
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Large-scale preparation of graphene by high temperature insertion of hydrogen into graphite†
Ali Reza
Kamali
* and
Derek J.
Fray
Department of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge CB3 0FS, UK. E-mail: ark42@cam.ac.uk; alirezakam@yahoo.com
First published on 14th May 2015
Abstract
Experimental evidence for high temperature diffusion of hydrogen into the interlayer space of graphite is provided. This process is discussed as a possible method for the rapid production of high-quality, inexpensive graphene in large quantities, which could lead to the widespread application of graphene. It was found that hydrogen cations, dissolved in molten LiCl, can be discharged on cathodically polarized graphite rods, which then intercalate into the graphite structure, leading to the peeling of graphite to produce graphene. The graphene nanosheets produced displayed a single-crystalline structure with a lateral size of several hundred nanometers and a high degree of crystallinity and thermal stability. The method introduced could be scaled up to produce industrial quantities of high-quality graphene.
Introduction
Diffusion of hydrogen in graphite can be important in many diverse scientific and technological fields including hydrogen storage systems,1 fusion nuclear reactors,2 hydrogen production,3 evolution of interstellar and protoplanetary clouds,4 fuel cells5 and the physical properties of graphene.6Three possible paths for the diffusion of hydrogen in graphite are crystallite surfaces and boundaries, voids and the interlayer space between graphene layers.7 Diffusion of hydrogen by the first two mechanisms have been theoretically8,9 and experimentally7,10 studied. However, the diffusion of hydrogen in the interlayer space has only been theoretically examined,8,9,11 which is mainly due to the lack of experimental methods.11 Therefore, the information available in the literature concerning the diffusion of hydrogen in the interlayer space of graphite mainly comes from theoretical calculations.11
To the best of our knowledge, this paper for the first time provides the experimental evidence of diffusion of hydrogen in the interlayer space of graphite. It was interestingly found that the diffusion of hydrogen in graphite crystallites can lead to the exfoliation of graphite into graphene nanosheets, a phenomenon which has not been reported in the past. Hence, this finding can be of importance in theoretical studies concerning the diffusion of hydrogen in the graphite lattice. Results presented in this paper can also be of special interest in the field of graphene production.
Carbon nanostructures include fullerenes, carbon nanotubes, nanofibres and graphene. Particularly, graphene possesses many extraordinary properties such as high ballistic electron mobility, thermal conductivity, Young's modulus, fracture strength, and high specific surface area.12–16 Recently, graphene-based nanomaterials called carbon nanoflakes,17 nanoflowers,18 nanohorns,19 nanowalls20 or graphene nanosheets21 have attracted extensive attention due to their unique dimensions, structure, and electronic properties which make them promising candidates for many applications such as electron field emitters,22 electrochemical capacitors,23 electrode material for capacitive deionisation,24 anode materials for lithium-ion batteries,25,26 catalyst supports,27 biosensors,28 electrodes for fuel cells,29 photocatalytic applications,30 nanocontactors31 and field-effect transistors.32 Also, graphene nanosheets can be used as templates for the fabrication of other nanostructured materials.33
Graphene was initially produced in small amounts by the “up to bottom” approach of micromechanical cleavage of highly oriented pyrolytic graphite (HOPG).34 Then, relatively larger amounts of chemically modified graphene were produced by a number of methods.34–42 Many of these methods are based on chemical oxidization of graphite. The subsequent exfoliation of the graphite oxide formed leads to the formation of individual layers of graphene oxide, which are electrically insulating. Although graphene oxide sheets can be chemically reduced to remove most of the oxides, the reduction processes induce structural defects, resulting in deterioration of the product quality.42–45
In order to overcome this problem, non-chemical, solution-phase exfoliation of graphite in organic solvents has been investigated.46–49 However, these methods resulted in low-concentration dispersions, which can be as low as 0.01 mg ml−1.46 Unfortunately, water which is the most useful solvent cannot exfoliate graphene42 and all of these techniques make use of HOPG as the starting material and involve labour-intensive preparation.
Zhang et al. reported on the preparation of graphene flakes using carbonization and calcination of glucose and FeCl3 mixtures.50 Although some success was achieved with this method, it was still a multi-step and time consuming process, and its scalability is yet to be evaluated.
Paton et al.51 reported the exfoliation of graphite powder to graphene flakes in solvents N-methyl-2-pyrrolidone, aqueous surfactant solution of sodium cholate, or polymer polyvinyl alcohol by the application of shear forces. In N-methyl-2-pyrrolidone, graphene flakes of a mean thickness of about 7 nm with a yield of about 70% could be produced.
In an attempt towards the development of a green process avoiding the use of toxic organic solvents/surfactants, Liu et al.52 reported the exfoliation of graphite in a solution comprising of water, FeCl2 and sodium dodecylbenzenesulfonate with a weight ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :300:5:2, respectively, under the hydrothermal conditions of 240 °C, 3 MPa and 4–10 h. Although single or few-layered graphene could be produced, the yield of exfoliation was only 10%, which could potentially be improved up to 35%.
:300:5:2, respectively, under the hydrothermal conditions of 240 °C, 3 MPa and 4–10 h. Although single or few-layered graphene could be produced, the yield of exfoliation was only 10%, which could potentially be improved up to 35%.
It should also be mentioned that horizontally53 or vertically54 oriented graphene films can be grown on various substrates by chemical vapour deposition (CVD) techniques and also by pyrolysis-based methods.55,56 These techniques, however, have a number of drawbacks related to the low production rates which can be as low as 32 nm min−1,57 low yields and the complexity of the equipment required.
As detailed above, the available methods of producing graphene suffer from one or more disadvantages such as a low rate of production, the low quality of graphene product and the use of hazardous organic solvents. Therefore, currently, there is no process available that can economically produce large amounts of graphene. Furthermore, alternative methods such as electrochemical exfoliation of graphite in ionic liquids58–61 require further investigation of their potential. Hence, the development of efficient production routes is considered a key element for widespread application of graphene. Here, for the first time, we report a unique process for rapid exfoliation of bulk graphite to graphene nanosheets based on high temperature intercalation of graphite with hydrogen in molten lithium chloride.
In the solid form, LiCl shows no considerable affinity for hydrolysis because of the energy barrier involved.
| LiCl + H2O(g) = LiOH + HCl(g) ΔG°(T<600 °C) > 78 kJ | (1) |
In the molten state, however, the hydrolysis of LiCl becomes much more significant because of the fact that the hydrolysis products are readily soluble in molten lithium chloride. At higher temperatures, the hydroxide can be transferred into oxide.62,63
The occurrence of the hydrolysis reaction leads to the formation of oxygen anions in the molten salt and hence is considered to be undesirable in the electrochemical applications in which molten LiCl serves as the electrolyte such as the electrolytic production of lithium metal,64 the reprocessing of spent nuclear fuels,65 and thermal batteries.66,67
In this paper, however, the hydrolysis of molten LiCl is promoted in order to enhance the formation of hydrogen cations in the molten salt at 800 °C. It is reported that the cathodic polarization of graphite electrodes exposed to the molten LiCl containing hydrogen cations leads to the exfoliation of the graphite cathodes into graphene nanosheets. This process offers a high production rate and product quality. Moreover, the process only uses industrial grade graphite, lithium chloride and water to produce graphene and lithium carbonate (as the by-product) and hence is environmentally acceptable. Lithium carbonate by-product could be removed from graphene nanosheets by a simple heat treatment.
