have not been used to direct the self-assembly of multiblock copolymers into MCNs. If not assembled under bulk conditions, the choice of solvent controls the structure, ultimately decided by either kinetic trapping or thermodynamic equilibration. The slow addition of non-solvents on appropriate time scales facilitates reorganization of polymer chains and allows for sequential collapse and hierarchical self-assembly (in case of three or more blocks). On the contrary, deliberate kinetic trapping through rapid addition of non-solvents with high χAS reduces polymer chain mobility and freezes the structure.56 Polymer blocks with high glass transition temperature, Tg, or high molecular weights, Mn, are especially effective for kinetic control. Environmental triggers further induce shape changes to the superstructure,57 an effect that becomes especially relevant when self-assembling multiphase particles on several hierarchies.
 | ||
| Fig. 1 Structuring concepts for the formation of MCNs. | ||
3 Multicompartment nanostructures (MCNs)
Diblock copolymer vesicles (or polymersomes) may be considered as the simplest MCN able to encapsulate solvophilic cargo in the hollow, solvent-filled interior while storing a solvophobic substance within the membrane. Vesicles made from polymers or polypeptides58 are attractive for drug delivery,59 as artificial cell mimics (hybrid protein/polymer prototypes),60,61 nanoreactors62 or for biomedical applications (see also section 4.4).63–67 Besides vesicles, several other compartmentalized particles were created from one single diblock copolymer. The interaction dynamics between polymer and solvent are delicate and allow targeting a variety of kinetically trapped intermediates with complex internal morphology, e.g., concentric multilamellar vesicles (onion-like), bicontinuous networks (“plumber's nightmare”) and vesicles with tubular channels inside the membrane.68–70 Although these structures only appear in a narrow window of parameters, they draw the picture of a broad spectrum of intermediate morphologies between thermodynamic equilibration and kinetic trapping. To reach higher structural complexity, eventually three polymer blocks need to be confined into one nanoparticle, either by supramolecular blending of two diblock copolymers or using covalently linked triblock terpolymers. The number of possible block arrangements and morphologies increases considerably as predicted by theory and verified experimentally.71–76 The combined research efforts on block copolymer self-assembly in solution led to a zoo of exotic, but versatile and multifunctional superstructures. Fig. 2 summarizes a small selection of core- and/or corona- compartmentalized MCNs, but draws by all means not the complete morphological picture.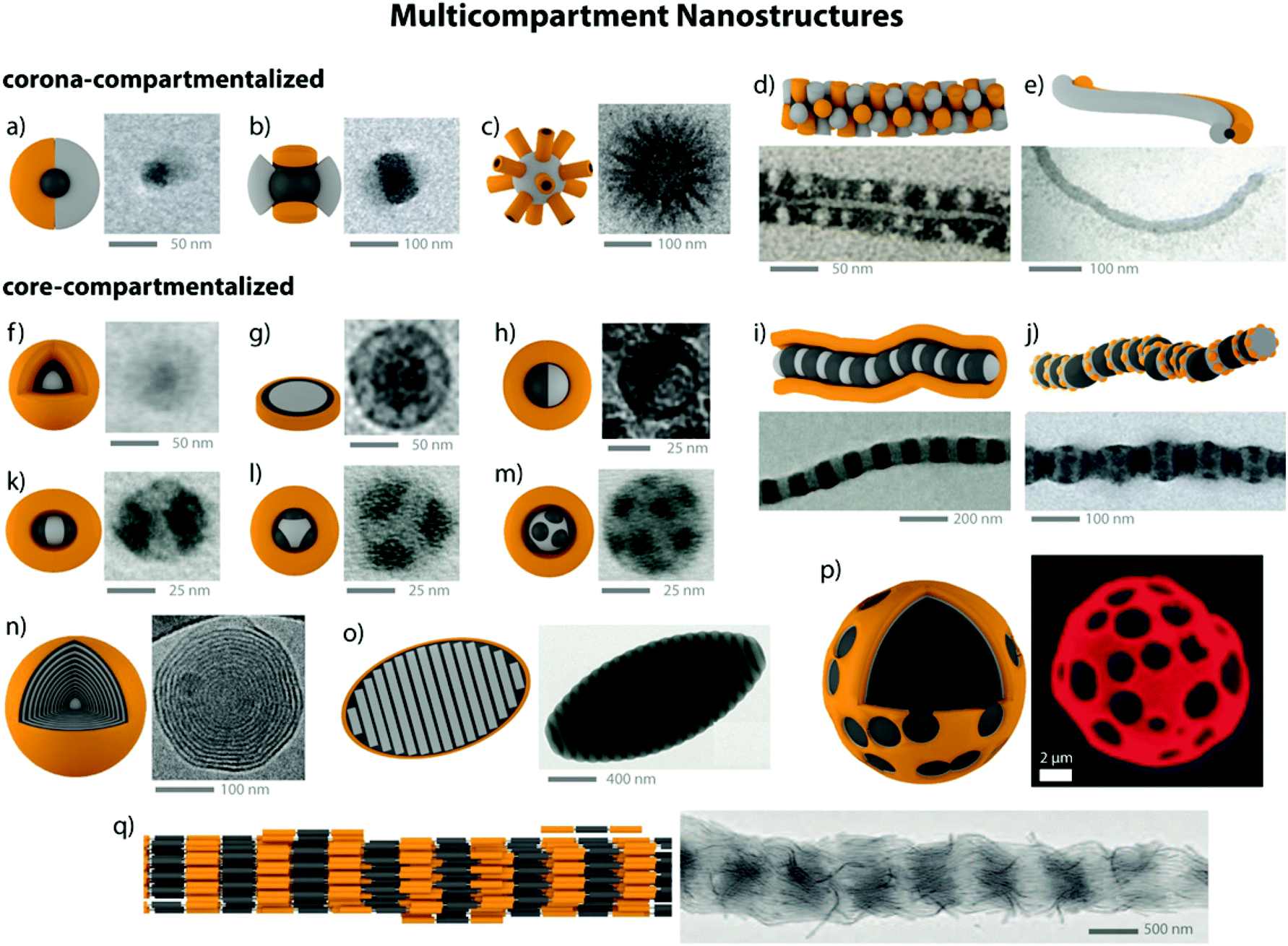 | ||
| Fig. 2 Collection of MCNs divided into corona-compartmentalized: (a) Janus micelle;77 (b) Inverse “hamburger” micelle;77 (c) spherical micelle with cylindrical interpolyelectrolyte complex brushes;78 (d) patchy core-crystalline cylinder;79 (e) Janus cylinder.80Core-compartmentalized: (f) core–shell–corona micelles;81 (g) compartmentalized disc;82 (h) micelle with Janus core;83 (i) cylinder with alternating core segments;77 (j) cylinder with complex arrangement of multiple core segments;77 (k–m) homogeneous MCNs with two, three and multiple core compartments;84 (n) concentric lamella (onion-like) assembly;85 (o) ellipsoidal particle with stacked internal lamellae;86 (p) polymersome with compartmentalized membrane.87 (q) Cylindrical core-segmented supermicelle of block co-micelles.88 Images reprinted with permission from the references as indicated. Copyright, as appropriate, American Association for the Advancement of Science, American Chemical Society, National Academy of Sciences, Nature Publishing Group, Wiley-VCH. | ||
Janus particles feature two strictly separated hemispheres with distinct difference in physical properties and are the simplest corona-compartmentalized particles. Soft Janus nanoparticles are of particular interest for the solution self-assembly of triblock terpolymers, because they may be an essential transient building block in many documented self-assembly processes.84 Section 3.4 and 4.2 emphasize on the synthesis of polymeric Janus nanoparticles and exemplify their interfacial properties and self-assembly capabilities. A comprehensive review covering most aspects of Janus particle synthesis and self-assembly has been publish recently.89
3.1 Self-assembly of AB diblock copolymers
Solvophobic interactions are amongst the most important forces in self-assembly and despite limitations in directionality, very effective for the selective compartmentalization of polymer segments.90 If one diblock copolymer is supposed to form two collapsed polymer compartments, additives (e.g. surfactants) are necessary to stabilize the interface and to prevent uncontrolled coagulation and macroscopic precipitation.3D confinement. Concepts that microphase separate block copolymers under 3D confinement, e.g., inside nano-droplets, make use of co-solvents and additives (emulsifiers) to kinetically control the development of MCNs with a rich ensemble of internal morphologies (Fig. 3).91–97 The dimension of the bulk phase is reduced to a spherical droplet containing an immiscible homopolymer blend, diblock copolymers or a variation of both.98,99 Molecular or polymeric surfactants stabilize the energetically unfavorable blocks–solvent interface that is particularly large for nano-droplets as compared to the bulk state.100 Deviations from the bulk behavior have been observed especially under strong confinement conditions, i.e., when the droplet diameter approaches the periodicity of the block copolymer.101 Although this concept was demonstrated for triblock terpolymers102 and dentritic linear block copolymers,103 diblock copolymers are by far the most reported, most likely because they are easier accessible and block orientation can be predicted with high certainty. Polymer-rich droplets are produced by drop-wise addition of water-immiscible polymer solutions to water, e.g., polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) in chloroform), under vigorous stirring or sonication (Fig. 3a). The organic solvent is allowed to evaporate inducing polymer concentration and microphase separation within the droplet. This method gives access to nanostructures with block arrangements that are hardly accessible otherwise, e.g., Janus particles composed of two homopolymer hemispheres (held together solely by the non-solvent)99,104 or axially stacked lamellae as exemplified on PS-b-P4VP particles in Fig. 3b.100 Beyond that, internal organization into spherical, cylindrical and lamella morphologies with lateral (or centrosymmetric) orientation were found for polystyrene-b-polybutadiene (Fig. 3c)105 (or polyisoprene),97 but also rings, helices and branched helices have been reported.106 Stacked lamellae of glassy PS-b-P4VP undergo dynamic unidirectional swelling along the stack normal when partially cross-linked P4VP layers are selectively swollen. The expanding P4VP phase is covalently connected to the glassy PS discs of the stack, a constraint that allows particle stretching exclusively into one direction.86 Cross-linking or high Tg domains give access to well-controlled nanoparticles through transfer into a solvent selective for only one of the blocks while keeping the other collapsed. The compartmentalized droplets break-up into anisotropic particles with a geometry complementary to the microphase inside the droplet, e.g., hemispheres, nano-disks or short rods with narrow length distribution.97 The lateral microphase separation of PS-b-P4VP into stacked lamellae (Fig. 3b) served as source for the preparation of near-monodisperse Janus nanodiscs.107 The stacked lamellar morphology was separated in selective solvent (here ethanol) into discs with a kinetically frozen PS core and P4VP corona on both sides of the disc. Co-surfactants covered the lateral surface of the PS discs, separating the P4VP corona to exclusively locate on top and bottom of the PS disc (else P4VP would envelope the entire particle). Cross-linking of the P4VP phase with 1,5-diiodopentane in ethanol fixated the P4VP-b-PS/PS-b-P4VP arrangement and PS/P4VP Janus nanodiscs with cross-linked P4VP core were liberated after redispersion in THF, dissolving the supramolecularly bound PS chains.
 | ||
| Fig. 3 Diblock copolymer self-assembly under 3D confinement. (a) Typical procedure involving emulsification of polymer solutions stabilized by surfactants followed by evaporation of organic solvent. (b) TEM images of striped ellipsoids with low dispersity consisting of axially stacked PS-b-P4VP lamellae (discs). Tilted image of ellipsoid, where the P4VP phase was infiltrated with gold nanoparticles to clarify the spatial distribution of stacked lamellae. (c) TEM and SEM of oblate ellipsoids with axially oriented PS-b-PB cylinder morphology stabilized by PEO-b-PB/PEO-b-PS surfactant mixtures in water. (a, b) Reprinted from ref. 100 with permission from American Chemical Society and (c) from ref. 105 with permission from Wiley VCH. | ||
Aerosol particles are a variation of block copolymer self-assembly under confinement requiring no solvent as the surrounding medium (often nitrogen gas) and thus, this method is scalable with lower environmental impact.108,109 Exemplified on PS-b-P4VP, high polymer content droplets develop during chloroform evaporation at elevated temperatures. The process is conducted in a chimney where a hot gas stream accelerates drying droplets towards a collector. Microphase separation evolves in a similar manner as in dispersion, yet at temperatures above the Tg of the blocks and with a non-polar interface towards nitrogen gas.110 This method was combined with cholesteryl hemisuccinate mesogens hydrogen-bonded to the P4VP block to give PS-b-(P4VP/mesogen) concentric lamellae, where every P4VP lamella contains a nanometer-sized sub phase of parallelized mesogens.111
Liquid crystalline and semicrystalline rod-coil block co-polymers. Block copolymers carrying rigid moieties show strong tendency for dense chain packing inducing microphase separation from any solvent.112–114 The nanostructures often inherit frustrated core morphologies and unusual shapes have been reported with nanoscale liquid crystalline features, some of which are exemplified in Fig. 4. There, the mesogen is not supramolecularly bound, but polymerized as side group of monomeric units or as part of the main chain backbone. Proper design of block lengths/ratios results in the classical solution geometries, yet, due to the liquid crystalline block, the spheres,115 cylinders116 and vesicles,117 display an additional, lateral striped pattern. The series in Fig. 4a shows self-assemblies of poly(ethylene oxide)-block-p(mesogen) block copolymers with a variety of mesogen monomers, e.g. 4-methoxyphenyl 4-(6-acryloyloxy-hexyloxy)-benzoate or cholesteryl-based moieties. The spacing within the microphases corresponds to the length of the stacking mesogen. Especially in case of bilayer capsules, the liquid crystalline order of the membrane-forming block leads to the conflicting situation of a bending shell competing with the rigid packing of the mesogen. As a result, vesicles deform to polyhedral shapes, where topological defect locate at the pronounced high curvature tips.118
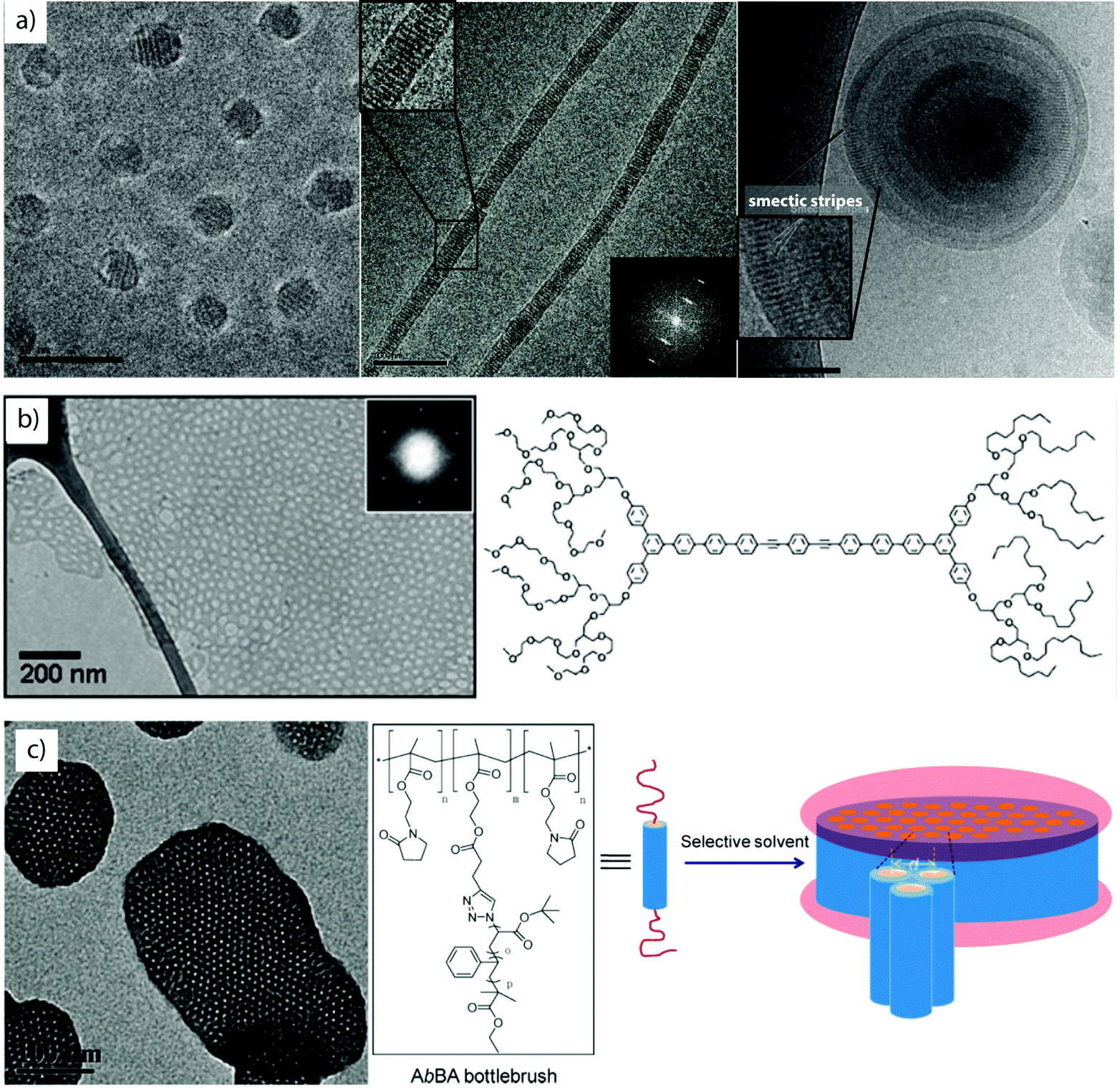 | ||
| Fig. 4 Rod-coil copolymers. (a) Cryo-TEM series of smectic spheres, cylinders and capsules of PEO-b-P(liquid crystalline) block copolymers in water/THF mixture. (b, c) Coil-rod-coil and coil-brush-coil triblock copolymer self-assembly into mesh-like sheets and hexagonally perforated discs. (a) Reprinted from ref. 115 and 116 with permission from Elsevier and American Chemical Society, (b) from ref. 114 with permission from Wiley CH and (c) from ref. 119 with permission from American Chemical Society. | ||
As alternative to covalent bonding, hydrogen-bonded mesogens offer a versatile route to functional and responsive particles as demonstrated on UV-responsive vesicles self-assembled from rod-coil polymers.120 Light-responsive chromophores (e.g. azobenzene derivatives) are known for their soluble/insoluble transitions upon irradiation.50 Absorbed photons induce cis–trans conformational changes of the rod-like side groups and alter polarity and packing behavior. The light-triggered release of cargo from hydrophobic azo-polymer domains is one application of these transitions.121,122 Mesogens often exert geometrical constraints on polymer chain packing promoting two-dimensional growth into planar and disc-like shapes.123 The anisotropy then strongly favors axial stacking into columns resembling segmented cylinders or columnar phases.124 In-plane aggregation into extended sheets was also observed for coil-rod-coil (ABA) liquid crystalline block copolymers (Fig. 4b).114,125 The sheet-like growth into extensive hexagonally perforated meshes involves several intermediate structures from continuous sheets, hole formation, equilibration to long-range ordered array of hexagonal pores and finally, roll-up into perforated capsules. Growth and structural transitions were followed with cryogenic transmission electron microscopy (cryo-TEM) in minute detail. Cryo-TEM is not to be confused with the method of freeze fracturing. In the latter, samples are frozen, fractured and a replica of the topology is prepared by vapor deposition of e.g., gold or platinum, whereas in cryo-TEM the sample is rapidly vitrified to obtain amorphous ice.126,127 Keeping the sample constantly at low temperature (e.g. −150 °C) preserves the vitrified sample and dispersed particles can be visualized in almost native state. Coil-brush-coil (ABA) block copolymers are structural analogues with similar aggregation behavior, yet on larger scale (Fig. 4c).119 Here, self-assembly is triggered after collapse of the cylindrical core–shell polymer brush resulting in hexagonal packing and 2D growth. Related are AB amphiphilic bottlebrushes with volume fractions designed in accordance to known parameters for diblock copolymer self-assembly that give rise to sphere and cylinder bottlebrush micelles as well as bottlebrush capsules.128 These examples impressively demonstrate the commonalities in self-assembly of amphiphiles on multiple lengths scales, i.e., amphiphilic molecules, block copolymers, bottlebrushes and even microscopic colloids.
Besides liquid crystalline moieties, also semicrystalline block copolymers display diverse self-assembly behavior. Poly(ethylene oxide)-block-poly(octadecyl methacrylate) block copolymers, for instance, self-assemble to internally structured bicontinuous particles in water.129 The semicrystalline block adds kinetic obstacles during self-assembly due to the contribution of the crystallization-driven phase separation, slowed kinetics and fixation of phases. The bicontinuous internal network was resolved in sophisticated detail by means of cryogenic transmission electron tomography (cryo-ET) of vitrified samples and 3D reconstruction, underlining the complexity achieved with one single diblock copolymer. Cryo-ET adds even more detail than cryo-TEM by capturing a series of images of an individual particle (or area) at varying tilt angles followed by reconstruction of a 3D model.130 This method is greatly appreciated in biological particle analysis, but also advances to aid characterization of soft matter nanostructures. Cryo-ET becomes especially relevant when resolving internal overlapping features that are otherwise indistinguishable or challenging to interpret in superimposed cryo-TEM projections.130–132 It is desirable that in the future, cryo-TEM and cryo-ET become state-of-the-art techniques for the characterization of self-assembled solution structures so that drying artifacts can be excluded and morphologies interpreted with high confidence.
Polypeptide blocks have been intensively investigated for their kinetic constraints that cause polymer chains to twist and turn. Several works demonstrated sophisticated peptide block self-assembly into single, double or triple stranded helices, tubes, fibers, bundles and even shapes reminiscent of nano-flasks, just to name a few.133–138 Despite the intriguing complexity of these self-assemblies, polypeptides are beyond the scope of this review, owing to the fact that mostly oligomeric or diblock copolymer self-assemblies are reported. There are comprehensive works that review supramolecular self-assembly of peptide oligomers and amphiphilic block copolypeptides.139–143
Crystallization-driven self-assembly. The epitaxial growth of crystalline main chain polymers requires distinction from the previous cases. The crystallization-driven growth of block copolymers has become a well-established approach to create complex patterns and nanoobjects comprehensively studied by e.g., Manners and Winnik et al.144,145 Exemplified on poly(ferrocenylsilane) (PFS), the semicrystalline nature of the main chain shows extraordinarily high incompatibility with other (amorphous) blocks and adds remarkable control over length and polydispersity of the created self-assembly governed by crystallization kinetics. This concept led to a impressive library of unusual and functional solution geometries.146–149 The PFS block re-crystallizes upon changing the solvent conditions and nucleates grow to core–crystalline micelles protected by a stabilizing corona block (e.g., polydimethylsiloxane, PDMS). The facetted core of these “seed micelles” allows continuous growth into cylindrical micelles with precise control over cylinder length and length distribution (close to mono-disperse) simply by varying the block copolymer feed. This process even tolerates slight variations in chemistry of the crystalline blocks giving rise to block co-micelles with e.g. poly(ferrocenyldimethylgermane). Re-initiation and sequential growth of various crystal core segments in a controlled/living manner fabricates unique core/corona micelles where the distribution and length of the core segments is identical to the corona segments (Fig. 5). The requirements for chemical composition of the corona block are less strict, because its sole purpose is to provide stability in the target medium and therefore, the corona can be modified with any desired function as long as it does not interfere with the crystallization process (see also section 4.3). Based on this concept, a variety of functional structures were grown such as fibers, ellipsoids, sheets, and inverse photonic opals.150–153 More recently, multi-arm micelles were generated by growing a defined number of crystalline poly(ferrocenyldimethylsilane) (PFDMS) fibers off crystalline PFDMS cores or other colloidal objects pre-coated with seed micelles.154,155 The multifaceted crystalline cores act as “polyfunctional” initiator (crystalline analogue of spherical polymer brushes), where the number of arms is controlled by the size of the crystalline core and the number of facets. Since one end of the arms is tethered to the core, growth of sequential block co-micelle arms to the other end is possible and results in well-defined multiblock co-micelle arm particles.
 | ||
| Fig. 5 Crystallization-driven self-assembly of cylindrical micelles. (a) Schematic of growth mechanism to alternating core and corona compartments. (b) TEM image of pentablock co-micelle with bifunctional crystalline nucleus (orange) of PFS-b-PDMS, extended with PFG-b-PI (red) and again with PFS-b-PDMS. Reprinted from ref. 146 with permission from Nature Publishing Group. | ||
3.2 Supramolecular blending of AB diblock copolymers
To expand the number of geometries and internal morphologies, blending of diblock copolymers (AB + AC or AB + CD) is a prominent pursuit that led to MCNs with polymer/polymer compartments in core or corona. Here, the central challenge lies in confining multiple phases that are immiscible but not covalently linked into the same nanodomain. Incompatible core blocks spontaneously microphase separate or do not even assemble within the same particle to begin with. Over the years, several supramolecular strategies emerged to approach this obstacle and to achieve co-assembly of multiple diblock copolymers by use of non-covalent interactions. The controlled collapse of polymer segments from the liquid phase by means of supramolecular approaches requires directing agents (co-solvents, multivalent ions) or blocks equipped with orthogonal function (polyions, hydrogen bonding).156–161Blending through non-solvent. There are examples for the successful co-assembly of AB + BC copolymers where the addition of a non-solvent for B results in homogeneous B core particles “welded” together by solvophobic interactions stabilized by a compartmentalized A/C corona.162,163 The incompatibility, χA/C, drives demixing of the corona from multipatch for weak repulsion to complete Janus separation for strong repulsion. However, such non-directional forces will lead to statistical distributions of MCNs together with regular micelles with B core and A (or C) shell and size-disperse corona-MCNs.
