
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of hexagonal-molybdenum trioxide (h-MoO3) nanostructures and their pseudocapacitive behavior†
Vipin
Kumar
,
Xu
Wang
and
Pooi See
Lee
*
School of Materials Science and Engineering, 50 Nanyang Avenue. and Nanyang Technological University, 639798, Singapore. E-mail: pslee@ntu.edu.sg
First published on 24th June 2015
Abstract
The crystallographic structure and morphology of redox active transition metal oxides have a pronounced effect on their electrochemical properties. In this work, h-MoO3 nanostructures with three distinct morphologies, i.e., pyramidal nanorod, prismatic nanorod and hexagonal nanoplate, were synthesized by a facile solvothermal method. The morphologies of h-MoO3 nanostructures were tailored by a controlled amount of hexamethylenetetramine. An enhanced specific capacitance about 230 F g−1 at an applied current density of 0.25 A g−1 was achieved in h-MoO3 pyramidal nanorods. Electrochemical studies confirmed that the h-MoO3 pyramidal nanorods exhibit superior charge-storage ability. This improved performance can be ascribed to the coexistence of its well exposed crystallographic planes with abundant active sites, i.e., hexagonal window (HW), trigonal cavity (TC) and four-coordinated square window (SW). The mechanism of charge-storage is likely facilitated by the vehicle mechanism of proton transportation due to the availability of the vehicles, i.e., NH4+ and H2O. The promising, distinct and unexploited features of h-MoO3 nanostructures reveal a strong candidate for pseudocapacitive electrode materials.
1. Introduction
The physicochemical properties and electronic structure of transition metal oxides have been a subject of intense fundamental research.1,2 Transition metal oxides are considered promising candidates for electrochemical energy-storage (supercapacitors and batteries) applications due to their excellent reduction–oxidation (redox) properties (Mn+ + e− → Mn±1).3,4 Recently, supercapacitors have emerged as promising electrochemical energy-storage devices due to their spectacular features such as higher power density compared to batteries (∼104 W kg−1 for supercapacitors and ∼100 W kg−1 for batteries), quick charging/discharging (ms–min) and excellent cycling stability (104–106) in aqueous as well as non-aqueous electrolytes.5 Supercapacitors have attracted considerable attention because of their potential use in vehicle start/stop/start operation and micro-electro-mechanical and portable electronic devices.6,7 To date, binary transition metal oxides (MnO2, CoO3, V2O5, etc.)8,9 have gained enormous attention as prospective electrode materials for supercapacitor applications (based on intercalation pseudocapacitance and surface pseudocapacitance). The capacitive behavior can be improved by tuning the state of crystallinity of the materials, e.g., amorphous or crystalline. However, this approach is found to be strongly materials system dependent, for example Zheng et al.10,11 and Farsi et al.12 showed that the amorphous RuO2·xH2O and amorphous MoO3 nanostructures store more charge than their crystalline counterparts, respectively, while recently Brezesinski et al.13 and Kim et al.14 found that the crystalline MoO3 and Nb2O5 are superior to their amorphous counterparts as charge-storage carriers.Nanostructuring of the binary transition metal oxides in various crystallographic structures has also been considered an effective and feasible approach to improve the electrochemical performance of the electrode materials.15 To date, layered binary transition metal oxides have been studied frequently due to their two dimensional crystallographic structure, which provide effective paths for the conduction of electrolyte ions. However, binary transition metal oxides with three dimensional (3D) crystallographic structures (open-structure), which can provide 3D conduction paths for electrolyte ions have not been explored. The 3D crystallographic structure primarily exists in the metastable phase of binary transition metal oxides such as spinel-MnO2, beta-V2O5, MoO3, etc.9,10,13,15
Among them MoO3 is considered an interesting material due to the high electrochemical activity of its stable as well as metastable polymorphs.16–18
The thermodynamic stable phase of MoO3, i.e., α-MoO3 possesses a layered (ABA) framework structure (i.e., 2D), while its metastable polymorphs possess an open-structure (i.e., 3D) in the hexagonal phase (h-MoO3), a layered (AAA) framework structure in the MoO3-II phase and rutile or distorted rutile structure in the β-MoO3 phase.19 To date, the electrochemical properties of α-MoO3 have been investigated. Various strategies have been employed to enhance the electrochemical performance of α-MoO3, for example nanowire arrays20 core/shell architecture21 decorations of conducting supports (polymers)22 and incorporation of CNT and nanocomposites with other oxides.23,24 However, these efforts did not improve the specific capacitance significantly. Recently, a kinetically favoured intercalation mechanism was proposed, benefiting from the mesoporous and iso-oriented nanocrystalline domain of the layered α-MoO3,13 for improved Li+ ions storage (605 C g−1). In addition to all the efforts made to date, several computational and experimental studies have shown that the crystal plane structure of the electrode materials has a substantial influence on the electrochemical performance of materials.25–27 Therefore, size and shape controlled nanomaterials are essential in unleashing and understanding their intrinsic properties, which are critical in electrochemical applications. More recently, the highest achievable specific capacitance of 326 F g−1 (at 0.25 A g−1) using α-MoO3 nanobelts has been reported, but the loading mass tends to be low (∼0.51 mg).28 Also, the specific capacitance of α-MoO3 nanoplates as high as 280 F g−1 (calculated from non-rectangular CV curves) has been achieved, benefiting from the small diffusion length of the ions into the nanosheets.29 Most recently, Chang et al. introduced the concept of the work function difference to enhance the working potential window of an asymmetric supercapacitor device based on rGO-α-MoO3, in an aqueous electrolyte solution.30 Due to an inter-layer spacing (6 Å) between the two layers of α-MoO3, it can accommodate a massive amount of electrolyte ions (0.28 Å for H+), but its poor conductivity limits the diffusion of the ions. Compared to α-MoO3 that has intercalation sites along one dimension only, the open-structure of h-MoO3 offers an increase in the degree of freedom (from 1 to 3) for electrolyte ions to intercalate in all possible directions. The hexagonal phase of MoO3 (h-MoO3) can be considered a promising host material for electrolyte ions because of the availability of the various intercalation sites, i.e., hexagonal window (HW), trigonal cavity (TC) and four-coordinated square window (SW). The open-structure of h-MoO3 could facilitate intercalation as well as diffusion of ions to realize improved supercapacitor performance.
