
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphology-dependent photocatalytic activity of octahedral anatase particles prepared by ultrasonication–hydrothermal reaction of titanates†
Zhishun
Wei
a,
Ewa
Kowalska
*a,
Jonathan
Verrett
b,
Christophe
Colbeau-Justin
b,
Hynd
Remita
bc and
Bunsho
Ohtani
a
aCatalysis Research Center, Hokkaido University, Sapporo 001-0021, Japan. E-mail: kowalska@cat.hokudai.ac.jp; Fax: +81-11-706-9133; Tel: +81-11-706-9130
bLaboratory of Physical Chemistry, UMR 8000, Paris-Sud University, 91405 Orsay, France
cCNRS, Laboratory of Physical Chemistry, UMR 8000, 91405 Orsay, France
First published on 18th May 2015
Abstract
Octahedral anatase particles (OAPs) were prepared by an ultrasonication (US)–hydrothermal (HT) reaction of partially proton-exchanged potassium titanate nanowires (TNWs). The structural/physical properties of OAP-containing samples, including specific surface area, crystallinity, crystallite size, particle aspect ratio, composition and total OAP content, were analyzed. Photocatalytic activities of samples were measured under irradiation (>290 nm) for oxidative decomposition of acetic acid (CO2 system) and dehydrogenation of methanol (H2 system) under aerobic and deaerated conditions, respectively. Total density of electron traps (ETs) was measured by double-beam photoacoustic spectroscopy (DB-PAS). Mobility and lifetime of charge carriers (electrons) were investigated by the time-resolved microwave conductivity (TRMC) method. The effects of synthesis parameters, i.e., HT duration, HT temperature and US duration, on properties and photocatalytic activities of final products were examined in detail. The sample prepared with 1 h US duration and 6 h HT duration at 433 K using 267 mg of TNWs in 80 mL of Milli-Q water exhibited the highest photocatalytic activity. It was found that change in HT duration or HT temperature while keeping the other conditions the same resulted in changes in all properties and photocatalytic activity. On the other hand, duration of US treatment, before HT reaction, influenced the morphology of both the reagent (by TNWs breaking) and final products (change in total OAP content); samples prepared with various US durations exhibited almost the same structural/physical properties evaluated in this study but were different in morphology and photocatalytic activity. This enabled clarification of the correlation between morphology and photocatalytic activity, i.e., the higher the total OAP content was, the higher was the level of photocatalytic activity, especially in the CO2 system. Although the decay after maximum TRMC signal intensity (Imax) was almost constant for all samples used in this study, photocatalytic activities were roughly proportional to Imax, which tended to be proportional to total OAP content. Assuming that Imax corresponds to the product of density of electrons in mobile shallow ETs and their mobility, the results suggest that OAP particles have beneficial shallow ETs in higher density and thereby the OAP content governs the photocatalytic activities. Thus, morphology-dependent photocatalytic activity of OAP-containing particles was reasonably interpreted by density of ETs presumably located on the exposed {101} facets.
1 Introduction
Titanium(IV) oxide (titania) has been widely used for many years as a photocatalyst, presumably due to its advantages including high redox ability, stability and nontoxicity.1–3 Another hidden reason for the popular use of titania as a photocatalyst is that titania shows appreciable (detectable) photocatalytic activity when purchased from any supplier or prepared through any procedure. Although fundamental mechanisms and widespread applications of titania photocatalysis, e.g., for environmental purification and solar energy conversion,4,5 have been studied for at least 40 years, there are still many issues which should be explained and clarified.6,7 The key property of high photocatalytic activity is one of those non-clarified issues, but there seems to be no effective way to clarify this issue since it is almost impossible to obtain a series of photocatalyst samples with a difference in only one structural/physical property and with no change in the other properties. A large number of papers showing correlations between structural/physical properties and photocatalytic activities have been published.8–10 For example, it has been suggested that larger specific surface area, smaller crystalline size and higher crystallinity lead to higher photocatalytic activity.11–13 However, as stated above, a direct correlation between one structural/physical property and photocatalytic activity cannot be proved since all structural properties change simultaneously when conditions of photocatalyst preparation and/or treatment are changed. As a result, the balance between different properties has often been suggested as a reason for the high level of photocatalytic activity.Morphology-dependent or exposed facet-dependent activity of photocatalysts,14 which has been widely studied for reactions occurring on titania single crystal (mainly rutile {110}),15 has recently been investigated also for particulate photocatalysts using facetted polyhedral particles. To study the morphology-dependent photocatalytic activity of titania photocatalysts, octahedral anatase particles (OAPs) seem to be appropriate since they expose eight equivalent most thermodynamically stable {101} facets, the same as natural minerals of anatase titania.16 We have recently developed an ultrasonication (US)–hydrothermal (HT) reaction to prepare OAPs, which exhibited high levels of photocatalytic activity for oxidation of acetic acid in comparison with the photocatalytic activities of many commercial titania samples with similar properties.16,17
An HT process has been widely used for the preparation of various materials with controlled morphology.18–21 Conditions for the HT process, including HT duration, HT temperature, and pre-treatment and post-treatment operations, may influence the physical properties, morphology and photocatalytic activities of the resulting products. Generally speaking, a higher temperature and longer duration of the HT process result in increases in crystallinity22 and crystalline size.23–25 Thus, similar products may be obtained either by prolonging the HT duration26 or increasing the HT temperature.26 Prolonged HT duration, as well as post-treatment operations, can change the morphology of a product due to particle sintering.25
Although many papers showing the influence of morphology on photocatalytic activities have already been published,27,28 there has been no report, except for our recent letter on OAPs,29 showing a direct correlation between morphology and photocatalytic activity. We have found that OAP-containing products obtained with various durations of US prior to HT treatment showed variations in morphology and photocatalytic activity with all of the other properties remaining almost unchanged, and we concluded that their photocatalytic activities depend solely on morphology; the higher the OAP content is, the higher is the photocatalytic activity.
In this paper, we present results of detailed studies on structural/physical properties and photocatalytic activities of OAPs prepared under various conditions of US–HT processes. The aim of this study was to clarify the intrinsic reason for morphology-dependent photocatalytic activity of samples containing OAPs through photocatalytic-activity tests and analyses of structural/physical properties including time-resolved microwave conductivity (TRMC) measurements.