Electrostatic attraction. Blending of two unlike blocks within one core, i.e. AB + C, requires some finesse as described for self-assembly under 3D confinement. An alternative route is to establish short-range attraction. Co-assembly of polyionic blocks to the point of charge neutrality, Z+/− = ncationic/nanionic ≈ 1, was demonstrated for several systems.164–168 Polyionic blocks form hydrophobic chelate complexes with multivalent counterions41,169,170 or interpolyelectrolyte complexes with complementarily charged polyvalent polymers (Fig. 6).51 The driving force for aggregation is the release of monovalent counterions that increase the entropy of the system.
 | ||
| Fig. 6 MCNs formed through electrostatic attraction. (a) Corona-compartmentalized Janus micelles formed by PAA-b-PAAm and P2MVP-b-PEO with a complex co-acervate core of PAA/P2MVP and PEO/PAAm hemispheres. (b) Lateral microphase separation of PEO-b-PB and PAA-b-PB vesicle membranes induced by gelation of PAA with divalent cations, Ca2+ and Cu2+. Spot size develops with blend ratio from 25% PAA-b-PB (patchy) to 50% (Janus) and 75% (inverse patches) (scale bar is 2 μm). Segmented cylinders are obtained at high pH and 50% PAA-b-PB (scale bar is 4 μm). (a) Reprinted from ref. 171 with permission from Wiley VCH. (b) Reprinted from ref. 87 with permission from Nature Publishing Group. | ||
In the context of MCN formation from diblock copolymers (AB + CD), co-acervate core Janus micelles were formed in aqueous solution through complexation of poly(acrylic acid)-block-poly(acrylamide) (PAA-b-PAAm) with poly(2-methylvinylpyridinium iodide)-block-poly(ethylene oxide) (PM2VP-b-PEO) under charge stoichiometric conditions.171–173 The block-block immiscibility of PEO and PAAm corona chains overcomes the entropic penalty of demixing on the nano-scale and microphase separates into two hemispheres. The absence of PEO/PAAm interaction was verified with 1H-NMR 2D NOESY (Nuclear Overhauser Enhanced) magnetic resonance spectroscopy that showed no indication of cross coupling between the corona blocks. The PAA−/PM2VP+ core of these Janus micelles adapts the shape of a slightly deformed prolate ellipsoid as concluded from cryo-TEM analysis of the circular (axial) and elongated (lateral) particles (Fig. 6a). Structural optimization among the repulsive PEO/PAAm corona blocks exerts stress on the PAA−/PM2VP+ core that is covalently bound to the corona. The shape deformation is then a result of the energetic balance between attractive forces in the core, repulsive forces in the corona and minimization of interfacial energies. Solution-based approaches to Janus micelles are very rare as de-symmetrization is challenging without the use of templates. Other approaches will be revisited in section 3.4 and 4.2.
Controlling the Coulomb interaction of polyionic blocks with multivalent counterions was utilized to control the lateral microphase separation of vesicle membranes with nanoscale thickness from spotted pattern to entirely phase-separated (Janus distribution) (Fig. 6b).87 Blends of poly(ethylene oxide)-block-polybutadiene (PEO-b-PB) and poly(acrylic acid)-block-polybutadiene (PAA-b-PB) form a soft (almost fluidic) PB membrane in water protected by a mixed corona of PEO and PAA. Addition of Ca2+ or Cu2+ ions induces gelation of PAA/M2+ complexes and phase separation from the PEO corona. The increased hydrophobicity collapses the chelate complexes onto the likewise incompatible PB membrane. Depending on the mixing ratio of the block copolymers, the membrane morphology evolves from small spots to entirely separated Janus hemispheres to inverse spots (PB labeled with fluorescent dye). The mixing ratio and the spot size alters transport kinetics as well as mechanical properties of the capsule membrane given the difference in mechanical properties of the soft PB phase and the cross-linked (more rigid) PAA/M2+ ion complex. With this simple ion trigger, highly complex, tough and nano-structured vesicles were generated at low pH and segmented, cylindrical superstructures at high pH. Aside from the studied ions, a whole range of functional salts are conceivable (including redox or photo responsive ions) adding further properties to the membrane, where electrochemical response could be of particular interest towards redox-controlled pore stimulation (and gating). Vesicles with nano-structured membrane could demonstrate distinct release profiles due to persistent channels connecting the interior with the surrounding medium, which would be a desirable ability of delivery vehicles or nanoreactors.174 The high stability of vesicles at almost physiological conditions is of additional benefit for in vivo applications.
Once counterions are released, strong electrostatic forces tightly bind and immobilize complementary polymer chains with limited re-organization capabilities. Considerable salt concentrations are required to screen polyion charges and break up these complexes. Metal coordinations175–177 on the other hand, are less tight orthogonal supramolecular connectors for polymer blocks much like hydrogen bonding motifs178–180 and host–guest complexes181,182 to name a few. Although examples for MCN formation through these motifs are sparse, they are essential structuring tools in the more general context of polymer modification and self-assembly. Strong hydrogen bonder are for instance complementary units of poly(4-vinylpyridine)/alkylphenol with one,183,184 self-coordinating ureidopyrimidinone motifs with four185 and Hamilton wedge with six binding sites per unit.186 Prominent host–guest systems comprise cur[8]cubituril/viologen/naphtalene187 and cyclodextrin/ferrocene188 (also azobenzene, adamantyl).189 These motifs find widespread appreciation in diblock copolymer self-assembly and although they pose valuable structuring tools, they are less explored with respect to MCNs. There still is plenty of ground to cover and room for developments in this direction with the prospect of responsive MCNs and reversible hierarchical self-assembly.
3.3 One-step self-assembly of ABC triblock terpolymers
From a synthetic point of view, diblock copolymers are clearly advantageous over triblock terpolymers, yet the necessity to equip one block with orthogonal motifs to direct self-assembly into well-ordered structures limits their applicability in MCN formation. Although orthogonal motifs are an emerging field157 and diblock copolymer morphologies can compete in complexity with triblock terpolymers as shown in the previous section, covalently linked triblock terpolymers cannot evade each other and will microphase-separate to MCNs more readily and reliably.37,190–196Miktoarm star terpolymers. The miktoarm star terpolymer architecture with block chemistries above the super-strong segregation limit (SSSL) facilitates control over phase separation to form core-MCNs almost independent of the solvent condition. Fig. 7 summarizes the self-assembly of miktoarm star terpolymers with perfluorinated and equally hydrophobic hydrocarbon core segments stabilized by a hydrophilic corona block exemplified on studies pioneered by Hillmyer and Lodge.197 This systems is one of the most prominent and best understood approaches.198 The miktoarm star architecture of μ-(poly(ethyl ethylene)/poly(ethylene oxide)/poly(perfluoropropylene oxide) (μ-EOF) does not allow the hydrophobic blocks to arrange into a core–shell–corona sequence, as observed for linear triblock terpolymers,199,200 and thus, a core-segmented structure develops. The highly incompatible polymer blocks self-assemble to geometries usually not found for amorphous polymers. The extraordinary high chain stretching minimizes the equilibrium interfacial area per chain and approaches the cross-section of the repeating unit; planar aggregates such as discs and sheets become stable. If the corona does not provide sufficient protection for the frustrated disc-like segments, micelles stack via mutually attractive patches into core-compartmentalized superstructures to minimize unfavorable core/water interface. Depending on the block fractions spherical clusters were identified with internal morphologies reminiscent of “raspberries” and “hamburgers” or anisotropic one-dimensional micelles with alternating core-segmentation. These works were among the first to visualize internal morphologies of multicompartment micelles in situ using cryo-TEM. The shape and morphology of miktoarm star terpolymers depends on many parameters, which was summarized in a comprehensive ternary phase diagram with a rich repertoire of MCNs (Fig. 7b).201 The phase diagram demonstrates that the volume fractions of the hydrophobic blocks control morphology of the core segments and that the corona volume controls the degree of association of the “hamburger” micelles.202 Mixing a EO diblock copolymer with a long corona block into solutions containing worm-like micelles of μ-EOF gave an interesting mechanistic insight into the morphological evolution from cylinders back to spheres. The additional stabilizing corona volume and the smaller hydrophobic to hydrophilic ratio induced depolymerisation into small fragments. Beyond these early mechanistic insights, over the years, several other morphologies were examined such as polygonal sheets and raspberry-like or laterally nanostructured vesicles.203–208
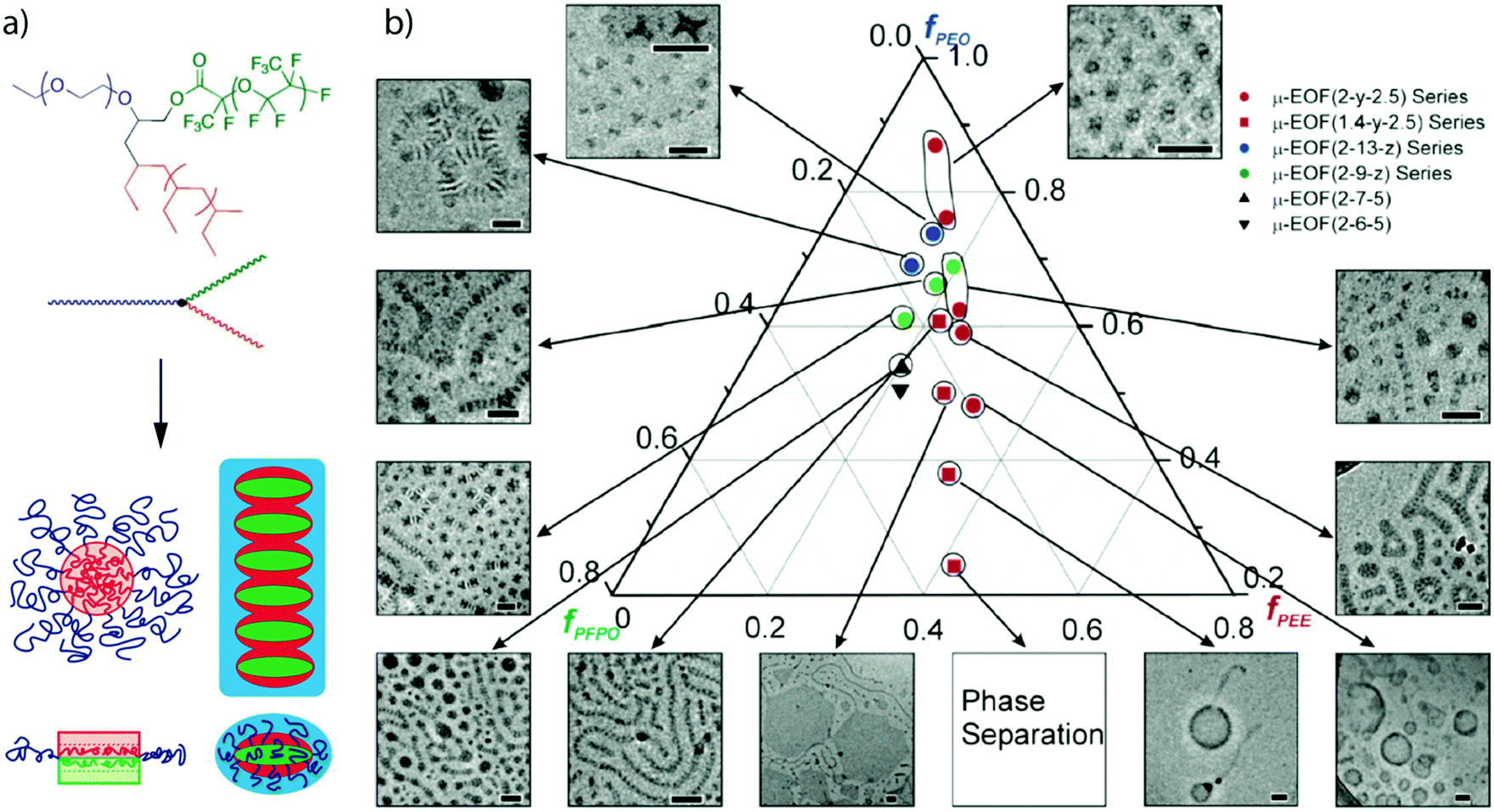 | ||
| Fig. 7 Miktoarm star terpolymers in water. (a) Schematic of the miktoarm star terpolymer architecture and proposed aggregation into laterally microphase-separated discs. (b) Comprehensive phase diagram of one type of miktoarm star terpolymer encompassing spherical and worm-like micelles, laterally structured hexagons and vesicles. Reprint of (a) from ref. 197 with permission from American Association for the Advancement of Sciences and (b) from ref. 201 with permission from American Chemical Society. | ||
Miktoarm star terpolymers with less repulsive arms but larger overall molecular weights display similar phase behavior (χN). For instance, μ-polystyrene/polybutadiene/poly(2-vinyl pyridine) (μ-SBV) with pH-responsive P2VP, glassy PS and soft PB self-assemble to patchy micelles in acidic water.209 The soft PB compartments reside on the spherical PS core as spherical patches and increase with core size to minimize interfacial energy. The MCNs originate from transient Janus unimers and grow to multimer clusters over time. During the growth process, the PB patches at the core/solvent interface draw fiber-like hydrophobic bridges between particles most likely as a result of particle collision. This kinetic phenomenon can be traced back to the stickiness of the PB patches that connect cluster fragments after merging. The bridge formation requires sufficient chain dynamics in water and is a possible explanation for the observed growth of cluster size by merging and reorganization of core components.
Linear ABC triblock terpolymers. The synthesis of linear ABC triblock terpolymers has become state-of-the-art and is accomplished by several polymerization techniques.210–213 In fact, quite a number of linear triblock terpolymers has been reported so far offering an enormous playground for self-assembly endeavors. Several studies demonstrated that linear sequences of triblock terpolymers are capable of forming MCNs such as patchy micelles,215 going beyond the expected core–shell–corona arrangement,.81,214 Both structures are similar except that the “shell” of the latter has dewetted on the spherical substrate. Stadler et al. observed the spheres-on-sphere morphology in bulk films of polystyrene-block-polybutadiene-block-poly(methyl methacrylate) and later Ritzenthaler et al. also in epoxy resins.216,217 In solution, linear block terpolymers with a water-soluble PEO corona and hydrocarbon/fluorocarbon core blocks showed similar self-assembly behavior as the already described miktoarm star architecture. The strong segregation and the pronounced interfacial energies between the highly immiscible hydrocarbon/fluorocarbon blocks cause de-wetting of the minority phase from the core and induce a morphological transition from core–shell–corona to patchy spheres.218,219 PEO as the middle block in combination with poly(2-ethylhexyl acrylate) and poly(1H,1H,2H,2H-perfluorodecyl acrylate) end blocks still led to raspberry-like micelles (Fig. 8a).218,219 Varying the block sequence appeared to have only minor influence on the overall fraction of MCNs, as structural analogues of spherical MCN were also found when changing the block sequence from ABC to ACB and BAC. Cryo-ET further gave unique insight into the complex core architecture of the particles.220 Beyond hydrocarbon and fluorocarbon polymer blocks, the direct dispersion of polystyrene-block-polybutadiene-block-poly(2-vinylpyridine) in methanol/toluene mixtures likewise resulted in patchy micelles with very homogeneous size and core segments (Fig. 8b).221 The patch number and patch location almost corresponds to geometrically arranged clusters. In another example, MCNs with Janus distribution of the core as well as the corona were fabricated by direct dispersion of bishydrophilic poly(ethylene oxide)-block-poly(ε-caprolactone)-block-poly(2-aminoethyl methacrylate hydrochloride) (PEO-b-PCL-b-PAMA) in water at temperatures close to the Tg of the hydrophobic PCL segment to give thermodynamically equilibrated PCL cores and a mixed PEO/PAMA corona.83 The cationic corona chains catalytically induce a sol–gel transition of added tetramethyl orthosilicate. With progressing reaction, the PAMA corona chains microphase separate into patches due to increasing incompatibility. At the same time selective diffusion of silica precursor into the core drives separation into a Janus-like core. Silicified compartments can be clearly distinguished in TEM by the higher electron contrast (compare Fig. 2h).
 | ||
| Fig. 8 Spheres-on-sphere morphology by direct dispersion of linear ABC triblock terpolymers. (a) Cryo-TEM of MCNs with fluorocarbon/hydrocarbon core and PEO corona; inset shows employed polymer blocks. (b) TEM of PS-b-PB-b-P2VP dispersed in methanol/toluene mixtures with PB patches arranged on the PS core with almost geometrical precision. (c) Schematic and TEM image of PB-b-P2VP-b-PtBMA in acetone with grey PB core and dark P2VP spherical patches. (a) Reprinted from ref. 218 with permission from Royal Chemical Society and (b, c) from ref. 221 and 222 with permission from American Chemical Society. | ||
Spontaneous self-assembly into patchy MCNs in organic solvents suggests a more generic and more widely applicable mechanism for the aggregation behavior of triblock terpolymers.222 The high χB/V in polybutadiene-block-poly(2-vinylpyridine)-block-poly(tert-butyl methacrylate) (PB-b-P2VP-b-PtBMA) induces de-wetting of the P2VP shell into a defined number of spherical patches residing on the spherical PB core stabilized by the PtBMA corona that in turn emanates from the P2VP patches (Fig. 8c). The multicompartment character was unambiguously visualized in TEM after selective staining of P2VP with iodine. Although the polymer was directly dispersed in acetone, MCNs were exclusively spherical, with low size dispersity and with homogeneous internal morphology. The fast microphase separation into homogeneous species was an advancement compared to dispersed blocks that require annealing for days or even weeks to overcome kinetic barriers. Acetone is a plasticizing solvent for P2VP compartments and in combination with the low Tg of the PB core allows for fast structural rearrangements during equilibration. Polymer blocks are able to adapt rapidly to the solvent and interfacial energies are minimized instantly preventing the formation of ill-defined or kinetically trapped intermediate structures.
In related works, PB-b-P2VP-b-PtBMA was converted to a zwitterionic polyelectrolyte through polymer analogue reactions, i.e., quaternization of P2VP to P2VPq (Vq) and hydrolysis of PtBMA to PMAA. The resulting PB-b-P2VPq-b-PMAA (BVqMAA) features complementarily charged blocks for mutual electrostatic attraction under proper solvent conditions. The polymer was dispersed in dioxane as a common solvent for all blocks and dialyzed into water pH > 6 to trigger self-assembly into MCNs with hydrophobic PB core and complex co-acervate patches of complementarily charged blocks P2VPq/PMAA (Fig. 9a).223 The self-assembly concept is equivalent to the electrostatic blending of diblock copolymers (compare section 3.2), although here, complexation occurs among blocks that belong to the same polymer chain.51 These intramicellar interpolyelectrolyte complexes (IPECs) become hydrophobic and collapse onto the hydrophobic PB core, where the nano-domains phase separate because of the extraordinarily high χB/IPEC. MCNs are stabilized by excess of charged PMAA corona that retain their pH responsiveness as demonstrated by DLS and TEM measurements at pH 10 (expanded corona) and pH 4 (collapsed corona (pKa (PMAA) = 5). The core can be cross-linked to stabilize the particles and IPECs may act as storage compartment for gold nanoparticles towards catalytic and traceable carrier systems.224
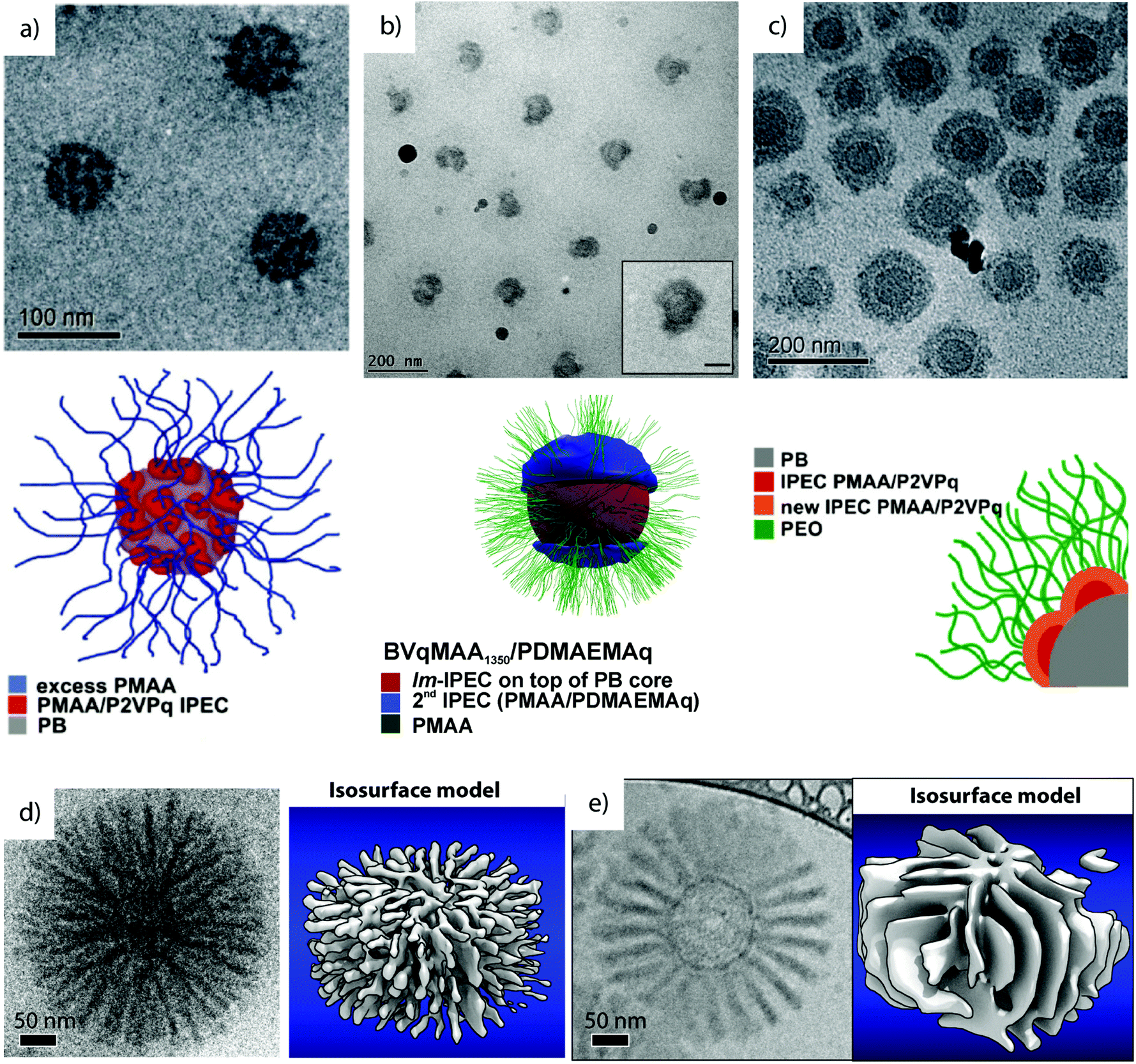 | ||
| Fig. 9 Cryo-TEM characterization of complex MCNs prepared via IPEC formation. (a) BVqMAA IPECs with Vq/MAA patches on a B core in water pH 6.223 (b) Patchy IPECs of BVqMAA coordinated with polycationic PDAMA.224 (c) Core–shell–shell–corona particles of BVqMAA and polycationic bishydrophilic PEO-b-P2VP.225 (d) “Sea-urchin” micelles with cylindrical IPEC morphology of BVqMAA with PEO-b-PLL and tomographic reconstruction.226 (e) “Turbine” micelle with lamellar IPEC morphology of BVqMAA with PEO-b-PDq and tomographic reconstruction.226 (a–e) Reprinted with permission from American Chemical Society. | ||
Several works are dedicated to study the effect of PMAA chain length on complexation with polycationic polymers, e.g., poly(2-((2-(dimethylamino)ethyl)methylamino) ethyl methacrylate) homopolymers (Fig. 9b).225 Here, a thick compartmentalized IPEC shell is the energetically favorable structure. On the contrary, the complexation with polycationic bishydrophilic poly(ethylene oxide)-block-poly(2-vinylpyridine) leads to the collapse of the entire PMAA corona as homogeneous IPEC shell, while the new PEO corona now stabilizes the particle irrespective of pH (Fig. 9c).224 The system was further extended to core–shell–shell–corona micelles via IPEC formation between the PMAA corona and poly(ethylene oxide)-block-poly(2-(dimethylamino)ethyl methacrylate) (PEO-b-PDq; the second block was quaternized), where the second PMAA/PDq IPEC was distinguishable from the first PMAA/PVq IPEC.227 Cryo-ET and 3D reconstruction of these and other IPEC particles revealed subtle structural differences in the arrangement of corona compartments.226 For instance, BVqMAA coordinated with poly(ethylene glycol)-block-poly(L-lysine) (PEG-b-PLL) with predetermined chain length mismatch among the polyions (PLL/PMAA) conceptually yielding ionically grafted PMAA chains with short polycationic diblocks towards bottlebrush-on-sphere particles. The degree of ionic grafting correlates to the chain length mismatches of polyions and stabilizes anisotropic IPEC morphologies. In analogy to microphase separation in bulk, the volume requirement of PEO and IPEC gave lamellae (“paddle wheel”) or hexagonally arranged cylinders (“sea-urchin”) residing perpendicular to the particle core (Fig. 9d and e). The architecture of these particles is fairly complex and features 5 distinct phases: (i) the soft PB core, (ii) intramicellar IPEC patches of PMAA/PVq, (iii) cylindrical (PMAA/PLL) or lamellar (PMAA/PDq) IPEC domains extending from the core, (iv) a dense PEO brush intercalated between the IPECs and (v) an outer PEO corona brush surrounding and stabilizing the entire particle. These IPEC MCNs progressed beyond mere academic interest to become promising agents for non-viral gene transfection228 and advanced delivery systems for photodynamic therapy.78 The latter consisting of BVqMAA/PLL-b-PEG grafts with tunable degree of PEGylation (see also section 4.4.). Such modular systems are of great importance to understand the effect of particle shape, size and topology on cellular uptake.229–232
Polymerization-induced self-assembly (PISA) is a technique that relies on the block extension of a soluble macroinitiator with solvophobic blocks.233–237 With progressing polymerization and increasing length of the second block, unfavorable block/solvent interactions induce microphase separation into a variety of micellar shapes. The final morphology is conveniently controlled by initial monomer concentration that directly translates into the block length ratio after full conversion. Morphological transitions of diblock copolymers were followed with unprecedented detail with special emphasis on direct visualization of the evolutionary steps from cylinders to sheets and vesicles.34 Transitions involve network formation of worm-like micelles, filling of voids to perforated bilayers with jellyfish morphology and, in the end, closing of voids and roll-up into vesicles.