Taking into account all these factors, we have synthesized h-MoO3 nanostructures using a facile solvothermal method. The synthesis of h-MoO3 nanostructures with diverse morphologies has been rarely found; this is related to the difficulty in phase formation due to thermodynamic (formation enthalpy and entropy of constituents) barriers.31–33 We have previously attempted to tune the morphology of h-MoO3 using thiourea as the source of ammonium ions. With the increase in the loading amount of the ammonium ions, a predominant formation of a flower-like morphology was observed. In this work, we are able to circumvent the challenges limiting pseudocapacitive behaviour by judicious control of the synthesis in tuning the physical structure of the h-MoO3 nanostructures.
2. Experimental
2.1. Chemical used
All chemicals, Mo powder 99.9%, H2O2 (30%), HNO3 (conc.), potassium hydroxide (KOH) flakes and hexamethylenetetramine (HMTA) were used as received from Alpha Aesar and Sigma Aldrich, respectively, without further purification.2.2. Preparation of h-MoO3 nanostructures
To synthesize h-MoO3 with pyramidal nanorod, prismatic nanorod and hexagonal nanoplate-like morphology, a simple yet effective solvothermal approach was employed, modified from our previous report32 with the use of hexamethylenetetramine (HMTA) as a structure directing agent instead of thiourea (CS(NH2)2). In brief, peroxomolybdic solution was prepared by the addition of hydrogen peroxide (H2O2) into a 20 ml glass vial containing 0.50 g of Mo powder. In a typical synthesis procedure, 100 mg, 150 mg and 250 mg of HMTA in 10 ml of DI water were used. The solution of HMTA was slowly added into peroxomolybdic solution and left for stirring. The reaction times to prepare h-MoO3 are compared with the other synthesis routes, and presented in the ESI, Table S1.† The solvothermal reaction was carried out at 100 °C for 18 h. Finally, the precipitates were collected and washed thoroughly with ethanol and DI water to remove loosely bound or bulk particles. The as-obtained precipitates were dried at 70 °C to obtain the final product.2.3. Electrode preparation for electrochemical testing
The working electrodes were prepared by mixing the as-prepared h-MoO3 powder (85%), carbon black (10%) and polyvinylidene fluoride (PVDF) (5%) into a 5 ml glass vial containing 500 μl of N-methyl-2-pyrrolidone (NMP), under magnetic stirring to make a slurry. 20 μl of this slurry was coated onto a current collector (graphite paper, 1 cm2) and dried in a laboratory oven at 100 °C. The loading mass was measured to be 0.95 ± 0.05 mg, for all three samples. Electrochemical testing was carried out in an aqueous solution of H2SO4 (1 M) in a potential window of 0.05 V to 0.65 V vs. Ag/AgCl.2.4. Structural characterization
The samples were characterized using powder X-ray diffractometer (Shimadzu XRD – 6000, Cu Kα radiation λ = 1.54 Å; power 2 kW) at a scan rate of 1° min−1 in the 2θ range of 10°–60°. Field emission scanning electron microscopy (FESEM; JEOL, JSM 7600F thermal FEG, JSM 6340F cold cathode FEG) and transmission electron microscopy (TEM; JEOL, JEM 2010 and JEM 2100F) was used to evaluate the morphology of the samples. Thermal analysis was done using thermal gravimetric analysis (TGA) (Q 500, max. temperature 900 °C), and IR spectroscopy (Perkin Elmer, Model Spectrum GX) was used to obtain information about the state of the bonds present. X-ray photoelectron measurements (XPS) were carried out using a VG ESCALAB 220I-XL spectrometer with a twin monochromated Al Kα X-ray source (1486.6 eV) at a constant dwell time of 50 ms and a pass-energy of 50 eV. CasaXPS peak fitting software was used to perform curve fitting. The physical surface area (Brunauer–Emmett–Teller) and pore size distribution were measured using a Tristar-II surface area and porosity analyser. Electrochemical measurements were carried out using Solartron, S1470E electrochemical interface and AutoLab PGSTAT 30 potentiostat. A three electrode cell was used for electrochemical testing, in which graphite paper coated with active materials, platinum sheet and saturated Ag/AgCl were employed as the working, counter and reference electrodes, respectively, in an aqueous electrolyte (1 M H2SO4). Electrochemical impedance spectroscopy (EIS) measurements were carried out by applying an AC voltage with 1 mV amplitude in a frequency range from 100 mHz to 100 kHz at open circuit potential (OCP).3. Results and discussion
3.1. Structural characterization
The crystallographic phases of the as-prepared samples were evaluated using X-ray diffraction (XRD), as shown in Fig. 1. All the XRD peaks in the patterns were matched exclusively with the hexagonal phase of MoO3 (ICDD # 15-5332) with lattice parameters a = 10.54 Å, c = 3.72 Å and cell volume V = 359.37 Å3.34 A few impurity peaks (◆) were observed in the pattern of hexagonal nanoplates (probably due to the residue of the by-product formed by Mo5+ ions, which made complex ammonium hydroxides of Mo, i.e., (NH4)8Mo10O34). The major peaks positioned at 2θ ∼ 25.5° and 19.2° are characteristic of the hexagonal phase of MoO3. | ||
| Fig. 1 X-Ray diffraction patterns of pyramidal nanorods (1), prismatic nanorods (2) and hexagonal nanoplates (3). The nanoplates show some impurity peaks labelled in blue diamond (◆). | ||