Results and discussion
The structural and microstructural changes in an industrial synthetic graphite rod occurring by high temperature diffusion of hydrogen into its crystallites are investigated in this paper.A schematic representation of the set-up used is shown in Fig. 1. A graphite rod was immersed in molten LiCl at 800 °C and cathodically polarized for 30 min at an electric current of 33 A in a moist Ar flow. The average potential difference between the graphite rod and a Mo pseudo-reference electrode immersed in molten LiCl was measured to be about −2.8 V (Fig. 1S†). After the molten salt process, the cell was cooled to room temperature. It was observed that the graphite rod served as the cathode was eroded and the erosion product is thoroughly mixed with the salt. Fig. 2 shows the photographs of the graphite rod which was used as the cathode in the molten salt process indicating considerable erosion of the graphite rod. A black powder (about 10 g) was retrieved from the solidified salt mixture by washing with copious amounts of distilled water followed by vacuum filtering and drying at 150 °C for 2 h. As discussed later in this article, the disintegration of the cathodically polarized graphite rod in molten LiCl under a moist atmosphere is attributed to the insertion of hydrogen into the crystalline lattice of the graphite material.
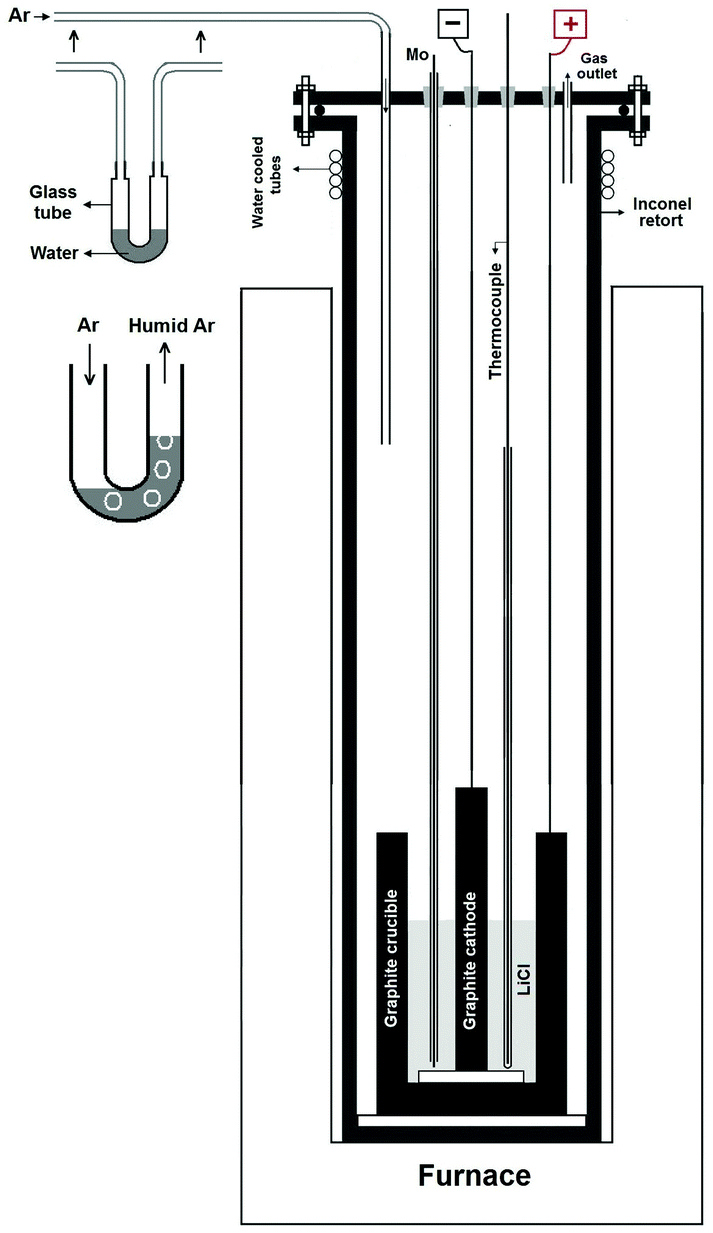 | ||
| Fig. 1 A schematic presentation of the reactor used for the molten salt process. The apparatus comprised of a vertical tubular Inconel reactor, which is positioned inside a resistance furnace. The upper end of the reactor is closed with a stainless steel lid sealed with an O-ring and compression fittings. The lid is equipped with feedthroughs for electrodes leads and the thermocouple as well as with steel pipes for a gas inlet and an outlet. The gas inlet was coupled to a gas canister containing argon. The argon could bubble through the water within a U-bend tube before entering the reactor at the gas inlet. The U-bend tube is shown more clearly in the inset of the figure. On passing through the U-bend tube, the dry argon absorbed the water vapour and becomes moist argon. Thus, the atmosphere within the reactor above the molten lithium chloride was moist argon that enters through the gas inlet and exits through the gas outlet. | ||
 | ||
| Fig. 2 The photographs of (a) the graphite rod which was used as the cathode in the molten salt process conducted under moist gas flow, (b) the graphite cathode after the molten salt process, and (c) the graphene product stored in a jar. | ||
The structure and morphology of the as-received graphite material and the erosion product were characterized by various techniques. The X-ray diffraction pattern of the as-received graphite material used in the 2θ range from 20° to 40° is exhibited in Fig. 3a. The prominent and sharp peak in the profile at 2θ = 26.441° corresponds to the (002) peak of graphite with an interlayer distance of 0.337 nm.
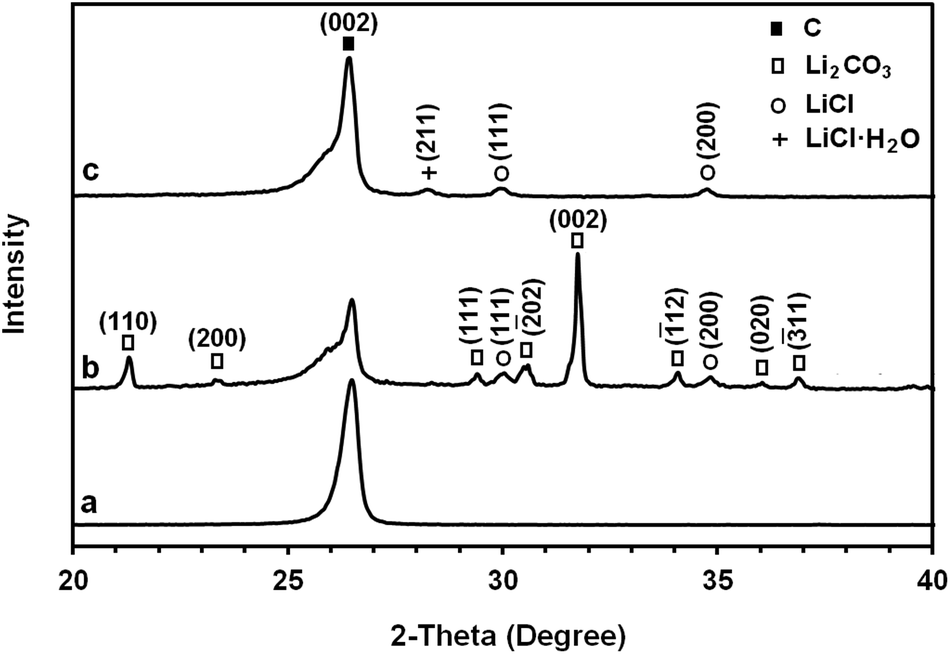 | ||
| Fig. 3 The X-ray diffraction patterns of (a) the as-received graphite material used as the cathode during the molten salt process, (b) as-synthesised carbonaceous material and (c) graphene nanosheets produced by heating the as-synthesised carbonaceous material to 1300 °C. | ||
Fig. 4 shows the SEM morphology of the as-received graphite material. It consists of flake-like grains of different sizes ranging from 1 to 10 μm with smooth or jagged edges. The grains, which are separated by submicrometer-sized voids, consist of graphite crystallites with an average height of 33.2 nm, calculated from the XRD data of the (002) peak shown in Fig. 3 (see Table 1S†). The as-received graphite is prepared by mixing petroleum coke with a coal tar binder, followed by extrusion and baking to carbonize the binder. It was finally graphitized by heating at high temperatures, at which point the carbon atoms arrange into graphite. Owing to the manufacturing process, it contains porosity within its structure, as a consequence of mismatch between grains and gas evolution. The pore density in synthetic graphite materials can be as high as 109 pores per cm3 with pore diameters varying between 1 nm and 1 mm.7,68
 | ||
| Fig. 4 SEM morphology of the as-received graphite material used as the cathode during the electrolysis process. | ||
As explained above, a graphite rod of this material disintegrated during the molten salt process. Fig. 3b shows the XRD profile of the carbonaceous materials obtained. The spectrum contains the (002) peak of graphite at 2θ = 26.485° corresponding to an interlayer distance of 0.336 nm. It also contains additional peaks that are due to the Li2CO3 and LiCl phases. The results suggest that some quantity of lithium chloride can be trapped in the microstructure of the carbon materials produced during the molten LiCl process. The trapped salt in the sample may not be removed completely by washing with water. The possible mechanism of the formation of Li2CO3 is discussed in this paper.