2 Experimental
2.1 Preparation of samples containing OAPs
OAP-containing samples were fabricated by US–HT processes using titanate nanowires (TNWs; Earthclean Tohoku Co. Ltd)30 as a precursor, prepared by HT treatment of Evonik P25 titania (Nippon Aerosil) with a potassium hydroxide solution (17 mol L−1) at 383 K for 20 h. In brief, the following procedure was used. TNWs (267 mg) were dispersed in 40 mL of Milli-Q water and ultrasonicated for 0–4 h at 298 K (US process). The suspension was then placed in a sealed Teflon bottle (100 mL) into which an additional 40 mL of Milli-Q water was added. Then the bottle was heated for 3–48 h at 413–473 K in an oven without agitation (HT process). Products were collected by centrifugation, washed with Milli-Q water, and dried under vacuum (353 K, 12 h).2.2 Characterization
Specific surface areas of samples were estimated by nitrogen adsorption at 77 K using the Brunauer–Emmett–Teller (BET) equation. The morphology was studied by scanning electron microscopy (SEM; JEOL JSM-7400F), scanning transmission electron microscopy (STEM, HITACHI HD-2000) and transmission electron microscopy (TEM, JEOL JEM-2100F). Particles were classified by morphology into three groups based on the SEM analysis, as shown in Fig. 1, i.e., (a) OAP: an octahedral particle without observable defects, (b) semi-OAP: an octahedral particle with a defect (defects), and (c) others: an irregular shaped non-octahedral particle. The content of these particles in each sample was measured by counting at least 200 particles in several SEM images. However, there was no guarantee that the OAPs had no defects in their back side or in a part overlapped with another particle, and defects may have been too small to be detected. As reported in the ESI of our previous paper,29 the ratio of OAPs to semi-OAPs was ca. 20–25%, and the sum of OAPs and semi-OAPs will hereafter be shown as “total OAP content”. | ||
| Fig. 1 Representative SEM images with respective models of (a) OAPs, (b) semi-OAPs and (c) others. | ||
Powder X-ray diffraction (XRD) analysis was performed using a Rigaku SmartLab diffractometer equipped with a sealed tube X-ray generator (a copper target; operated at 40 kV and 30 mA), a D/teX high-speed position-sensitive detector system and an ASC-10 automatic sample changer. All of the XRD analyses were performed under the following conditions: 2θ range, 10–90°; scan speed, 1.00° min−1; and scan step, 0.008°. The obtained XRD patterns were analyzed by Rigaku PDXL, a crystal structure analysis package including Rietveld analysis,31 installed in a computer controlling the diffractometer. Crystallite size was estimated from the corrected width of an anatase 101 diffraction peak using the Scherrer equation. Crystallinity of a sample was estimated using highly crystalline nickel oxide (NiO) as an internal standard. The standard (20.0 wt%) was mixed thoroughly with a sample (80.0 wt%) by braying in an agate mortar before XRD analysis. The crystallinity determination was based on the assumption that Rietveld analysis provides the weight fraction of each crystal among total crystalline components, and the weight fraction of an internal standard is overestimated if a sample mixture contains a non-crystalline component. Aspect ratio is defined as a ratio of average crystallite size (depth) vertical to {001} and {101} planes estimated by using a peak width of 004 and 101 diffraction peaks with the Scherrer equation.
The chemical formulas and atomic compositions of TNWs and HT products were determined by crystallinity, content of water, surface atomic content and bulk atomic content, which were examined by X-ray diffractometry (XRD), thermogravimetry (TG; ULVAC-RIKO TGD-9700), X-ray photoelectron spectroscopy (XPS; JEOL JPC-9010MC) and scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM/EDS, JEOL JSM-6360LA), respectively. The content of water was determined by TG based on the assumption that no volatile components other than water were included in the samples and thereby loss of weight was solely caused by removal of water. The SEM/EDS measurements were performed with an accelerating voltage higher than 15.0 kV and a working distance of 10 mm. Five different areas were analyzed for each sample and average data were used for determination of composition.
Photoabsorption properties of samples were measured by diffuse reflectance spectroscopy on a JASCO V-670 spectrophotometer equipped with a PIN-757 integrating sphere. Barium sulfate was used as the reference.
2.3 Photocatalytic activity test
Photocatalytic activities of samples were evaluated by measuring the rate of liberation of carbon dioxide (CO2) or hydrogen (H2) from a continuously stirred (1000 rpm) suspension of a sample (50 mg) in an aerated aqueous solution of acetic acid (5.0 mL, 5.0 vol%) (CO2 system) or in a deaerated aqueous methanol solution (5.0 mL, 50 vol%) containing chloroplatinic acid (corresponding to 2.0 wt% (as platinum) of a sample) for in situ platinum photodeposition (H2 system), respectively. Photoirradiation (>290 nm) was performed with a 400 W high-pressure mercury lamp (Eiko-sha) at 298 K under magnetic stirring (1000 rpm). Amounts of liberated CO2 and H2 in a gas phase were measured by gas chromatography (TCD-GC). The photocatalytic activities were presented with reference to those of a commercial titania photocatalyst, FP-6 (Showa Denko Ceramic; anatase, specific surface area of ca. 100 m2 g−1 and crystalline size of 15 nm), of high-level photocatalytic activities similar to those of the well-known Evonik (Degussa) P25.322.4 Photoacoustic spectroscopy measurements
Double-beam photoacoustic spectroscopy (PAS) was used for estimation of total density of electron traps (ETs) in titania samples.33 A dry sample powder was placed in a cell equipped with a quartz window. Two light beams were introduced into the cell through the window: a modulated visible-light beam to detect photoabsorption of trapped electrons by PAS and a continuous ultraviolet-light beam to excite titania and generate electron–hole pairs. The holes are consumed (removed) by oxidation of methanol introduced into the cell as a vapor, and electrons are captured by ETs. Upon irradiation with ultraviolet light, photoabsorption by trapped electrons increased to the saturated level, which corresponded to total density of ETs.2.5 Time-resolved microwave conductivity measurements
Charge-carrier dynamics was studied by measuring time-resolved microwave conductivity (TRMC). The incident microwave of 36.8 GHz was generated by a Gunn diode of the Ka band, and UV laser pulses were created by the third harmonic of a 1064 nm Nd:YAG laser (10 Hz) with full width at half maximum of ca. 10 ns. Details of measurement and data processing have been reported elsewhere.34,353 Results and discussion
3.1 Characterization of a precursor
Structural/physical properties and photocatalytic activities of samples prepared in this study as well as precursor TNWs are summarized in Table 1. Fig. 2 shows the XRD pattern of TNWs, which resembles the pattern of potassium titanate, K2Ti8O17 (International Centre for Diffraction Data, Powder Diffraction File 9874), but the peaks were less intense and broadened presumably due to the low crystallinity of TNWs, i.e., presumably including amorphous titanates. No peaks attributed to anatase or rutile titania were observed. The composition of TNWs was determined to be H1.32K0.68Ti8O17·4.0H2O, corresponding to partially (ca. two-thirds) proton-exchanged potassium titanate (PEPT), by calculation based on the assumption that TNWs consisted of H2−xKxTi8O17·yH2O without any titania (TiO2) component using water content (11.1% corresponding to 18.02(y + (2 − x)/2)/{1.01(2 − x) + 39.10x + 8 × 47.87 + 17 × 16.00 + 18.02y}) and K/Ti ratio (0.085 corresponding to x/8) estimated by TG and EDS, respectively. | ||