The morphology of the as-prepared h-MoO3 nanostructures was evaluated using FESEM, as shown in Fig. 2. Fig. 2(a)–(c) depict pyramidal nanorods, prismatic nanorods and the hexagonal nanoplate-like morphology of the samples prepared using 100 mg, 150 mg and 250 mg of HMTA, respectively. The typical diameter of pyramidal nanorods was about 400 and 100 nm at the base and tip, respectively, while the diameter of prismatic nanorods was about 350 nm. The length and thickness of the hexagonal nanoplates were about 1 μm and 80 nm, respectively. The effect of the concentration of ammonium ions on the nucleation and growth is critical in controlling the morphology of the as prepared nanostructures, as explained in our previous report using thiourea as the source of the ammonium ions.32
 | ||
| Fig. 2 FESEM micrographs of the samples prepared using (a) 100 mg, (b) 150 mg and (c) 250 mg of HMTA in peroxomolybdic acid solution. | ||
The concentration of HMTA or ammonium (NH4+) ions is essential to control the morphology of the final products. HMTA is a non-ionic, hetero-cyclic organic compound with the chemical formula (CH2)6N4, which eventually dissociates into NH4+ and OH− ions upon hydrolysis under the ambient conditions.35 The reaction of molybdenum (Mo) powder with the hydrogen peroxide (H2O2) generates a variety of species attached to 1 to 6 peroxide groups.36,37 The attachment of the peroxide groups to the molybdic units strongly depends on the pH of the medium. A yellow peroxomolybdic solution was obtained in the acidic medium (pH ∼ 1–1.5), which mainly consists of monomer- and dimer-peroxo species, i.e., [MoO(O2)2(H2O)2], [O{MoO(O2)2(H2O)2}]2− and other even more complex poly-peroxo species.38
The chemistry of the HMTA with the peroxomolybdic acid solution generates the nuclei and forms the lattice of molybdenum trioxide (MoO3) in the hexagonal phase. The interaction of NH4+ and OH− ions (hydrolysed product of HMTA) with the peroxomolybdic species can be expressed according to the following reaction (1):
| [Mo2(O)3(O2)4(H2O)2]2− + 3NH4+ + OH− → 2[MoO3−x(NH4)x(H2O)y] | (1) |
When a small amount (100 mg) of HMTA was incorporated into the peroxomolybdic acid solution, a pyramidal or pencil-like morphology of h-MoO3 predominates. This morphology is due to the depletion of the NH4+ ions during the synthesis reaction. The NH4+ ions continuously alter the interaction of the growth species [Mo8O26]4− on the crystal surfaces. At the instant NH4+ ions are depleted, the growth of the low-energy planes at the tip of the pyramidal nanorods leads to crystal structure stability. As the loading amount of HMTA was increased (150 mg), a perfect prismatic or hexagonal morphology could be realized. It is interesting to note that the hexagonal nanoplates were obtained with a relatively high amount (250 mg) of HMTA. The as-obtained NH4+ ions are pivotal in the formation of the hexagonal framework and simultaneously form partially-reduced molybdenum cations (Mo6+ to Mo6−x). Further growth of the hexagonal framework is hindered by the dissolution of the compound formed by Mo6−x cations in the vicinity (due to excessive ammonium ions), as the course of reaction proceeds. The surfaces of the as-prepared hexagonal nanoplates were found to be extremely rough or defective, which could probably due to the dissolution of the product formed by Mo6−x ions. This phenomenon was not prevalent during the formation of pyramidal and prismatic nanorods due to a relatively low amount of NH4+ ions in the reaction.
The morphologies of the as-obtained nanostructures were further characterized by TEM. Fig. 3(a)–(c), unambiguously depict the pyramidal nanorods, prismatic nanorods and hexagonal nanoplate-like morphology, respectively, which are consistent with FESEM analysis.
 | ||
| Fig. 3 TEM micrographs (a)–(c), HRTEM micrographs (d)–(f) and SAED and FFT patterns (g)–(i) of the samples prepared using 100 mg, 150 mg and 250 mg of HMTA, respectively, are shown. The red and yellow spots in SAED images show standard patterns of h-MoO3. | ||
Fig. 3(d)–(f) show the HRTEM micrographs of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, respectively. The spacings between two neighbouring fringes were found to be 0.21 nm and 0.18 nm, as can be seen in Fig. 3(d) and (e), corresponding to the d-spacing of the (221) plane and (002) plane of h-MoO3, respectively. The growth direction of the bulk of pyramidal nanorods was found similar to the prismatic nanorods (ESI, Fig. S1†). The interplanar spacings in hexagonal nanoplates were related to the (101) and (210) planes of h-MoO3, as shown in Fig. 3(f). Furthermore, the selected area electron diffraction (SAED) and fast Fourier transformation (FFT) patterns (Fig. 3(g)–(i)) show the diffraction spots along the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10], [100] and [1−2−1] zone axis for the tip of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, respectively.
10], [100] and [1−2−1] zone axis for the tip of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, respectively.
The chemical purity and thermal stability of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates were evaluated using TGA. Fig. 4 shows the typical characteristics of the ammonium ion-assisted formation of h-MoO3.32 The amount of water and ammonia molecules was calculated to be 3.4%, 1.3% for pyramidal nanorods, 4.5%, 2.5% for prismatic nanorods and 3.35%, 4.95% for hexagonal nanoplates, respectively, taking their weight loss characteristics into account. The weight losses in the temperature range of 150 °C–300 °C and 300 °C–450 °C for all three samples correspond to the removal of water and ammonium molecules, respectively.