Although still mostly employed for diblock copolymer synthesis/self-assembly, more recently the extension to ABC triblock terpolymer was reported. Polymerization of a fluorinated monomer (SSSL) as the second insoluble block that is also immiscible with the first core-forming block shows the tendency to microphase separate within the core towards MCNs (Fig. 10).238 This method shows particular potential to overcome the current limitation of scalability and shape anisotropy for MCNs going beyond spherical objects or core-segmented cylinders.
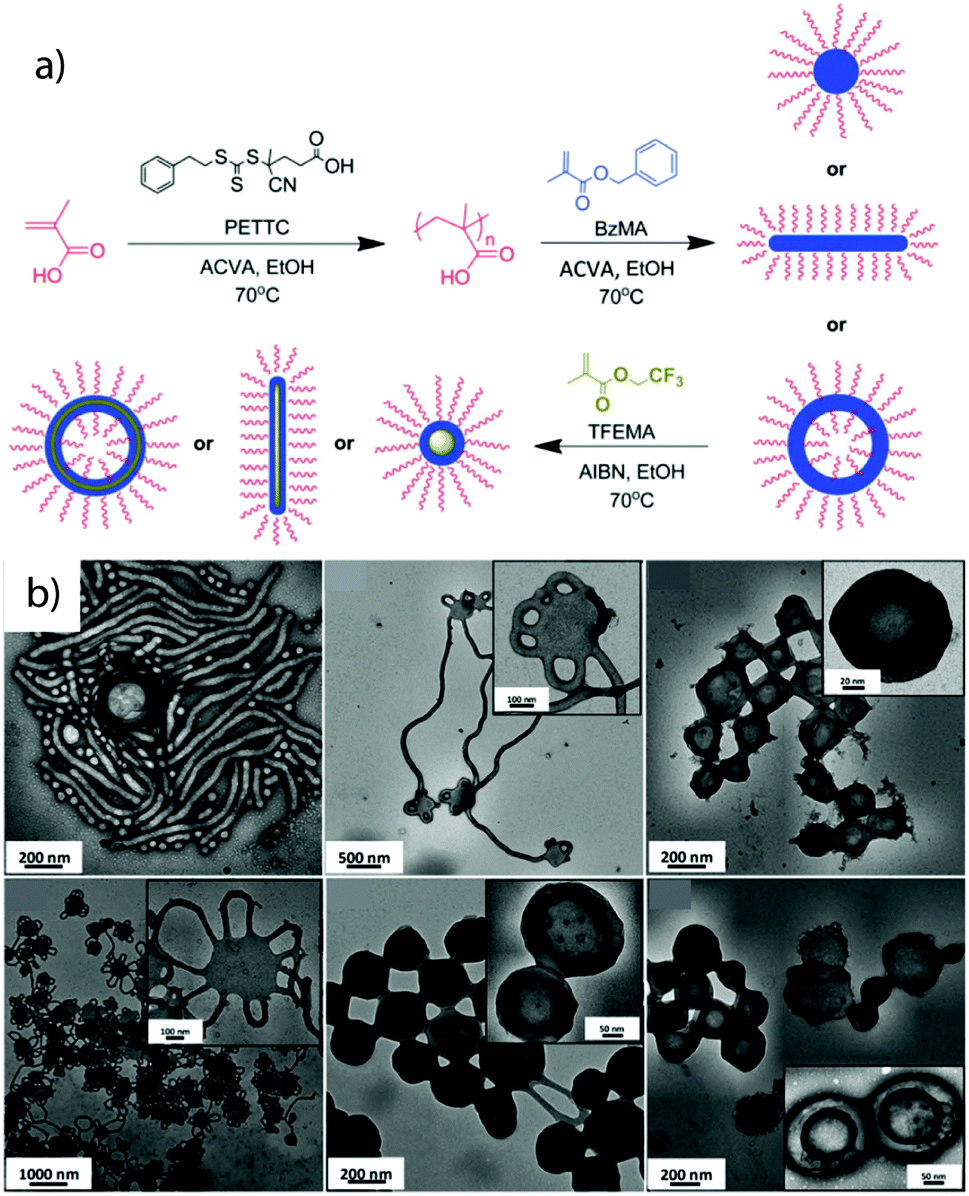 | ||
| Fig. 10 Polymerization-induced self-assembly. (a) Schematic of sequential RAFT polymerization to poly(methacrylic acid) in water followed by emulsion polymerization of poly(benzyl methacrylate) and poly(2,2,2-trifluoroethyl methacrylate). (b) Morphologies after PISA of the three blocks and structural evolution from spheres/cylinders to cylinders and vesicles. Reprinted from ref. 238 with permission from Royal Society of Chemistry. | ||
3.4 Re-dispersion of ABC triblock terpolymer bulk morphologies
The structuring via bulk morphologies can, by itself, not be considered as a directed or templated process, because the effects of the substrate immediately abate after a few nano-domain repetitions. From there on, the interfacial energies are exclusively balanced between the blocks. However, polymer chains require mobility to rearrange into the thermodynamically favorable microphase and slight variations in block–solvent interactions during film formation may still influence the development of the morphology by premature assembly to nano-domains. We will only briefly introduce bulk morphologies as structuring tool for MCNs and refer the interested reader to recent and established reviews covering this vast subject in more detail.239–241 The bulk phase diagram of triblock terpolymers is particularly rich in complex morphologies, whose stabilities depend on the Flory–Huggins interaction parameter, χAB, χAC, χBC, and the block volume fractions ϕA, ϕB. Owing to the dedicated efforts of Stadler et al. and many others, today quite a number of triblock terpolymer morphologies are well-characterized.242–250 The arrangement of three blocks offers the possibility to extract well-defined nanoparticles with narrow size/length distribution after fixation of one of the phases, e.g., through cross-linking strategies or re-hydration/re-dispersion in selective non-solvents that physically bind the core phase (as long as the chosen phase is not continuous).Crosslinking strategies. To fixate a particular microphase, monomers with reactive groups are required that are susceptible to irradiation, thermal cross-linking or reactive additives delivered through chemical vapor deposition or admixing before film formation. For instance, poly(2-cinnamoyl ethyl methacrylate) (PCEMA) was shown to photodimerize under UV irradiation effectively cross-linking the PCEMA microphase.251 In case of the cylinder morphology, nanofibers could be generated with near-monodisperse diameter and micrometer length that is merely limited by the grain size (usually of the order 10 μm). Using PS-b-PCEMA-b-PtBA triblock terpolymers that form core–shell cylinders in a PS matrix, photo-crosslinking results in hybrid iron oxide core/polymer shell fibers after hydrolysis of PtBMA to PMAA and loading and oxidation of iron chloride.252 Vinylic groups that are not capable of self-crosslinking require admixing of photoinitiators in form of small organic molecules. In that regard, properly tuned BVT triblock terpolymers with a P2VP double helix wrapped around PB cylinders embedded within a PtBMA matrix provide a high concentration of double bonds confined to the PB cylinder phase accessible for cross-linking reactions.253 Here, cross-linking was realized by UV initiation of admixed photoinitiator and subsequent radical polymerization in the cylinder phase. After cross-linking and re-dispersion the double helix retained its structure as to the covalent linkage to the cylinder substrate.254 In another example, μ-SBV miktoarm star terpolymers underwent microphase separation into complex hexagonal pattern.255 Cross-linking of the PB phase and redispersion transformed these pattern into cylindrical corona-MCNs (Fig. 11a).256 P2VP patches flank the ribbon-like PB core in plane and PS patches sit on top and bottom as visualized by TEM tilt series (Fig. 11b). In selective solvents, the opposing PS and P2VP patches expand and contract inversely, mechanistically reminiscent of agonists and antagonists. When the P2VP phase is loaded with inorganic nanoparticles, the selective swelling dynamically adjusts compartment volumes and alters internanoparticle spacing. The system opens interesting directions regarding dynamic modulation of plasmon resonance of gold nanoparticles or as conductive fibers.
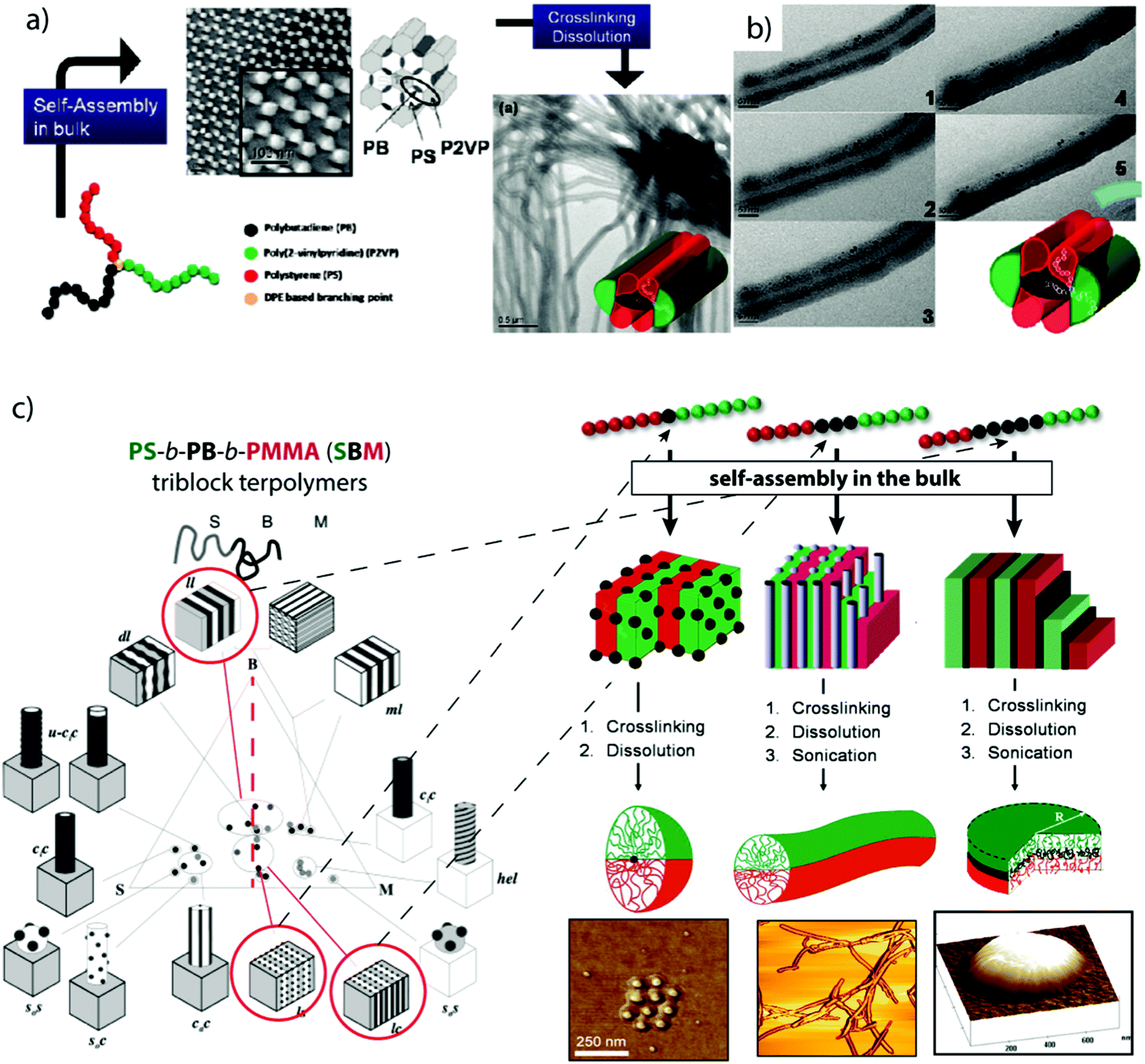 | ||
| Fig. 11 Compartmentalized superstructures via bulk morphologies. (a) Hexagonal bulk morphology of a miktoarm star terpolymer with polystyrene, polybutadiene and poly(2-vinylpyridine) arms and cylindrical corona-MCNs after cross-linking and re-dispersion in THF. (b) Tilt-angle cryo-TEM image series (0°–60°) after selective loading of P2VP corona compartment with inorganic particles. (c) Comprehensive phase diagram of SBM triblock terpolymers; lamella–sphere, lamella–cylinder and lamella–lamella morphologies (red circles) are the source for Janus spheres, cylinders and discs after cross-linking of B (black phase). (a, b) Reprinted from ref. 256 and (c) from ref. 257–259 with permission from American Chemical Society. | ||
Lamellar triblock terpolymer morphologies are particularly attractive for cross-linking, because they are the main source for polymeric Janus nanoparticles of various geometries (Fig. 11c). The lamella–sphere, lamella–cylinder and the lamella–lamella morphology give access to Janus micelles,257 cylinders258 and discs259via cross-linking of the spherical, cylindrical or likewise lamellar PB phase sandwiched between the other two lamellae (black phase in Fig. 11c).
Terpolymer-based Janus nanoparticles are colloidal amphiphiles and feature two hemispheres differing in chemical and/or physical properties, which is the origin of their extraordinary high interfacial activity and their versatile self-assembly behavior in selective solvents.260–264 Amphiphilic polystyrene-block-polybutadiene-block-poly(methacrylic acid) (SBMAA) Janus nanoparticles self-assemble into spherical clusters of defined size after reducing the solvent quality for the PS hemisphere (e.g. water).265 However, also much larger superclusters could be identified and were thus termed supermicelles. Janus cylinders on the other hand could be controlled in length through mild sonication treatment and their self-assembly led to 1D fibers, because the overlap area of individual cylinders does not match perfectly (unlikely) and thus, multiple cylinders come together to grow 1D supracolloidal fibers while minimizing the unfavorable interfaces.266 Janus discs showed similar self-assembly behavior, where either two discs stacked on top of each other or one individual disc folded into ill-defined crumbled sheets to cover one side.267
The rehydration of bulk morphologies in selective solvents to generate vesicles has received much attention and in case of three polymer blocks may lead to non-uniform membranes as shown for PEO-b-PS-b-PB-b-PEO ABCA tetrablock terpolymers.268 Here, vesicles are stabilized by PEO corona chains on the in- and outside, while PS and PB microphase separate either to an inner PB and outer PS layer if the overall weight fraction of PEO is below fPEO = 50 wt% or in plane to hexagonal ordered PS domains in a continuous PB membrane (alternatively bicontinuous).
With a slight alteration of the experimental setup, vesicles with a mesh-like morphology were obtained from polyisoprene-block-polystyrene-block-poly(2-vinyl pyridine) (PI-b-PS-b-P2VP) in the confined space of anodic aluminium oxide (AAO) channels (Fig. 12a).269 A moderately concentrated polymer/toluene solution (0.5 wt%) was spread on a glass substrate and picked up by the tubular channels (d ≈ 100–200 nm) of the AAO through capillary forces. During solvent evaporation a polymer film forms inside the channels and the arising shear forces on the curved walls enhance and direct microphase separation into the mesh-like morphology confined to the membrane of the hollow tubular particles. The P2VP corona block comprises less than 15 mol% and the structure stabilizes between the cylinders and vesicles region. After annealing in ethylene glycol vapor at 150 °C (selective solvent for P2VP), a basic aqueous solution dissolves the AAO template and releases mostly closed, spherical vesicles of narrow size dispersity with diameter in the range of the AAO channel diameter. Rayleigh instabilities break up the tubular geometry into spheres followed by reshaping with respect to surface energy minimization. The final vesicles are composed of a P2VP corona protecting the hydrophobic polyisoprene mesh interpenetrating the PS membrane (Fig. 12b and c). Controlling the morphology of vesicle membranes is very attractive, because permanent responsive nano-channels could result in advanced gating and release profiles, (inorganic) meshes could reinforce capsules towards hybrid ultrastrong nanoreactors and onion-like perforated vesicles could serve as a model systems for artificial cells.