SEM studies (Fig. 2S†) have demonstrated that the as-synthesised carbonaceous material consisted of graphene nanosheets mixed with irregular structures originating from non-conductive Li2CO3 and LiCl. The graphene nanosheets were separated from the lithium compounds by heating the as-synthesised carbonaceous material.
It is worth noting that the large difference between the physical properties of carbon with the sublimation point of about 3640 °C and those of Li2CO3 and LiCl with the evaporation/decomposition point of about 1300 °C makes it possible to reduce the amounts of Li2CO3 and LiCl in the as-synthesised carbonaceous material by an appropriate heat treatment. The heat treatment must be carried out in an atmosphere with a low oxygen concentration to avoid the high temperature oxidation of carbon. For this, the material was heated to 1300 °C with a dwell time of 30 min in a reducing atmosphere of 80% N2–15% H2. The XRD profile of the purified graphene nanosheets is shown in Fig. 3c. As it can be seen, the Li2CO3 diffraction peaks are absent from the XRD pattern, which indicates its removal during the heat treatment. However, as indicated in Fig. 3c, very weak diffraction peaks of LiCl and LiCl·H2O could still be detected in the pattern.
It is noteworthy that the SEM analysis of the heat-treated product, unlike that of the as-synthesised one, was possible without a need for coating with gold, demonstrating the removal of Li2CO3 and LiCl which acted as non-conductive impurities. The morphology of the material obtained is presented in the SEM micrographs shown in Fig. 5 (left panels), demonstrating the fabrication of high yield graphene nanosheets with the lateral size of several hundred nanometers. The upper panel in Fig. 6 shows a bright field TEM micrograph of the graphene nanosheets. The right-down panel of Fig. 6 exhibits a HRTEM image showing the presence of single-layered graphene sheets. A typical selected-area electron diffraction pattern taken from the edge of a nanosheet with the peaks labelled by Miller–Bravais indices can also be seen in Fig. 6 (left-down panel) revealing the distinctive hexagonal structure of graphene. The diffraction intensity ratio I(1–210)/I(0–110) was analysed to be 0.8 indicating the presence of single-layered graphene.69 SEM and TEM images confirmed the preparation of high quality single- or few layered graphene.
 | ||
| Fig. 5 Secondary electron SEM images of the graphene nanosheets, produced by heating the as-synthesised carbonaceous material to (left panels) 1300 °C and (right-upper panel) 1450 °C at different magnifications. The right-down panel shows a backscattered electron SEM micrograph of graphene nanosheets obtained at 1450 °C. The products exhibited nanosheets morphology with a high yield. | ||
 | ||
| Fig. 6 The upper panel shows a TEM micrograph of the graphene nanosheets produced by heating the as-synthesised carbonaceous material to 1300 °C. The left-down panel shows a typical electron diffraction pattern recorded at a relatively flat edge of a graphene sheet, with the peaks labelled by Miller–Bravais indices. The right-down panel exhibits a HRETM image showing a single-layer of graphene. | ||
Raman spectroscopy is a powerful technique to study the structural properties of carbon based materials.70 The raw Raman spectrum of the graphene nanosheets in the wavenumber range 200–3000 cm−1 is presented in Fig. 7b (the upper panel). For comparison, the raw Raman spectrum of natural flake graphite is shown in Fig. 7a. Both spectra are characterized by the presence of the so-called G band at 1575–1581 cm−1 and the D band at 1327 cm−1. The G-band is related to the vibration of sp2 bonded carbon atoms in a two-dimensional hexagonal lattice while the D-band is associated with structural defects and partially disordered carbon structures. The intensity ratio of the G and D bands, IG/ID, is an index corresponding to the crystallinity of graphitic carbons.