 | ||
| Fig. 4 TGA analyses of (1) pyramidal nanorods, (2) prismatic nanorods and (3) hexagonal nanoplates. The step-wise weight loss of the materials corresponds to removal of water and ammonium molecules from the lattice of h-MoO3. | ||
The major weight losses of the material occur at 760 °C, 698 °C and 670 °C for pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, respectively. The weight loss at different temperatures suggests that the stability of the MoO6 octahedra decreases from pyramidal nanorods to prismatic nanorods to hexagonal nanoplates. Approximate formula units (NH4)0.11(H2O)0.28MoO2.89, (NH4)0.23(H2O)0.39MoO2.77 and (NH4)0.44(H2O)0.30MoO2.56 are derived for pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, respectively, taking thermal analysis into consideration. Effects of the concentration of NH4+ ions on the bonding between Mo and O was further evaluated using IR spectroscopy, as shown in the ESI, Fig. S2 and Table S2.†
XPS measurements were performed to analyse the valance state and chemical shift of Mo and N atoms in all three types of samples. Fig. 5(a)–(c) show the XPS spectra of Mo atoms in pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, respectively. Fig. 5(a)–(c) consist of two peaks which correspond to spin orbit doublets of Mo6+ (3d5/2) and Mo6+ (3d3/2).40 The Mo6+ (3d5/2) and Mo6+ (3d3/2) peaks are centred at 235.4 eV and 238.5 eV for pyramidal nanorods, 234.9 eV and 238.2 eV for prismatic nanorods, and 234.2 eV and 238 eV for hexagonal nanoplates. With respect to the binding energies of the deconvoluted Mo6+ peaks in pyramidal nanorods, a negative shift of about 0.5 eV and 0.7 eV in the binding energies is identified for prismatic nanorods and hexagonal nanoplates, respectively. This shift in the binding energies indicates that the valance state of Mo decreases from pyramidal nanorods to prismatic nanorods to hexagonal nanoplates. Besides a shift in binding energies, the integrated intensity of the deconvoluted Mo6+ peaks decreases from pyramidal nanorods to hexagonal nanoplates, which suggests that the pyramidal nanorods possess a higher content of Mo6+. The change in the binding energies can be ascribed to the change in the NH4+ content in all three samples. To verify this change in NH4+ content, XPS analyses of N 1s atoms were performed, as depicted in Fig. 5(d–f). The N 1s peak for all three samples centred at 401 eV originates from NH4+ ions.41 The integrated intensity of N 1s increases comparing the spectra from pyramidal nanorods to prismatic nanorods to hexagonal nanoplates, which indicates that the hexagonal nanoplates possess a higher loading amount of NH4+ ions. Due to the relatively high loading amount of NH4+ ions in hexagonal nanoplates, the binding energies of deconvoluted Mo6+ peaks are found less positive than that of the pyramidal nanorods. A shake-up satellite peak for all three samples is identified at about 404 eV that could be due to nonstoichiometric NH4+ ions generated by N–O complexes, but not much is known about them.42 The relative amount of these oxidized nitrogen complexes is found to be much smaller than that of the N–H complexes. XPS analyses show the same trend in the amount of NH4+ ions as revealed by TGA and IR analyses.
 | ||
| Fig. 5 XPS analysis of Mo components in (a) pyramidal nanorods, (b) prismatic nanorods and (c) hexagonal nanoplates, and analysis of N components in (c) pyramidal nanorods, (d) prismatic nanorods and (e) hexagonal nanoplates. | ||
3.2. Electrochemical testing of the samples
It is widely accepted that the electrochemical properties of the materials can be altered by tuning the morphology and size of the materials.43–45 The electrochemical measurements of h-MoO3 with three different morphologies, i.e., pyramidal nanorods, prismatic nanorods and hexagonal nanoplates, for supercapacitor application, were carried out in a three electrode cell configuration using 1 M H2SO4 solution. Cyclic voltammograms (CVs) of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates were carried out at scan rates of 1–50 mV s−1, as shown in Fig. 6(a) and (b), respectively. The strong redox peaks in CVs of h-MoO3 with different morphologies indicate that the charges are stored in association with the faradic redox reactions.46,47 A slight shift in the peak potential signifies the effect of morphologies on the electrochemical activities of the electrode materials.43,48 The origin of this polarization lies in the fact that the reaction potential (over potential) is extremely sensitive towards the activity of the surfaces. From the electrochemical measurements, we found that the pyramidal nanorods exhibit superior charge-storage capacity compared to prismatic nanorods or hexagonal nanoplates. In the case of hexagonal nanoplates, there is one set of strong redox peaks at 0.22 V/0.39 V (ΔE = 170 mV) and another set of faint peaks appearing at 0.45/0.49 (ΔE = 40 mV). For the pyramidal nanorods there are two sets of strong redox peaks positioned at 0.24 V/0.29 V (ΔE = 50 mV) and 0.46 V/0.50 V (ΔE = 40 mV), respectively. These peaks correspond to the reversible ingress/digress of H+ ions into/out of the framework of h-MoO3, according to reaction (2):| MoO3(NH4)x(H2O)y + zH+ + ze− → HzMoVzMoVI1−zO3(NH4)x(H2O)y | (2) |
 | ||
| Fig. 6 Cyclic voltammograms (CV) of (1) pyramidal nanorods, (2) prismatic nanorods and (3) hexagonal nanoplates are carried out at scan rate of (a) 1 mV s−1 and (b) 50 mV s−1. The redox peaks in CV analysis indicate reversible ingress and degrees of electrolyte ions, and (c) peak current response of the as-prepared electrodes with respect to the square root of the scan rates indicates diffusion controlled charge transfer process. The dotted lines in (c) indicate fitting profiles according to equation y = a + bx. | ||
The intercalation of H+ ions into the h-MoO3 is favoured by the redox centres (Mo6+ ions), which are located at the various intercalation sites, i.e., hexagonal window (HW), hexagonal cavity (TC) and square window (SW), in the framework of h-MoO3. These intercalation sites are available in the range of potentials, i.e., 0.1 ≤ V ≤ 0.3 (SW), 0.3 ≤ V ≤ 0.6 (TC) and V ≥ 0.7 (HW).
It is commonly believed that the proton conduction in hydrated compounds occur via the Grotthuss (H2O, H5O2+, H3O+, OH−etc.) or Vehicle (H2O, NH4+, N2H5+, CH3NH3+, etc.) type mechanism facilitated by the water molecules or proton carriers.10,49 In our case, due to the availability of proton carriers (Vehicle), i.e., NH4+ and H2O molecules, the charge-storage mechanism may favour the Vehicle mechanism of proton transportation. Vehicle mechanism is readily encountered in aqueous solutions, liquid/melts and solids. In solids, Vehicle mechanism is usually limited to the materials with layered or open-structures.50 It is believed that the compounds with a smaller amount of water molecules (<2 H2O) conduct protons by Vehicle mechanism.51 Here, we considered that the charge-storage mechanism in h-MoO3 does not rely on the Grotthuss mechanism of proton transportation due to the open-structure of h-MoO3, a small amount of water molecules and the availability of proton carriers (NH4+ and H2O). Additionally, in the crystal structure of h-MoO3 (shown in the ESI, Fig. S3†), water molecules are coordinatively attached with the octahedral units, which do not favour the formation of Zundel (H5O2+) cations (essential for the Grotthuss mechanism).52 Therefore, the charge-storage mechanism in h-MoO3 is more likely facilitated by Vehicle mechanism of proton transportation.