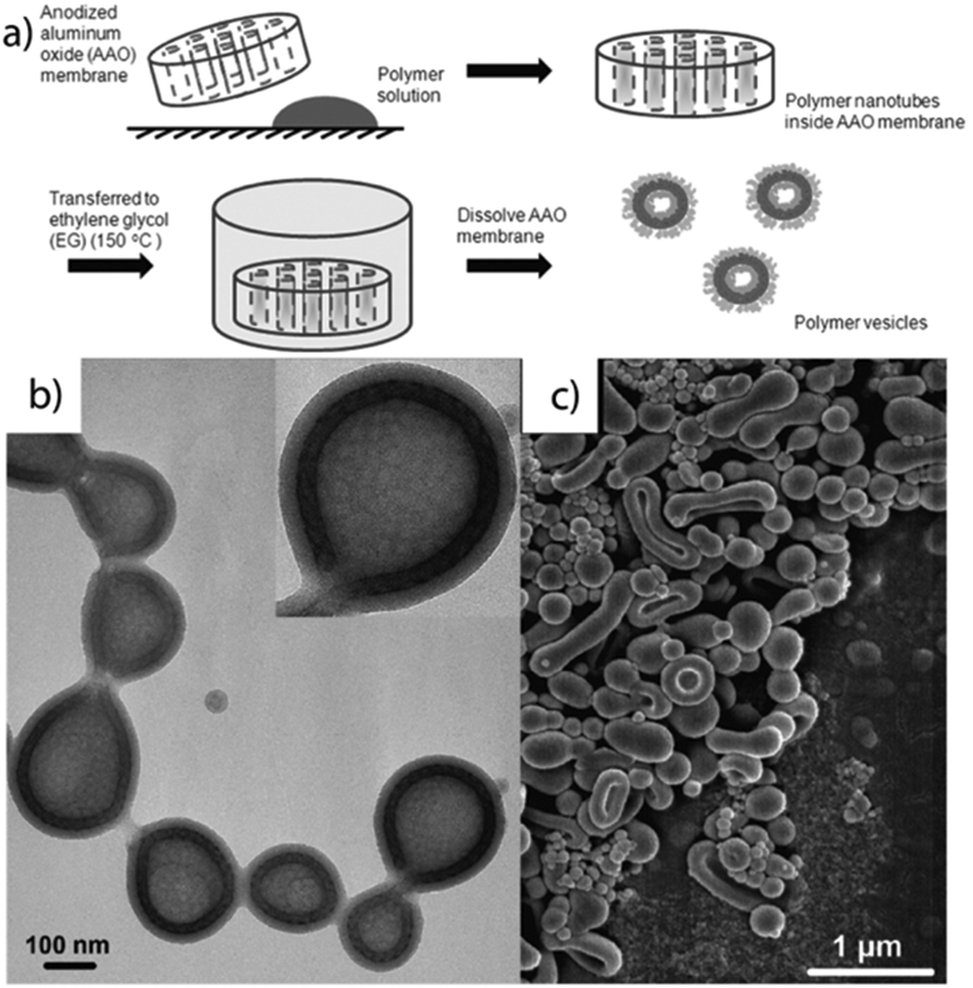 | ||
| Fig. 12 Rehydration of bulk films in AAO channels. (a) Processing steps comprise: AAO template-assisted shearing of PI-b-PS-b-P2VP, annealing in selective solvents at elevated temperatures followed by rehydration and release of vesicles. (b, c) TEM and SEM characterization of vesicles with mesh-like membrane morphology. Reprinted from ref. 269 with permission from American Chemical Society. | ||
3.5 Two-step self- and co-assembly of ABC triblock terpolymers
A larger number of polymer blocks open the way to direct self-assembly across hierarchies. Multiblock copolymers with N blocks give access to N − 1 hierarchies, if the stimulus is selective and sequential self-assembly possible.36 Structuring of macroscopic objects from nanoscale building blocks unlocks unforeseen combinatorial potential considering permutations of trigger sequences and co-assembly of multiple building blocks. There are several possibilities to collapse a polymer block and if done in controlled way, self-assembly of multiblock copolymers gives rise to an entirely new range of structures with low size dispersity. Although this section seems to be yet another chapter on triblock terpolymer self-assembly, the preparation and underlying mechanisms are entirely different from those discussed so far. In recent years, a step-wise self-assembly mechanism emerged as reported independently in several works involving trigger sequences and precursor particles with patchy corona (Fig. 13). It is very difficult to identify the underlying self-assembly mechanism for a given MCN in its final form. For instance, spheres-on-sphere MCNs may be fabricated by de-wetting of core–shell–corona micelles as discussed before (see also Fig. 13a) or through clustering of patchy precursor micelles (Fig. 13b upper path). These two pathways are conceptually very different, yet result in the same MCN indistinguishable after self-assembly is completed. Although hierarchical self-assembly strategies may not have been deliberately implemented or used without further discussion in works related to triblock terpolymer self-assembly, the greatly improved homogeneity of some reported MCNs could point towards mechanistic similarities.270–274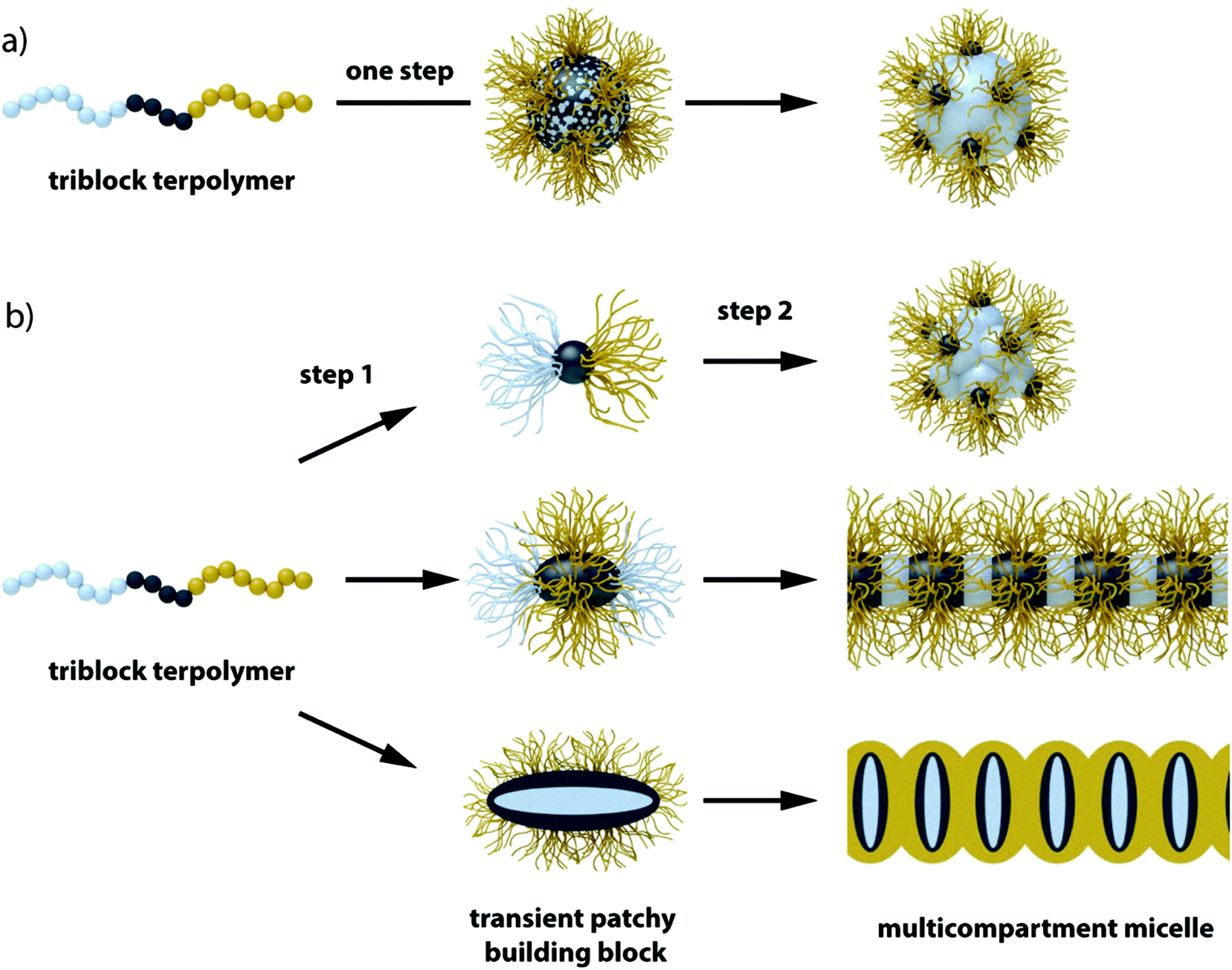 | ||
| Fig. 13 One-step self-assembly vs. two-step self-assembly. (a) Direct assembly may progress via particle formation and de-wetting of the shell to spherical patches. (b) Step 1: formation of transient precursor particles with patchy corona followed by step 2: clustering and equilibration gives conceptually the same particle with spherical patches. Triblock terpolymers self-assemble to transient patchy particles in step 1. These building blocks further assembly to the final superstructure in step 2. | ||
A step-wise procedure may appear more laborious at first, but the control over structure, size distribution and positioning of the blocks is often unparalleled.275 First, the triblock terpolymer is either directly dispersed in a non-solvent for one of the blocks (mostly the middle block) or the non-solvent is added in portions. Slow addition of non-solvent or use of low Tg blocks ensures thermodynamically equilibrated precursor micelles crucial to fabricate uniform MCNs, because irregularities will amplify throughout the hierarchies. In a subsequent step, these building blocks self-assemble via a second trigger for one of the remaining corona blocks, A or C, e.g., with temperature, a second non-solvent, hydrogen-bonding, electrostatic attraction or others. The collapsing corona block forms a new core compartment with an unfavorable polymer/solvent interface that serves as mutual contact point for the patchy nanoparticles to merge with each other. The newly formed MCNs then consist of pre-shaped and refined building blocks and with narrow size distribution and almost identical internal morphology. All examples discussed in this section follow this basic principle one way or the other utilizing specific combinations of structuring tools.
Non-solvent and temperature. Considering possible interactions, temperature-induced self-assembly is among the most dynamic, soft and reversible. A system responding to temperature and non-solvent comprises poly(ethylene oxide)-block-poly(n-butyl acrylate)-block-poly(N-isopropylacrylamide) (PEO-b-PnBA-b-PNIPAAm) in aqueous solution.273 Directly dispersed in water as non-solvent for PnBA the triblock terpolymer forms micelles with bishydrophilic and patchy PEO/PNIPAAm corona at ambient temperature. Consecutive heating/cooling cycles above the lower critical solution temperature of PNIPAAm repeatedly collapse the PNIPAAm patches and induce aggregation into linear and branched colloidal chains. Several repetitions gradually enhance the quality of the MCNs and increase colloidal chain length. Structure formation was followed in situ by dynamic light scattering during the heating cycles as well as cryo-TEM of samples vitrified from elevated temperatures.276 Temperature-responsive bishydrophilic building blocks for hierarchical self-assembly are still very rare and even trishydrophilic responsive polymers could be feasible,277 which illustrates the capabilities of reversible (gentle) manipulation for block copolymer structuring in aqueous solution.278–280
Hydrogen bonding and non-solvent. To produce stable and reversible self-assemblies, hydrogen bonding and non-solvents give direct control of blend composition and nanodomain orientation. Spherical MCNs are generated combining two structuring tools: first two diblock copolymers are bound together by multiple hydrogen bonding sites followed by change of the solvent quality for sequential collapse of the blocks. For that, the second block of poly(tert-butyl acrylate)-block-poly(2-cinnamoyloxyethyl methacrylate) (PtBA-b-PCEMA) was randomly copolymerized with complementary base pair derivatives 2-(1′-thyminyl-acetoxyethyl methacrylate) (T) or 2-(1′-adeninylacetoxyethyl methacrylate) (A) (Fig. 14a).281–283 The complementary diblock copolymers are “chemically stitched” together via hydrogen-bonding in chloroform and yield a non-covalently linked triblock terpolymer analogue, PS-b-P(CEMA-A/T)-b-PtBA. Addition of a specific amount of non-solvent (hexane) for PS and P(CEMA-A/T) triggers self-assembly to MCNs with very good control over geometry and number of compartments (“molecular model-like micelles”, Fig. 14b). During addition of hexane, the solvent mixture passes through several compositions and blocks respond individually according to their miscibility with chloroform or hexane. Structuring thus possibly follows an in situ hierarchical self-assembly process while crossing these solvent compositions. First, the stitched middle block could collapse into precursor particles with a P(CEMA-A/T) core carrying a patchy corona of PS/PtBA. With progressing increase of hexane concentration to a critical solvent composition of chloroform/hexane 20/80 (v/v), the PS corona patches also start to collapse and aggregate into the PS core decorated with P(CEMA-A/T) core patches and stabilized by the PtBA corona. Following this logic, the triblock terpolymer analogue could go through a step-wise hierarchical structuring process that evolves from molecularly dissolved polymer to patchy micelles and further to spherical MCNs.
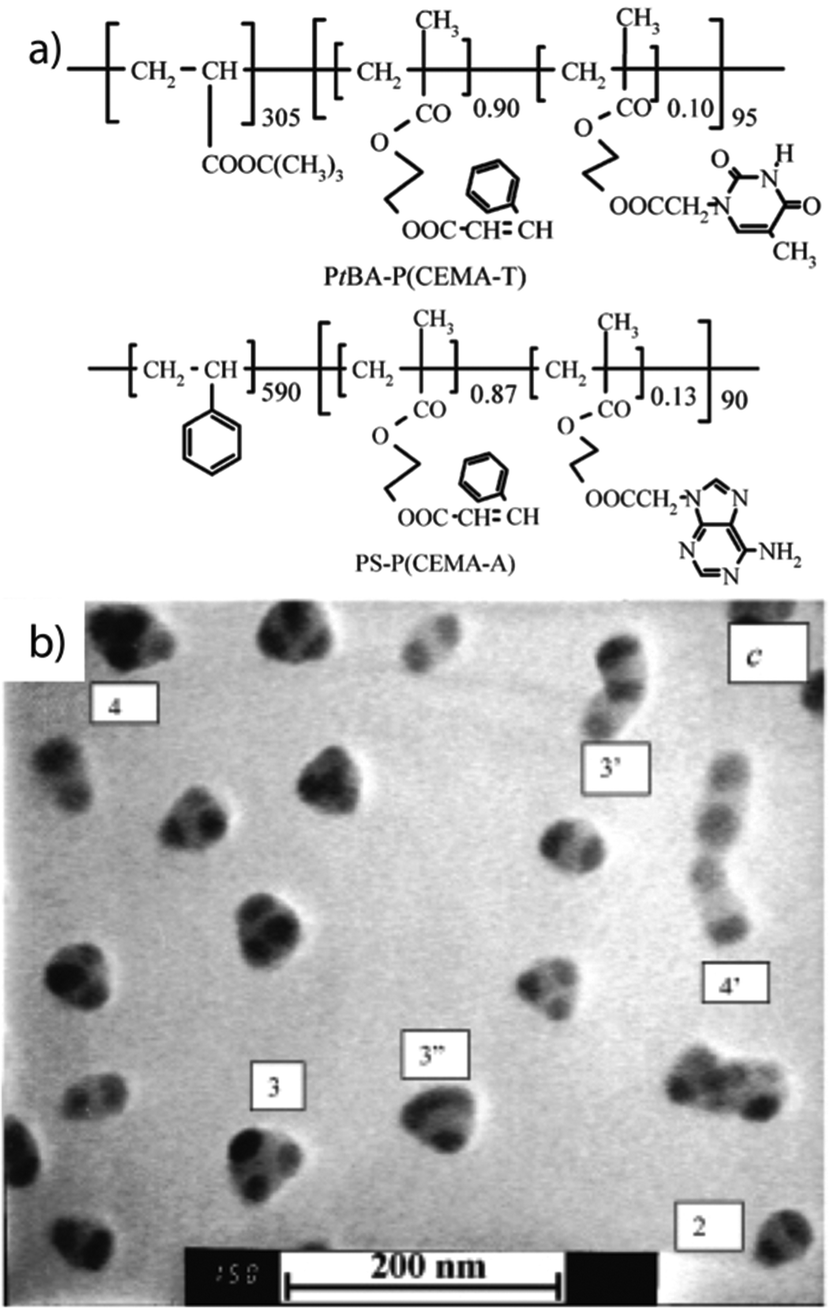 | ||
| Fig. 14 MCNs of chemically stitched triblock terpolymer. (a) Diblock copolymers modified with complementary DNA base-pair derivatives. (b) MCNs with a PS core, two to four compartments of chemically stitched P(CEMA-A/T) stabilized by a PtBA corona. Reprinted from ref. 281 with permission from American Chemical Society. | ||
Electrostatic attraction and non-solvent. One of the most prominent examples for block copolymer self-assembly to MCNs relies on a combination of electrostatic attraction and addition of non-solvent for kinetic trapping.284 Therefore, polystyrene-block-poly(methyl acrylate)-block-poly(acrylic acid) (PS-b-PMA-b-PAA) is first molecularly dissolved in THF together with the divalent organic base, 2,2′-(ethylenedioxy) diethylamine (EDDA). The addition of water triggers aggregation of the hydrophobic block into particles with a PS core, PMA shell and PAA corona, while simultaneously the increasing polarity of the THF/water mixture augments complexation between PAA/diamine. These micelles experience anisotropic deformation into discs controlled by the delicate THF/water composition (Fig. 15a). At a mixing ratio of PAA/diamine 1/1, one EDDA molecule per two AA units of the core-shell-corona disc results in equally cationic as well as anionically charged shells. The discs stack during balancing of the THF/water content into columns or core-segmented one-dimensional MCNs with high quality regarding shape and internal morphology due to the preferential overlap of the large planar disc surface (Fig. 15b and c). The disc-like micelles are interlocked through electrostatic attraction of the cationic amine groups of neighboring PAA coronas. Here, the block sequence within the micellar building blocks as well as the final MCN corresponds to ABCBA where A = PAA/EDDA, B = PMA and C = PS. Since PAA is part of the core, incorporation of cationic gold nanoparticles by complexation places the nanoparticles into every PAA segment with equidistant spacing. The self-assembly process can be further tuned by many external forces that result in great structural diversity of the same polymer by slight variations of system parameters.285–288
 | ||
| Fig. 15 Linear superstructures based on step-wise self-assembly. (a) Self-assembled disc-like PS-b-PMA-b-PAA micelles and (b) stacking into (c) extended segmented cylinders directed by solvent mixtures and additives. (d) The polyionic complex allows for selective storage of cargo, here gold particles. Reprinted from ref. 284 with permission from American Association for the Advancement of Sciences. | ||
Similar strategies of kinetic trapping target intermediate morphologies of diblock copolymer blends and display good control over core composition and particle geometry. As an example, rapid addition of water “electrostatically stitches” poly(acrylic acid)-block-polybutadiene (PAA-b-PB) to poly(acrylic acid)-block-polystyrene (PAA-b-PS) and confines the two hydrophobic blocks into the same MCN core.289 The ionic linking of diblock copolymers via PAA block formally creates ternary patchy particles that self-assemble on the next level. However here, the stitched middle block acts as the corona, which has a pronounced effect on the self-assembly outcome. The mixing ratio of both diblocks determines the core composition, while the volume fractions of the individual hydrophobic blocks (ϕS and ϕB) control particle geometry. The hydrophilic to hydrophobic ratio of PAA-b-PS corresponds to cylinder micelles and that of PAA-b-PB to spherical micelles. The blend ratio then tunes the core composition between these extremes. At higher fractions of the glassy, cylinder-forming PAA-b-PS, particles grow into elongated, anisotropic cylinders, while at higher PAA-b-PB contents spherical or “hamburger” MCNs dominate. When forming the PS/PB interface, the enthalpic penalty between PS and PB works against co-assembly. To overcome this penalty, rapid addition of water to PAA/EDDA and fast kinetic trapping of the core blocks becomes necessary.290 This results in a size and composition distribution of MCNs, while populations of particles with a homogeneous core are equally probable (e.g., linking of only PAA-b-PB). In a similar approach, electrostatic stitching of PAA-b-PS to PAA-b-PI both with proper hydrophilic/hydrophobic balance led to aggregation into trigonal planar and polygonal shapes with three to six distinctly visible edges.291 The sheet-forming PS would locate towards the inner, i.e., the low-curvature part of the disc, whereas the sphere forming PI blocks exclusively locate at the disc edges with high curvature (selective staining with osmium tetroxide). This work clearly demonstrates that the information about particle geometry encoded into the diblock copolymers (block lengths) manifests in blended superstructures as an intermediate geometry or rather as a compromise between the geometries of both underlying block copolymers.
Metal ligation and non-solvent. Metal ligation of supramolecularly linked segments to triblock terpolymers adds more flexibility for transformations to a given system.292,293 Terpyridine end-tethered polymers show strong complexation with divalent transition metal cations and thereby connect block copolymers, e.g., the supramolecular linkage of polystyrene-block-poly(2-vinylpyridinde)-[Ru]-poly(ethylene oxide). Here, the terpyridine-modified PS-b-P2VP diblock copolymer first coordinates to Ru(III) ions forging stable mono-complexes that link with terpyridine-modified PEO chains under reductive conditions (Ru(III) → Ru(II)). After complexation, non-solvents direct terpolymer self-assembly into core–shell–corona micelles with redox-responsive, detachable corona. Besides terpyridines, pincer ligand Pd(II) complexes led to the formation of hollow nanostructures by supramolecular connection of poly(acrylic acid)-block-poly(methyl methacrylate)-[Pd]-polystyrene.294 Although there has been great progress in metal ligation of block copolymers also towards functional materials, they have not been used much for hierarchical or programmable self-assembly of MCNs.
pH and non-solvent. A system responsive to pH and non-solvent comprises the self-assembly of poly((sulfamate-carboxylate)isoprene)-block-polystyrene-block-poly(ethylene oxide) (PISC-b-PS-b-PEO) with a pH-responsive PISC polyelectrolyte endblock.295 The bishydrophilic triblock terpolymer was synthesized from chlorosulfonyl isocyanate and polyisoprene in a polymer analogue reaction and dissolves in THF, while the addition of acidic water triggers self-assembly into PS core micelles with patchy PICS/PEO corona. Due to the acidic conditions the patchy micelles further cluster into MCNs with a PICS core, a defined number of PS patches and a PEO corona. The pH-responsive PICS block contains carboxylic acid (–COOH) as well as anionic sulfonate (–SO3−) that form an intermolecular polyelectrolyte complex with the cationic amine (–NH2+) at pH < 5. In pure water (pH > 5) the core swells due to electrostatic repulsion of the ionized carboxylate groups. Final break-up of the MCNs into (patchy) micellar fragments occurs under alkaline conditions (pH > 8), where all groups of the PICS block are deprotonated and repulsive. Static light scattering experiments revealed a three-fold lower mass of the released micellar fragments as compared to the MCNs, which coincides to the cluster number. This transition appears to be irreversible attributed to strong hydrogen-bonding between PEO and PISC, which prevents controlled re-aggregation of the PICS patches upon decreasing the pH back to acidic conditions.
Counterions and non-solvent. In terms of electrostatic attraction, monovalent counterions have specific interactions with the polyion chains and the nature of the ion determines its location along the polymer chain.296 Close contact ion pairs are moderately hydrophobic as compared to the more hydrophilic loose ion pairs. This delicate counterion/polyion interaction can be utilized to tune the solubility parameter of the polyion chains in small increments from swollen to random coil to collapsed state. Non-solvents in combination with counterions were used on μ-polybutadiene/poly(N-methyl-2-vinylpyridinium iodide)/poly(tert-butyl methacrylate) miktoarm star terpolymers (μ-(BVqT)+/I−)297 where the type and concentration of the counterion controlled the self-assembly behavior (Fig. 16a).298
 | ||
| Fig. 16 Counterion-mediated structuring of μ-(BVqT)+/I− miktoarm star terpolymers in water. (a) Hierarchical self-assembly mechanism and morphological evolution with iodine content. (b) Spherical micelles, worm-like micelles and axially stacked lamellae in ellipsoidal MCNs. Reprinted from ref. 299 with permission from American Chemical Society. | ||
Dispersion of μ-(BVqT)+/I− in dioxane and dialysis into water gives molecularly dissolved μ-(BVqT)+/I− at a specific dioxane/water ratio during solvent exchange. With progressing dialysis micelles with a mixed B/T core and Vq+/I− corona emerge. Addition of defined molar amounts of iodine (I2) changes the counterion to triiodide (I3−) with increased hydrophobicity and lower hydration energy. Both factors cause de-swelling of the corona and induce further hierarchical self-assembly of spherical micelles to cylindrical micelles, folding of cylinders to sheets and finally axial stacking of sheets to lamellar ellipsoidal MCNs with pronounced elongated shape (Fig. 16b).299 This straightforward trigger creates highly complex MCNs through multistep self-assembly following delicate balancing of interfacial energies. The final material is reminiscent of the axially stacked lamellae obtained from 3D confinement in section 3.1, but has progressed through an entirely different formation mechanism. This example again teaches the importance of kinetic studies and thorough characterization of intermediate steps both supporting interpretation of structure and its formation. Following and deciphering the mechanism will become increasingly relevant in the future, where identification of three, four or even more phases becomes extremely challenging by just evaluating the final structure.
Crystallization-driven self-assembly and non-solvent. Nucleation and growth are crucial steps in crystallization-driven self-assembly in selective solvents and might be considered as refinement steps of a hierarchical process. In that regard, anisotropic worm-like micelles grow with a compartmentalized corona and a crystalline core from polystyrene-block-polyethylene-block-poly(methyl methacrylate) (PS-b-PE-b-PMMA) (Fig. 17).79,300,301 To erase thermal history, the crystalline PE blocks are molecularly dissolved in toluene or THF and heated above the melting point of PE. Cooling below the crystallization temperature then triggers self-assembly into either cylindrical micelles in THF (good solvent for PE at T > Tcryst) with a crystalline PE core (Fig. 17a) and incompatible corona patches of PS/PMMA or hockey puck patchy particles in toluene (poor solvent for PE) (Fig. 17b).
 | ||
| Fig. 17 Patchy, core-crystalline micelles. Schematics and TEM images of crystalline micelles with segregated corona of PS-b-PE-b-PMMA with (a) cylindrical core in good solvent for PE, and (b) hockey puck core in poor solvent for PE. Reprinted from ref. 79 with permission from American Chemical Society. | ||
The micellar seeds exclusively undergo linear epitaxial growth via crystalline facets. The linear growth of the PE middle block forces the PS and PMMA blocks into patchy corona-compartments with alternating pattern. The visualization of such patches is usually a challenging task, but in this example, the dense packing of chains mediated by the crystalline core draws a clear picture of the compartment distribution (contrast enhanced by selective staining with ruthenium tetroxide). Epitaxial growth not only occurs on the crystalline facets of seed micelles, but other surfaces that offer attractive surface patter on the length scale of the crystallizing unit. Strong attachment of the PS-b-PE-b-PMMA triblock terpolymer to the surface of multi-walled carbon nanotubes with the PE middle block creates hybrid materials with very similar alternating patch configuration of PS/PMMA.302 The starting materials are simply mixed in organic solvents and dispersed through mild sonication. This reversible non-covalent grafting approach is further attractive due to the non-invasive stabilization of the carbon nanotubes in organic solvents at comparably high weight fractions of up to 3 wt%.
The combination of crystallization and non-solvent was also explored for the self-assembly of crystalline micelles with ABC sequence of micellar segments, i.e., ABC block ter-micelles. First, bifunctional cylindrical seed micelles of poly(ferrocenylsilane)-block-poly(dimethylsiloxane) (PFS-b-PDMS) were fabricated and extended on both ends with polyisoprene-b-poly(ferrocenylsilane) (PI-b-PFS) to symmetric BAB block co-micelles (Fig. 18a).303 After cross-linking of the PI corona of the micellar end blocks, the terminal facets are inaccessible for further epitaxial growth. Dissolving the A connecting micellar segment yields B seed micelles with only one “reactive” facet. The sequential crystallization of a second and a third micellar segment, gives cylindrical ABC block ter-micelles with different corona for all three micellar blocks corresponding to a corona sequence of PI/PDMS/P2VP. The block ter-micelles are reminiscent of bottlebrushes except that they are supramolecularly bound and not single molecules. Also the mass is considerably higher owing to the much larger core diameter. Nevertheless, they are able to self-assemble into supermicelles as known for ABC triblock terpolymers (Fig. 18b and c). This higher hierarchical level is reached the same way as for block copolymers chains. By reducing the solvent quality for PI, the corona collapses and block ter-micelles minimize the interface by self-assembly into supermicelles with a core of PFS/PI that is stabilized by micellar arms with a soluble corona of PDMS and P2VP. What has started with nano-sized block copolymers ends in a hierarchical, micron-sized superstructure even visible in optical microscopy (Fig. 18d). The effect of micelle segment length on the hierarchical self-assembly capabilities of block co-micelles were further studied on micelles resembling ABA block copolymer sequences in selective solvents.88,304 Much like described above, BAB triblock co-micelles were generated by crystallization-driven growth of A = PFS-b-PDMS in hexane with varying length, A = 110, 260 and 505 nm. The second block, B = PFS-b-P2VP, was initiated by the A seed micelles to BAB block co-micelles in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 hexane/isopropanol mixture. Dialysis into pure isopropanol triggers aggregating of the hydrophobic PDMS corona blocks of the A mid segment into crisscrossed or hexagonally crossed supermicelles with narrow number distributions for A = 110 nm, larger star-like supermicelles for A = 260 nm and elongated worm-like brushes for A = 505 nm.