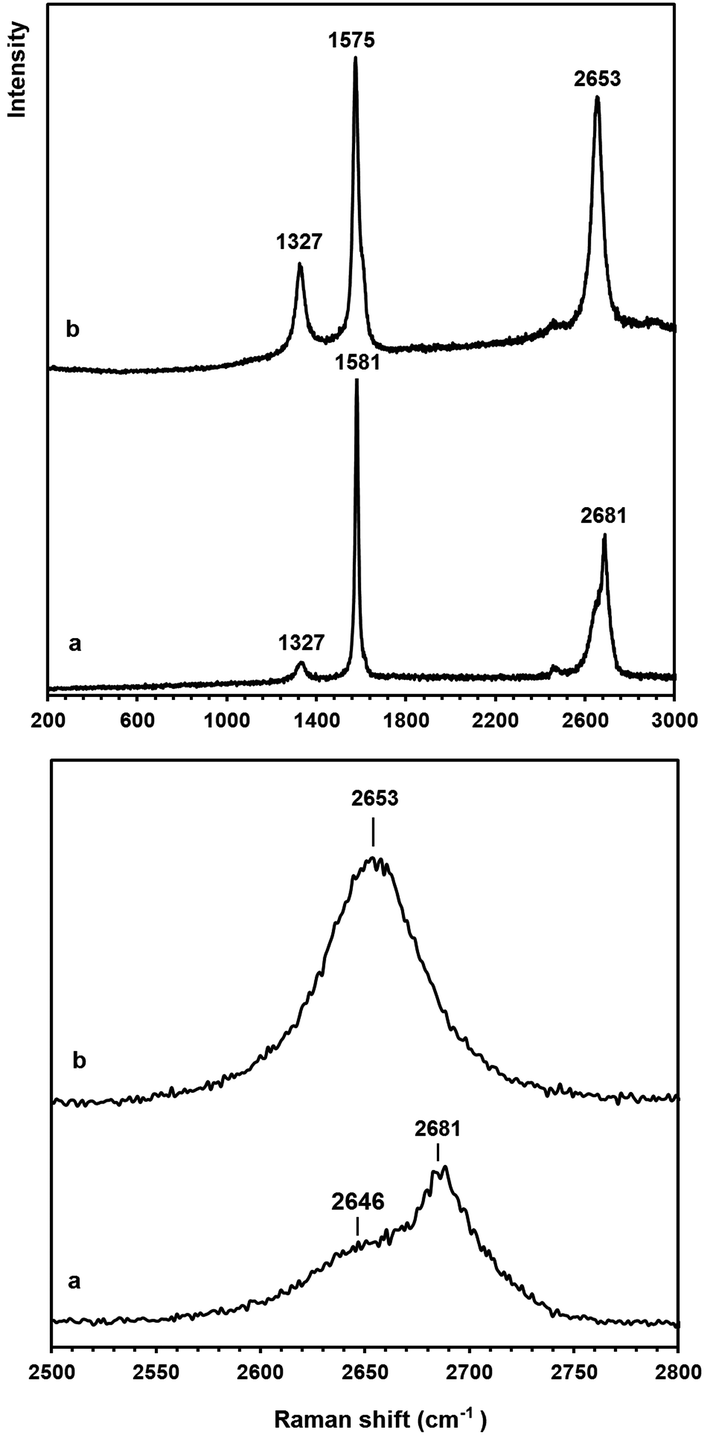 | ||
| Fig. 7 Raman spectra of (a) natural graphite flakes and (b) graphene nanosheets which were produced in molten LiCl, and then heated to 1300 °C in the range of (upper panel) 200–3000 and (down panel) 2500–2800 cm−1 at a 633 nm wavelength. | ||
The IG/ID ratio of the natural flake graphite and the graphene nanosheets were calculated from the Raman spectra, and these values were 8.8 and 3.5, respectively. Taking into account that the D peak is also induced by the edge of the graphene sheets, the smaller value of the IG/ID ratio in the graphene nanosheets, therefore, is attributed to their higher density of graphene edges. However, the IG/ID ratio in the graphene nanosheets is still significantly high and suggests that the nanosheets produced are composed of carbon crystallites with a large degree of crystallinity. Cançado et al.71 found that the average crystallite size (La) of graphene is proportional to (IG/ID), and can be calculated as:
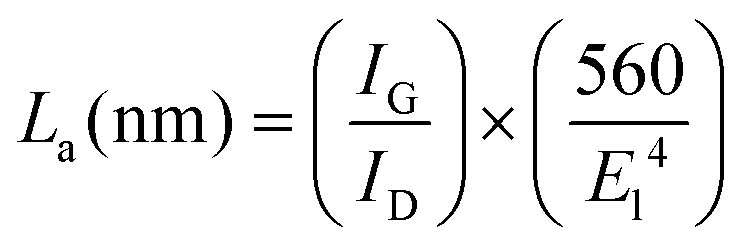 | (1) |
The down panel of Fig. 7 shows the feature of 2D peaks at a higher resolution. It is known that the 2D peak of graphitic materials is extremely sensitive to the number of layers. The 2D peak of graphite is asymmetric consisting of two components of 2D1 and 2D2, whilst the 2D peak of single-layered graphene is composed of a single peak.72 The comparison of the shape of the 2D peak recorded on the natural graphite with that of the graphene nanosheets suggests that the graphene nanosheets produced have the Raman characteristics of single layered graphene. Thermal analysis studies (see Fig. 3S†) confirmed that the graphene nanosheets prepared are thermally stable below 450 °C.
It should be noted that the graphene nanosheets which were synthesised by heating the as-synthesised carbonaceous material to 1300 °C contained a small amount of lithium chloride, as shown in Fig. 3c. The residual lithium chloride in the graphene product could be eliminated by heating the as-synthesised carbonaceous material to a higher temperature of 1450 °C in an atmosphere of Ar containing 4% H2, as shown in Fig. 8 (upper panel). The Raman spectrum of the sample is displayed in Fig. 8 (down panel), from which the ratio of IG/ID was determined to be 3.4. From this, the value of La was calculated, according to eqn (1), to be 129 nm. A comparison of Fig. 3 and 8 indicates that increasing the heat treatment temperature from 1300 to 1450 °C led to the elimination of the residual LiCl, but did not change the average crystallite size of the graphene nanosheets. It confirms that the residual LiCl in graphene nanosheets did not contribute to the structural defects of the nanosheets.
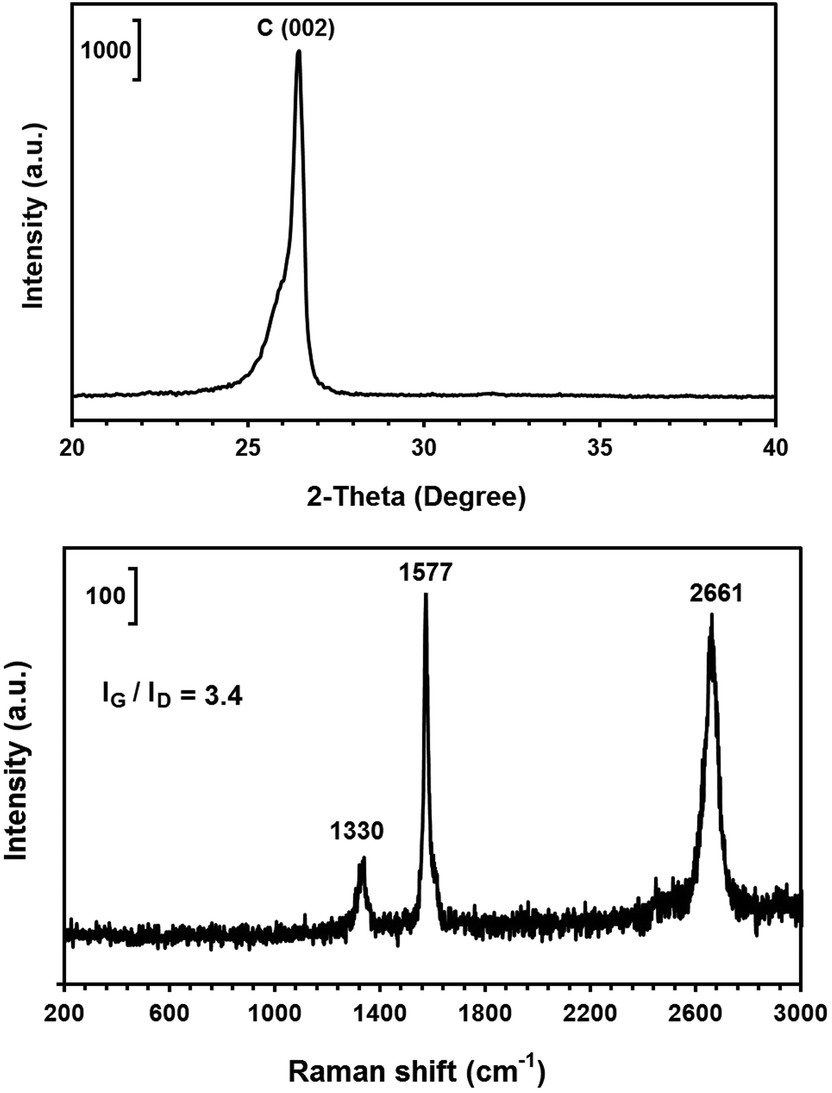 | ||
| Fig. 8 The X-ray diffraction pattern (upper panel) and Raman spectra (down panel) of the graphene nanosheets produced by heating the as-synthesised carbonaceous material to 1450 °C. | ||
It is worth noting that under ambient conditions, a low concentration of oxygen, as much as for example 3.56%73 or 3.70%,74 is always present in graphite in the form of oxygen molecules chemisorbed on the graphite edge planes and also oxygen atoms intercalated between basal planes.75
The C/O atomic ratio of the as-received graphite material used as the cathode during the electrolysis process and graphene nanosheets was determined by elemental analysis and found to be 29.96 and 21.01, respectively. The higher C/O value in graphite in comparison with that of graphene is attributed to the higher edge density of graphene nanosheets. The BET specific surface area of the graphene sample obtained was measured by nitrogen adsorption and found to be 235 m2 g−1. Prior to the analysis, a sample of about 0.1 g was degassed at 300 °C under a vacuum of 10−3 Pa for 24 h in order to remove the adsorbed species.