The scan rate response of the as-prepared electrodes was also carried out at different scan rates (1 mV s−1 to 100 mV s−1), as depicted in Fig. 6(c). The anodic and cathodic peak currents (in a potential window of 0.4 V to 0.6 V) increase linearly with the increase in scan rates (CVs are shown in the ESI, Fig. S4†). Additionally, linear dependence of the peak currents (Icathodic and Ianodic) on the square root of the scan rate (ν1/2) describes the process as a diffusion controlled electron transfer process.46,47 The slope of curves, shown in Fig. 6(c), can be used to estimate the diffusion coefficient (D) of the redox-species (H+ ions) using the Randle Sevcik equation (3),46 and this relationship can also be used to evaluate the electrochemical surface area of the electrode:54
| Ip = 2.69 × 105n3/2ACD1/2ν1/2 | (3) |
Fig. 7(a) shows the galvanic charge/discharge curves of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates at an applied current density of 0.25 A g−1 (charge/discharge curves at higher current densities are shown in the ESI, Fig. S6†). The non-linearity associated with the charge/discharge curves further confirms the pseudocapacitive response of the as-prepared h-MoO3 nanostructures. The specific capacitance (Csp) of the as-prepared electrodes can be calculated from charge/discharge curves using eqn (4):
| Csp = 2I∫Vdt/AΔV2 | (4) |
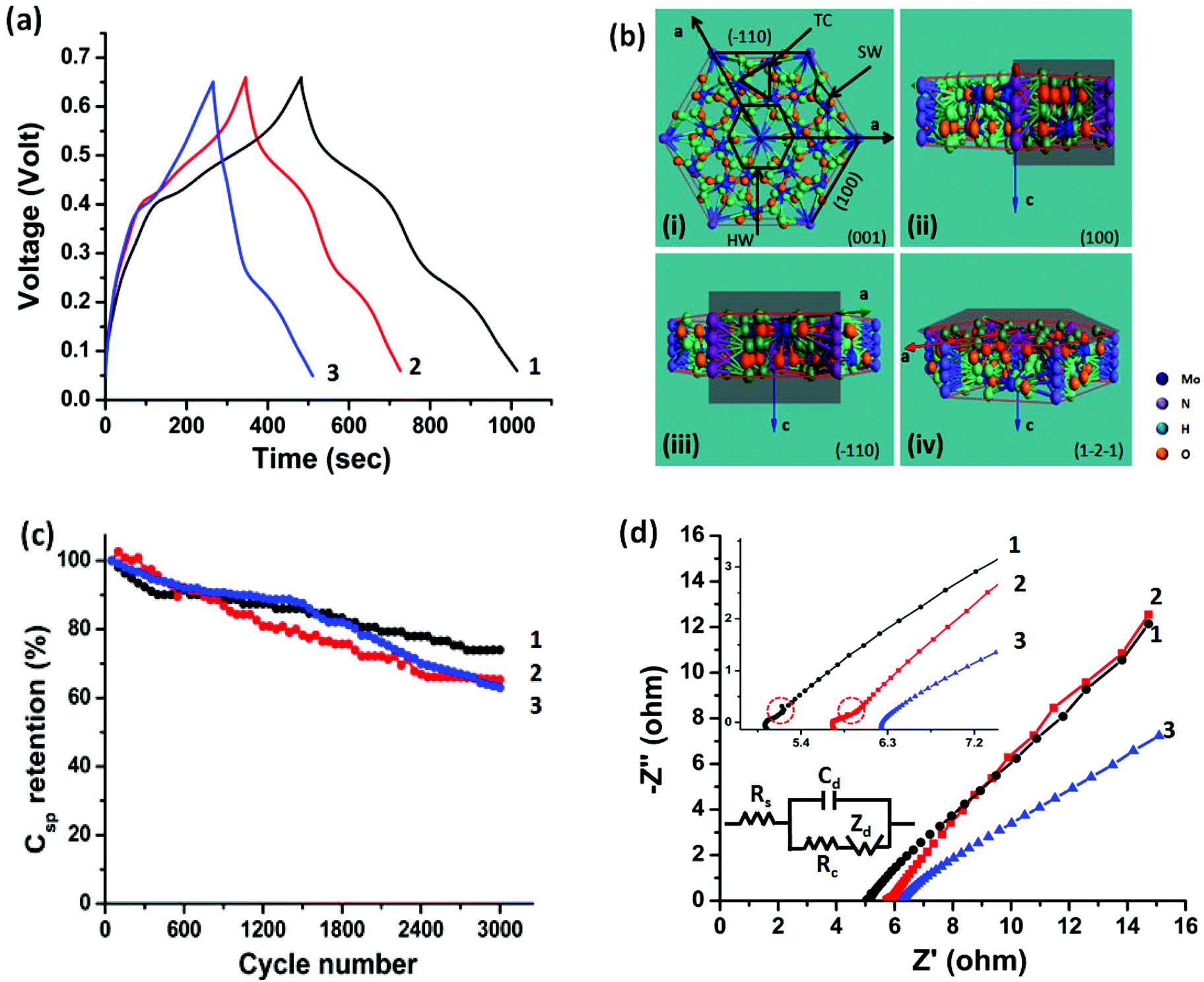 | ||
| Fig. 7 Charge/discharge profiles of (1) pyramidal nanorods, (2) prismatic nanorods and (3) hexagonal nanoplates are performed at an applied current density of (a) 0.25 A g−1, (b) atomic representation of hexagonal framework in the various plane configurations, (c) cycling test of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates using cyclic voltammetry (CV) at a scan rate of 50 mV s−1 and (d) Nyquist plots of pyramidal nanorods (1), prismatic nanorods (2) and hexagonal nanoplates (3) in a frequency bandwidth of 100 mHz to 100 kHz at open circuit potential (OCP). The inset shows the response of the as-prepared electrodes (pyramidal and prismatic nanorods) in the high frequency region (100 kHz–10 mHz). In the inset of figure (d) (Randles circuit), Rs, Zd, Rc and Cdl indicates solution resistance, diffusion resistance or Warburg resistance, charge transfer resistance and double layer capacitance, respectively. | ||
We assert that the exposure of desirable crystallographic planes is of paramount importance. The hexagonal framework of MoO3 resembles the hexagonal framework of WO3.59 The amount of the stabilizing ions, i.e., ammonium ions, occupancies in the hexagonal framework affects the available intercalation sites and therefore the electrochemical performance. The h-MoO3 framework is constructed by MoO6 octahedra sharing the equatorial oxygen in the ab plane and stacks along the c axis by sharing MoO6 octahedra. The hexagonal framework contains three different intercalation sites, i.e., hexagonal window (HW), trigonal cavity (TC) and four-coordinated square window (SW) running along the c axis, and these intercalation sites are available for the guest ions,60 as shown in Fig. 7(b)-(i). Among the various available intercalation sites, most of the HW sites were partially occupied by the NH4+ ions to stabilize the hexagonal framework, and exposed along the [001] direction of h-MoO3. On the other hand, TC and SW are positioned along the [001] and [100] directions, respectively. It is believed that the TC sites can accommodate a large number of electrolyte ions.60 In our case, pyramidal nanorods are constructed by the (100) and (10) planes (shadow plane in the atomic representation, Fig. 7(b)-(ii) and (iii)), representing their base and tip plane, respectively, while prismatic nanorods possess only (100) planes (shadow plane in the representation, Fig. 7(b)-(ii)). The