:3 hexane/isopropanol mixture. Dialysis into pure isopropanol triggers aggregating of the hydrophobic PDMS corona blocks of the A mid segment into crisscrossed or hexagonally crossed supermicelles with narrow number distributions for A = 110 nm, larger star-like supermicelles for A = 260 nm and elongated worm-like brushes for A = 505 nm.
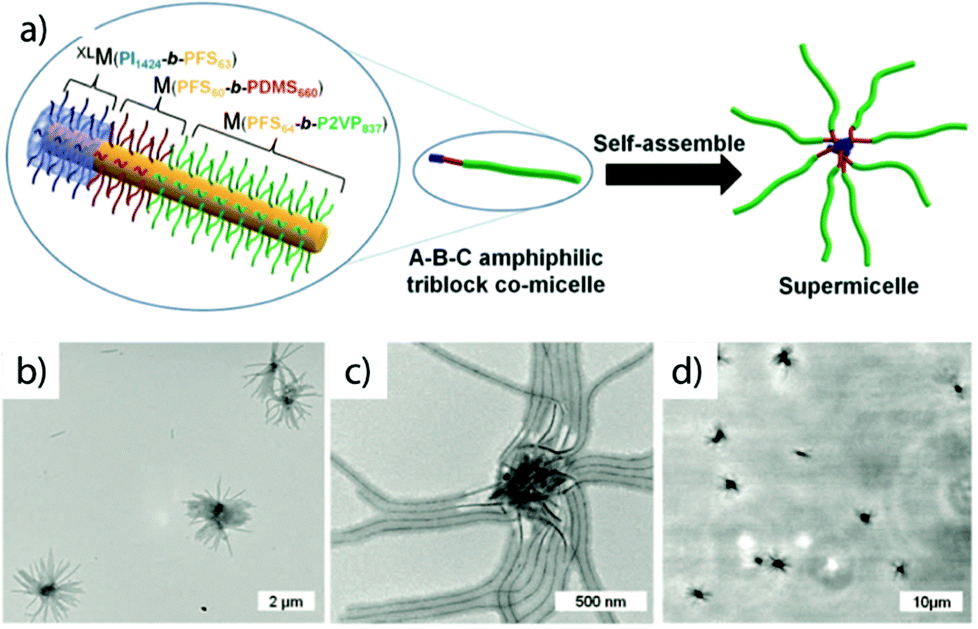 | ||
| Fig. 18 Hierarchical, crystallization-driven growth of MCNs. (a) Schematic of the sequential block crystallization to ABC block ter-micelles. (b, c) TEMs of the supermicelles obtained from higher level self-assembly using ABC block ter-micelles as building blocks. (d) Optical microscopy image of the micron-sized supermicelles. Reprinted from ref. 303 with permission from American Association for the Advancement of Sciences. | ||
SSSL and non-solvent. The combination of low entropy polymer blocks above the SSSL and non-solvents allows for long-range ordered superstructure formation of segmented bamboo-like MCNs from poly(4-tert-butoxystyrene)-block-polybutadiene-block-poly(tert-butyl methacrylate) (PtS-b-PB-b-PtBMA) (Fig. 19).305 To enhance microphase separation, the remaining PB double bonds were first modified to PfB with 1-mercapto-1H,1H,2H,2H-perfluorooctane via thiol–ene polymer analogue reaction. The fluorinated block aggregates in dioxane to give precursor micelles with PfB core and a patchy corona of PtS/PtBMA. The patches and patch distribution was visualized by selective staining of the PtS phase with ruthenium tetroxide (Fig. 19a). Subsequent dialysis of precursor micelles into ethanol triggers self-assembly of insoluble PtS patches into undulated bamboo-like MCN networks of several micrometers in length. The final superstructure resembles a complex network of linear undulating sections, branching points and terminal positions originating from particles of varying patchiness. The linear segments emerge from particles with PtS patches on opposing sides of the core undergoing stacking or step-growth polymerization, while terminal positions correspond to particles with a Janus distribution of the corona patches blocking further growth (Fig. 19b and c). On some occasions the stabilizing PtBMA corona cannot sufficiently protect the PtS compartment and branches grow from such undulated sections. It should be noted that although blocks above the SSSL are very well capable of spontaneous microphase separation in any solvent, the step-wise procedure employed here certainly aided in refining the structure before final aggregation. Since blocks are collapsed into compartments one at a time, structural optimization processes face less kinetic obstacles.
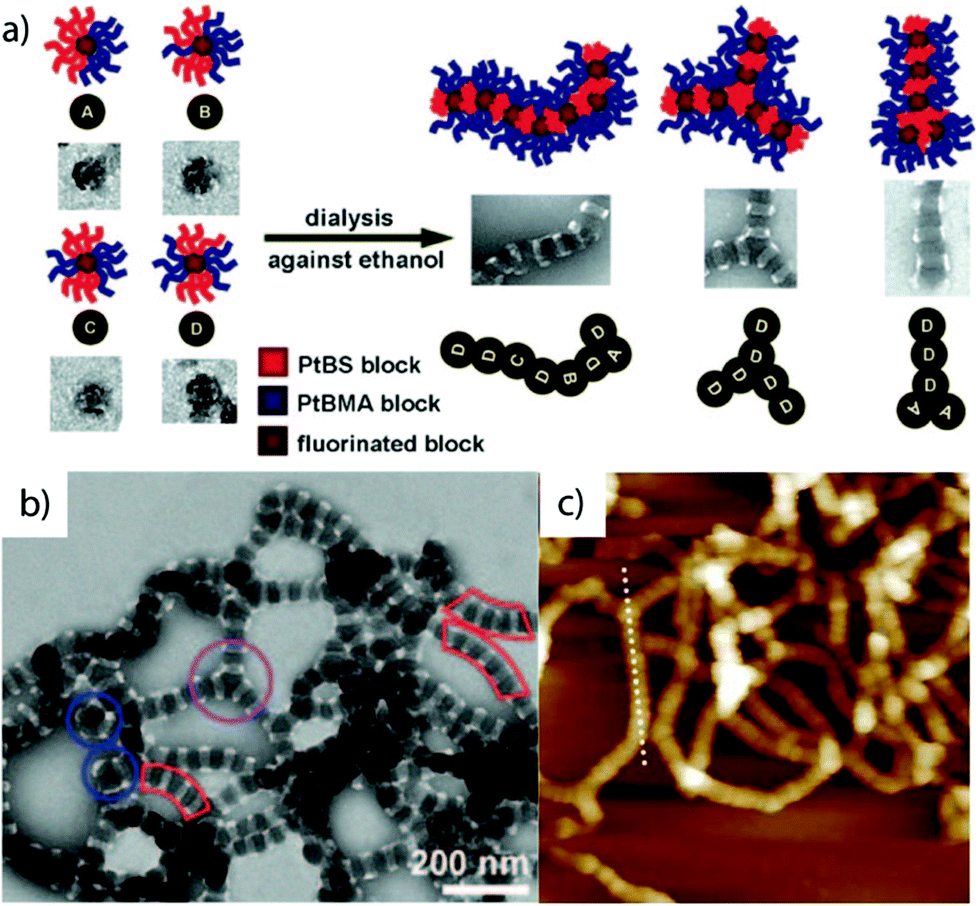 | ||
| Fig. 19 Hierarchical self-assembly of patchy micelles. (a) Step-wise self-assembly involving first the collapse of PfB middle block of PtS-b-PfB-b-PtBMA followed by stacking of patchy micelles to linear undulated MCNs. (b, c) TEM image illustrating the internal segmentation of cylindrical micelles into PfB (bright) and PtS (dark) compartments and AFM showing topography of the undulated cylinders. Reprinted from ref. 305 with permission from Wiley-VCH. | ||
Non-solvent and non-solvent. A more generalized, step-wise approach to direct triblock terpolymers into predictable and near-monodisperse MCNs relies on sequences of non-solvents. Since differences in solubility are an inherent feature of block copolymers, for every monomer combination there should be a solvent or solvent mixture that selectively addresses only one of the blocks. The basic principle of properly tuned non-solvent sequences of specific quality was demonstrated on polystyrene-block-polybutadiene-block-poly(methyl methacrylate) (PS-b-PB-b-PMMA) (Fig. 20).84 The first non-solvent precipitates the middle block of the triblock terpolymer, creating PB core micelles with a patchy PS/PMMA corona. The resulting soft nanoparticles are imparted with molecule-like directionality of repulsive (PMMA) and attractive patches (PS). The self-assembly of the PS patches is triggered in a subsequent step by transferring the patchy micelles into a non-solvent for both PB and PS. The solvophobic PS patches develop attractive interactions and cluster into a variety of core-compartmentalized MCNs with surprisingly narrow dispersity of particle size and compartment number (aggregation number of patchy micelles). The MCNs are always composed of a segmented PS/PB core stabilized by the PMMA corona. Since the triblock terpolymer was directed step-by-step into the final superstructure the position of the participating blocks within the MCNs is precisely controlled. The volume ratios of the core forming blocks VPS/VPB govern self-assembly into either spherical or one-dimensional MCNs (also colloidal polymers). With long PS blocks and a high VPS/VPB > 1, the transient subunits display a “monovalent” Janus distribution of corona patches (SB Janus particles) and aggregate into spherical MCNs. Higher block asymmetries VPS/VPB increase the aggregation numbers of transient SB particles and thus the number of core compartments. Lowering VPS/VPB < 1, a “divalent” SBS patch distribution of the transient building blocks becomes favorable, which grow into one-dimensional MCNs. The block length and/or solvent quality for the corona controls the degree of colloidal polymerization through shielding or exposure of the associating terminal patches (same mechanism controls the cluster size for the spherical MCNs). The corona patches are capable of adjusting their volume dynamically with respect to the solvent quality and the resulting reversible switching between corona- and core-compartments is a unique feature of these soft patchy nanoparticles. The separation of single self-assembly events into individual steps revealed the underlying mechanism and highlighted the necessity of a gradual change of environmental conditions to reach high structural homogeneity. The direct dispersion of the triblock terpolymer in the final solvent composition only resulted in less defined und unpredictable arrangement of all blocks (within the core) and kinetic trapping of intermediates.
 | ||
| Fig. 20 Schematic of MCN formation using non-solvent sequences for linear triblock terpolymers. The series of TEM images shows the underlying self-assembly mechanism involving dispersion of SBM triblock terpolymer in a non-solvent for PB to patchy micelles and subsequent collapse of PS patches into spherical or cylindrical MCNs depending on VPS/VPB. Reprinted from ref. 84 with permission Nature Publishing Group. | ||
A step-wise approach not only allows predicting the block location, but also reduces kinetic barriers for equilibration. The general applicability of this concept was demonstrated for a series of triblock terpolymers with widely different physical/chemical properties. Especially poly(tert-butyl acrylate)-block-poly(2-cinnamoyloxyethyl methacrylate)-block-poly(2-(dimethylamino)ethyl methacrylate) (TCD) demonstrated sophisticated self-assembly capabilities. The pH-programmable corona of the building blocks permits reversible self-assembly/disassembly into extended chains and back to unimers. Besides the possibility to generate a plethora of MCNs from the same type of triblock terpolymer, spherical MCNs were obtained in extraordinary homogeneity. Exemplified on PS-b-PB-b-PMMA, “hamburger” (dimer) MCNs were obtained at a VPS/VPB ≈ 1.5 and “clover” (trimer) MCNs at VPS/VPB ≈ 1.7 in selective solvents for PMMA (acetone/isopropanol). Evaluation of 500 MCNs peaked in over 92% identical internal morphologies, i.e., dimers with two compartments geometrically positioned in an angle of α ≈ 180° and trimers with three compartments in an angle of α ≈ 120°. The remaining 8% of MCNs exhibited ± 1 compartment, adding up to an extremely narrow distribution of core segments per MCN sample.
Going one step further, the co-assembly of the monovalent and divalent structuring units was aiming at the goal of predictive multicomponent self-assembly that requires a sophisticated level of control in many aspects. Using the above-described method to create narrowly-dispersed MCNs, multiple colloidal building blocks were co-assembled into well-ordered supracolloidal structures with nanoscale periodicities.77 The SB Janus nanoparticles were assembled from PS-b-PB-b-PMMA and linearly aggregating SDS particles from PS-b-PD-b-PMMA obtained after post modification of PB with dodecanethiol to give poly(3-butenyl (dodecyl)sulphane) (PD). Fig. 21 summarizes the mechanism of hierarchical co-assembly of the SB/SDS system. Both triblock terpolymers were dispersed separately in a non-solvent for PB and PD into the SB and SDS particles, then mixed in the desired particle ratio and co-dialyzed into a selective solvent for the common PMMA corona. The PS blocks start to collapse and particles aggregate. Surprisingly, the SB particles do not self-assemble to spherical clusters, but exclusively decorate the newly formed PS compartment of the linearly growing [SDS]x core-segmented cylinders. The size ratio of the building blocks SB/SDS controls the number of SB particles per PS segment of [SDS]x. Increasing the SB particles above a critical size, excludes compartment decoration and promotes localization at the end of the one-dimensional assemblies to act as terminal “end-cappers”. The mixing ratio of end-capper per SDS unit allows controlling the overall length of the colloidal polymer. End-cappers that feature a corona with different solubility as the SDS building blocks, result in telechelic oligomers (bifunctional colloidal polymer chain) to bridge the third hierarchical level: (1) triblock terpolymers self-assemble to the soft patchy nanoparticles that (2) co-assemble to telechelic oligomers that (3) aggregate end-to-end when the solvent for the corona of the end-capper is switched solvophobic. This concept demonstrates that instead of synthesizing ever-more complex (or complicated) building blocks, combinations or properly designed, simple building blocks can create highly complex self-assemblies. In the future this modular approach may be extended to particle combinations of diverse origin, i.e., co-assemblies constructed from various sources (biological or inorganic) given mutually attractive surface patches are provided.
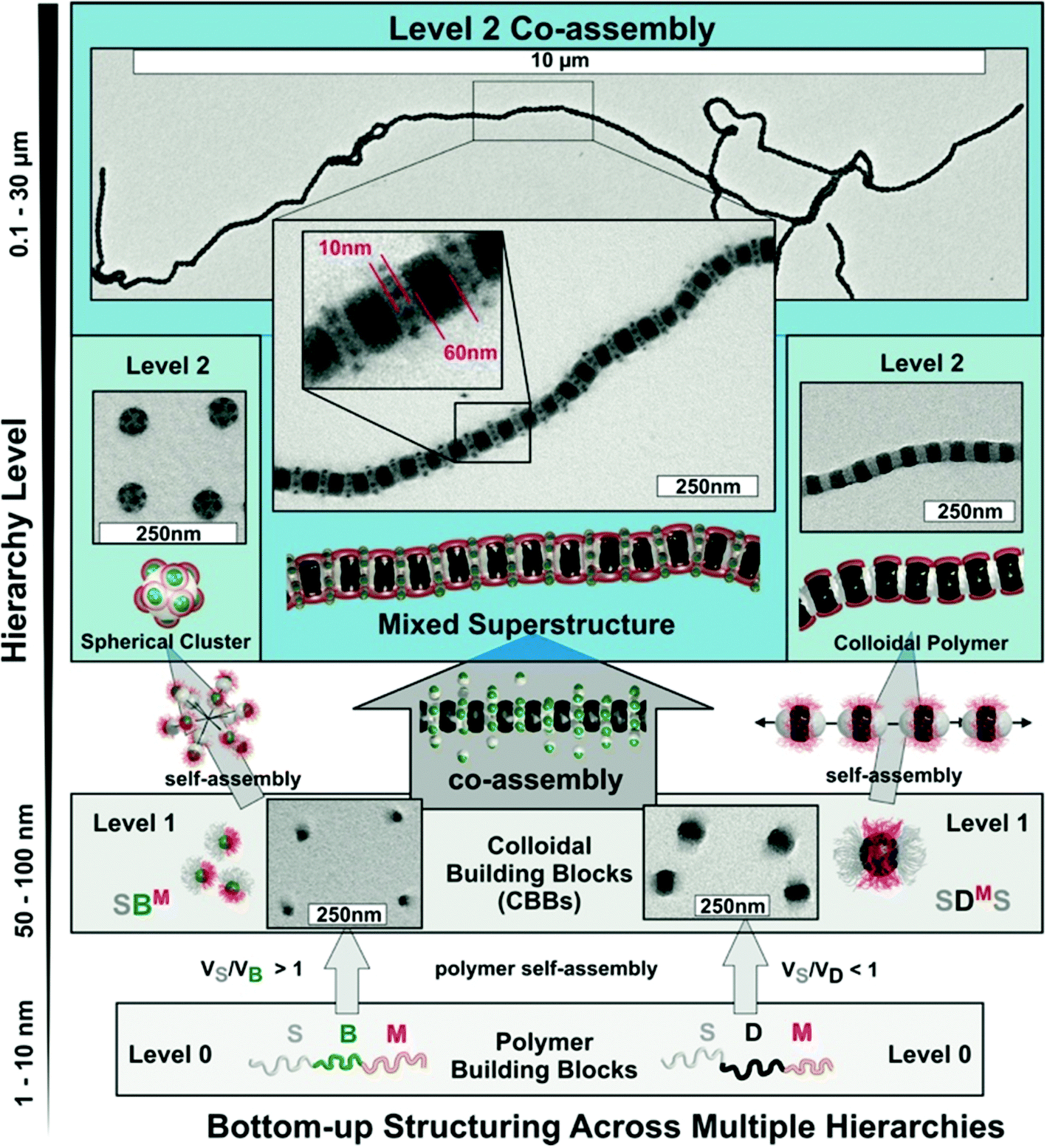 | ||
| Fig. 21 Hierarchical co-assembly of soft patchy nanoparticles. SBM and SDM triblock terpolymers self-assemble into monovalent SB and divalent SDS building blocks (depends on the volume ratios of the core forming blocks), which self-assemble to spherical SB clusters and linear [SDS]x supracolloidal polymers if kept separately. When mixed, SB and SDS building blocks co-assemble with their mutually attracting PS patches into mixed superstructures stabilized by the common PMMA corona. (OsO4 staining: PS segments grey, PB cores dark grey, PD segments black and PMMA not visible due to e-beam degradation). Reprinted from ref. 77 with permission from Nature Publishing Group. | ||
4 Application of terpolymer-analogue systems
To create superstructures with ever-smaller feature size, refined structural homogeneity or controlled location of components require optimization steps towards potential applications and technological relevance. Hierarchical self-assembly plays an important role in most natural structuring processes and teaches how clever building block design allows combination of usually incompatible properties. The following examples are an excerpt of compartmentalized nanomaterials generated via self-assembly of smaller building blocks resulting in spatially separated environments with distinct properties encompassing hybrid materials with tunable plasmonics, precisely patterned fluorescent probes, advanced colloidal surfactants and drug delivery.4.1 Self-assembly of hybrid block copolymer analogue building blocks
Hybrid materials combine otherwise incompatible matter (e.g. organic and inorganic), provide stabilization for insoluble particles and allow for tunable interparticle distance in superstructures306 and polymer matrices.307 Nanoscale hybrids promise novel opto-electronic and magnetic effects and may find application in high-density magnetic storage, catalytic systems or as plasmon-based sensors.308–311 The precise design of the self-assembling motifs of hybrid building blocks is quintessential for ordered particle aggregation with direct relation to the quality of physical effect, transition or property.54 The clustering behavior is predominantly regulated by the soft interaction potential of the organic surface modification of the hybrid particles.For instance, amphiphilic inorganic nanoparticles were prepared by ligand exchange on gold nanoparticle surfaces with thiol-endcapped poly(ethylene oxide)-block-polystyrene.312 The core–shell–corona structure of these particles conceptually yields ABCBA penta blocks of PEO-[PS(gold)PS]-PEO that spontaneously self-assemble into vesicular and tubular geometry with the nanoparticles selectively located within the vesicle membrane in a hexagonal packing (Fig. 22a). The vesicles are prepared by rehydration of dried thin films of hybrid particles, which is a well-established method for block copolymers as already discussed in section 3.4. The asymmetric volume fractions in favor of the hydrophobic component directs self-assembly into hollow capsules, whereas the molecular weight of the PS shell controls lateral spacing. The hollow character of the vesicle and the hexagonal pattern in the membrane were confirmed with electron tomography. The interparticle spacing determines the surface plasmon resonance of the gold particles and the absorption peak for single particles at λ = 518 nm shifts to λ = 560 nm after clustering. The approach is modular and allows facile tuning of the hybrid capsules through synthetic variation of the building blocks. The properties and size of the inorganic particle can be exchanged with any other particle reactive to thiol–ene chemistry, the thickness of the vesicle membrane is equal to the particle diameter and interparticle spacing scales with block molecular weight. Heterogeneous and Janus-like microphase separation of particles within the vesicle membrane was tailored by entropic equilibration of diblock copolymer/core–shell–corona particle mixtures.315 Both components were dissolved in THF as common solvent and drop-wise addition of water triggered co-assembly of block copolymer and inorganic nanoparticles. The blending ratio determined clustering behavior of nanoparticles by maximizing the entropy of block copolymer chains (conformational), which drives microphase separation into polymer rich and particle rich domains. The chain length mismatch between free and tethered polymer chains was found to be an essential design criterion for this effect to take place.