Table 1 compares the properties of graphene nanosheets produced in this paper with selected graphene materials fabricated by other methods. From an application point of view, a graphene material with a higher degree of crystallinity and lower oxygen content is preferable as the structural defects and oxygen content of graphene have an adverse effect on the thermoelectric performance of graphene composites.103 It is also known that graphene with lower oxygen content shows lower hemolytic activity.97 The data displayed in Table 1 indicate that graphene nanosheets fabricated in this paper possess an interesting combination of high crystallinity and low oxygen content together with a high production rate.
| Method of fabrication | |||||
|---|---|---|---|---|---|
| CVD | Reduction of graphene oxide | Solution-phase exfoliation of graphite | Solution-phase methods using non-graphitic precursors | High temperature insertion of hydrogen in graphite [current study] | |
| Product | Graphene film | Graphene powder/dispersion | Graphene powder/dispersion | Graphene powder/dispersion | Graphene powder |
| I G/ID | 1,76 2,77 2.9,78 3.679 | 0.4,86 0.6,82 1,83,88 0.8,84,87,89,91 0.9,85 1.1,90 1.284 | 1.4,94 1.1,95 1.396 | 0.8,101 1–3102 | 3.4 |
| C/O (at%) | 13.7,80 2081 | 3.3,85 7.4,88 8.9,92 11.99,93 9.6,90 3.891 | 7,91 9.2,95 1.5–297 | — | 21.5 |
| Yield | 32 nm min−1 (ref. 57) | 52 mg,83 <30 mg,86 <20 mg,89 <0.5 g,84 <50 mg91 | 10 wt%,52 <38%,94 <0.5 g,95 0.05–0.3 mg mL−1,98 2 mg, 12%99 | 0.1 mg ml−1 (ref. 101) | 50 kg per m2 of electrode per day, 80% |
| 8.6–24.4%,100 0.39 mg mL−1 (ref. 97) | |||||
Here we discuss the mechanisms involved in the formation of graphene nanosheets. It is known that molten LiCl63,105 and LiCl-based mixtures106–108 react under a humid atmosphere to form lithium oxide and hydrogen chloride according to reaction (1) with ΔG°800 °C = 85.3 kJ.
Also it is known that the lithium hydroxide formed by the hydrolysis of LiCl can decompose to form lithium oxide, according to reaction (2).63
| 2LiOH = Li2O + H2O(g) ΔG°800 °C = 11.3 kJ | (2) |
Although the Gibbs free energy of reactions (1) and (2) is positive, yet the reactions can proceed at a finite rate as a result of dissolution of products in the molten salt. It should be mentioned that lithium chloride is unique among the alkali chlorides in forming solid hydrates under ordinary conditions. Dehydration of lithium hydrates occurs at temperatures below 200 °C to form anhydrous lithium chloride. However, the tendency of LiCl to react with water (through hydrolysis) becomes more pronounced once the melting point has been exceeded, since both LiOH and HCl formed can dissolve in the molten LiCl.63,108
HCl is soluble in LiCl-based molten salts and the dissolved HCl is dissociated into protons and chloride ions. The diffusion coefficient of the proton arising from the dissolved HCl is an order of magnitude higher than most other ions in molten salts.102–112 Also it is known that the solubility of Li2O in molten LiCl can be more than 11 mol%.113
As a conclusion, reactions (1) and (2) lead to the formation of hydrogen cations (H+) and oxygen anions (O2−) in molten LiCl. Therefore, the processes that occur during the cathodic polarization of the graphite rod in molten LiCl can be described as follow:
| 2H+ + 2e = H2 (at the cathode) | (3) |
| O2− + 1/2C = 1/2 CO2 + 2e (at the anode) | (4) |
In fact, H2 (and no Cl2) could be detected in the gas stream outlet of the reactor shown in Fig. 1, confirming the occurrence of reaction (3). The structural disintegration of the graphite cathode to graphene sheets, therefore, can be attributed to the interaction of hydrogen formed with graphite. Hence, we should consider the hydrogen electrode reactions in more detail.
During cathodic polarisation, the reduction of proton from chloride molten salts on carbon cathodes is known to proceed via a reversible one electron charge transfer.109–112 Atomic hydrogen formed can be chemisorbed on the surface of graphite by binding to a carbon atom, leading to the formation of an additional σ bond and the rehybridization from sp2 to sp3.114–119
Therefore, the first step of the cathodic reaction is the formation of adsorbed atomic hydrogen on graphite (Had):
| H+ + e → Had | (5) |
The subsequent step is the formation of molecular hydrogen:112–125
| Had + H+ + e → H2 | (6) |
Diffusion of atomic and molecular hydrogen in graphite has been theoretically studied. It was found that the diffusion of the chemisorbed hydrogen atoms on the surface of graphite crystallites is rather short and direct,11 and involves the breaking of the C–H bond and forming another bond with a nearby carbon atom in the same or in an adjacent sheet, which requires an activation energy of 0.38–0.5 eV.9,11,116
The temperature dependency of diffusion coefficient of atomic hydrogen on graphite sheets (DH, cm2 s−1) can be concluded from ref. 11 to be:
| DH = 2.0 × 10−3 exp (−6.09 × 10−20/kBT) (300–1700 °C) | (7) |
The hydrogen atoms can then diffuse in the bulk of graphite through its porosity.7 In the as-received graphite electrode material, the graphite flakes are separated by micrometer sized-voids. Moreover, it is known that in a graphite flake, crystallites are usually separated by nanometre sized-voids which are a few Å in size, providing chemically reacting internal surfaces on which hydrogen atoms can diffuse and combine to form hydrogen molecules in the bulk of the graphite.7 The penetration of atomic hydrogen in graphite porosity is strongly affected by temperature and the hydrogen exposure rate. The typical time taken for hydrogen atoms to penetrate several Å into graphite is found to be about 15 days at 25 °C, less than a millisecond at 600 °C and around a microsecond at 1200 °C.7 Causey et al. reported that when graphite is exposed to hydrogen atoms or ions, the retention of hydrogen considerably rises as the exposure rate exceeds 5 × 1020 atoms per cm2, which is attributed to the fact that a greater number of atoms reach the internal porosity.68
In our experiments, the molten salt process was conducted at 800 °C and a current of 33 A, corresponding to the cathode current density of about 1 A cm−2. It was found that graphite cathodes are visually eroded in molten LiCl under a humid Ar atmosphere if the cathode current density exceeds about 0.5 A cm−2, and that the rate of erosion increases with the current density. It is, therefore, straightforward to assume that more Had (formed on the graphite cathode according to reaction (5)) can penetrate deep into the porosity of graphite material at higher current densities.
Under these conditions, the combination of hydrogen atoms to form hydrogen molecules (reaction (6)) is also likely to occur in the porosity of graphite. Therefore, the molecules formed have a reduced chance of escaping from the graphite electrode, and thus progressively dissolve in graphite.
Theoretical calculations show that H2 with a size of 2.5 Å can diffuse in the interlayer space of graphite faster than atomic hydrogen.10,11,115,116 The energy activation for diffusion of atomic hydrogen in the interlayer space between graphene sheets in the hexagonal structure of graphite can be calculated to be very high (5 eV) owing to its ability to bind to carbon atoms, and therefore, the likelihood of hydrogen intercalation in graphite is low.11
There is no direct bonding between molecular hydrogen and graphite. The diffusion coefficient of molecular hydrogen in graphite does not follow a single straight Arrhenius line (such as eqn (7)). At room temperature, the molecular diffusion proceeds via jumps between the nearest-neighbour adsorption sites in a random walk of the H2 molecule in the interlayer space. At temperatures greater than about 200 °C, however, the hydrogen molecules jump about twice longer and also mostly one-directional, enhancing the effective diffusion length of H2 in graphite.11 At 25 and 800 °C, the diffusion coefficient of H2 in the interlayer space of graphite (DH2) can be extracted from ref. 11 to have the values of 6.9 × 10−6 and 3.5 × 10−4 cm2 s−1, respectively. The activation energy of the later was calculated to be only 0.19 eV.11
The thermal behaviour of hydrogen between graphite layers has been addressed in a number of studies.8,9,11 Molecular-dynamics simulations have shown that the kinetic energy of a H2 molecule in the interlayer space of graphite increases as the temperature rises, so that it was calculated to be 22.9 and 32.7 kJ mol−1 at 25 and about 800 °C, respectively.11
The interlayer binding energy of highly oriented pyrolytic graphite was measured to be about 0.19 J m−2. However, the exfoliation energy, the energy required to remove one graphene layer from single-crystalline graphite, was experimentally measured and the values reported were 0.26126 and 0.32 J m−2.127 The exfoliation energy was also theoretically calculated to be 0.31 J m−2.128
Considering the density and the interlayer space of the as-received graphite to be 2.7 g cm−3 and 0.337 nm, respectively, and using the above values, it can be concluded that a local concentration of more than 2 wt% H2 in graphite is energetically enough to exfoliate graphite into graphene, and thus to the surface erosion of the graphite cathode. Exceeding this critical concentration should be possible during the cathodic polarization of the graphite electrode, considering the fact that the interlayer space in graphite can theoretically accommodate hydrogen molecules up to 6 wt%.129 A higher hydrogen concentration of 7.4 wt% has also been reported in some specific conditions.130 Giving these figures, the exfoliation of graphite cathodes in our experiment can be attributed to the diffusion of hydrogen into the interlayer space of graphite.