coexistence of the (10) plane and the (100) plane in the case of pyramidal nanorods offers an additional degree of freedom to electrolyte ions for intercalation into the TC and SW sites.60 The existence of two distinct redox peaks in the CV analysis signifies the intercalation into the SW (0.1 ≤ V ≤ 0.3) and TC (0.3 ≤ V ≤ 0.6) sites; similar behavior was also identified in structurally identical Na doped V2O5.9 The improved current with distinct redox peaks in the case of pyramidal nanorods can be considered a consequence of this process, as can be seen in Fig. 6(a). The persistence of redox peaks even at a higher scan rate (50 mV s−1), Fig. 6(b), indicates the ease of intercalation into pyramidal nanorods due to the reasons discussed above. A pitiable capacitive charge-storage performance of hexagonal nanoplates originates from not only a higher content of ammonium ions within the framework but also the plane exposed by hexagonal nanoplates. The (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) plane of hexagonal nanoplates predominantly exposes HW and TC sites, which are partially occupied by NH4+ ions. The shadow plane in Fig. 7(b)–(iv) shows the (1) plane of h-MoO3. The availability of SW sites in hexagonal nanoplates gives rise to the strong redox peaks (0.22 V/0.39 V) and is attributed to the lack of appropriate TC sites, a set of faint or depressed redox peaks appeared at 0.45 V/0.49 V, as can be seen in Fig. 6(a).
) plane of hexagonal nanoplates predominantly exposes HW and TC sites, which are partially occupied by NH4+ ions. The shadow plane in Fig. 7(b)–(iv) shows the (1) plane of h-MoO3. The availability of SW sites in hexagonal nanoplates gives rise to the strong redox peaks (0.22 V/0.39 V) and is attributed to the lack of appropriate TC sites, a set of faint or depressed redox peaks appeared at 0.45 V/0.49 V, as can be seen in Fig. 6(a).
The cycling stability of h-MoO3 with various morphologies (pyramidal nanorods, prismatic nanorods and hexagonal nanoplates) is evaluated by the cyclic voltammetry (CV) test, at a scan rate of 50 mV s−1 in 1 M H2SO4 electrolyte solution, as shown in Fig. 7(c). A capacitance of 74%, 65% and 62% of the initial value is retained for pyramidal nanorods, prismatic nanorods and hexagonal nanoplates after 3000 cycles of continuous charge/discharge. This is attributable to the increase in the contact resistance that comes from poor contact (between the current collector and active materials) upon expansion/contraction of the active material (due to ingress/digress of electrolyte ions).54,56 Despite achieving good cycling stability, an early stage (∼300 cycles) degradation (12%) of capacitance is observed in pyramidal nanorods. To address the early stage degradation in the capacitance of pyramidal nanorods, an h-MoO3 electrode without addition of carbon black and PVDF was subjected to 300 cycles. It is evident from the FESEM micrographs (ESI, Fig. S7(a) and (b)†) that the initial stage degradation is caused by breakage of pyramidal nanorods. The framework of metal oxides may collapse during ingress/digress of electrolyte ions.57 Due to a relatively high intake of electrolyte ions in pyramidal nanorods, a small number of Mo5+ ions comes out from the framework (caused by the mobile Mo5+ ions). The excess negative charge of the system (produced by the removal of Mo5+ ions) is compensated by electrolyte ions (H+) and results in the framework being subjected to stress. Eventually, the framework of the nanorods breaks at the low energy site (neck of the nanorods) and isolates the tip and base of pyramidal nanorods. Due to formation of the abundant active sites at the tip as well as at the base of the nanorods (broken pyramidal nanorods), a long term cycling stability of the sample was realized, as shown in Fig. 7(c).