| Fig. 2 An XRD pattern of TNWs. Arrows show the characteristic peaks of potassium titanate (K2Ti8O17). | ||
| Code | US duration (h) | HT temperature (K) | HT duration (h) | Crystallinity (%) | Size (nm) | SSAb (m2 g−1) | Total OAP content (%) | d 001/d101 | Water (%) | K/Ti | K recoveryc (%) | Compositiond |
d
ET![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e (μmol g−1) e (μmol g−1) |
TRMCf | Activityg | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | x | I max |
I
40/Imaxf |
I
4000/Imaxg |
CO2 (%) | H2 (%) | |||||||||||||
| a Samples HD6, HT433 and UD1 were all prepared with 1 h US duration, 433 K HT temperature and 6 h HT duration. b Specific surface area estimated by the BET equation. c Recovery of potassium calculated using the pH value of supernatant solutions after HT treatment and K/Ti ratio of products (see text). d Composition of products estimated using the formula aTiO2·H2−xKxTi8O17·bH2O. e Total density of electron traps (see text). f Results of time-resolved microwave conductivity measurements. Imax, I40 and I4000 are intensities (arbitrary) at maximum, at 40 nm and 4000 nm after a 355 nm laser pulse, respectively. g Relative photocatalytic activity with reference to that of Showa Denko Ceramics FP-6. CO2 and H2 correspond to systems of oxidative decomposition of acetic acid and methanol dehydrogenation with in situ deposited platinum (2 wt%), respectively. | ||||||||||||||||||||
| TNWs | 0.0 | — | 0.0 | 0.0 | — | 360 | — | 11.1 | 0.085 | 0 | 4.0 | 0.7 | ||||||||
| HD3 | 1.0 | 433 | 3.0 | 28.0 | 13 | 250 | 7 | 1.58 | 9.3 | 0.070 | 0.5 | 4 | 4.4 | 0.8 | 239 | 42 | 0.86 | 0.16 | 84 | 22 |
| HD4.5 | 1.0 | 433 | 4.5 | 60.0 | 16 | 170 | 35 | 1.73 | 6.5 | 0.044 | 29 | 15 | 6.2 | 1.0 | 158 | 122 | 0.82 | 0.22 | 110 | 33 |
| HD6a | 1.0 | 433 | 6.0 | 78.0 | 17 | 124 | 64 | 1.71 | 4.5 | 0.025 | 105 | 37 | 8.6 | 1.1 | 114 | 139 | 0.86 | 0.20 | 145 | 37 |
| HD9 | 1.0 | 433 | 9.0 | 83.0 | 19 | 85 | 62 | 1.66 | 135 | 124 | 0.84 | 0.18 | 102 | 30 | ||||||
| HD12 | 1.0 | 433 | 12.0 | 83.0 | 19 | 81 | 61 | 1.65 | 3.2 | 0.011 | 74 | 49 | 7.5 | 0.6 | 98 | 103 | 0.92 | 0.22 | 87 | 26 |
| HD24 | 1.0 | 433 | 24.0 | 81.0 | 19 | 80 | 62 | 1.64 | 2.4 | 0.011 | 209 | 40 | 4.2 | 0.5 | 65 | 108 | 0.85 | 0.19 | 88 | 28 |
| HD48 | 1.0 | 433 | 48.0 | 82.0 | 20 | 78 | 60 | 1.63 | 85 | 32 | ||||||||||
| HT413 | 1.0 | 413 | 6.0 | 38.0 | 14 | 255 | 7 | 1.57 | 8.5 | 0.059 | 0.3 | 6 | 4.8 | 0.8 | 32 | 0.77 | 0.14 | 75 | 22 | |
| HT423 | 1.0 | 423 | 6.0 | 54.0 | 15 | 209 | 39 | 1.71 | 7.9 | 0.060 | 30 | 12 | 6.7 | 1.2 | 66 | 0.83 | 0.15 | 98 | 30 | |
| HT433a | 1.0 | 433 | 6.0 | 78.0 | 17 | 124 | 64 | 1.71 | 4.5 | 0.025 | 105 | 37 | 8.6 | 1.1 | 114 | 139 | 0.86 | 0.20 | 145 | 37 |
| HT443 | 1.0 | 443 | 6.0 | 79.0 | 19 | 84 | 61 | 1.66 | 113 | 34 | ||||||||||
| HT453 | 1.0 | 453 | 6.0 | 88.0 | 19 | 87 | 66 | 1.61 | 2.8 | 0.011 | 190 | 80 | 10.3 | 1.0 | 91 | 0.78 | 0.21 | 109 | 32 | |
| HT463 | 1.0 | 463 | 6.0 | 79.0 | 19 | 82 | 61 | 1.58 | 99 | 35 | ||||||||||
| HT473 | 1.0 | 473 | 6.0 | 87.0 | 19 | 81 | 60 | 1.61 | 2.5 | 0.010 | 179 | 69 | 7.8 | 0.8 | 91 | 0.84 | 0.21 | 110 | 31 | |
| UD0 | 0.0 | 433 | 6.0 | 80.0 | 17 | 131 | 51 | 1.80 | 3.7 | 0.026 | 42 | 7.6 | 1.3 | 118 | 118 | 0.81 | 0.20 | 104 | 32 | |
| UD0.5 | 0.5 | 433 | 6.0 | 79.0 | 17 | 127 | 58 | 1.72 | 4.1 | 0.017 | 39 | 7.9 | 0.8 | 111 | 103 | 0.85 | 0.20 | 129 | 36 | |
| UD1a | 1.0 | 433 | 6.0 | 78.0 | 17 | 124 | 64 | 1.71 | 4.5 | 0.025 | 105 | 37 | 8.6 | 1.1 | 114 | 139 | 0.86 | 0.20 | 145 | 37 |
| UD1.5 | 1.5 | 433 | 6.0 | 75.0 | 17 | 129 | 56 | 1.66 | 3.7 | 0.021 | 29 | 5.4 | 0.8 | 108 | 116 | 0.85 | 0.22 | 127 | 36 | |
| UD2 | 2.0 | 433 | 6.0 | 80.0 | 17 | 130 | 53 | 1.77 | 3.9 | 0.030 | 43 | 8.2 | 1.5 | 108 | 113 | 0.84 | 0.19 | 121 | 35 | |
| UD3 | 3.0 | 433 | 6.0 | 76.0 | 17 | 122 | 50 | 1.70 | 4.1 | 0.022 | 32 | 6.6 | 0.9 | 113 | 100 | 0.89 | 0.24 | 121 | 33 | |
| UD4 | 4.0 | 433 | 6.0 | 78.0 | 17 | 129 | 45 | 1.73 | 4.1 | 0.021 | 36 | 7.5 | 0.9 | 111 | 88 | 0.84 | 0.20 | 101 | 27 | |
3.2 Composition of samples prepared under various US–HT reaction conditions
First, the influence of HT duration on the properties of products was examined using samples prepared with different HT durations (3–48 h), keeping the other synthesis parameters constant, i.e., 433 K HT temperature, 1 h US duration and precursor concentration of 267 mg TNWs in 80 mL Milli-Q water. The XRD patterns of samples are shown in Fig. 3 (left panels). The wide range of patterns (left upper panel) indicates that the starting titanate is converted into anatase titania by HT treatment; a characteristic anatase peak at ca. 25.3° appeared with 3 h HT treatment and grew with prolonged HT duration (left upper and lower panels). An expanded view (left middle panel) at around 30° suggested that PEPT was still included in the samples even after 6 h HT treatment. Intensity of anatase peaks seemed to be almost saturated by 9 h HT treatment with disappearance of peaks attributed to titanate, suggesting complete crystallization with this HT duration. However, as shown in Table 1, Rietveld analysis of XRD patterns of samples mixed with an internal standard (nickel oxide) indicated that crystalline content of anatase, the sole crystalline component, reached a constant level of ca. 83% with 9–12 h HT treatment, and the 12 h-HT sample still contained an appreciable amount of potassium (ca. 12% of potassium in the precursor). The composition of products was estimated using the formula aTiO2·H2−xKxTi8O17·bH2O based on the assumption that only anatase (a part of “aTiO2”) is crystalline giving XRD peaks; coefficients a, b and x were calculated using the data of crystallinity (79.87 × a/[total molar mass]), K/Ti ratio (x/(a + 8)) and water content (18.02 × {b + (2 − x)/2}/[total molar mass]) as shown in Table 1. The potassium content in the amorphous part of samples, x, did not greatly change during HT treatment, but it increased slightly by prolonging HT duration (and by higher HT temperature as discussed later) and then decreased. This suggests that the precursor TNWs were homogeneous PEPT without phase separation and were converted into anatase titania with dissolution of the potassium component as potassium hydroxide, i.e.,| H1.32K0.68Ti8O17·4.0H2O → 8TiO2 + 4.32H2O + 0.68KOH. | (1) |
 | ||
| Fig. 3 XRD patterns of the precursor (TNWs) and products obtained with (left) various HT durations and (right) HT temperatures in various 2θ ranges. | ||
In fact, the pH of the reaction mixture after HT treatment increased, being consistent with the stoichiometry. Assuming that the pH change was solely responsible for potassium-hydroxide release and that the titanium component was not dissolved in the reaction mixture by HT treatment, molar amounts of potassium ion (i) in a supernatant solution and (ii) expected to be released from TNWs could be calculated by using the pH value of supernatant solutions and K/Ti ratio of products. The recovery of potassium ion, (i)/(ii) (Table 1) seemed reasonable to support the potassium release, though some of them exceed 100% possibly due to a relatively large error in (i) especially higher pH and the fact that titanium component might be dissolved by HT.