According to Fig. 3b, the as-synthesized carbonaceous material contained Li2CO3. The formation of lithium carbonate during the molten salt process can be attributed to the reaction between CO2 formed at the anode (reaction (4)) and the Li2O dissolved in the molten salt (formed by reaction (2)):
| CO2 + Li2O = Li2CO3 ΔG°800 °C = −66.0 kJ | (8) |
As explained, Li2CO3 can be removed from the graphene product by an appropriate heat treatment.
This paper provides insights into the feasibility of large scale preparation of inexpensive but high quality graphene nanosheets by a simple molten salt method, exhibiting at least a 10-fold higher production rate in comparison with that of the currently available methods, such as CVD and wet chemistry techniques. Moreover, industrial grade graphite electrodes which could be used as the raw material for the preparation of graphene by this method are readily available on a large scale with reasonably low costs. Such electrodes are primarily used as the electrodes of electric arc and ladle furnaces in the steel industry and therefore are readily available in different sizes up to about 3 m long and 0.7 m in diameter. The graphene production rate by this method is more than 50 kg per m2 of electrode per day.
Conclusions
The application of a cathodic potential to graphite electrodes immersed in molten LiCl in a moist argon gas flow leads to the formation of a high yield of graphene nanosheets mixed with lithium carbonate. The Li2CO3 was then removed by heating the product at a high temperature, leaving graphene nanosheets. The formation of high quality graphene produced was attributed to the intercalation of hydrogen into the interlayer space of graphite crystallites at the graphite electrode. The graphene nanosheets produced possessed a lateral size of several hundred nanometers and a hexagonal structure of graphene. The process proposed is anticipated to be a simple and efficient method for the large-scale production of graphene nanomaterials.Experimental
Preparation of graphene nanosheets
Anhydrous lithium chloride powder (Sigma Aldrich), 250 g, was placed in a graphite crucible with an internal diameter of 60 mm and a height of 150 mm. The graphite crucible and a graphite rod (Morgan Advanced Materials, industrial grade synthetic graphite) with a diameter of 15 mm and a length of 120 mm were employed as the anode and the cathode, respectively, during the electrochemical process. The temperature was measured by the thermocouple placed inside the graphite crucible. At first, the temperature was raised to about 800 °C, above the melting point of LiCl, by a ramp of 5 °C min−1, under an argon flow of 20 cm3 min−1. At this temperature, a water bath was placed in the path of the argon gas and the flow of the gas was set at 100 cm3 min−1 (see Fig. 1). Then a constant direct current of 33.0 A was applied between the cathode and the anode for about 30 min.The potential difference between both the graphite electrodes and a Mo pseudo-reference electrode immersed in molten LiCl was measured during the electrochemical process. After the molten salt process, the cell was cooled to room temperature, and the product obtained retrieved from the solidified salt by washing with copious amounts of distilled water and vacuum filtering. A black powder was obtained and dried at 150 °C for 2 h. In the second step, the dried material (about 10 g) was heat treated in a horizontal tube furnace under the atmosphere of an inert gas containing hydrogen. The tube furnace was equipped with a thermocouple as well as hydrogen detection and fire detection systems. For this, the temperature was raised to 1300 or 1450 °C at a heating rate of 15 °C min−1 and held at this temperature for 30 min, before cooling down to room temperature. The final product (about 6 g) was a black fluffy powder which was characterized to be graphene nanosheets.
Characterization methods
A JEOL 6340F field emission scanning electron microscope (SEM), a 200 kV JEOL 2000FX analytical transmission electron microscope (TEM) equipped with electron diffraction, and a 200 kV FEI Tecnai F20 field emission gun high resolution TEM (HRTEM) were used for electron microscopy evaluations. A Philips 1710 X-ray diffractometer (XRD) with Cu-Kα radiation (k = 1.54 A°) was used to record the diffraction patterns with a step size and a dwell time of 0.05 2θ and 5 s, respectively. The diffraction patterns recorded were analyzed using the X'Pert High Score Plus program. Raman data were collected using a Renishaw 1000 Ramanscope with a He–Ne ion laser of a wavelength of 633 nm (red, 1.96 eV). The thermal analysis study including thermal gravimetry (TG) and differential scanning calorimetry (DSC) was carried out with 3.3 mg of the as-synthesized carbonaceous material, 3.3 mg graphene nanosheets and 10 mg as-received graphite powder using a thermal analyser model SDT-Q600 equipped with alumina crucibles. The difference in the mass of the materials used was due to the fluffy nature of carbon nanomaterials in comparison with the graphite powder. The thermal analysis was conducted at a heating rate of 40 °C min−1 under a constant air flow rate of 100 mL min−1 through the sample chamber. Elemental analysis was carried out using a CHN/O Analyzer, Model Perkin-Elmer 2400. Brunauer–Emmett–Teller (BET) surface area analysis was performed by recording nitrogen adsorption/desorption isotherms using a static volumetric technique with a Micromeritics TriStar 3000 V6.04 A analyser at −196 °C.Notes and references
- S. Konstantinos, D. Gournis and P. Rudolf, ECS J. Solid State Sci. Technol., 2013, 2, M3160 CrossRef PubMed.
- S. L. Kanashenko, A. E. Gorodetsky, V. N. Chemikov, A. V. Markin, A. P. Zakharov, B. L. Doyle and W. R. Wampler, J. Nucl. Mater., 1996, 233–237, 1207 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Computational high-throughput screening of electrocatalyticmaterials for hydrogen evolution, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed.
- V. L. Page, T. P. Snow and V. M. Bierbaum, Astrophys. J., 2009, 704, 274 CrossRef.
- O. E. Haas, J. M. Simon and S. Kjelstrup, J. Phys. Chem. C, 2009, 113, 20281 CAS.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS PubMed.
- M. Warrier, R. Schneider, E. Salonen and K. Nordlund, Nucl. Fusion, 2007, 47, 1656 CrossRef CAS.
- M. Warrier, R. Schneider, E. Salonen and K. Nordlund, Phys. Scr., 2004, T108, 85 CrossRef CAS.
- A. Shimizu and H. Tachikawa, J. Phys. Chem. Solids, 2003, 64, 419 CrossRef CAS.
- H. Atsumi, J. Nucl. Mater., 2002, 307–311, 1466 CrossRef CAS.
- C. P. Herrero and R. Ramirez, J. Phys. D: Appl. Phys., 2010, 43, 255402 CrossRef.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312 CrossRef CAS PubMed.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191 CrossRef CAS PubMed.
- X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han, D. Chen, R. D. Piner, L. Colombo and R. S. Ruoff, Nano Lett., 2009, 9, 4359 CrossRef CAS PubMed.
- W. Cai, Y. Zhu, X. Li, R. D. Piner and R. S. Ruoff, Appl. Phys. Lett., 2009, 95, 123115 CrossRef PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS PubMed.
- Y. NuLi, P. Zhang, Z. Guo, H. Liu, J. Yang and J. J. Wang, J. Mater. Res. Bull, 2009, 44, 140 CrossRef CAS PubMed.
- X. Ma and B. Yuan, Appl. Surf. Sci., 2009, 255, 7846 CrossRef CAS PubMed.
- B. Aïssa, Z. Hamoudi, H. Takahashi, K. Tohji, M. Mohamedi and M. A. E. Khakani, Electrochem. Commun., 2009, 11, 862 CrossRef PubMed.
- S. Shimada, K. Teii and M. Nakashima, Diam. Relat. Mater., 2010, 19, 956 CrossRef CAS PubMed.
- Q. Kuang, S. Y. Xie, Z. Y. Jiang, X. H. Zhang, Z. X. Xie, R. B. Huang and L. S. Zheng, Carbon, 2004, 42, 1737 CrossRef CAS PubMed.
- M. Y. Zhu, R. A. Outlaw, M. Bagge-Hansen, H. J. Chen and D. M. Manos, Carbon, 2011, 49, 2526 CrossRef CAS PubMed.
- L. Hou, L. Lian, D. Li, G. Pang, J. Li, X. Zhang, S. Xiong and C. Yuan, Carbon, 2013, 64, 149 CrossRef PubMed.
- B. Jia and L. Zou, Carbon, 2012, 50, 2315 CrossRef CAS PubMed.
- A. R. Kamali and D. J. Fray, J. New Mater. Electrochem. Syst., 2010, 13, 147 CAS.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049 CrossRef CAS PubMed.
- S. Yang, C. Shen, X. Lu, H. Tong, J. Zhu, X. Zhang and H. J. Gao, Electrochim. Acta, 2012, 62, 242 CrossRef CAS PubMed.
- Z. Wang, M. Shoji and H. Ogata, Talanta, 2012, 99, 487 CrossRef CAS PubMed.
- Z. Wang, M. Shoji and H. Ogata, Appl. Surf. Sci., 2012, 259, 219 CrossRef CAS PubMed.
- S. Ghasemi, S. R. Setayesh, A. Habibi-Yangjeh, M. R. Hormozi-Nezhad and M. R. Gholami, J. Hazard. Mater., 2012, 199–200, 170 CrossRef CAS PubMed.
- C. Zhang, Y. Wang and L. Huang, Appl. Phys. Lett., 2010, 97, 062102 CrossRef PubMed.
- Q. He, S. Wu, Z. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764 RSC.
- H. Wanga, Y. Su, S. Chen and X. Quan, Mater. Res. Bull., 2013, 48, 1304 CrossRef PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS PubMed.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229 CrossRef CAS PubMed.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912 CrossRef CAS.
- B. Puangbuppha, P. Limsuwan and P. Asanithi, Procedia Eng., 2012, 32, 1094 CrossRef CAS PubMed.
- D. Li, M. Muller, S. Gilje, R. Kaner and G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS PubMed.
- S. Gilje, S. Han, M. Wang, K. Wang and R. Kaner, Nano Lett., 2007, 7, 3394 CrossRef CAS PubMed.
- G. Compagnini, M. Sinatra, P. Russo, G. C. Messina, O. Puglisi and S. Scalese, Carbon, 2012, 50, 2347 CrossRef PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- Y. Hong, Z. Wang and X. Jin, Sci. Rep., 2013, 3, 3439 Search PubMed.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. GunKo, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. O. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- J. S. Bunch, Y. Yaish, M. Brink, K. Bolotin and P. L. McEuen, Nano Lett., 2005, 5, 287 CrossRef CAS PubMed.
- P. Blake, P. D. Brimicombe, P. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704 CrossRef PubMed.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14 CrossRef CAS PubMed.
- B. Zhang, J. Song, G. Yang and B. Han, Chem. Sci., 2014, 5, 4656 RSC.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill1, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed.
- X. Liu, M. Zheng, K. Xiao, Y. Xiao, C. He, H. Dong, B. Lei and Y. Liu, Nanoscale, 2014, 6, 4598 RSC.
- Y. Zhang, L. Zhang and C. Zhou, Acc. Chem. Res., 2013, 46, 2329 CrossRef CAS PubMed.
- Z. Bo, Y. Yang, J. Chen, K. Yu, J. Yan and K. Cen, Nanoscale, 2013, 5, 5180 RSC.
- J. Zhang, I. Khatri, N. Kishi, S. M. Mominuzzaman, T. Soga and T. Jimbo, Thin Solid Films, 2011, 519, 4162 CrossRef CAS PubMed.
- M. Choucair, P. Thordarson and J. A. Stride, Nat. Nanotechnol., 2009, 4, 30 CrossRef CAS PubMed.
- T. Mori, M. Hiramatsu, K. Yamakawa, K. Takeda and M. Hori, Diam. Relat. Mater., 2008, 17, 1513 CrossRef CAS PubMed.
- J. Lu, J. X. Yang, J. Z. Wang, A. L. Lim, S. Wang and K. P. Loh, ACS Nano, 2009, 3, 2367 CrossRef CAS PubMed.
- G. X. Wang, B. Wang, J. Park, Y. Wang, B. Sun and J. Yao, Carbon, 2009, 47, 3242 CrossRef CAS PubMed.
- M. Mao, M. Wang, J. Hu, G. Lei, S. Chen and H. Liu, Chem. Commun., 2013, 49, 5301 RSC.
- H. Huang, Y. Xia, X. Tao, J. Du, J. Fang, Y. Gan and W. Zhang, J. Mater. Chem., 2012, 22, 10452 RSC.
- P. Masset, ECS Trans., 2010, 25, 155 CAS.
- A. R. Kamali, D. J. Fray and C. Schwandt, J. Therm. Anal. Calorim., 2011, 104, 619 CrossRef CAS PubMed.
- O. Takeda, M. Li, T. Toma, K. Sugiyama, M. Hoshi and Y. Sato, J. Electrochem. Soc., 2014, 161, D820 CrossRef CAS PubMed.
- Y. Castrillejo, C. de la Fuente, M. Vega1, F. de la Rosa, R. Pardo and E. Barrado, Electrochim. Acta, 2013, 97, 120 CrossRef CAS PubMed.
- P. Masset and R. A. Guidotti, J. Power Sources, 2007, 164, 397 CrossRef CAS PubMed.
- P. Masset, J. Y. Poinso and J. C. Poignet, J. Power Sources, 2004, 137, 140 CrossRef CAS PubMed.
- R. A. Causey, J. Nucl. Mater., 1989, 162, 151 CrossRef.
- C. Shih, A. Vijayaraghavan, R. Krishnan, R. Sharma, J. H. Han, M. H. Ham, Z. Jin, S. Lin, G. L. C. Paulus, N. F. Reuel, Q. H. Wang, D. Blankschtein and M. l. S. Strano, Nat. Nanotechnol., 2011, 6, 439 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio and R. Saito, Annu. Rev. Condens. Matter. Phys., 2010, 89, 108 Search PubMed.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, A. Jorio, L. N. Coelho, R. Magalhães-Paniago and M. A. Pimenta, Appl. Phys. Lett., 2006, 88, 163106 CrossRef PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef CAS PubMed.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CAS.
- F. Wah Low, C. W. Lai and S. B. Abd Hamid, Ceram. Int., 2015, 41, 5798 CrossRef PubMed.
- Z. Pan and R. T. Yang, Ind. Eng. Chem. Res., 1992, 31, 2675–2680 CrossRef CAS.
- X. Song, J. Liu, L. Yu, J. Yang, L. Fang, H. Shi, C. Du and D. Wei, Mater. Lett., 2014, 137, 25 CrossRef CAS PubMed.
- Y. Tzeng, W. L. Chen, C. Wu, J. Y. Lo and C. Y. Li, Carbon, 2013, 53, 120 CrossRef CAS PubMed.
- E. Dervishi, Z. Li, F. Watanabe, A. Biswas, Y. Xu, A. R. Biris, V. Saini and A. S. Biris, Chem. Commun., 2009, 4061 RSC.
- C. H. Tu, W. Chen, H. C. Fang, Y. Tzeng and C. P. Liu, Carbon, 2013, 54, 234 CrossRef CAS PubMed.
- D. A. C. Brownson, M. Gómez-Mingot and C. E. Banks, Phys. Chem. Chem. Phys., 2011, 13, 20284 RSC.
- D. A. C. Brownson, S. A. Varey, F. Hussain, S. J. Haigh and C. E. Banks, Nanoscale, 2014, 6, 1607 RSC.
- J. Chen, Y. Li, L. Huang, C. Li and G. Shi, Carbon, 2015, 81, 826 CrossRef CAS PubMed.
- F. T. Johra, J. W. Lee, W. G. Jung and F. Tuz, J. Ind. Eng. Chem., 2014, 20, 2883 CrossRef CAS PubMed.
- Z. Chen, T. Huang, B. C. Jin, J. Hu, H. Lu and S. Nutt, Carbon, 2014, 68, 167 CrossRef CAS PubMed.
- X. Mei, X. Meng and F. Wu, Physica E, 2015, 68, 81 CrossRef CAS PubMed.
- W. Yoon, Y. Lee, H. Jang, M. Jang, J. S. Kim, H. S. Lee, S. Im, D. W. Boo, J. Parkc and S. Y. Jua, Carbon, 2015, 81, 629 CrossRef CAS PubMed.
- Y. Zhang, H. L. Ma, Q. Zhang, J. Peng, J. Li, M. Zhai and Z. Z. Yu, J. Mater. Chem., 2012, 22, 13064 RSC.
- B. Shen, D. Lu, W. Zhai and W. Zheng, J. Mater. Chem. C, 2013, 1, 50 RSC.
- S. Some, S. Kim, K. Samanta, Y. Kim, Y. Yoon, Y. Park, S. M. Lee, K. Lee and H. Lee, Nanoscale, 2014, 6, 11322 RSC.
- Y. Liu, Y. Ma, Y. Jin, G. Chen and X. Zhang, J. Electroanal. Chem., 2015, 739, 172 CrossRef CAS PubMed.
- S. V. Rodil, J. I. Paredes, A. M. Alonso and J. M. D. Tascón, J. Mater. Chem., 2009, 19, 3591 RSC.
- L. Feng, G. Gao, P. Huang, X. Wang, C. Zhang, J. Zhang, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6, 551 CrossRef PubMed.
- F. W. Low, C. W. Lai and S. B. Abd Hamid, Ceram. Int., 2015, 41, 5798 CrossRef CAS PubMed.
- Z. Ji, J. Chen, L. Huang and G. Shi, Chem. Commun., 2015, 51, 2806 RSC.
- B. Subramanya and D. Krishna Bhat, J. Power Sources, 2015, 275, 90 CrossRef CAS PubMed.
- H. G. Xu and K. S. Suslick, J. Am. Chem. Soc., 2011, 133, 9148 CrossRef CAS PubMed.
- J. Li, F. Ye, S. Vaziri, M. Muhammed, M. C. Lemme and M. Ostling, Carbon, 2012, 50, 3092 CrossRef PubMed.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155 CrossRef CAS PubMed.
- G. S. Bang, H. M. So, M. J. Lee and C. W. Ahn, J. Mater. Chem., 2012, 22, 4806 RSC.
- W. Liu, R. Zhou, D. Zhou, G. Ding, J. M. Soah, C. Y. Yue and X. Lu, Carbon, 2015, 83, 188 CrossRef CAS PubMed . http://www.sciencedirect.com/science/article/pii/S0008622314011087.
- M. Choucair, P. Thordarson and J. A. Stride, Nat. Nanotechnol., 2006, 4, 30 CrossRef PubMed.
- B. Zhang, J. Song, G. Yang and B. Han, Chem. Sci., 2014, 5, 4656 RSC.
- L. Wang, Q. Yao, H. Bi, F. Huang, Q. Wang and L. Chen, J. Mater. Chem. A, 2014, 2, 11107 CAS.
- K. H. Liao, Y. S. Lin, C. W. Macosko and C. L. Haynes, Appl. Mater. Interfaces, 2011, 3, 2607 CrossRef CAS PubMed.
- A. R. Kamali and D. J. Fray, Ceram. Int., 2014, 40, 1835 CrossRef CAS PubMed.
- H. A. Laitinen, Y. Yamamura and I. Uchida, J. Electrochem. Soc., 1978, 125, 1450 CrossRef CAS PubMed.
- J. B. Raynor, Ber. Bunsen-Ges. Phys. Chem., 1963, 67, 360 CrossRef CAS PubMed.
- W. J. Burkhard and J. D. Corbett, J. Am. Chem. Soc., 1957, 79, 6361 CrossRef CAS.
- N. Q. Minh and B. J. Welch, Aust. J. Chem., 1975, 28, 965 CrossRef.
- N. Q. Minh and B. J. Welch, Aust. J. Chem., 1975, 28, 2579 CrossRef.
- N. Q. Minh and B. J. Welch, J. Electroanal. Chem., 1978, 92, 179 CrossRef.
- J. D. Van Norman and R. J. Tivers, J. Electrochem. Soc., 1971, 118, 258 CrossRef CAS PubMed.
- Y. Sakamura, J. Electrochem. Soc., 2010, 157, E135 CrossRef CAS PubMed.
- M. H. F. Sluiter and Y. Kawazoe, Phys. Rev. B: Condens. Matter, 2003, 68, 085410 CrossRef.
- W. A. Dino, Y. Miura, H. Nakanishi, H. Kasai and T. Sugimoto, J. Phys. Soc. Jpn., 2003, 72, 1867 CrossRef CAS.
- Y. Ferro, F. Marinelli and A. Allouche, Chem. Phys. Lett., 2003, 368, 609 CrossRef CAS.
- S. Casolo, O. M. Lovvik, R. Martinazzo and G. F. Tantardini, J. Chem. Phys., 2009, 130, 054704 CrossRef PubMed.
- D. W. Boukhvalov, M. I. Katsnelson and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter, 2008, 77, 035427 CrossRef.
- C. P. Herrero and R. Ramirez, Phys. Rev. B: Condens. Matter, 2009, 79, 115429 CrossRef.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850 CrossRef CAS PubMed.
- J. C. F. Boodts and S. Trasatiti, J. Appl. Electrochem., 1989, 19, 255 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- J. Bonde, P. G. Moses, T. F. Jaramillo, J. K. Nørskov and I. Chorkendorff, Faraday Discuss., 2008, 140, 219 RSC.
- L. A. Girifalco and R. A. Lad, J. Chem. Phys., 1956, 25, 693 CrossRef CAS PubMed.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter, 2004, 69, 155406 CrossRef.
- T. Gould, S. Lebègue and J. Dobson, J. Phys.: Condens. Matter., 2013, 25, 445010 CrossRef PubMed.
- R. Strobel, J. Garche, P. T. Moseley, L. Jorissen and G. Wolf, J. Power Sources, 2006, 159, 781 CrossRef PubMed.
- S. Orimo, G. Majer, T. Fukunaga, A. Zuttel, L. Schlapbach and H. Fujii, Appl. Phys. Lett., 1999, 75, 3093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr01132a |
| This journal is © The Royal Society of Chemistry 2015 |