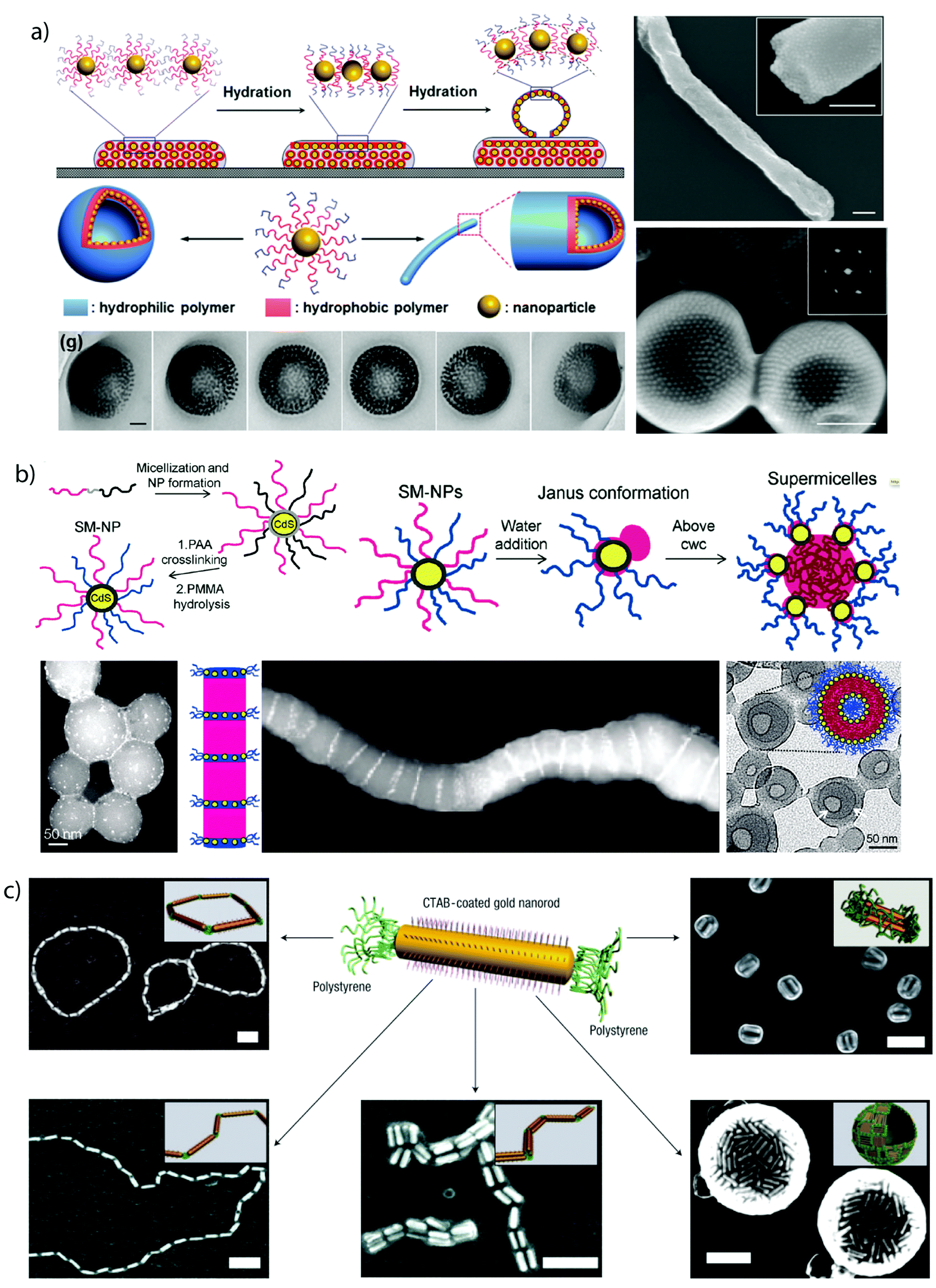 | ||
| Fig. 22 Hybrid materials based on block copolymer self-assembly in solution. (a) PEO-[PS(gold)PS]-PEO amphiphilic building blocks to nanoparticle capsules and hollow tubes. (b) In situ formation of CdS nanoparticles in PS-b-PMAA-b-PMMA micelles with PMAA/CdS core and amphiphilic PS/PMMA patches; higher level self-assembly of patchy CdS nanoparticles in water (PS collapses) into hybrid large compound micelles, segmented cylinders and capsules depending on block volume fractions. (c) Amphiphilic gold nanorod self-assembly in ternary solvent mixtures to linear (multi) strings, solid and hollow spheres. (a) and (b) reprinted from ref. 312 and 313 with permission from American Chemical Society and (c) from ref. 314 with permission from Nature Publishing Group. | ||
Responsive and fluorescent particles with AB Janus distribution of the assembling patches were prepared in situ by CdS nanoparticle formation within the PMAA core of PS-b-PMAA-b-PMMA micelles (Fig. 22b).313 For that, the PMAA middle block is loaded with precursor salt and after sulfidation to CdS nanoparticles, inorganic block copolymer analogues are generated. Prior to the next self-assembly step, the PMMA corona was hydrolyzed to PMAA (the PMAA middle block was derived from PtBMA) and self-assembly was triggered by water as the selective solvent. A Janus character was ascribed to the transient configuration of these inorganic block copolymer mimics. Aggregation of the collapsed PS blocks results in diverse solution geometries comprising hybrid large-compound micelles, segmented one-dimensional micelles and vesicles. As a special feature of this approach the nanoparticles are exclusively located in spatially separated domains with precise interparticle distance on two lengths, i.e., the particles are separated by a polymer coating of defined thickness (∼nm) and on a second level by the domain spacing of the superstructure (>10 nm). In a similar approach, gold nanoparticles were bound to poly(ethylene oxide)-block-poly(lipoic acid 2-hydroxy-3-(methacryloyloxy)propyl ester-co-glycidyl methacrylate)-block-polystyrene with 1,2-dithiolane-functionalized middle block.316 The amphiphilic PEO/PS endblocks provide the means for hierarchical self-assembly in selective solvents. The combined volume fraction of the water-insoluble segment directs self-assembly predominantly into nanoparticle vesicles.
Difunctional polymer/inorganic hybrid nanoparticles with ABA distribution of responsive patches are another well-described system. The aggregation behavior of such difunctional ABA colloids has been reported for a number of inorganic nanoparticles and inorganic–polymer hybrids.317–322 Here, two attractive patches are located at opposing sides of the anisotropic (rod-like) particle core covered by a repulsive patch. This setup represents the advancement of the Janus distribution by an additional attractive patch, thus altering the growth direction from spherical clustering to extended linear end-to-end addition via a step-growth polymerization analogue (equivalent to the ABA patchy micelles). The ABA configuration was achieved by selective end-decoration of rod-like gold nanoparticles with PS polymer chains selectively attached to the “arrowheads”. This was possible due to the different reactivity of etched {111} facets of the arrowheads of the gold particles as compared to the {000} facets of the longitudinal sides.314 The longitudinal side of gold rods is covered by a bilayer of cationic surfactant, cetyl trimethyl ammonium bromide (CTAB), equaling a lateral repulsive (charged) interaction potential (under self-assembly conditions for the terminal patches). Colloidal polymerization was realized with PS-gold-PS (ABA) colloids and each steps of the growth kinetics visualized at specific time intervals.323 The recorded growth kinetics of the nanoparticles shows striking similarities to well-known step-growth polymerization kinetics of molecules. Besides end-to-end aggregation into chains and rings, the selectivity of the solvent to the surface chemistries causes lateral self-assembly into spheres, vesicles and multi-string chains (Fig. 22c).314 The amphiphilic nature of the gold rods allows fine-tuning of the self-assembly conditions according to solvent polarity adjusted by DMF/THF/water ternary solvent mixtures. Thereby, DMF is an equally good solvent for both PS and CTAB patches, THF is only selective for PS, and water for CTAB.324 The building blocks are dispersed in DMF as unimers at fixed water contents triggering pre-assembly into linear colloidal polymer chains. The gradual addition of THF to this solvent mixture creates additional unfavorable CTAB/THF interface and side-by-side aggregation into multistring chains at low THF contents as well as clustering to solid and hollow spheres in the final THF/water composition.
4.2 Soft Janus nanoparticles
Bulk morphologies have long been a necessary tool to break the symmetry for the synthesis of polymer-based Janus nanoparticles (see also section 3.4), because usually corona chains do not spontaneously separate into two hemispheres due to entropic penalties upon total de-mixing of the corona chains. An alternative solution-based route for the preparation of soft Janus nanoparticles makes use of the block arrangement of spherical MCNs (Fig. 23).325 This process is general, scalable and does not require templates, because symmetry breaking occurs during MCN formation. Exemplified on PS-b-PB-b-PMMA crosslinking of the PB patches after self-assembly to MCNs and transfer into a common solvent for all blocks separates MCNs into individual Janus nanoparticles. Besides of being a scalable process, this method also gives control over the Janus balance, i.e. the ratio of the two corona volumes, through tuning of block fractions and overall particle size through molecular weights of the underlying triblock terpolymer. Janus nanoparticles are not only of academic interest, e.g., as delivery vehicles able to undergo reversible self-assembly/disassembly processes,326 but also move towards technological relevance as colloidal surfactants with exceptional interfacial activity. The combination of amphiphilicity and colloidal character increase the energy for interfacial desorption about 2–3 fold as compared to molecular surfactants or isotropic particles.260 Several theoretical and empirical studies investigated the relation between size and geometry of Janus particles and the energy to desorb particles from the liquid–liquid, liquid–air and solid–liquid interface.261–263 Thus, applications that require interfacial stabilization may in the future benefit from the superior activity of Janus nanoparticles. Their potential use has been demonstrated recently in the compatibilization of homopolymer blends in lab- and industry-scale extrusion equipment,327,328 as colloidal dispersants for carbon nanotubes329 and in emulsion polymerization.330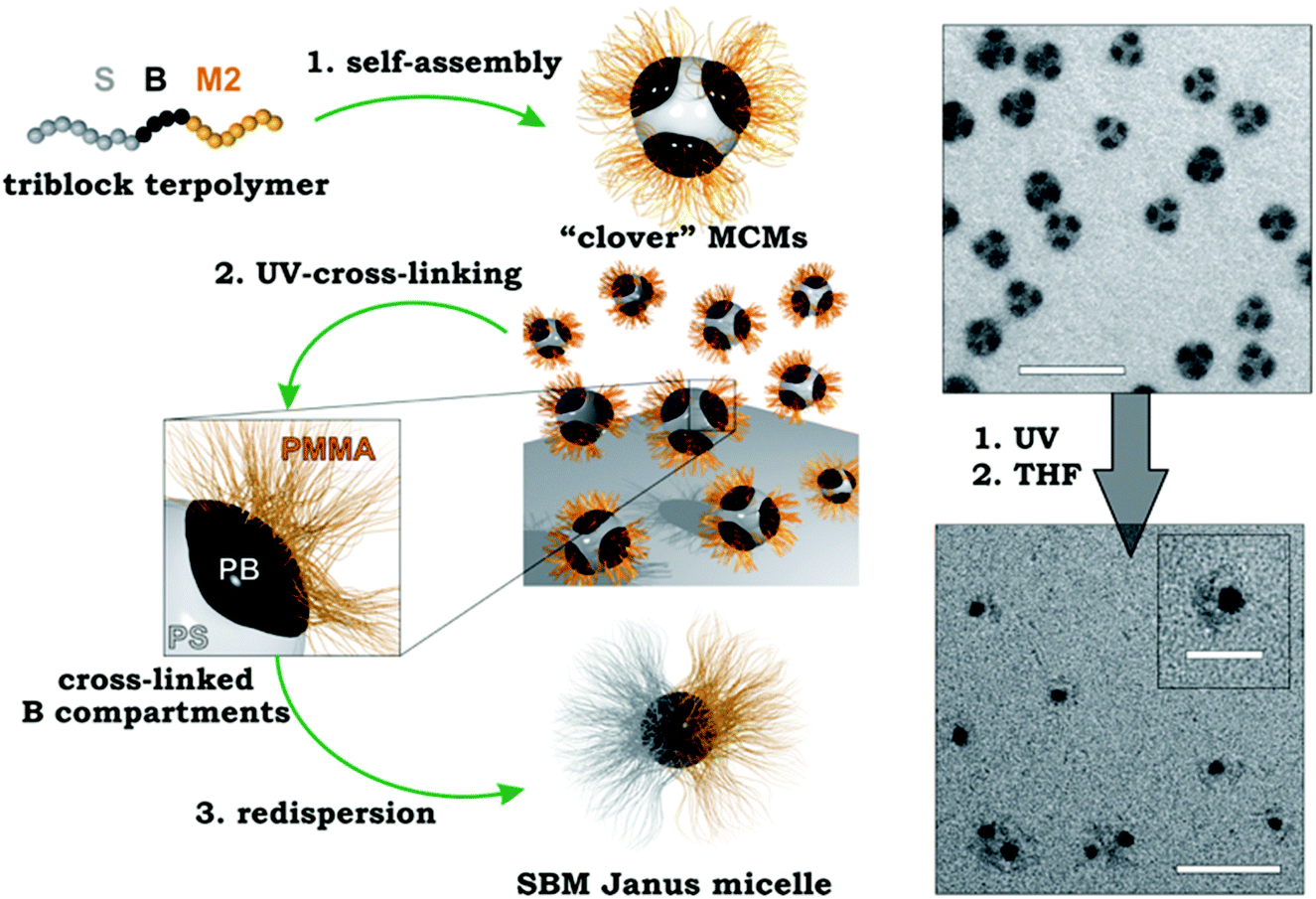 | ||
| Fig. 23 Solution-based approach to produce SBM Janus nanoparticles. Self-assembly of SBM triblock terpolymer into MCNs, crosslinking of PB compartments and re-dispersion in good solvent for all blocks releases Janus nanoparticles. TEM images show MCNs before cross-linking and individual Janus nanoparticles after re-dispersion. Reprinted from ref. 325 with permission from American Chemical Society. | ||
4.3 Fluorescent nanosticks
The capabilities of crystallization-driven growth to one-dimensional micelles hold great promise for the implementation of functional groups at very precise locations. Despite the ability of functional blocks to crystallize into near-monodisperse fiber-like micelles (conducting or redox active),331–333 the epitaxial crystallization process itself is sensitive to slight changes in molecular structure of the repeating units. Modification of the corona blocks on the other hand is more practical, because the only task of the corona is to provide stabilization. Functional groups copolymerized into the corona or attached in polymer analogue reactions may range from coordination sites for metal ions or nanoparticles,334,335 reversible cross-linking sites336 or optically active moieties.52,337,338 The latter was demonstrated by attachment of light-emitting fluorophores to the corona bock followed by co-crystallization of various emitter sequences with exceptional precision regarding the location. In one example, poly(ferrocenyl dimethylsilane)-block-poly(2-vinylpyridine) (PFDMS-b-P2VP) diblock copolymers were fluorescently labeled by chain extension with oligomeric 2,5-di-(2′-ethylhexyloxy)-1,4-phenylvinylene giving a labeled and unlabeled version of otherwise fully compatible block copolymers for consecutive epitaxial growth. The length and spatial separation of fluorescent and “blank” segments are thereby controlled by feed ratio of block copolymer to seed micelles. Extremely thin (about 40 nm) wires were produced with up to 9 alternating segments fluorescent and non-fluorescent that may find application in sensing, diagnostics or display technology. This concept was further extended to the full color spectrum building a powerful platform for the modular construction of nano-sized pixels.338 Red, green and blue fluorescent dyes based on 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene were incorporated into the poly(dimethylsiloxane-ran-vinylmethylsiloxane) corona block by thiol–ene modification (Fig. 24a). Each polymer by itself self-assembles into short, narrowly length-dispersed cylinder micelles with red, green or blue fluorescence when irradiated with λ = 365 nm. The fiber-like, glowing objects in Fig. 24b are imaged with confocal laser scanning fluorescence spectroscopy. Mixing all three of the labeled block copolymers in proper molar ratios creates emitters with a predictable mixed color. The spatial confinement of multiple fluorescent dyes without coupling or quenching is by itself a profound achievement and the ability to target the full RGB color spectrum (including emission of white light) by straightforward combinatorial co-mixing is equally striking. As final demonstration of control, block sequences were self-assembled reminiscent of nanoscale RGB color pixels (Fig. 24c).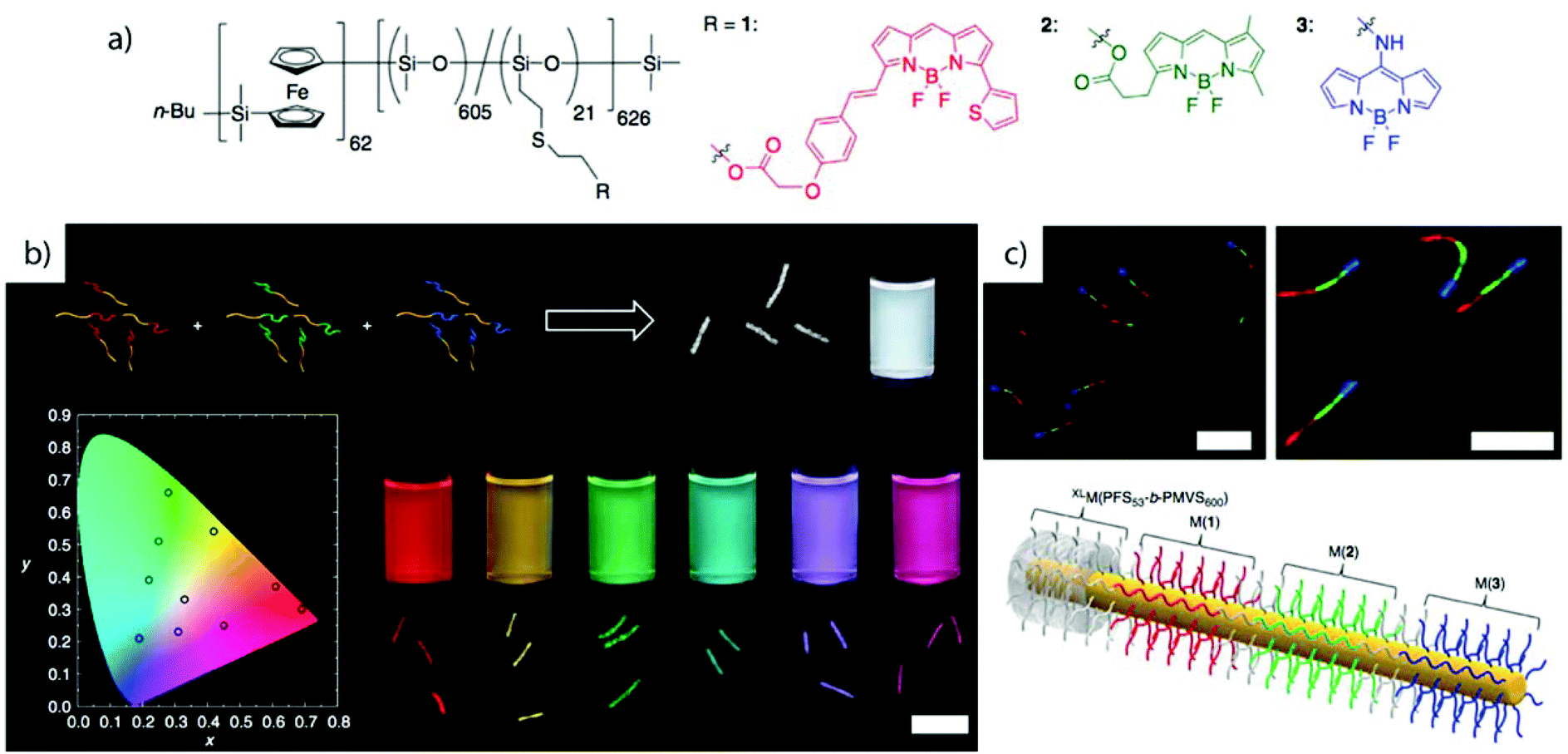 | ||
| Fig. 24 Crystallization-driven statistical and sequential co-assembly of corona-segmented fluorescent block co-micelles. (a) PFS62-b-(PDMS605-r-PMVS21) copolymer is the basis for thiol–ene click modification with red, green and blue dyes. (b) Combinatorial co-crystallization approach for controlled RGB color targeting in a CIE 1931 chromaticity diagram. Solutions are excited with UV at λ = 365 nm. (c) Sequential crystallization of RGB segments. Scale bars in laser confocal fluorescence microscopy images is 3 μm. Reprinted from ref. 338 with permission from Nature Publishing Group. | ||
4.4 Storage, release and delivery capabilities
When dealing with block copolymer self-assembly and compartmentalized nanostructures, drug delivery and nanoreactors are evident applications.174,339–342 The structural configuration of MCNs is ideally suited to deliver multiple cargo separately stored in incompatible compartments.343Storage. Selective and simultaneous loading of payloads was conceptually demonstrated on the incorporation of a red and a green fluorescent dye into the compartments of MCNs.344 “Hamburger” micelles of μ-poly(ethylene oxide)/poly(ethylethylene)/poly(fluorethylene) (μ-EOF) miktoarm star terpolymers with E/F core compartments were selectively loaded with pyrene (into E) and 1-naphthyl perfluoroheptanyl ketone (into F). UV absorbance spectra confirmed the activity of both compounds in agreement with theoretical models. The naphthalene derivative does not show significant intensity when located in hydrocarbon environment and thus, the synergetic absorbance of both substances should originate from the dyes located separately in the respective compartments.
Release. Equally important to studies about storing cargo are the capabilities of MCNs to again release the payload preferably with specificity to target, release profile and stimulus. Regarding triblock terpolymers, the pseudo miktoarm star μ-poly(tert-butyl acrylate/poly(2-cinnamoyloxyethyl acrylate)/poly(ethylene oxide) self-assembled into vesicles with a laterally structured membrane (pseudo because PEO was clicked to a central block with 1.14 chains on average).345 The channels or pores of the membrane are decorated with polyacrylic acid (after hydrolysis of PtBA) responsive to changes of pH. Pyrene-labeled PEO was encapsulated during self-assembly into the vesicles and its release followed recording the UV signal intensity. Since the pKa of PAA is around 5, release increases rapidly when the pH changes from pH ≈ 3 (collapsed chains block channels) to pH ≈ 11 (chains dissolved). Using a different concept, the pH-dependent release of a water-soluble dye was demonstrated for core–shell–corona vesicles formed by poly(ethylene oxide)-block-polystyrene-block-poly(2-(diethyl-amino)ethyl methacrylate) (PEO-b-PS-b-PDEA). At high pH values the vesicles feature an inner PDEA membrane covered with a thin layer of PS outer membrane. Decreasing the pH value protonates the PDEA block and the swelling phase disrupts the PS layer at a certain point and the dye is released. Vesicles with a nanostructured membrane are rare, but desirable delivery vehicles with promising advanced gating capabilities.
Delivery. Towards more hands-on applications, the BVqMAA IPEC particles introduced in section 3.2 were recently studied as drug delivery system for cancer treatment in photodynamic therapy.78 The hydrophobic PB micellar core incorporates the likewise hydrophobic porphyrazine derivate (photosensitizer) (Fig. 25) and the anionic PMAA corona is coordinated with polycationic and bishydrophilic poly(ethylene glycol)-block-poly(L-lysine) (PEG-b-PLL). Complexation replaces the former anionic corona with a neutral, biocompatible PEG brush. The ratio of PMAA to PLL repeating units conveniently controls the degree of PEGylation relevant for studies on the effect of corona composition on cell uptake (and circulation time). The resulting particles display narrow size-dispersity and the IPEC brush structure is reminiscent of virus particles. The modular nature of this multicomponent system gives facile access to functional bioarrays by altering the corona composition with various charged and functional block copolymers. In vitro and in vivo studies corroborated the potential of these MCNs as carrier systems in photodynamic therapy. A similar modular triblock terpolymer-based system was constructed from a poly(L-lysine)/DNA polyion complex core, a dendritic photosensitizer loaded into the poly{N-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} shell and a stabilizing PEG corona.63 The rod-shaped core–shell–corona polyplex showed high transfection efficiency and was successfully studied for the application in photochemical internalization processes with good in vivo gene expression after light activation (λ = 680 nm).