The benefit of h-MoO3 with pyramidal shape becomes more apparent when Electrochemical Impedance Spectroscopy (EIS) is used to evaluate the charge transfer and electrode kinetics of h-MoO3. The Nyquist plot in Fig. 7(d) shows the charge transfer characteristics of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates in the higher and lower frequency regions. The semicircle in the high frequency region (as shown in the inset of Fig. 7(d)) signifies the charge transfer resistance associated with the faradic reactions. To determine the parameters (Rs, Cdl, Rc and Zd) of EIS, a Randles circuit diagram was used, as presented in the inset of Fig. 7(d). The values of the parameters are listed in Table 1. Pyramidal nanorods give a low intercept value (5.03 Ω) on the real axis (X-axis), which indicates its low internal resistance (solution resistance and uncompensated resistance) in comparison to prismatic nanorods (5.79 Ω) and hexagonal nanoplates (6.31 Ω). The slight kinks (dotted circle in the inset of Fig. 7(d)) in the high frequency region of the Nyquist plot (for pyramidal and prismatic nanorods) are likely due to the adsorption of electrolyte ions with the surface of the nanorods. Due to the adsorption of electrolyte ions with the surface of the nanorods, generation of a new resistive element in the series with the charge transfer resistance takes place. In the low frequency region, the sloping lines are related to the diffusion resistance (Warburg resistance), when the ions enter into the interior of the host material. However, in the very low frequency region (f < 10 Hz) the resistance of h-MoO3 in pyramidal morphology increased slightly (due to sluggish redox reactions), as evaluated by Bode plot (shown in the ESI, Fig. S8†).
| Sample | R s/ohm | R c/ohm | Z d | C dl/mF |
|---|---|---|---|---|
| 1-Pyramidal nanorods | 5.03 | 0.10 | 9.71 | 2.31 |
| 2-Prismatic nanorods | 5.79 | 0.16 | 10.2 | 2.73 |
| 3-Hexagonal nanoplates | 6.31 | 0.45 | 11.71 | 3.01 |
4. Conclusions
In summary, we have demonstrated a facile approach to control the desirable morphology of h-MoO3 nanostructures. By rationally controlling the amount of HMTA, h-MoO3 with pyramidal nanorod, prismatic nanorod and hexagonal nanoplate-like morphologies are achieved, with an approximate molecular formula unit of (NH4)0.11(H2O)0.28MoO2.89, (NH4)0.23(H2O)0.39MoO2.77 and (NH4)0.44(H2O)0.30MoO2.56, respectively. The as-synthesized h-MoO3 in pyramidal morphology exhibited high specific capacitance (230 F g−1) compared to prismatic nanorods (160 F g−1) or hexagonal nanoplate morphology (103 F g−1) at an applied current density of 0.25 A g−1. The coexistence of the (100) and (10) planes with pyramidal morphology exposes various intercalation sites (HW, SW and TC) for the intercalation of electrolyte ions. The ammonium ions not only stabilize the hexagonal framework, but also facilitate the mechanism of charge-storage (by the Vehicle mechanism of proton transportation). This unique strategy to enhance the electro-kinetics leads to increase in the charge-storage capacity of MoO3. This represents the first attempt to prepare h-MoO3 for electrochemical capacitors, offering possibilities in catalytic, sensing and solar applications.
Acknowledgements
Vipin Kumar acknowledges the research scholarship provided by NTU and Temasek Laboratories @ NTU in Singapore, under project TLSP13-02. The work is also partly funded by the NTU-A*STAR Silicon Technologies Centre of Excellence under the program (Grant No. 112 3510 0003).Notes and references
- S. Li and D. A. Dixon, J. Phys. Chem. A, 2006, 110, 6231–44 CrossRef CAS PubMed.
- F. Zhou, M. Cococcioni, C. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter, 2004, 70, 235121 CrossRef.
- P. S. Directorate and F. Monmouth, J. Electrochem. Soc., 1995, 142, 6–8 CrossRef PubMed.
- H. Y. Lee and J. B. Goodenough, J. Solid State Chem., 1999, 144, 220–223 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- M. S. Gordon, D. Dabdub and R. B. Gerber, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16889–16889 CrossRef PubMed.
- X. Wang, B. D. Myers, J. Yan, G. Shekhawat, V. Dravid and P. S. Lee, Nanoscale, 2013, 5, 4119–22 RSC.
- E. Khoo, J. Wang, J. Ma and P. S. Lee, J. Mater. Chem., 2010, 20, 8368 RSC.
- J. P. Zheng, P. J. Cygan and T. R. Jow, J. Electrochem. Soc., 1995, 142, 2699 CrossRef CAS PubMed.
- J. P. Zheng and T. R. Jow, J. Electrochem. Soc., 1995, 142, L6 CrossRef CAS PubMed.
- H. Farsi, F. Gobal, H. Raissi and S. Moghiminia, J. Solid State Electrochem., 2009, 14, 643–650 CrossRef.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–51 CrossRef CAS PubMed.
- J. W. Kim, V. Augustyn and B. Dunn, Adv. Energy Mater., 2012, 2, 141–148 CrossRef CAS PubMed.
- O. Ghodbane, J. L. Pascal and F. Favier, ACS Appl. Mater. Interfaces, 2009, 1, 1130–1139 CAS.
- R. L. Smith and G. S. Rohrer, J. Solid State Chem., 1996, 115, 104–115 CrossRef.
- E. M. McCarron III, J. Chem. Soc., Chem. Commun., 1986, 336–338 RSC.
- J. C. Calabrese and E. M. McCarron, J. Solid State Chem., 1991, 125, 121–125 Search PubMed.
- V. Kumar, A. Sumboja, J. Wang, V. Bhavanasi, V. C. Nguyen and P. S. Lee, Chem. Mater., 2014, 26, 5533–5539 CrossRef CAS.
- T. Tao, Q. Chen, H. Hu and Y. Chen, Mater. Lett., 2012, 66, 102–105 CrossRef CAS PubMed.
- G. R. Li, Z. L. Wang, F. L. Zheng, Y. N. Ou and Y. X. Tong, J. Mater. Chem., 2011, 21, 4217 RSC.
- V. Kumar and P. S. Lee, J. Phys. Chem. C, 2015, 119, 9041–9049 CAS.
- I. Shakir, M. Shahid, S. Cherevko, C.-H. Chung and D. J. Kang, Electrochim. Acta, 2011, 58, 76–80 CrossRef CAS PubMed.
- I. Shakir, M. Shahid, M. Nadeem and D. J. Kang, Electrochim. Acta, 2012, 72, 134–137 CrossRef CAS PubMed.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher, P. R. Slater, M. C. Group, C. Di V, V. Uni, G. Gu and U. Kingdom, Chem. Mater., 2005, 5085–5092 CrossRef CAS.
- C. Nan, J. Lu, C. Chen, Q. Peng and Y. Li, J. Mater. Chem., 2011, 21, 9994 RSC.