The above-described results do not exclude the possibility of the presence of amorphous titania, i.e., the titanate part (H2−xKxTi8O17) is a mixture of amorphous titania and potassium titanate ((8 − 4x)TiO2·0.5xK2Ti8O17·(1 − 0.5x)H2O). However, our recent study has shown that post-calcination of samples prepared in the same way even at 1173 K does not change the crystallinity or cause phase transition from anatase to rutile.36 This suggests the absence of amorphous titania in the samples prepared with longer HT durations. Thus, it is concluded that HT treatment converted TNWs into a mixture of anatase crystallites and amorphous PEPT.
The presence of PEPT in the final product is not surprising since the HT process proceeds to reach an equilibrium in the reaction mixture, i.e., between PEPT and anatase with a sodium hydroxide solution (eqn (1)): TNWs were prepared through the backward reaction of eqn (1) using an anatase–rutile mixture with a much higher concentration of potassium hydroxide. Similar equilibria have already been reported for the synthesis of anatase nanowires,37 nanotubes,38,39 microspheres40 and rutile nanorod films.41 A study on the preparation through a HT process using diluted acid instead of water or a two-step HT process in water to shift the equilibrium to the titania side is now in progress to obtain purer OAP samples.
The structural/physical properties of the products, i.e., crystallite size, crystallinity and specific surface area, prepared with different durations are summarized in Fig. 4 (upper panel). As expected, prolonged HT duration caused larger crystallite size, lower specific surface area and higher crystallinity reaching plateaus after 9 h HT duration of ca. 19 nm, 80 m2 g−1 and 83%, respectively. Thus, under the conditions of 433 K HT temperature and 1 h US duration, 9 h HT duration was sufficient to convert TNWs into crystallized anatase titania.
 | ||
| Fig. 4 Crystallinity, crystalline size, specific surface area and total OAP content as functions of (upper) HT duration, (middle) HT temperature and (lower) US duration. | ||
It has been reported that properties of HT products strongly depended on HT temperature for various HT processes, e.g., synthesis of square bismuth tungstate (Bi2WO6) nanoplates,42 reduction of graphene oxide to graphene,43 preparation of nanosize rutile and anatase,23 synthesis of manganese(IV) oxide nanowires/nanorods,44 and phase transition of amorphous titania to anatase.45 Therefore, the influence of HT temperature on properties of samples was investigated for samples prepared at different HT temperatures (413–473 K) with no change in the other synthesis parameters, i.e., 6 h HT duration, 1 h US duration and precursor concentration of 267 mg TNWs in 80 mL Milli-Q water. Table 1 and Fig. 3 (right panels) also show the influence of HT temperature on products prepared with 1 h US and 6 h HT durations; change in XRD patterns in Fig. 3 seemed similar to that with different HT durations, and the samples prepared even at higher HT temperatures still contained amorphous PEPT. The composition of products changed with elevation of HT temperature, the trend of which was similar to that with prolonged HT duration; the higher the HT temperature was, the higher, lower and larger were the crystallinity, specific surface area and crystallite size, which reached plateaus of ca. 80%, 80 m2 g−1 and 19 nm, respectively, at a HT temperature of 433 K (Fig. 4, middle panel). This finding is consistent with the above-mentioned reaction mechanism considering that the equilibrium between PEPT and anatase titania is almost constant regardless of the HT temperature, and HT temperature of 433 K may be sufficient to reach the equilibrium.
On the other hand, US duration is expected to have no influence on composition of the product under the given conditions of HT temperature and duration since the US process for breaking TNWs is unlikely to affect the equilibrium in the HT reaction (eqn (1)). In fact, as shown in Fig. 4 (lower panel), the influence of US duration (before HT treatment at 433 K for 6 h) on the composition of products seemed negligible, i.e., crystallinity, specific surface area and crystallite size were almost constant at ca. 80%, 125 m2 g−1 and 17 nm, respectively, as was reported previously29 (as there was a negligible difference in XRD patterns as shown in Fig. S1 in the ESI†). The significant exception is morphology, which depended on US duration, as discussed in the next section.
3.3 Particle morphology of samples prepared under various US–HT conditions
Fig. 5 shows representative SEM and TEM images of the product prepared by 1 h US and 6 h HT treatment at 433 K, which was one of the samples with the highest crystallinity (78%) and total OAP content (64%). As clearly seen in the SEM images, many of the particles had an octahedral morphology (see also Fig. S2–4 in the ESI†). The inset TEM image (Fig. 5, middle panel) shows diamond-shaped particles with lower contrast tops, consistent with the decreasing thickness in the direction vertical to the electron beam in TEM near the apical ends of an OAP. A high-resolution image (Fig. 5, lower panel) showed fringes corresponding to (101) and (001) lattice spacings and the angle (68.3°) between them, as has already been reported previously.16 | ||
| Fig. 5 (upper/middle) Typical SEM images of a product prepared by 6 h HT (433 K and 1 h US). Scale bars correspond to 50 nm. (inset) A TEM image of OAPs. (lower) The corresponding high-resolution TEM image. | ||
As shown in Fig. 4, total OAP content was increased and almost saturated when HT duration was prolonged (upper) or HT temperature was raised (middle), indicating that the HT process led to the production of OAPs from TNWs. A slight decrease in total OAP content after saturation at 6 h and at 433 K might be due to transformation of OAPs under more severe conditions as has been reported for HT synthesis of hematite; dissolution of the spindle-like precursor started from the apexes with new {001} facet formation.46 The total OAP content was, roughly speaking, almost proportional to crystallinity for all of the samples prepared with the same US duration (1 h) as shown in Fig. 4. Fig. 6 shows plots of total OAP content and aspect ratio d001/d101 as a function of crystallinity. Higher HT temperature and longer HT duration (closed and half-closed circles, respectively) caused higher crystallinity and, at the same time, higher total OAP content, suggesting a constant ratio of total OAP to total anatase particles independent of HT duration and temperature when the same US duration was used. Extrapolation of a possible linear line to 100% crystallinity suggests a maximum total OAP content of ca. 85%, not 100%, presumably due to the existence of the potassium component, which cannot be converted into anatase. On the other hand, samples prepared with different US durations (6 h HT duration, 433 K HT temperature, and 267 mg TNWs suspended in 80 mL Milli-Q water) showed differences in total OAP content, though the crystallinity was almost constant (see Fig. 4, lower panel), i.e., particle morphology was highly dependent on the initial structural properties, mainly the morphology, of the precursor TNWs. Ultrasonication (US) has generally been used as a pre-treatment process to obtain a homogeneous suspension for reaction to make products uniform.47,48 In the present study, however, another function of US was suggested, i.e., US changed morphology of substrates by breaking TNWs. The morphological change caused by the US process is shown in Fig. S5 (ESI†); nanowire bundles seemed to be broken by US, but the extent of breakage could not be evaluated quantitatively.