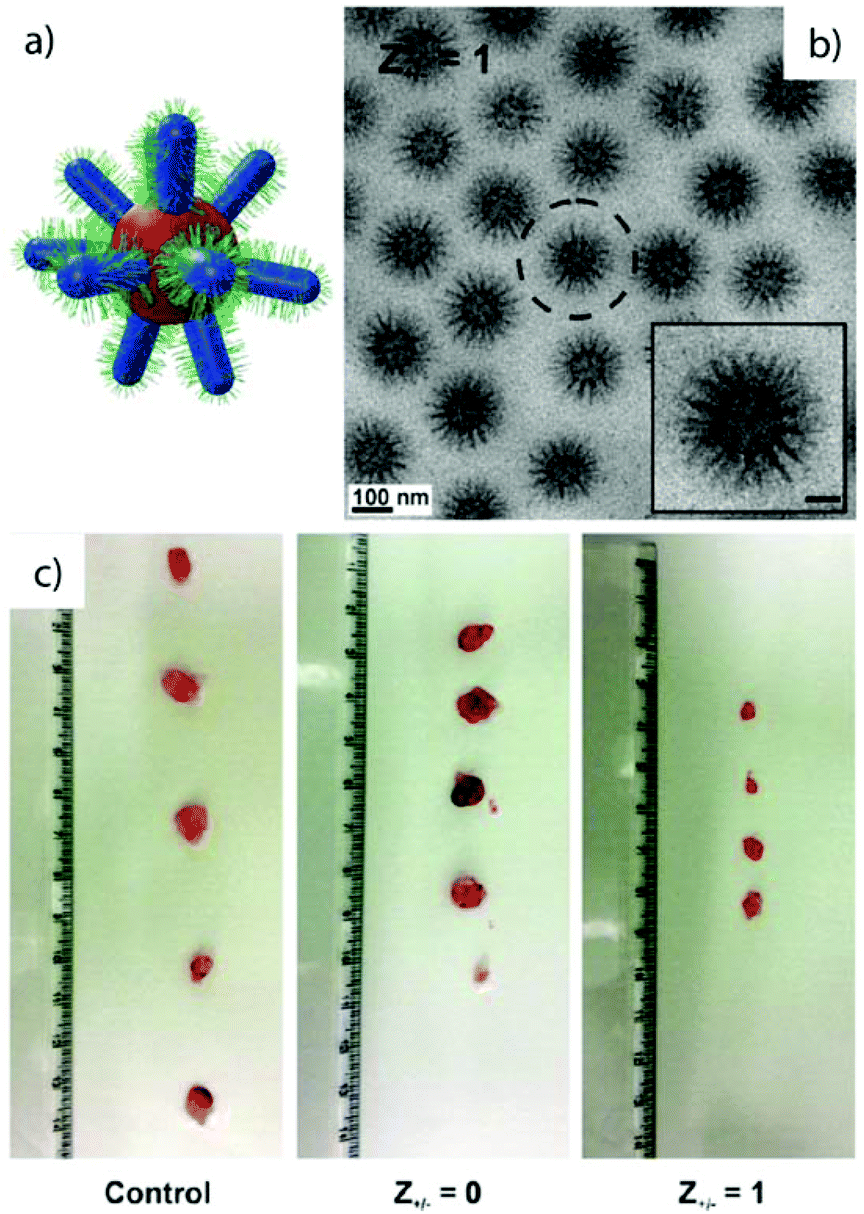 | ||
| Fig. 25 Multicompartment micelles for tumor targeting. (a) Schematic of the BVqMAA/PLL-b-PEG IPEC. (b) Cryo-TEM image of brush-like IPEC particles with narrow size-distribution. (c) Size of tumors of lung cancer cells grown to much less extent applying fully PEGylated IPEC micelles (Z+/− = 1). Reprinted from ref. 78 with permission from American Chemical Society. | ||
5 Future perspective and challenges
The concept of multicompartment micelles was proposed by Ringsdorf346,347 in the mid 90s and immediately attracted the interest of the polymer science community. Much like controlled/“living” polymerization techniques allow the spatial arrangement of polymer blocks, directed self-assembly of block copolymers may today allow controlled positioning of entire compartments on larger length scales. It would be desirable to gain a more comprehensive understanding about how to target known particle geometries also with ABC triblock terpolymers and introduce complexity to compartmentalized cylinders and vesicles. Multiblock copolymers should be able to undergo the classical morphological transitions known for diblock copolymers, i.e., from sphere to cylinder to vesicle. The established nomenclature for morphologies where two or more immiscible blocks form a microphase-separated solvophobic core will require some extension in the near future. Indeed, a number of multiblock copolymer nanostructures has already been verified experimentally, whereas spheres-on-sphere and core–shell–corona are most documented. To find a unifying concept for the complete description of this morphological puzzle would facilitate access of complex structures to diverse interdisciplinary research fields. One major objective securing the future interest in compartmentalized nanostructures is to reproducibly manufacture particles with utmost homogeneity. So far only few reports demonstrate self-assembly approaches that reliably yield particles with narrow size distribution and homogeneous internal compartment arrangement. Most processes lead to a broad distribution of superstructures within the same sample considerably limiting further use as building blocks, templating material or for biomedical applications. It is still one of the major challenges to master the parameter space in solution to understand and manipulate nano-domain formation kinetics. If chain mobility and diffusion can be controlled by design and shallow energy minima targeted with high probability, we will gain access to narrowly dispersed, internally structured architectures of unprecedented homogeneity. Efforts to produce monodisperse materials close to nature's capabilities will be greatly rewarding and unlock new technologies such as artificial model cells, long-range three-dimensional templates with complex internal symmetries or soft components for nanorobotics. Another powerful argument for compartmentalized nanostructures in solution is drug delivery and although the most advanced clinical trials underway rely on simpler core–corona micelles,348 recent developments in block copolymer self-assembly encourage the use of more complex aggregates as delivery vehicles. Reproducibility is then the motor driving efforts towards commercialization and scalable production approaching industrial scales. On a more practical level, a better understanding of underlying self-assembly paths is required to be able to simplify the synthesis of multicompartment particles and to make them more attractive for a broader audience. Accessibility is essential to qualify for future technologies and to forge links between rising technologies more effectively. Patchy particles come in all shapes, sizes and are constructed from many sources and may be readily combined to functional multicomponent systems. Many structures are still considered exotic and obscure, without any direct relation to applications or interdisciplinary fundamental research. Monodisperse patchy particles with tetragonal geometry may readily self-assemble into long-range ordered superstructures with diamond lattice and potentially interesting physical properties; co-assemblies comprising multiple particle species and geometries from all sorts of scientific disciplines may be unified within one superstructure with unprecedented complexity and response to biological and environmental triggers; adding a dynamic component to self-assembly, predicting and controlling morphological changes over time raises self-assembly to a completely new level. Many of the available stimuli have been used to induce self-assembly, but so far not in the context of hierarchies or multicompartment structures. Magnetic and electric fields are widely implemented to direct the self-assembly of hard colloids and similar concepts emerge only slowly for smaller length scales where soft polymer particles operate. These manipulators might be the solution to finally realize dynamic self-assembly even for polymer-based systems. The building blocks are in our hands and paths are laid and understood in its basics, yet theoretical as well as practical issues (or opportunities) are plenty. The near future will show what novel materials can be devised from the concepts that are developed today.List of Acronyms
| 1D, 2D, 3D | One-, two-, three-dimensional |
| Cryo-ET | Cryogenic transmission electron tomography |
| Cryo-TEM | Cryogenic transmission electron microscopy |
| EDDA | 2,2′-(Ethylenedioxy)diethylamine |
| IPEC | Interpolyelectrolyte complex |
| JP(s) | Janus particle(s) |
| MCN(s) | Multicompartment nanostructure(s) |
| N | Block length |
| p | Packing parameter |
| PB | Polybutadiene |
| PCEMA | Poly(2-(cinnamoyloxy)ethyl methacrylate) |
| PD | Poly(3-butenyl (dodecyl)sulphane) |
| PEO | Polyethylene oxide |
| PFS | Poly(ferrocenyldimethyl silane) |
| PI | Polyisoprene |
| PISA | Polymerization-induced self-assembly |
| PMA | Poly(methyl acrylate) |
| PMAA | Poly(methacrylic acid) |
| PMMA | Poly(methyl methacrylate) |
| PS | Polystyrene |
| PSGMA | Poly(sucinnated glyceryl monomethacrylate) |
| PtBA | Poly(tert-butyl acrylate) |
| PtBMA | Poly(tert-butyl methacrylate) |
| PtBS | Poly(4-tert-butoxy-styrene) |
| P2VP/P4VP | Poly(2-vinylpyridine)/poly(4-vinylpyridine) |
| P2VPq | Poly(1-methyl-2-vinylpyridinium) |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| T g | Glass transition temperature |
| THF | Tetrahydrofuran |
| V X | Molar volume of block X |
| Z | Aggregation number |
| χ XY | Flory–Huggins-interaction parameter |
| ϕ X | Block volume fraction of block X |
References
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- R. D. Kamien, Science, 2003, 299, 1671–1673 CrossRef CAS PubMed.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS PubMed.
- J. A. Pelesko, Self-Assembly: The science of things that put themselves together, Chapman & Hall/CRC, 2007 Search PubMed.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef CAS PubMed.
- Z. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203–3224 CrossRef CAS PubMed.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- D. H. Gracias, J. Tien, T. L. Breen, C. Hsu and G. M. Whitesides, Science, 2000, 289, 1170–1172 CrossRef CAS.
- F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 8–11 Search PubMed.
- Y. Kang, J. J. Walish, T. Gorishnyy and E. L. Thomas, Nat. Mater., 2007, 6, 957–960 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- S. Sengupta, M. E. Ibele and A. Sen, Angew. Chem., Int. Ed., 2012, 51, 8434–8345 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers: Self-Assembly and Applications, Elsevier, Amsterdam, 2000 Search PubMed.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- S. Förster, N. Hermsdorf, C. Böttcher and P. Lindner, Macromolecules, 2002, 35, 4096–4105 CrossRef.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2009, 5, 1269–1278 RSC.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429–4440 CrossRef CAS.
- S. Förster, B. Berton, H.-P. Hentze, E. Krämer and M. Antonietti, Macromolecules, 2001, 34, 4610–4623 CrossRef.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- S. Förster, M. Zisenis, E. Wenz and M. Antonietti, J. Chem. Phys., 1996, 104, 9956 CrossRef PubMed.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 9297, 1144–1154 CrossRef.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- P. Lim Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef PubMed.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
- J.-F. Gohy, Adv. Polym. Sci., 2005, 190, 65–136 CrossRef CAS.
- E. B. Zhulina, M. Adam, I. Larue, S. S. Sheiko and M. Rubinstein, Macromolecules, 2005, 38, 5330–5351 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- J. Rzayev, ACS Macro Lett., 2012, 1, 1146–1149 CrossRef CAS.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS PubMed.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- M. Müllner, T. Lunkenbein, M. Schieder, A. H. Gröschel, N. Miyajima, M. Fo, J. Breu, F. Caruso and A. H. E. Müller, Macromolecules, 2012, 45, 6981–6988 CrossRef.
- J. Bolton, T. S. Bailey and J. Rzayev, Nano Lett., 2011, 11, 998–1001 CrossRef CAS PubMed.
- G. M. Miyake, V. A. Piunova, R. A. Weitekamp and R. H. Grubbs, Angew. Chem., Int. Ed., 2012, 51, 11246–11248 CrossRef CAS PubMed.
- M. Danial, C. M.-N. Tran, P. G. Young, S. Perrier and K. A. Jolliffe, Nat. Commun., 2013, 4, 2780 Search PubMed.
- D. Yan, Y. Zhou and J. Hou, Science, 2004, 303, 65–67 CrossRef CAS PubMed.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512–1515 CrossRef CAS PubMed.
- J. Burdyńska, Y. Li, A. V. Aggarwal, S. Höger, S. S. Sheiko and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 12762–12770 CrossRef PubMed.
- R. J. Williams, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2015, 6, 2998–3008 RSC.
- J. J. Crassous, P. Schurtenberger, M. Ballauff and A. M. Mihut, Polymer, 2015, 62, A1–A13 CrossRef CAS PubMed.
- D. Wang and X. Wang, Prog. Polym. Sci., 2013, 38, 271–301 CrossRef CAS PubMed.
- D. V. Pergushov, A. H. E. Müller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- A. Priimagi and A. Shevchenko, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 163–182 CrossRef CAS PubMed.
- R. C. Hayward and D. J. Pochan, Macromolecules, 2010, 43, 3577–3584 CrossRef CAS.
- R. B. Grubbs and Z. Sun, Chem. Soc. Rev., 2013, 42, 7436–7445 RSC.
- S. Egli, H. Schlaad, N. Bruns and W. Meier, Polymers, 2011, 3, 252–280 CrossRef CAS PubMed.
- S. De Koker, R. Hoogenboom and B. G. De Geest, Chem. Soc. Rev., 2012, 41, 2867–2884 RSC.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- S. F. Fenz and K. Sengupta, Integr. Biol., 2012, 4, 982–995 RSC.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- T. Nomoto, S. Fukushima, M. Kumagai, K. Machitani, Y. Matsumoto, M. Oba, K. Miyata, K. Osada, N. Nishiyama and K. Kataoka, Nat. Commun., 2014, 5, 3545 Search PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS PubMed.
- G. Fuks, R. Mayap Talom and F. Gauffre, Chem. Soc. Rev., 2011, 40, 2475–2493 RSC.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS PubMed.
- K. Yu and A. Eisenberg, Macromolecules, 1998, 31, 3509–3518 CrossRef CAS.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf. B, 1999, 16, 3–27 CrossRef CAS.
- S. Burke, H. Shen and A. Eisenberg, Macromol. Symp., 2001, 175, 273–283 CrossRef CAS.
- L. Wang and J. Lin, Soft Matter, 2011, 7, 3383–3391 RSC.
- Y. Zhu, H. Yu, Y. Wang, J. Cui, W. Kong and W. Jiang, Soft Matter, 2012, 8, 4695–4707 RSC.
- R. M. Holmes and D. R. M. Williams, Macromolecules, 2011, 44, 6172–6181 CrossRef CAS.
- T. Jiang, L. Wang, S. Lin, J. Lin and Y. Li, Langmuir, 2011, 27, 6440–6448 CrossRef CAS PubMed.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS.
- J.-F. Gohy, E. Khousakoun, N. Willet, S. K. Varshney and R. Jérôme, Macromol. Rapid Commun., 2004, 25, 1536–1539 CrossRef CAS PubMed.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247–251 Search PubMed.
- C. V. Synatschke, T. Nomoto, H. Cabral, M. Förtsch, K. Toh, Y. Matsumoto, K. Miyazaki, A. Hanisch, F. H. Schacher, A. Kishimura, N. Nishiyama, A. H. E. Müller and K. Kataoka, ACS Nano, 2014, 8, 1161–1172 CrossRef CAS PubMed.
- J. Schmelz, M. Karg, T. Hellweg and H. Schmalz, ACS Nano, 2011, 5, 9523–9534 CrossRef CAS PubMed.
- A. Wolf, A. Walther and A. H. E. Müller, Macromolecules, 2011, 44, 9221–9229 CrossRef CAS.
- J.-F. Gohy, N. Willet, S. Varshney, J. Zhang and R. Jérome, Angew. Chem., Int. Ed., 2001, 113, 3214–3216 CrossRef.
- J. Zhu, S. Zhang, K. Zhang, X. Wang, J. W. Mays, K. L. Wooley and D. J. Pochan, Nat. Commun., 2013, 4, 2297 Search PubMed.
- J. Du and S. P. Armes, Soft Matter, 2010, 6, 4851–4857 RSC.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- S. Zhang, H.-J. Sun, A. D. Hughes, R.-O. Moussodia, A. Bertin, Y. Chen, D. J. Pochan, P. A. Heiney, M. L. Klein and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9058–9063 CrossRef CAS PubMed.
- D. Klinger, C. X. Wang, L. A. Connal, D. J. Audus, S. G. Jang, S. Kraemer, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2014, 57, 7018–7022 CrossRef PubMed.
- D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Nat. Mater., 2009, 8, 843–849 CrossRef CAS PubMed.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS PubMed.
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- Y. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS PubMed.
- H. Yabu, Polym. J., 2013, 45, 261–268 CrossRef CAS PubMed.
- Z. Lu, G. Liu and F. Liu, Macromolecules, 2001, 34, 8814–8817 CrossRef CAS.
- I. Wyman, G. Njikang and G. Liu, Prog. Polym. Sci., 2011, 36, 1152–1183 CrossRef CAS PubMed.
- S. Qin, H. Li, W. Z. Yuan and Y. Zhang, Soft Matter, 2012, 8, 2471–2476 RSC.
- R. Deng, F. Liang, W. Li, S. Liu, R. Liang, M. Cai, Z. Yang and J. Zhu, Small, 2013, 9, 4099–4103 CrossRef CAS PubMed.
- R. Deng, F. Liang, W. Li, Z. Yang and J. Zhu, Macromolecules, 2013, 46, 7012–7017 CrossRef CAS.
- T. Higuchi, A. Tajima, K. Motoyoshi, H. Yabu and M. Shimomura, Angew. Chem., Int. Ed., 2009, 48, 5125–5128 CrossRef CAS PubMed.
- H. Yabu, T. Higuchi and H. Jinnai, Soft Matter, 2014, 10, 2919–2931 RSC.
- T. Higuchi, A. Tajima, H. Yabu and M. Shimomura, Soft Matter, 2008, 4, 1302–1305 RSC.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S.-W. Kim, K. T. Delaney, S.-M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS PubMed.
- T. Higuchi, K. Motoyoshi, H. Sugimori, H. Jinnai, H. Yabu and M. Shimomura, Soft Matter, 2012, 8, 3791–3797 RSC.
- J. Xu, K. Wang, J. Li, H. Zhou, X. Xie and J. Zhu, Macromolecules, 2015, 48, 2628–2636 CrossRef CAS.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed.
- H. Yabu, M. Kanahara, M. Shimomura, T. Arita, K. Harano, E. Nakamura, T. Higuchi and H. Jinnai, ACS Appl. Mater. Interfaces, 2013, 5, 3262–3266 CAS.
- S.-J. Jeon, G.-R. Yi and S.-M. Yang, Adv. Mater., 2008, 20, 4103–4108 CrossRef CAS PubMed.
- R. Deng, S. Liu, J. Li, Y. Liao, J. Tao and J. Zhu, Adv. Mater., 2012, 24, 1889–1893 CrossRef CAS PubMed.
- R. Deng, F. Liang, P. Zhou, C. Zhang, X. Qu, Q. Wang, J. Li, J. Zhu and Z. Yang, Adv. Mater., 2014, 26, 4469–4472 CrossRef CAS PubMed.
- A. Rahikkala, A. J. Soininen, J. Ruokolainen, R. Mezzenga, J. Raula and E. I. Kauppinen, Soft Matter, 2013, 9, 1492–1499 RSC.
- K. Zhang, L. Gao, Y. Chen and Z. Yang, J. Colloid Interface Sci., 2010, 346, 48–53 CrossRef CAS PubMed.
- A. Nykänen, A. Rahikkala, S.-P. Hirvonen, V. Aseyev, H. Tenhu, R. Mezzenga, J. Raula, E. Kauppinen and J. Ruokolainen, Macromolecules, 2012, 45, 8401–8411 CrossRef PubMed.
- A. J. Soininen, A. Rahikkala, J. T. Korhonen, E. I. Kauppinen, R. Mezzenga, J. Raula and J. Ruokolainen, Macromolecules, 2012, 45, 8743–8751 CrossRef CAS.
- J. Zhang, X.-F. Chen, H.-B. Wei and X.-H. Wan, Chem. Soc. Rev., 2013, 42, 9127–9154 RSC.
- C. Tschierske, Angew. Chem., Int. Ed., 2013, 52, 8828–8878 CrossRef CAS PubMed.
- J.-K. Kim, E. Lee, Y. Lim and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 4662–4666 CrossRef CAS PubMed.
- H. Yang, L. Jia, C. Zhu, A. Di-cicco, D. Levy, P. Albouy, M. Li and P. Keller, Macromolecules, 2010, 43, 10442–10451 CrossRef CAS.
- L. Jia, P.-A. Albouy, A. Di Cicco, A. Cao and M.-H. Li, Polymer, 2011, 52, 2565–2575 CrossRef CAS PubMed.
- L. Jia, A. Cao, D. Lévy, B. Xu, P.-A. Albouy, X. Xing, M. J. Bowick and M.-H. Li, Soft Matter, 2009, 5, 3446–3451 RSC.
- X. Xing, H. Shin, M. J. Bowick, Z. Yao, L. Jia and M.-H. Li, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5202–5206 CrossRef CAS PubMed.
- Y. Shi, W. Zhu, D. Yao, M. Long, B. Peng, K. Zhang and Y. Chen, ACS Macro Lett., 2014, 3, 70–73 CrossRef CAS.
- L. Li, M. Rosenthal, H. Zhang, J. J. Hernandez, M. Drechsler, K. H. Phan, S. Rütten, X. Zhu, D. a. Ivanov and M. Möller, Angew. Chem., Int. Ed., 2012, 51, 11616–11619 CrossRef CAS PubMed.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911–8917 CrossRef CAS.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS PubMed.
- S. Venkataraman, A. L. Lee, H. T. Maune, J. L. Hedrick, V. M. Prabhu and Y. Y. Yang, Macromolecules, 2013, 46, 4839–4846 CrossRef CAS.
- P. A. Beales, N. Geerts, K. K. Inampudi, H. Shigematsu, C. J. Wilson and T. K. Vanderlick, J. Am. Chem. Soc., 2013, 135, 3335–3338 CrossRef CAS PubMed.
- J.-K. Kim, E. Lee, Y.-H. Jeong, J.-K. Lee, W.-C. Zin and M. Lee, J. Am. Chem. Soc., 2007, 129, 6082–6083 CrossRef CAS PubMed.
- B. E. McKenzie, S. J. Holder and N. a. J. M. Sommerdijk, Curr. Opin. Colloid Interface Sci., 2012, 17, 343–349 CrossRef PubMed.
- D. Danino, Curr. Opin. Colloid Interface Sci., 2012, 17, 316–329 CrossRef CAS PubMed.
- R. Fenyves, M. Schmutz, I. J. Horner, F. V. Bright and J. Rzayev, J. Am. Chem. Soc., 2014, 136, 7762–7770 CrossRef CAS PubMed.
- B. E. McKenzie, F. Nudelman, P. H. H. Bomans, S. J. Holder and N. A. J. M. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed.
- F. Nudelman, G. de With and N. A. J. M. Sommerdijk, Soft Matter, 2011, 7, 17–24 RSC.
- A. L. Parry, P. H. H. Bomans, S. J. Holder, N. A. J. M. Sommerdijk and S. C. G. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8859–8862 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef CAS PubMed.
- Y. Geng, D. E. Discher, J. Justynska and H. Schlaad, Angew. Chem., Int. Ed., 2006, 45, 7578–7581 CrossRef CAS PubMed.
- J. J. L. M. Cornelissen, M. Fischer, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 1998, 280, 1427–1430 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS PubMed.
- M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Soft Matter, 2011, 7, 4143–4146 RSC.
- N. Houbenov, A. Nykänen, H. Iatrou, N. Hadjichristidis, J. Ruokolainen, C. F. J. Faul and O. Ikkala, Adv. Funct. Mater., 2008, 18, 2041–2047 CrossRef CAS PubMed.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- S. Lecommandoux, H.-A. Klok and H. Schlaad, in Block Copolymers in Nanoscience, ed. M. Lazzari, G. Liu and S. Lecommandoux, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, pp. 117–150 Search PubMed.
- X. Wang, M. A. Winnik and I. Manners, in Block Copolymers in Nanoscience, eds. P. D. M. Lazzari, P. D. G. Liu and P. D. S. Lecommandoux, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 151–168 Search PubMed.
- J. Schmelz, F. H. Schacher and H. Schmalz, Soft Matter, 2013, 9, 2101–2107 RSC.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef PubMed.
- J. G. Son, J. Gwyther, J.-B. Chang, K. K. Berggren, I. Manners and C. A. Ross, Nano Lett., 2011, 11, 2849–2855 CrossRef CAS PubMed.
- R. Ahmed, S. K. Patra, I. W. Hamley, I. Manners and C. F. J. Faul, J. Am. Chem. Soc., 2013, 135, 2455–2458 CrossRef CAS PubMed.
- A. Nunns, C. A. Ross and I. Manners, Macromolecules, 2013, 46, 2628–2635 CrossRef CAS.
- A. C. Arsenault, D. P. Puzzo, I. Manners and G. A. Ozin, Nat. Photonics, 2007, 1, 468–472 CrossRef CAS PubMed.
- A. P. Soto, J. B. Gilroy, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2010, 49, 8220–8223 CrossRef PubMed.
- J.-C. Eloi, D. A. Rider, G. Cambridge, G. R. Whittell, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8903–8913 CrossRef CAS PubMed.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef CAS PubMed.
- H. Qiu, G. Cambridge, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2013, 135, 12180–12183 CrossRef CAS PubMed.
- L. Jia, G. Zhao, W. Shi, N. Coombs, I. Gourevich, G. C. Walker, G. Guerin, I. Manners and M. A. Winnik, Nat. Commun., 2014, 5, 3882 CAS.
- S.-L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950–5968 RSC.
- M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860–6909 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. A. Cohen Stuart, Colloid Polym. Sci., 2008, 286, 855–864 CAS.
- G. ten Brinke, K. Loos, I. Vukovic and G. G. du Sart, J. Phys.: Condens. Matter, 2011, 23, 284110 CrossRef PubMed.
- V. V. Palyulin and I. I. Potemkin, Macromolecules, 2008, 41, 4459–4463 CrossRef CAS.
- G. Li, L. Shi, R. Ma, Y. An and N. Huang, Angew. Chem., Int. Ed., 2006, 45, 4959–4962 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Science, 1999, 283, 65–67 CrossRef CAS.
- M. Harada-Shiba, K. Yamauchi, A. Harada, I. Takamisawa, K. Shimokado and K. Kataoka, Gene Ther., 2002, 9, 407–414 CrossRef CAS PubMed.