- G.-Z. Wei, X. Lu, F.-S. Ke, L. Huang, J.-T. Li, Z.-X. Wang, Z.-Y. Zhou and S.-G. Sun, Adv. Mater., 2010, 22, 4364–7 CrossRef CAS PubMed.
- J. Jiang, J. Liu, S. Peng, D. Qian, D. Luo, Q. Wang, Z. Tian and Y. Liu, J. Mater. Chem. A, 2013, 1, 2588 CAS.
- W. Tang, L. Liu, S. Tian, L. Li, Y. Yue, Y. Wu and K. Zhu, Chem. Commun., 2011, 47, 10058–60 RSC.
- J. Chang, M. Jin, F. Yao, T. H. Kim, V. T. Le, H. Yue, F. Gunes, B. Li, A. Ghosh, S. Xie and Y. H. Lee, Adv. Funct. Mater., 2013, 23, 5074–5083 CrossRef CAS PubMed.
- L. V. Gurvisch, I. V. Veyts and C. B. Alocock, Thermodynamic Properties of Individual Substances, CRC Press, Boca Raton, 4th edn, 1994 Search PubMed.
- V. Kumar, X. Wang and P. S. Lee, CrystEngComm, 2013, 15, 7663 RSC.
- H. J. Lunk, H. Hartl, M. A. Hartl, M. J. G. Fait, I. G. Shenderovich, M. Feist, T. A. Frisk, L. L. Daemen, D. Mauder, R. Eckelt and A. A. Gurinov, Inorg. Chem., 2010, 49, 9400–8 CrossRef CAS PubMed.
- B. Rachid, Ann. Chim., 2007, 32, 277–282 Search PubMed.
- K. M. McPeak, T. P. Le, N. G. Britton, Z. S. Nickolov, Y. A. Elabd and J. B. Baxter, Langmuir, 2011, 27, 3672–7 CrossRef CAS PubMed.
- F. Taube, M. Hashimoto, I. Andersson and L. Pettersson, J. Chem. Soc., Dalton Trans., 2002, 1002–1008 RSC.
- V. Nardello, J. Marko, G. Vermeerch and J. M. Aubry, Inrog. Chem., 1995, 34, 4950–4957 CrossRef CAS.
- M. Cindric, Z. Veksli and B. Kamenar, Croat. Chem. Acta, 2009, 82, 345–367 CAS.
- I. M. Szilagyi, J. Madrasaz, G. Pokol, P. Kiraly, G. Tarkanyi, S. Saukko, J. Mizsei, A. L. Toth, A. Szabo and K. V. Josepovits, Chem. Mater., 2008, 20, 4116–4125 CrossRef CAS.
- Y. L. Leung, P. C. Wong, K. A. R. Mitchell and K. J. Smith, Appl. Surf. Sci., 1998, 136, 4–6 Search PubMed.
- R. Bacsa, J. Kiwi, T. Ohno, P. Albers and V. Nadtochenko, J. Phys. Chem. B, 2005, 109, 5994–6003 CrossRef CAS PubMed.
- P. Serp and J. L. Figueiredo, Carbon Materials For Catalysis, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2009 Search PubMed.
- H. Jiang, T. Zhao, C. Li and J. Ma, J. Mater. Chem., 2011, 21, 3818 RSC.
- J. S. Chen, M. F. Ng, H. Bin Wu, L. Zhang and X. W. (David) Lou, CrystEngComm, 2012, 14, 5133 RSC.
- D. Cai, D. Wang, B. Liu, Y. Wang, Y. Liu, L. Wang, H. Li, H. Huang, Q. Li and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12905–10 CAS.
- X. Wang, W. S. Liu, X. Lu and P. S. Lee, J. Mater. Chem., 2012, 22, 23114 RSC.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 2, 14925–14931 Search PubMed.
- S. M. Majhi, P. Rai, S. Raj, B. Chon, K. Park and Y. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 7491–7497 CAS.
- K. D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed. Engl., 1982, 21, 3 CrossRef PubMed.
- K. D. Kreuer, A. Rabenau and R. Messer, Appl. Phys. A: Solids Surf., 1983, 32, 45–53 CrossRef.
- R. Thakkar and U. Chudasama, J. Sci. Ind. Res., 2009, 68, 312–318 CAS.
- H. Lin, F. Zhou, C. P. Liu and V. Ozolins, J. Mater. Chem. A, 2014, 2, 12280–12288 CAS.
- S. S. Mahajan, S. H. Mujawar, P. S. Shinde, A. I. Inamdar and P. S. Patil, Int. J. Electrochem. Sci., 2008, 3, 953–960 CAS.
- X. Wang, A. Sumboja, M. Lin, J. Yan and P. S. Lee, Nanoscale, 2012, 4, 7266–72 RSC.
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765 CrossRef CAS PubMed.
- Q. Mahmood, W. S. Kim and H. S. Park, Nanoscale, 2012, 4, 7855–60 RSC.
- I. Shakir, M. Shahid, H. W. Yang and D. J. Kang, Electrochim. Acta, 2010, 56, 376–380 CrossRef CAS PubMed.
- R. Liang, H. Cao and D. Qian, Chem. Commun., 2011, 47, 10305–7 RSC.
- M. Hibino, W. Han and T. Kudo, Solid State Ionics, 2000, 135, 61–69 CrossRef CAS.
- S. Balaji, Y. Djaoued, R. Z. Ferguson and R. Bru, Chem. Mater., 2009, 21, 1381–1389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) of pyramidal nanorods; comparison of reaction times to prepared h-MoO3 nanostructures, FTIR spectrum of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates; atomic representation of hexagonal framework; Cyclic voltammograms (CVs) of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates at higher scan rates; BET isotherms of all three samples; charge/discharge profiles of pyramidal nanorods; prismatic nanorods and hexagonal nanoplates at the various current densities; FESEM images of pyramidal nanorods before and after cycling; Bode plot of pyramidal nanorods, prismatic nanorods and hexagonal nanoplates; electrochemical performance comparison table. See DOI: 10.1039/c5nr01505g |
| This journal is © The Royal Society of Chemistry 2015 |