 | ||
| Fig. 6 Correlations of total OAP content (circles) and aspect ratio (squares) with crystallinity. | ||
Aspect ratio (d001/d101; AR) was introduced as an alternative parameter to evaluate particle morphology. Since XRD patterns give averaged data on crystallites, AR may be a better parameter representing how particles spire; the higher the AR value is, the higher is the extent of OAPs. In a previous report on thermal treatment of OAP-containing particles,37 AR, which changed between ca. 0.9 and 1.7, was used to discuss morphology. For the samples used in the present study, low-crystallinity (<0.4) samples exhibited AR lower than 1.6, though AR values for the others were in a narrow range of 1.6–1.7, and it seemed difficult to discuss the difference between them. Actually, as can be seen in Fig. 6, plots of AR were scattered, presumably due to the possible relatively large error in estimation of d001 from a small 004 peak. On the basis of these considerations, total OAP content seems better than AR for evaluation of particle morphology in this study, but further discussion is still needed. In the following sections, total OAP content will be mainly used for discussion of morphology-dependent photocatalytic activities.
3.4 Electron-trap density and TRMC response of samples prepared under various US–HT conditions
One of the most important properties characterizing heterogeneous photocatalysts is the amount of crystalline defects on which either recombination of charge carriers (electrons and holes) or adsorption of reactants can occur, resulting in photocatalytic activity reduction or enhancement, respectively.49–51 In general, products obtained at a higher temperature exhibit higher crystallinity and thus have fewer lattice defects.33,52 The amounts of lattice defects in products obtained at different HT and US durations were studied by the DB-PAS method as density of electron traps (ET density), i.e., density of vacant electron-trapping sites was measured. It is presumed that such ETs may capture photoexcited electrons and induce recombination with holes, if the depth from the bottom of the conduction band (CB) is large so as not to allow thermal reexcitation to the CB. The results are shown in Fig. 7 as a function of specific surface area for samples prepared with different HT durations. The longer the HT duration was, the lower was the specific surface area, and ET density was decreased in proportion to specific surface area. This suggests that ETs are located mainly on the surface or near the surface, and their density is constant, ca. 0.6 nm−2, which is comparable to that of commercial titania samples.46 This is consistent with the fact that ET density of samples prepared under the conditions of different US durations with the HT duration (6 h) and temperature (433 K) kept constant was almost the same as shown in Fig. 4 (lower panel) since specific surface areas of those samples were negligibly changed with change in US duration. | ||
| Fig. 7 Electron trap (ET) density as a function of specific surface area. Closed and open circles show data for samples prepared with different HT and US durations, respectively. | ||
Thus, the samples prepared with different US durations had almost constant structural/physical properties, including crystallinity, crystallite size, specific surface area and ET density, except for particle morphology (total OAP content), which may predominantly govern their photocatalytic activity as will be discussed later.
As a measure of mobility and lifetime of charge carriers, time-resolved microwave conductivity (TRMC) was investigated for the samples as shown in Fig. 8. The TRMC signal (arbitrary) was increased with a 355 nm laser pulse (ca. 10 ns) which excites titania to produce photoexcited electrons and holes and then decayed after the laser pulse in a ns to μs time scale. The signal intensity is considered to be proportional to the summation of products of mobility and number (density) of each charge carrier, i.e., to overall mobility of charge carriers, and therefore, e.g., even when the number of charges is small, the intensity is enhanced if their mobility is high and vice versa. It has been suggested that holes, which are charge carriers, generated in irradiated titania particles are quickly trapped in certain sites to contribute negligibly to the TRMC signal. On the basis of these assumptions, the signal in Fig. 8 is attributable to the number and mobility of photoexcited electrons. In the analysis, two parameters were extracted. One is the maximum intensity of TRMC signal (Imax) corresponding to overall mobility of photoexcited electrons in relaxed states after laser excitation. Since it was suggested in a previous report on femtosecond-laser spectroscopic measurement that photoexcited electrons in photocatalysts are readily trapped by shallow ETs below the conduction band within a few ps,53Imax might reflect the migration of electrons being trapped in shallow traps and detrapped thermally to the conduction band, i.e., density of such shallow traps. The other parameter is the rate of signal decay. There should be at least two reasons for the signal decay: decrease in the number (density) of electrons due to recombination with trapped holes and decrease in mobility due to trapping in deeper ETs, leading to low mobility of electrons. Since the TRMC decay profiles could not be fitted with a single exponential function in general, the extent of decay was evaluated by decrease after 40 and 4000 ns as I40/Imax and I4000/Imax, respectively.
 | ||
| Fig. 8 (left) Time-course of TRMC signal and (right) parameters of samples prepared with different (upper) HT durations, (middle) HT temperatures and (lower) US durations. | ||
It is clear from Fig. 8 (right panels) that signal decay is independent of the preparation conditions; I40/Imax and I4000/Imax were ca. 0.8 and 0.2, respectively, for all of the samples in this study, indicating that the density of deep ETs reducing signal intensity in the present samples was not greatly different. Our recent study on post-treatment of OAPs showed appreciable effects of calcination and grinding on TRMC signal decay,36 suggesting that the change in particle shape from OAPs to sintered aggregates and round-edged particles resulted in a change in the distribution of the ETs in the particles. Consequently, the negligible difference in decay profiles in the present samples is attributable to OAP-shaped morphology without sintering and/or thermal blunting.
On the other hand, Imax changed significantly depending on the preparation conditions, HT duration, HT temperature and US duration, and the patterns of change in Imax shown in Fig. 8 (right panels) resembled those of total OAP content shown in Fig. 4. Fig. 9 shows the relation of Imax, as well as ET density, with total OAP content. Though the plots were rather scattered (presumably due to the fact that Imax also depends on other structural/physical properties), Imax seems to increase with increase in total OAP content (ET density decreased with increase in total OAP content). It should be noted that Imax was almost proportional to total OAP content when limited to a series of samples prepared with different US durations that exhibited almost constant structural/physical properties including ET density (Fig. 9). This finding indicates that particle morphology governs charge-carrier (photoexcited electrons) dynamics. A hypothesis is that {101} facets selectively exposed on OAPs possess ETs suitable for trapping with higher mobility on their surface structure. The results of a theoretical study on this hypothesis will be reported in the near future. In any case, a significant point is that US duration-controlled samples are structurally or physically different from each other only in total OAP content and charge carrier dynamics.
 | ||
| Fig. 9 ET density (closed) and Imax (open) as functions of total OAP content. | ||
3.5 Correlation between properties and photocatalytic activity
The correlation between properties and photocatalytic activities was studied using photocatalytic reactions of oxidative decomposition of acetic acid (CO2 system) and dehydrogenation of methanol (H2 system) as shown in Fig. 10. In both reaction systems, certain conditions, 6 h HT duration, 433 K HT temperature and 1 h US duration, gave maximum photocatalytic activities. The maximum activity in the CO2 system was ca. 1.5 times (150%) of that of commercial Showa Denko Ceramics FP-6, one of the most active photocatalysts, while the maximum activity in the H2 system was ca. 40% of that of FP-6. The reason for the low levels of photocatalytic activity in the H2 system is still unknown. It has been suggested that {001} facets with higher surface energy are necessary for efficient separation of charge carriers,54 and alcohol molecules could be preferentially adsorbed on {001} facets, leading to efficient charge separation. It was also reported that platinum particles were photodeposited preferentially on {101} facets of decahedral anatase particles (DAPs) exposing eight {101} and two {001} facets and showed a high level of photocatalytic activity but DAPs with platinum photodeposited on both facets showed much lower photocatalytic activity than that of DAP with platinum only on {101} facets.55 Thus, platinum deposition as a requisite for hydrogen evolution might be a key issue for activity in the H2 system, and further studies on OAP activity in the H2 system are presently being conducted.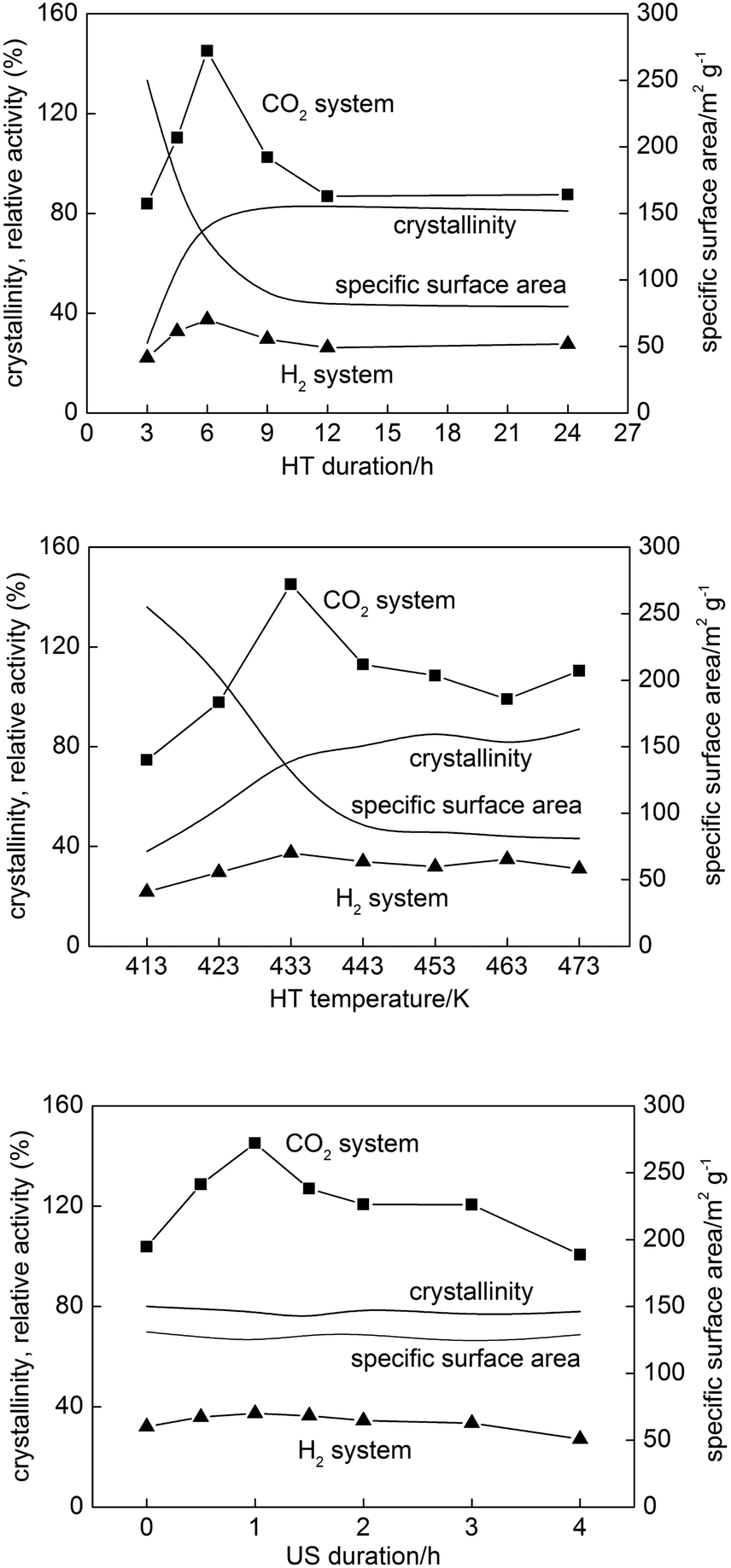 | ||
| Fig. 10 Photocatalytic activities of samples prepared with different (upper) HT durations, (middle) HT temperatures and (lower) US durations. Changes in crystallinity and specific surface area as representative conventional structural properties are also plotted. | ||
For samples prepared under the conditions with different HT durations and HT temperatures, it must be scientifically unacceptable to find a correlation between only one structural property and photocatalytic activity since all properties changed simultaneously with HT duration and HT temperature. In the conventional understanding, a balance between high crystallinity and sufficient specific surface area might be responsible for the highest photocatalytic activity of the sample prepared with 6 h HT at 433 K; in both cases, curves of plots of crystallinity and specific surface area crossed at this condition, 6 h and 433 K, though this coincidence should be occasional. It was reported that sufficient specific surface area resulting in a large amount of adsorbed substrates enhances the rate of capture of electrons and holes in them, while high crystallinity results in a lower density of lattice defects to reduce the rate of recombination of electrons and holes, and thus photocatalytic activity is enhanced.56,57 The differences in photoabsorption properties could be also considered as the reason for higher photocatalytic activity since with increase of HT duration particle size increased and more photons could be absorbed, as shown by diffuse reflectance spectra (DRS) in the left part of Fig. S6.† However, samples prepared at 4.5, 6 and 12 h possessed almost the same photoabsorption properties, but highly differed in photocatalytic activity, and increase of HT duration to 24 h resulted in a further increase in the photoabsorption range, but no change in crystalline size and photocatalytic activity was noticed. For samples prepared at various HT durations and HT temperatures all parameters changed simultaneously and thus various properties, i.e., balance between crystallinity and specific surface area, as well as photoabsorption properties and morphology influenced the resulting photocatalytic activity. Thus, the most important aspect was to prepare samples differing in only morphology, which we succeeded in by application of various US durations.
It should be noted that the maximum activity of the sample among those prepared with different US durations cannot be interpreted, at least for these samples, by the balance of specific surface area and crystallinity or photoabsorption properties, since those structural properties were almost the same (Fig. 10 and S6, bottom). In other words, the activity could be explained by the particle morphology, which was the sole structural parameter that varied depending on the US duration as discussed in the preceding section. Thus, our previous communication29 showed for the first time, as far as we know, morphology-dependent photocatalytic activity excluding the possible dependence on other structural/physical properties of photocatalyst particles such as crystallinity, composition, specific surface area and crystalline size. Fig. 11 (upper) shows the dependence of photocatalytic activities on total OAP content. A general trend of the activity being increased with increasing total OAP content was seen in both systems, but the plots were rather scattered except for the data for samples prepared with different US durations. Assuming that photocatalytic activities of OAPs and semi-OAPs are equal, the linear part of dependence on US duration-controlled samples in the CO2 system suggests that photocatalytic activities of pure OAPs should be ca. 230% of the photocatalytic activity of FP-6. Preparation of HT products with higher OAP content by modification of the present US–HT procedure is presently underway.
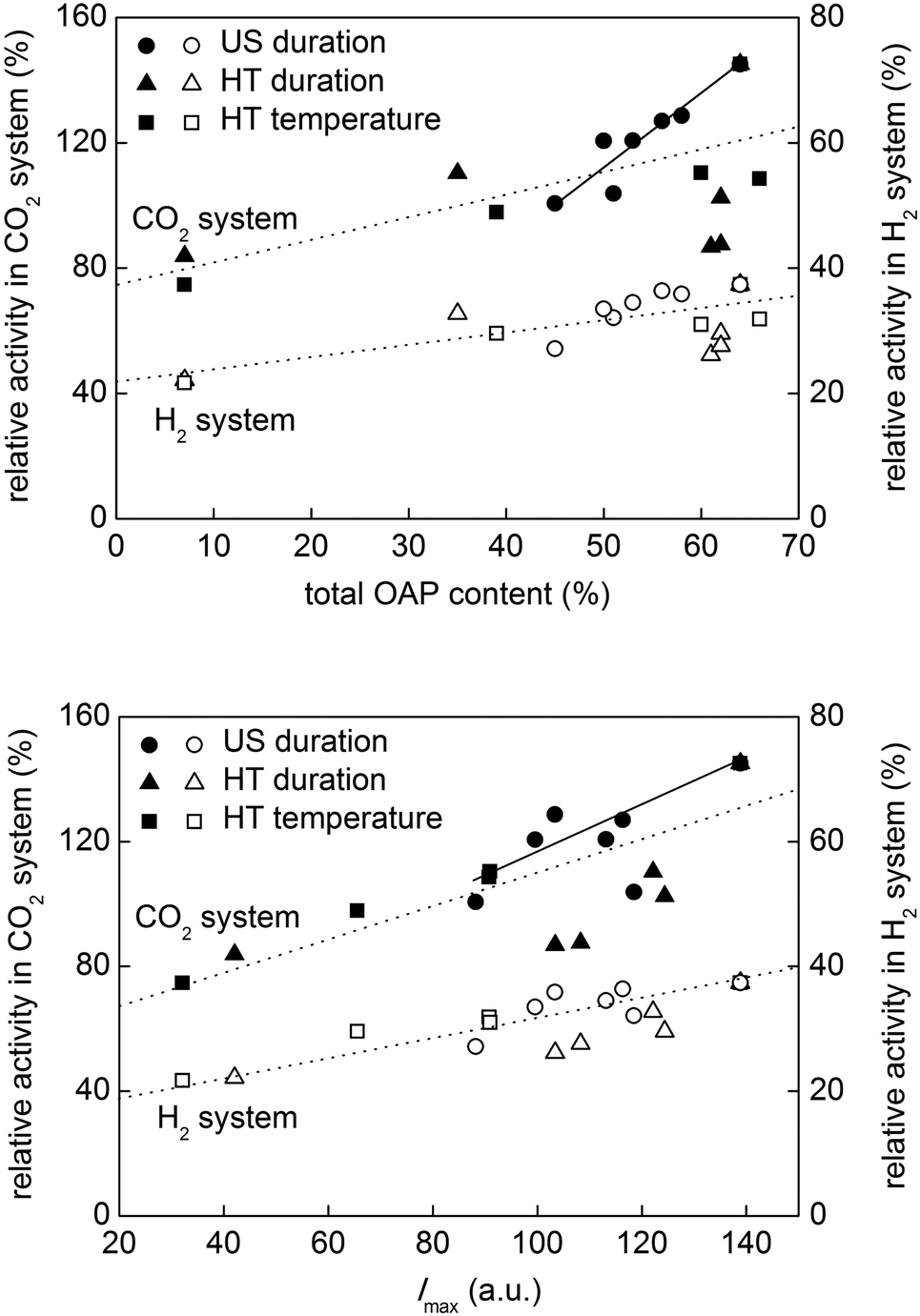 | ||
| Fig. 11 Photocatalytic activities of samples as functions of (upper) total OAP content and (lower) Imax. | ||
Then, how does particle morphology control the photocatalytic activity? A hint can be seen in Fig. 9 showing the correlation between Imax and total OAP content. The possible dependence of photocatalytic activities on Imax is shown in Fig. 11 (lower). Since, again, the samples prepared with different US durations have similar structural/physical properties except for total OAP content, the difference in the charge-carrier dynamics must be morphology-dependent. Therefore, a possible reason for the morphology-dependent photocatalytic activity may be related to ETs on the facets controlling the charge-carrier dynamics. As has been described in the preceding section, a hypothesis is that {101} facets selectively exposed on OAPs possess ETs suitable for trapping electrons loosely and thus keeping their mobility high on their surface structure, and the shallow traps might work as sites for one-electron reduction of molecular oxygen adsorbed on the surface but not for transfer of electrons to platinum deposited on the surface.
4 Conclusions
In summary, conditions of the ultrasonication–hydrothermal (US–HT) process significantly influenced structural/physical properties of OAP-containing products. Changes in HT duration or HT temperature resulted in simultaneous changes in all structural/physical properties, i.e., crystallinity, crystallite size, specific surface area, ET density and morphology. Therefore, a direct correlation between only one property and photocatalytic activity could not be shown. However, change in US duration resulted in the generation of HT products being different in morphology with all of the other structural properties being almost constant. Thus, the authors were able to conclude that morphology, represented empirically by total OAP content, governs the photocatalytic activity of OAP-containing samples, and the higher the OAP content is, the higher is the photocatalytic activity. The intrinsic reason for the morphology-dependent activity is still ambiguous at present, but the fact that TRMC-intensity maximum was almost proportional to total OAP content suggests that shallow ETs on the surface of exposed {101} facets are preferable especially for one-electron reduction of oxygen adsorbed on the surface to drive oxidative decomposition of organic compounds. On the basis of the above discussion, further studies are needed to clarify the intrinsic mechanism of titania photocatalysis. There is a need to prepare purer OAPs, to confirm their superior photocatalytic activity, by modification of the present US–HT process, which has been started in our laboratory, and analysis of the energy-resolved distribution of ETs on OAP-containing samples is also needed. A methodology to realize this has been developed, and preliminary results have been obtained and will be reported in the near future.Acknowledgements
This study was partly supported by CONCERT-Japan Program (Japan Science and Technology Agency) and a Grant-in-Aid (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (grant no. 2510750303). One of the authors (Z.W.) appreciates China Scholarship Council (CSC) for support. We also thank Kunlei Wang for his kind help on drawing cover picture.References
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- H. M. Zhang, Y. H. Han, X. L. Liu, P. R. Liu, H. Yu, S. Q. Zhang, X. D. Yao and H. J. Zhao, Chem. Commun., 2010, 46, 8395–8397 RSC.
- B. Ohtani, Chem. Lett., 2008, 37, 217–229 CrossRef CAS.
- B. Ohtani, Phys. Chem. Chem. Phys., 2014, 16, 1788–1797 RSC.
- O.-O. Prieto-Mahaney, N. Murakami, R. Abe and B. Ohtani, Chem. Lett., 2009, 38, 238–239 CrossRef CAS.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. Lu and H. M. Cheng, Chem. Commun., 2010, 46, 755–757 RSC.
- S. W. Liu, J. G. Yu and W. G. Wang, Phys. Chem. Chem. Phys., 2010, 12, 12308–12315 RSC.
- A. G. Agrios and P. Pichat, J. Photochem. Photobiol., A, 2006, 180, 130–135 CrossRef CAS PubMed.
- E. Kowalska, O. O. Prieto-Mahaney, R. Abe and B. Ohtani, Phys. Chem. Chem. Phys., 2010, 12, 2344–2355 RSC.
- D. Friedmann, C. Mendive and D. Bahnemann, Appl. Catal., B, 2010, 99, 398–406 CrossRef CAS PubMed.
- J. Z. Chen, H. Bin Yang, J. W. Miao, H. Y. Wang and B. Liu, J. Am. Chem. Soc., 2014, 136, 15310–15318 CrossRef CAS PubMed.
- M. Li, W. Hebenstreit, U. Diebold, A. M. Tyryshkin, M. K. Bowman, G. G. Dunham and M. A. Henderson, J. Phys. Chem. B, 2000, 104, 4944–4950 CrossRef CAS.
- F. Amano, T. Yasumoto, O. O. Prieto-Mahaney, S. Uchida, T. Shibayama and B. Ohtani, Chem. Commun., 2009, 2311–2313 RSC.
- F. Amano, T. Yasumoto, O. O. P. Mahaney, S. Uchida, T. Shibayama, Y. Terada and B. Ohtani, Top. Catal., 2010, 53, 455–461 CrossRef CAS.
- D. V. Bavykin, J. M. Friedrich and F. C. Walsh, Adv. Mater., 2006, 18, 2807–2824 CrossRef CAS PubMed.
- Z. Wei, Y. Liu, H. Wang, Z. Mei, J. Ye, X. Wen, L. Gu and Y. Xie, J. Nanosci. Nanotechnol., 2012, 12, 316–323 CrossRef CAS PubMed.
- X. G. Han, Q. Kuang, M. S. Jin, Z. X. Xie and L. S. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- C. C. Wang and J. Y. Ying, Chem. Mater., 1999, 11, 3113–3120 CrossRef CAS.
- H. M. Cheng, J. M. Ma, Z. G. Zhao and L. M. Qi, Chem. Mater., 1995, 7, 663–671 CrossRef CAS.
- M. Hirano and M. Takahashi, J. Mater. Sci., 2014, 49, 8163–8170 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549–1557 CrossRef CAS.
- D. C. Manfroi, A. dos Anjos, A. A. Cavalheiro, L. A. Perazolli, J. A. Varela and M. A. Zaghete, Ceram. Int., 2014, 40, 14483–14491 CrossRef CAS PubMed.
- N. Murakami, Y. Kurihara, T. Tsubota and T. Ohno, J. Phys. Chem. C, 2009, 113, 3062–3069 CAS.
- H. X. Li, Z. F. Bian, J. Zhu, D. Q. Zhang, G. S. Li, Y. N. Huo, H. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- Z. Wei, E. Kowalska and B. Ohtani, Chem. Lett., 2014, 43, 346–348 CrossRef CAS.
- S. Uchida and Y. Sanehira, Jpn. Pat., P2005-162584A, 2005 Search PubMed.
- B. Ohtani, R. M. Bowman, D. P. Colombo, H. Kominami, H. Noguchi and K. Uosaki, Chem. Lett., 1998, 579–580 CrossRef CAS.
- B. Ohtani, O. O. Prieto-Mahaney, D. Li and R. Abe, J. Photochem. Photobiol., A, 2010, 216, 179–182 CrossRef CAS PubMed.
- N. Murakami, O. O. P. Mahaney, R. Abe, T. Torimoto and B. Ohtani, J. Phys. Chem. C, 2007, 111, 11927–11935 CAS.
- E. Kowalska, H. Remita, C. Colbeau-Justin, J. Hupka and J. Belloni, J. Phys. Chem. C, 2008, 112, 1124–1131 CAS.
- Z. Hai, N. El Kolli, D. Uribe, P. Beaunier, M. Jose-Yacaman, J. Vigneron, A. Etcheberry, S. Sorgues, C. Colbeau-Justin, J. Chena and H. Remita, J. Mater. Chem. A, 2013, 1, 10829–10835 CAS.
- Z. Wei, E. Kowalska and B. Ohtani, Molecules, 2014, 19, 19573–19587 CrossRef CAS PubMed.
- H. B. Wu, H. H. Hng and X. W. Lou, Adv. Mater., 2012, 24, 2567–2571 CrossRef CAS PubMed.
- H. Yin, H. Liu and W. Z. Shen, Nanotechnology, 2010, 21 DOI:10.1088/0957-4484/21/3/035601.
- J. L. Tao, J. L. Zhao, C. C. Tang, Y. R. Kang and Y. X. Li, New J. Chem., 2008, 32, 2164–2168 RSC.
- X. Y. Hu, T. Zhang, Z. Jin, S. Z. Huang, M. Fang, Y. C. Wu and L. Zhang, Cryst. Growth Des., 2009, 9, 2324–2328 CAS.
- J. M. Wu and B. Qi, J. Am. Ceram. Soc., 2007, 90, 657–660 CrossRef CAS PubMed.
- C. Zhang and Y. F. Zhu, Chem. Mater., 2005, 17, 3537–3545 CrossRef CAS.
- Y. Zhou, Q. L. Bao, L. A. L. Tang, Y. L. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- X. Wang and Y. D. Li, Chem. – Eur. J., 2003, 9, 300–306 CrossRef CAS PubMed.
- K. Yanagisawa and J. Ovenstone, J. Phys. Chem. B, 1999, 103, 7781–7787 CrossRef CAS.
- C. J. Jia, L. D. Sun, Z. G. Yan, L. P. You, F. Luo, X. D. Han, Y. C. Pang, Z. Zhang and C. H. Yan, Angew. Chem., Int. Ed., 2005, 44, 4328–4333 CrossRef CAS PubMed.
- N. Sakai, R. Wang, A. Fujishima, T. Watanabe and K. Hashimoto, Langmuir, 1998, 14, 5918–5920 CrossRef CAS.
- Z. Zhong, F. Chen, A. S. Subramanian, J. Lin, J. Highfield and A. Gedanken, J. Mater. Chem., 2006, 16, 489–495 RSC.
- N. Murakami, O. O. P. Mahaney, T. Torimoto and B. Ohtani, Chem. Phys. Lett., 2006, 426, 204–208 CrossRef CAS PubMed.
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS PubMed.
- B. Ohtani, O. O. P. Mahaney, F. Amano, N. Murakami and R. Abe, J. Adv. Oxid. Technol., 2010, 13, 247–261 CAS.
- N. Murakami, R. Abe and B. Ohtani, Chem. Phys. Lett., 2008, 451, 316–320 CrossRef CAS PubMed.
- D. E. Skinner, D. P. Colombo, J. J. Cavaleri and R. M. Bowman, J. Phys. Chem., 1995, 99, 7853–7856 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167–1170 RSC.
- C. Liu, X. Han, S. Xie, Q. Kuang, X. Wang, M. Jin, Z. Xie and L. Zheng, Chem. – Asian J., 2013, 8, 282–289 CrossRef CAS PubMed.
- H. Kominami, T. Matsuura, K. Iwai, B. Ohtani, S. Nishimoto and Y. Kera, Chem. Lett., 1995, 693–694 CrossRef CAS.
- J. G. Yu, Q. J. Xiang, J. R. Ran and S. Mann, CrystEngComm, 2010, 12, 872–879 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr02386f |
| This journal is © The Royal Society of Chemistry 2015 |