- S. Schrage, R. Sigel and H. Schlaad, Macromolecules, 2003, 36, 1417–1420 CrossRef CAS.
- Y. Lu, A. Wittemann and M. Ballauff, Macromol. Rapid Commun., 2009, 30, 806–815 CrossRef CAS PubMed.
- M. Ballauff, Prog. Polym. Sci., 2007, 32, 1135–1151 CrossRef CAS PubMed.
- F. A. Plamper, A. Walther, A. H. E. Müller and M. Ballauff, Nano Lett., 2007, 7, 167–171 CrossRef CAS PubMed.
- A. Jusufi, O. Borisov and M. Ballauff, Polymer, 2013, 54, 2028–2035 CrossRef CAS PubMed.
- I. K. Voets, A. de Keizer, P. de Waard, P. M. Frederik, P. H. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. M. Leermakers and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2006, 45, 6673–6676 CrossRef CAS PubMed.
- I. K. Voets, A. de Keizer, M. A. Cohen Stuart, J. Justynska and H. Schlaad, Macromolecules, 2007, 40, 2158–2164 CrossRef CAS.
- I. K. Voets, F. A. Leermakers, A. De Keizer, M. Charlaganov and M. A. C. Stuart, Adv. Polym. Sci., 2011, 241, 163–185 CrossRef CAS.
- J.-F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E, 2011, 34, 14 CrossRef PubMed.
- G. R. Whittell and I. Manners, Adv. Mater., 2007, 21, 3439–3468 CrossRef PubMed.
- C.-A. Fustin, P. Guillet, U. S. Schubert and J.-F. Gohy, Adv. Mater., 2007, 19, 1665–1673 CrossRef CAS PubMed.
- Y. Yan and J. Huang, Coord. Chem. Rev., 2010, 254, 1072–1080 CrossRef CAS PubMed.
- S. Yang, X. Yu, L. Wang, Y. Tu, J. X. Zheng, J. Xu, R. M. Van Horn and S. Z. D. Cheng, Macromolecules, 2010, 43, 3018–3026 CrossRef CAS.
- Y. Han and W. Jiang, J. Phys. Chem. B, 2011, 115, 2167–2172 CrossRef CAS PubMed.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- X. Huan, D. Wang, R. Dong, C. Tu, B. Zhu, D. Yan and X. Zhu, Macromolecules, 2012, 45, 5941–5947 CrossRef CAS.
- T. M. Hermans, M. A. C. Broeren, N. Gomopoulos, P. van der Schoot, M. H. P. van Genderen, N. A. J. M. Sommerdijk, G. Fytas and E. W. Meijer, Nat. Nanotechnol., 2009, 4, 721–726 CrossRef CAS PubMed.
- J. Ruokolainen, G. Ten Brinke and O. Ikkala, Adv. Mater., 1999, 11, 777–780 CrossRef CAS.
- S. Valkama, H. Kosonen, J. Ruokolainen, T. Haatainen, M. Torkkeli, R. Serimaa, G. Ten Brinke and O. Ikkala, Nat. Mater., 2004, 3, 872–876 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. H. Hirschberg, R. F. Lange, J. K. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- S. Chang and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 1318–1319 CrossRef CAS.
- J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. a. Scherman and C. Abell, Science, 2012, 335, 690–694 CrossRef CAS PubMed.
- U. Rauwald and O. A. Scherman, Angew. Chem., Int. Ed., 2008, 47, 3950–3953 CrossRef CAS PubMed.
- Q. Yan, Y. Xin, R. Zhou, Y. Yin and J. Yuan, Chem. Commun., 2011, 47, 9594–9596 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. Avgeropoulos, Prog. Polym. Sci., 2005, 30, 725–782 CrossRef CAS PubMed.
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 RSC.
- J.-F. Lutz and A. Laschewsky, Macromol. Chem. Phys., 2005, 206, 813–817 CrossRef CAS PubMed.
- J.-F. Gohy, C. Ott, S. Hoeppener and U. S. Schubert, Chem. Commun., 2009, 6038–6040 RSC.
- Z. Wang and W. Jiang, Chem. Phys. Lett., 2010, 487, 84–87 CrossRef CAS PubMed.
- J. Zhu and W. Jiang, Macromolecules, 2005, 38, 9315–9323 CrossRef CAS.
- I. W. Wyman and G. Liu, Polymer, 2013, 54, 1950–1978 CrossRef CAS PubMed.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed.
- T. P. Lodge, M. a. Hillmyer, Z. Zhou and Y. Talmon, Macromolecules, 2004, 37, 6680–6682 CrossRef CAS.
- J.-F. Gohy, S. K. Varshney and R. Jérôme, Macromolecules, 2001, 34, 3361–3366 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409–9417 CrossRef CAS PubMed.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2008, 24, 12001–12009 CrossRef CAS PubMed.
- N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, ACS Nano, 2010, 4, 1907–1912 CrossRef CAS PubMed.
- N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2008, 41, 8815–8822 CrossRef CAS.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2009, 25, 13718–13725 CrossRef CAS PubMed.
- J. Xia and C. Zhong, Macromol. Rapid Commun., 2006, 27, 1110–1114 CrossRef CAS PubMed.
- S.-H. Chou, H.-K. Tsao and Y.-J. Sheng, J. Chem. Phys., 2006, 125, 194903 CrossRef PubMed.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245–1249 CrossRef CAS PubMed.
- A. Walther and A. H. E. Müller, Chem. Commun., 2009, 1127–1129 RSC.
- H. L. Hsieh and R. P. Quirk, Anionic Polymerization: Principles and Practical Applications, Marcel Dekker Inc., New York, NY, 1996 Search PubMed.
- K. Matyjaszewski and N. V Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- A. H. E. Müller and K. Matyjaszewski, Controlled and Living Polymerizations: From Mechanisms to Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2009 Search PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- M. Sasidharan and K. Nakashima, Acc. Chem. Res., 2014, 47, 157–167 CrossRef CAS PubMed.
- S. Kubowicz, J.-F. Baussard, J.-F. Lutz, A. F. Thünemann, H. von Berlepsch and A. Laschewsky, Angew. Chem., Int. Ed., 2005, 44, 5262–5265 CrossRef CAS PubMed.
- U. Breiner, U. Krappe, T. Jakob, V. Abetz and R. Stadler, Polym. Bull., 1998, 40, 219–226 CrossRef CAS.
- S. Ritzenthaler, F. Court, L. David, E. Girard-Reydet, L. Leibler and J. P. Pascault, Macromolecules, 2002, 35, 6245–6254 CrossRef CAS.
- K. Skrabania, H. V. Berlepsch, C. Böttcher and A. Laschewsky, Macromolecules, 2010, 43, 271–281 CrossRef CAS.
- J.-N. Marsat, M. Heydenreich, E. Kleinpeter, H. v. Berlepsch, B. Christoph and A. Laschewsky, Macromolecules, 2011, 44, 2092–2105 CrossRef CAS.
- H. V. Berlepsch, C. Böttcher, K. Skrabania and A. Laschewsky, Chem. Commun., 2009, 2290–2292 RSC.
- Z. Ma, H. Yu and W. Jiang, J. Phys. Chem. B, 2009, 113, 3333–3338 CrossRef CAS PubMed.
- F. Schacher, A. Walther, M. Ruppel, M. Drechsler and A. H. E. Müller, Macromolecules, 2009, 42, 3540–3548 CrossRef CAS.
- F. Schacher, A. Walther and A. H. E. Müller, Langmuir, 2009, 25, 10962–10969 CrossRef CAS PubMed.
- F. Schacher, E. Betthausen, A. Walther, H. Schmalz, D. V. Pergushov and A. H. E. Müller, ACS Nano, 2009, 3, 2095–2102 CrossRef CAS PubMed.
- C. V. Synatschke, T. I. Löbling, M. Förtsch, A. Hanisch, F. H. Schacher and A. H. E. Müller, Macromolecules, 2013, 46, 6466–6474 CrossRef CAS.
- T. I. Löbling, J. S. Haataja, C. V. Synatschke, F. H. Schacher, M. Müller, A. Hanisch, A. H. Gröschel and A. H. E. Müller, ACS Nano, 2014, 8, 11330–11340 CrossRef PubMed.
- C. V. Synatschke, F. H. Schacher, M. Förtsch, M. Drechsler and A. H. E. Müller, Soft Matter, 2011, 7, 1714–1725 RSC.
- A. C. Rinkenauer, A. Schallon, U. Günther, M. Wagner, E. Betthausen, U. S. Schubert and F. H. Schacher, ACS Nano, 2013, 7, 9621–9631 CrossRef CAS PubMed.
- Y. Bae, H. Cabral and K. Kataoka, in Block Copolymers in Nanoscience, ed. S. L. M. Lazzari and G. Liu, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, pp. 73–89 Search PubMed.
- M. Müllner, S. J. Dodds, T. Nguyen, D. Senyschyn and C. J. H. Porter, ACS Nano, 2015, 9, 1294 CrossRef PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- V. Abetz and P. F. W. Simon, Adv. Polym. Sci., 2005, 189, 125–212 CrossRef CAS.
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS PubMed.
- C. Auschrat and R. Stadler, Macromolecules, 1993, 26, 2171–2174 CrossRef.
- R. Stadler, C. Auschra, J. Beckmann, U. Krappe, I. Voigt-Martin and L. Leibler, Macromolecules, 1995, 28, 3080–3091 CrossRef CAS.
- U. Breiner, U. Krappe, V. Abetz and R. Stadler, Macromol. Chem. Phys., 1997, 198, 1051–1083 CrossRef CAS PubMed.
- S. Brinkmann, R. Stadler and E. L. Thomas, Macromolecules, 1998, 31, 6566–6572 CrossRef CAS.
- T. Goldacker, V. Abetz, R. Stadler, I. Erukhimovich and L. Leibler, Nature, 1999, 398, 137–139 CrossRef CAS.
- V. Castelletto and I. W. Hamley, Curr. Opin. Solid State Mater. Sci., 2004, 8, 426–438 CrossRef CAS PubMed.
- S. Ludwigs, A. Böker, V. Abetz, A. H. E. Müller and G. Krausch, Polymer, 2003, 44, 6815–6823 CrossRef CAS PubMed.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- T. I. Löbling, P. Hiekkataipale, A. Hanisch, F. Bennet, H. Schmalz, O. Ikkala, A. H. Gröschel and A. H. E. Müller, Polymer, 2015 DOI:10.1016/j.polymer.2015.02.025.
- G. Liu, L. Qiao and A. Guo, Macromolecules, 1996, 29, 5508–5510 CrossRef CAS.
- X. Yan, G. Liu, F. Liu and B. Tang, Angew. Chem., Int. Ed., 2001, 3593–3596 CrossRef CAS.
- F. Schacher, J. Yuan, H. G. Schoberth and A. H. E. Müller, Polymer, 2010, 51, 2021–2032 CrossRef CAS PubMed.
- F. H. Schacher, T. Rudolph, M. Drechsler and A. H. E. Müller, Nanoscale, 2011, 3, 288–297 RSC.
- H. Hückstadt, A. Gopfert and V. Abetz, Macromol. Chem. Phys., 2000, 201, 296–307 CrossRef.
- A. Walther, J. Yuan, V. Abetz and A. H. E. Müller, Nano Lett., 2009, 9, 2026–2030 CrossRef CAS PubMed.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-hintzen, G. Krausch, V. Abetz and A. H. E. Müller, Macromolecules, 2001, 34, 1069–1075 CrossRef CAS.
- Y. Liu, V. Abetz and A. H. E. Müller, Macromolecules, 2003, 36, 7894–7898 CrossRef CAS.
- A. Walther, X. André, M. Drechsler, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2007, 129, 6187–6198 CrossRef CAS PubMed.
- B. P. Binks and P. D. I. Fletcher, Langmuir, 2001, 5, 4708–4710 CrossRef.
- N. Glaser, D. J. Adams, A. Böker and G. Krausch, Langmuir, 2006, 22, 5227–5229 CrossRef CAS PubMed.
- T. M. Ruhland, A. H. Gröschel, A. Walther and A. H. E. Müller, Langmuir, 2011, 27, 9807–9814 CrossRef CAS PubMed.
- T. M. Ruhland, A. H. Gröschel, N. Ballard, T. S. Skelhon, A. Walther, A. H. E. Müller and S. A. F. Bon, Langmuir, 2013, 29, 1388–1394 CrossRef CAS PubMed.
- J. Schmelz, D. Pirner, M. Krekhova, T. M. Ruhland and H. Schmalz, Soft Matter, 2013, 9, 11173–11177 RSC.
- R. Erhardt, M. Zhang, A. Böker, H. Zettl, C. Abetz, P. Frederik, G. Krausch, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2003, 125, 3260–3267 CrossRef CAS PubMed.
- A. Walther, M. Drechsler, S. Rosenfeldt, L. Harnau, M. Ballauff, V. Abetz and A. H. E. Müller, J. Am. Chem. Soc., 2009, 131, 4720–4728 CrossRef CAS PubMed.
- A. Walther, M. Drechsler and A. H. E. Müller, Soft Matter, 2009, 5, 385–390 RSC.
- A. K. Brannan and F. S. Bates, Macromolecules, 2004, 37, 8816–8819 CrossRef CAS.
- W. Zhao, D. Chen, Y. Hu, G. M. Grason and T. P. Russell, ACS Nano, 2011, 5, 486–492 CrossRef CAS PubMed.
- J. Dupont, G. Liu, K. Niihara, R. Kimoto and H. Jinnai, Angew. Chem., Int. Ed., 2009, 48, 6144–6147 CrossRef CAS PubMed.
- S. Zhong, H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2008, 4, 90–93 RSC.
- J. Hu, G. Njikang and G. Liu, Macromolecules, 2008, 41, 7993–7999 CrossRef CAS.
- A. Walther, C. Barner-Kowollik and A. H. E. Müller, Langmuir, 2010, 26, 12237–12246 CrossRef CAS PubMed.
- R. Ma, B. Wang, Y. Xu, Y. An, W. Zhang, G. Li and L. Shi, Macromol. Rapid Commun., 2007, 28, 1062–1069 CrossRef CAS PubMed.
- L. Wang, J. Lin and X. Zhang, Polymer, 2013, 54, 3427–3442 CrossRef CAS PubMed.
- C. J. Newcomb, T. J. Moyer, S. S. Lee and S. I. Stupp, Curr. Opin. Colloid Interface Sci., 2012, 17, 350–359 CrossRef CAS PubMed.
- J. Weiss and A. Laschewsky, Langmuir, 2011, 27, 4465–4473 CrossRef CAS PubMed.
- C. Zhou, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2011, 44, 1635–1641 CrossRef CAS.
- M. J. Barthel, F. H. Schacher and U. S. Schubert, Polym. Chem., 2014, 5, 2647–2662 RSC.
- V. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2011, 242, 29–89 CrossRef CAS.
- X. Yan, G. Liu, J. Hu and C. G. Willson, Macromolecules, 2006, 39, 1906–1912 CrossRef CAS.
- R. Zheng, G. Liu and X. Yan, J. Am. Chem. Soc., 2005, 127, 15358–15359 CrossRef CAS PubMed.
- J. Hu and G. Liu, Macromolecules, 2005, 38, 8058–8065 CrossRef CAS.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS PubMed.
- K. Hales, Z. Chen, K. L. Wooley and D. J. Pochan, Nano Lett., 2008, 8, 2023–2026 CrossRef CAS PubMed.
- J. L. Sorrells, Y. Tsai and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4465–4472 CrossRef CAS PubMed.
- H. Cui, Z. Chen, K. L. Wooley, D. J. Pochan, R. V. April, V. Re, M. Recei and V. July, Macromolecules, 2006, 39, 6599–6607 CrossRef CAS.
- D. J. Pochan, J. Zhu, K. Zhang, K. L. Wooley, C. Miesch and T. Emrick, Soft Matter, 2011, 7, 2500–2506 RSC.
- J. Zhu, S. Zhang, F. Zhang, K. L. Wooley and D. J. Pochan, Adv. Funct. Mater., 2013, 23, 1767–1773 CrossRef CAS PubMed.
- J. Zhu, S. Zhang, K. Zhang, X. Wang, J. W. Mays, K. L. Wooley and D. J. Pochan, Nat. Commun., 2013, 4, 2297 Search PubMed.
- J.-F. Gohy, B. G. G. Lohmeijer, S. K. Varshney, B. Décamps, E. Leroy, S. Boileau and U. S. Schubert, Macromolecules, 2002, 35, 9748–9755 CrossRef CAS.
- A. Winter, M. D. Hager, G. R. Newkome and U. S. Schubert, Adv. Mater., 2011, 23, 5728–5748 CrossRef CAS PubMed.
- A. O. Moughton, K. Stubenrauch and R. K. O'Reilly, Soft Matter, 2009, 5, 2361–2370 RSC.
- M. Uchman, M. Štěpánek, K. Procházka, G. Mountrichas, S. Pispas, I. K. Voets and A. Walther, Macromolecules, 2009, 42, 5605–5613 CrossRef CAS.
- O. V. Borisov, E. B. Zhulina, F. A. M. Leermakers, M. Ballauff and A. H. E. Müller, Adv. Polym. Sci., 2011, 241, 1–55 CrossRef CAS.
- A. Hanisch, H. Schmalz and A. H. E. Müller, Macromolecules, 2012, 45, 8300–8309 CrossRef CAS.
- A. Hanisch, A. H. Gröschel, M. Förtsch, T. I. Löbling, F. H. Schacher and A. H. E. Müller, Polymer, 2013, 54, 4528–4537 CrossRef CAS PubMed.
- A. Hanisch, A. H. Gröschel, M. Förtsch, M. Drechsler, H. Jinnai, T. M. Ruhland, F. H. Schacher and A. H. E. Müller, ACS Nano, 2013, 7, 4030–4041 CrossRef CAS PubMed.
- J. Schmelz, A. E. Schedl, C. Steinlein, I. Manners and H. Schmalz, J. Am. Chem. Soc., 2012, 134, 14217–14225 CrossRef CAS PubMed.
- J. Schmelz and H. Schmalz, Polymer, 2012, 53, 4333–4337 CrossRef CAS PubMed.
- T. Gegenhuber, A. H. Gröschel, T. I. Löbling, M. Drechsler, S. Ehlert, S. Förster and H. Schmalz, Macromolecules, 2015, 48, 1767–1776 CrossRef CAS.
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed.
- H. Qiu, G. Russo, P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 11882–11885 CrossRef CAS PubMed.
- B. Fang, A. Walther, A. Wolf, Y. Xu, J. Yuan and A. H. E. Müller, Angew. Chem., Int. Ed., 2009, 48, 2877–2880 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Acc. Chem. Res., 2012, 45, 1657–1666 CrossRef CAS PubMed.
- S. Fischer, A. Salcher, A. Kornowski, H. Weller and S. Förster, Angew. Chem., Int. Ed., 2011, 50, 7811–7814 CrossRef CAS PubMed.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390–1247400 CrossRef PubMed.
- Y. Liu, Y. Liu, J.-J. Yin and Z. Nie, Macromol. Rapid Commun., 2015, 36, 711–725 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- J. He, Y. Liu, T. Babu, Z. Wei and Z. Nie, J. Am. Chem. Soc., 2012, 134, 11342–11345 CrossRef CAS PubMed.
- Y. Guo, S. Harirchian-Saei, C. M. S. Izumi and M. G. Moffitt, ACS Nano, 2011, 5, 3309–3318 CrossRef CAS PubMed.
- Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS PubMed.
- Y. Liu, Y. Li, J. He, K. J. Duelge, Z. Lu and Z. Nie, J. Am. Chem. Soc., 2014, 136, 2602–2610 CrossRef CAS PubMed.
- J. Hu, T. Wu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2012, 134, 7624–7627 CrossRef CAS PubMed.
- Z. Tang, N. A. Kotov and M. Giersig, Science, 2002, 297, 237–240 CrossRef CAS PubMed.
- L. Xu, H. Kuang, L. Wang and C. Xu, J. Mater. Chem., 2011, 21, 16759–16782 RSC.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS PubMed.
- J.-Y. Wang, Y. Wang, S. S. Sheiko, D. E. Betts and J. M. DeSimone, J. Am. Chem. Soc., 2012, 134, 5801–5806 CrossRef CAS PubMed.
- L. J. Hill, M. M. Bull, Y. Sung, A. G. Simmonds, P. T. Dirlam, N. E. Richey, S. E. DeRosa, I.-B. Shim, D. Guin, P. J. Costanzo, N. Pinna, M.-G. Willinger, W. Vogel, K. Char and J. Pyun, ACS Nano, 2012, 6, 8632–8645 CrossRef CAS PubMed.
- K. Liu, N. Zhao and E. Kumacheva, Chem. Soc. Rev., 2011, 40, 656–671 RSC.
- K. Liu, Z. Nie, N. Zhao, W. Li, M. Rubinstein and E. Kumacheva, Science, 2010, 329, 197–200 CrossRef CAS PubMed.
- D. Fava, Z. Nie, M. A. Winnik and E. Kumacheva, Adv. Mater., 2008, 20, 4318–4322 CrossRef CAS PubMed.
- A. H. Gröschel, A. Walther, T. I. Löbling, J. Schmelz, A. Hanisch, H. Schmalz and A. H. E. Müller, J. Am. Chem. Soc., 2012, 134, 13850–13860 CrossRef PubMed.
- P.-G. de Gennes, Angew. Chem., Int. Ed. Engl., 1992, 31, 842–845 CrossRef PubMed.
- A. Walther, K. Matussek and A. H. E. Müller, ACS Nano, 2008, 2, 1167–1178 CrossRef CAS PubMed.
- R. Bahrami, T. I. Löbling, A. H. Gröschel, H. Schmalz, A. H. E. Müller and V. Altstädt, ACS Nano, 2014, 8, 10048–10056 CrossRef CAS PubMed.
- A. H. Gröschel, T. I. Löbling, P. D. Petrov, M. Müllner, C. Kuttner, F. Wieberger and A. H. E. Müller, Angew. Chem., Int. Ed., 2013, 52, 3602–3606 CrossRef PubMed.
- A. Walther, M. Hoffmann and A. H. E. Müller, Angew. Chem., Int. Ed., 2008, 47, 711–714 CrossRef CAS PubMed.
- S. K. Patra, R. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842–8845 CrossRef CAS PubMed.
- N. McGrath, A. J. Patil, S. M. D. Watson, B. R. Horrocks, C. F. J. Faul, A. Houlton, M. A. Winnik, S. Mann and I. Manners, Chem. – Eur. J., 2013, 19, 13030–13039 CrossRef CAS PubMed.
- D. A. Rider, M. A. Winnik and I. Manners, Chem. Commun., 2007, 4483–4485 RSC.
- H. Wang, A. J. Patil, K. Liu, S. Petrov, S. Mann, M. A. Winnik and I. Manners, Adv. Mater., 2009, 21, 1805–1808 CrossRef CAS PubMed.
- H. Wang, W. Lin, K. P. Fritz, G. D. Scholes, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2007, 129, 12924–12925 CrossRef CAS PubMed.
- P. A. Rupar, G. Cambridge, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 16947–16957 CrossRef CAS PubMed.
- F. He, T. Gädt, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2011, 133, 9095–9103 CrossRef CAS PubMed.
- Z. M. Hudson, D. J. Lunn, M. A. Winnik and I. Manners, Nat. Commun., 2014, 5, 3372 Search PubMed.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- J. Rodriguezhernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS PubMed.
- H. M. De Hoog and N. Tomczak, Soft Matter, 2012, 8, 4552–4561 RSC.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yang, P. L. R. Ee, P. T. Hammond and Y. Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228–1246 CrossRef CAS PubMed.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS PubMed.
- T. P. Lodge, A. Rasdal, Z. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608–17609 CrossRef CAS PubMed.
- H. Hu and G. Liu, Macromolecules, 2014, 47, 5096–5103 CrossRef CAS.
- R. Weberskrich and H. Ringsdorf, in Meeting of HCM network, Patras, Greece, 1995.
- H. Ringsdorf, P. Lehman and R. Weberskirch, in 217th ACS National Meeting, Anaheim, CA, 1999.
- Y. Matsumura and K. Kataoka, Cancer Sci., 2009, 100, 572–579 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |