
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrarapid and ultrasensitive electrical detection of proteins in a three-dimensional biosensor with high capture efficiency†
Bo-Yeong
Kim
a,
Il-yung
Sohn
b,
Doowon
Lee
b,
Gill Sang
Han
b,
Won-Il
Lee
c,
Hyun Suk
Jung
b and
Nae-Eung
Lee
*abc
aSKKU Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, Suwon, Gyeonggi-do 440-746, South Korea
bDepartment of Advanced Materials Science & Engineering, Sungkyunkwan University, Suwon, Gyeonggi-do 440-746, South Korea
cSamsung Advanced Institute for Health Sciences and Technology (SAIHST), Sungkyunkwan University, Suwon, Gyeonggi-do 440-746, South Korea. E-mail: nelee@skku.edu
First published on 27th April 2015
Abstract
The realization of a high-throughput biosensor platform with ultrarapid detection of biomolecular interactions and an ultralow limit of detection in the femtomolar (fM) range or below has been retarded due to sluggish binding kinetics caused by the scarcity of probe molecules on the nanostructures and/or limited mass transport. Here, as a new method for the highly efficient capture of biomolecules at extremely low concentration, we tested a three-dimensional (3D) platform of a bioelectronic field-effect transistor (bio-FET) with vertically aligned and highly dense one-dimensional (1D) ZnO nanorods (NRs) as a sensing surface capped by an ultrathin TiO2 layer for improved electrolytic stability on a chemical-vapor-deposited graphene (Gr) channel. The ultrarapid detection capability with a very fast response time (∼1 min) at the fM level of proteins in the proposed 3D bio-FET is primarily attributed to the fast binding kinetics of the probe–target proteins due to the small diffusion length of the target molecules to reach the sensor surface and the substantial number of probe molecules available on the largely increased surface area of the vertical ZnO NRs. This new 3D electrical biosensor platform can be easily extended to other electrochemical nanobiosensors and has great potential for practical applications in miniaturized biosensor integrated systems.
Introduction
Extensive research effort has been made to develop fast, highly sensitive, and miniaturized biosensors for point-of-care diagnostics.1–5 In particular, the use of nanostructured materials in electrochemical6–16 and gravimetric biosensors17 has attracted great attention due to the possibility of obtaining high sensitivity, low limit of detection (LOD), and real-time and label-free detection, because nanostructured materials show extremely high sensitivity to bimolecular interactions. The detection of extremely dilute target molecules at the femtomolar (fM) level or below is of great interest for clinical diagnostics of diseases with low concentration biomarkers and for the detection of secreted biomolecules from a single cell.14,17–19 However, the development of these high-throughput nanobiosensor platforms with ultralow LODs at the fM level or below and with high sensitivity and ultrarapid detection (within a minute) of biomolecular interactions has not been achieved because the binding kinetics of probe–target molecules are limited by the small number of probe molecules on the small surface of the nanostructures and/or limited mass transport to the sensor's surface.1 To achieve ultrarapid and ultrasensitive detection in the minute range at target concentrations at the fM level, it is essential to develop engineered probe molecules with high affinity and selectivity, to increase the immobilized density of probe molecules in a given sensor area and/or to optimize the sensor geometry to increase mass transport for fast binding kinetics of target molecules with probe molecules.1,17,20–22The binding kinetics of the probes and target molecules on nanosensors or microsensors in lateral flow systems is generally described by the Damköhler number, Da, which is the ratio of reactive to diffusive flux.1,17 The Damköhler number can be expressed as Da = konbmAs/(JD/Co), where kon is the rate of association, bm is the surface density of probe molecules on the sensor surface, As is the surface area of the sensor (here, ZnO nanorods (NRs)), JD is the collection rate of target molecules reaching the sensor surface, and Co is the concentration of target molecules.1,17 For Da ≫ 1, the binding kinetics are entirely mass-transport limited, while for Da ≪ 1, the kinetics of capture become reaction limited. Experimentally, one-dimensional (1D) bioelectronic field-effect transistors (bio-FETs) based on a single Si nanowire (NW) showed ultrasensitive detection of proteins and deoxyribonucleic acids (DNAs) down to concentrations in the fM range and with response times of several tens of minutes.23–25 However, other experiments and numerical considerations on the protein binding kinetics on Si NW bio-FETs showed contradictory results with response times of several tens of minutes at pM levels or above.17,20,26,27 For example, numerical simulations on 1D NW nanosensors in the microfluidic channel indicated that the binding kinetics are reaction limited.1 It may take several days for one target molecule to be attached to the single NW surface at the fM level because binding events of the target and probe molecules rarely happen at extremely low Co due to the scarcity of probe molecules on the small area of Si NW giving a very small bmAs value.1 A lack of saturation in the sensing signal in 1D nanobiosensors can limit the LOD during detection of protein–protein interactions. To expedite the detection speed with fast binding kinetics at extremely low Co therefore, it is important to develop new approaches to simultaneously increase the bmAs value and the diffusion flux, J_D, simultaneously.20
Bio-FETs based on a two-dimensional (2D) nanomaterial channel with a microscale sensing area are capable of achieving ultrarapid detection of biomolecular interactions due to their relatively large bmAs values; they have a larger number of probe molecules on the sensing channel. In this sense, 2D bio-FETs based on graphene (Gr),9,11,12,28,29 reduced graphene oxide (rGO)30,31 and MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32,33 have potential as biosensors for the rapid detection of protein and DNA interactions. For example, a 2D bio-FET based on a large area rGO channel showed a response time of about 12 minutes for the detection of the prostate specific antigen/1-antichymotrypsin (PSA–ACT) complex, a protein biomarker for prostate cancer, at the 1 fM level.30
32,33 have potential as biosensors for the rapid detection of protein and DNA interactions. For example, a 2D bio-FET based on a large area rGO channel showed a response time of about 12 minutes for the detection of the prostate specific antigen/1-antichymotrypsin (PSA–ACT) complex, a protein biomarker for prostate cancer, at the 1 fM level.30
Even though there is still room for improvement in the detection speed of extremely dilute target molecules, there have been no such efforts for the development of bio-FETs having the capability of achieving ultrarapid detection with response time near one minute. As a new method, three-dimensional (3D) channel structures with vertical, highly dense 1D NWs or NRs on a microscale channel in bio-FETs are interesting because the surface area of the channels can be increased dramatically for the functionalization of a large number of probe molecules in a given device area, and the diffusion distance of probe molecules to reach the probe-functionalized surface of the vertical 1D nanostructures can be reduced to the nanoscale level for effective mass transport. However, to our knowledge, there have been no such studies on immunosensing using bio-FETs with 3D channel structures.
Herein, we demonstrate a 3D bio-FET with vertical, highly dense ZnO NR sensing materials on a chemical vapor-deposited (CVD) Gr channel which is an excellent substrate for directional growth of ZnO NRs and their ambipolar characteristics enable one to extract the sensing parameters more clearly. Furthermore, the conductance of the CVD Gr channel can be easily modulated by the electrical responses from biomolecular interactions on the surface of the ZnO NRs because of the high electrical coupling between Gr and ZnO NRs.34 A 3D bio-FET with vertically grown ZnO NRs on a Gr channel was successfully fabricated with ambipolar characteristics. The fabricated 3D bio-FET was used to detect the target analytes of the PSA–ACT complex. We obtained a sensitivity of ∼8 mV dec−1 from the charge neutrality point (CNP), a dynamic range of 107, a LOD of 100 fg ml−1 (∼1 fM), and a detection time of 64 s at a target concentration at the fM level. The 3D bio-FET showed significant improvements in sensitivity, dynamic range, and response time compared with other 1D or 2D bio-FETs.
Results and discussion
A schematic illustration of a 3D channel bio-FET with vertically grown ZnO NRs on the CVD Gr of the SiO2/Si wafer and biofunctionalization with PSA–ACT complex probe molecules is shown in Fig. 1a and b. The channel length between the source and drain electrodes of the 3D channel FET was set at 300 μm and the channel width was 6000 μm Details of the fabrication process of a 3D bio-FET (Fig. S1†) are explained in the Experimental section. The single layer graphene (SLG) on the SiO2/Si wafer as a transport channel was transferred using a gold (Au) transfer method previously developed by our group, which provided an ultraclean surface for further surface functionalization and high electrical performance.29,35 Growth of ZnO NRs on the Gr channel was achieved using a solution growth method.36–40 Details of the SLG synthesis and transfer process are provided in the Experimental section. Cross-sectional and top-view images of the grown ZnO NRs obtained by field-emission scanning electron microscopy (FE-SEM) indicated the average height (H) and diameter of ZnO NRs at ∼2.5 μm and ∼170 nm, respectively (Fig. 1c). Assuming a uniform distribution of ZnO NRs on the channel area, the ratio of the total sensing surface area of ZnO NRs (As) to the device channel area Ac, As/Ac, was estimated to be ∼23, which suggests a dramatic increase in the sensing area of 3D ZnO NRs compared to that of 2D Gr. Details of characterization of the CVD Gr and ZnO NRs are described in Fig. S2.† A further important feature of this fabrication method is the passivation of the source and drain electrodes using a photo-patternable SU-8 epoxy from the electrolyte to minimize the leakage current between the electrolyte and the electrodes and attachment of the probe molecules on the source–drain electrodes (see the details in the Experimental section). Due to the instability of ZnO NRs in aqueous environments,41 the surface of ZnO NRs was also capped by an ultrathin TiO2 layer (∼1 nm)42via atomic layer deposition (ALD) (Fig. S3†). The image of a TiO2-encapped ZnO NR obtained by high-resolution transmission electron microscopy (Fig. 1d) confirmed the ultrathin TiO2 layer on the ZnO NR surface. | ||
| Fig. 1 (a) Schematic illustration of a 3D channel FET biosensor. The ZnO NRs were vertically grown on the Gr channel area. The source/drain electrodes are electrically isolated from the analyte solution by passivation of a thin Al2O3 layer and SU-8 pattern. (b) Schematic showing the immobilization of antibody probe molecules on the surface of TiO2-encapped ZnO NR. (c) Cross-sectional FE-SEM image of ZnO NRs grown on CVD Gr (inset: top view FE-SEM image). ZnO NRs approximately 2.5 μm in length with a hexagonal shape were grown on Gr using a solution growth method. (d) Micrograph of a TiO2-encapped ZnO NR with the selected area diffraction pattern as an inset (left) and high-resolution image of the ultrathin TiO2 layer capping the ZnO NR (right) obtained by transmission electron microscopy. | ||
To understand the fundamental characteristics of solution-gated 3D channel FETs, their responses to solutions with different ionic concentrations were investigated. The transfer characteristics, the source–drain current (IDS) versus the gate bias voltage (VG), of 3D channel FETs and Gr FETs measured in sodium phosphate buffer solutions with various ionic concentrations at a fixed pH 7.4 are shown in Fig. S4a and S4b,† respectively. Interestingly, the 3D channel FET showed excellent ambipolar transfer characteristics similar to those of Gr FET. For both 3D and 2D Gr channel FETs, the transfer curve shifted to a more negative voltage when the ionic concentration of the buffer solutions was increased, and changes in the conductivity and CNP occurred at the same time.43 The electrical double layer (EDL) thickness decreased when the ionic concentration increased, accompanied by an increase of the EDL capacitance value, resulting in a reduced surface potential and, in turn, a shift of the charge neutrality point (CNP) in the negative direction.44 Similar behaviors were reported for electrolyte-gated SLG FETs, which were explained by electrostatic gating effects.43,45–47 The sensitivity of the 3D channel FET to ionic concentration was notably higher than that of Gr FET (Fig. S4c†). The increased responsivity of the 3D channel FET to ionic strength is attributed to the increased surface area of the 3D channel due to the high density, high aspect-ratio ZnO NRs on the Gr layer.
In bio-FETs used for immunosensing, having a high density of probe proteins on the channel surface is critical for enhancing the sensitivity, LOD, and dynamic range. Details of the immobilization procedure are described in Fig. S5.† To confirm the effective immobilization of the probe proteins on the surface of TiO2-encapped ZnO NRs at a high density before and after oxygen plasma treatments,48 biotin–streptavidin complexes were used as a test of protein interactions. For this purpose, streptavidin was first immobilized as a probe protein on TiO2-encapped ZnO NRs using the primary amine group in the protein, and the ZnO NRs were incubated with the biotin-AuNP solution for 30 min. After the binding of streptavidin molecules and biotin-AuNPs, the TiO2-encapped ZnO NRs with and without plasma treatments were characterized by FE-SEM (Fig. 2a and b). Clearly, the density of biotin-AuNPs is much higher after (Fig. 2b) oxygen plasma treatment than before (Fig. 2a), due to the increased density of streptavidin probe molecules immobilized onto the plasma-treated surface. The surface density of AuNPs on TiO2-capped ZnO NRs was ∼103 μm−2, which is comparable to that on the Gr (inset in Fig. 2b). The calculated density of AuNPs on the surface of the 3D channel for a given channel area of 1 μm2 was higher than that on the surface of the Gr channel by a factor of 32 (Fig. 2c). After efficient functionalization of the TiO2-encapped ZnO NRs by the streptavidin protein was confirmed, immunosensing of the prostate specific antigen/1-antichymotrypsin (PSA–ACT) complex antigen (Ag) as an analyte and the PSA–ACT antibody (Ab) as a probe was carried out. The PSA–ACT complex is widely used as a biomarker for the diagnosis of prostate cancer, because PSA is generated from prostatic tissue. Typically, men without prostate cancer have PSA levels below 4.0 ng ml−1, while men with prostate cancer have PSA concentrations above 20.0 ng ml−1.49 Early diagnosis is the best way to reduce patient mortality, and quantitative monitoring of PSA is important to patients. Detection of the PSA–ACT complex antigen (PSA–ACT complex Ag) was carried out by static and kinetic measurements. The PSA–ACT Ab was immobilized in the same way as shown in Fig. S5.† The transfer characteristics before and after immobilizing PSA–ACT Ab probe molecules on 3D and 2D channel FETs are shown in Fig. S6a and S6b,† respectively. Larger decreases in the IDS and CNP of 3D channel FET were observed compared to those of the 2D channel FET. These results obtained from the 3D channel FET (Fig. 3) may explain the high sensitivity and the wide dynamic range that are attributed to the largely increased density of immobilized Ab probe molecules over the same channel area. Since the charges of proteins are minimally detected by charge screening effects outside the Debye screening length, the analyte solution was prepared by controlling the amount of the PSA–ACT complex Ag in the diluted phosphate buffered saline (PBS) solution.30,31,50 Accordingly, we carried out immunosensing measurements by diluting analyte molecules in a 1 μm PBS solution, pH 7.4, with a Debye length of ∼70 nm, which is larger than that of the combined PSA–ACT Ab–Ag (∼15 nm). All measurements were carried out at a VG of 1 V to avoid electrolysis by a high gate voltage sweep and after blocking using ethanolamine at 100 mM to prevent interactions between unreacted −CHO groups of glutaraldehyde and the PSA–ACT complex Ag.
 | ||
| Fig. 2 The FE-SEM images were observed to confirm the binding of gold nanoparticles conjugated with biotin (biotin-AuNPs) and streptavidin functionalized on the TiO2 surface: (a) before and (b) after O2 plasma treatment of TiO2/ZnO NRs, respectively. The inset in Fig. 4b was obtained after binding of biotin-AuNPs with streptavidin functionalized on the Gr surface using PBSE linker molecules. (c) The density of biotin-AuNP biomolecules of 3D and Gr channel FETs at a channel surface area of 1 μm2. The density of biotin-AuNPs indirectly reflects the density at which probe biomolecules are immobilized on the sensing channels. | ||
 | ||
| Fig. 3 Transfer characteristics before and after adding different concentrations of PSA–ACT complex Ag molecules from 100 fg ml−1 to 100 ng ml−1 for (a) 3D channel Gr FETs (TiO2-encapped ZnO NRs/Gr) and (b) Gr FETs. (c) The shift in charge neutrality point (CNP) as a function of analyte concentration, Co. (d) Drain current (IDS) of 3D and Gr channel FETs as a function of analyte concentration, Co. The CNP was negatively shifted upon increasing the concentration of PSA–ACT complex Ag. Increased IDS and negative CNP shift (ΔVCNP) with increasing the concentration of PSA–ACT complex Ag molecules were obtained from transfer curves like the ones in (a) and (b) at a gate voltage (VG) of 0.6 V in the linear region. The data in (c) and (d) were averaging the values obtained from five bio-FETs. | ||
The transfer characteristics of 3D and 2D bio-FETs with the analyte concentration, Co, of the PSA–ACT complex Ag from 100 fg ml−1 (1.1 fM) to 100 ng ml−1 (1.1 nM) at VDS = 0.1 V, respectively, are shown in Fig. 3a and b. The transfer curves of both solution-gated bio-FETs were obtained over 10 min while simultaneously adding the analyte solution. For both 3D and 2D bio-FETs, the CNP in the transfer curves (VCNP) shifted to negative VG; the current gradually increased with the increase of the PSA–ACT complex Ag concentration. However, the transfer characteristics of the two solution-gated bio-FETs demonstrated a clear difference in the degree of shift of CNP. The VCNP in both FETs was quantified and plotted as a function of the PSA–ACT complex Ag concentration (Fig. 3c). The shift in the CNP (ΔVCNP) can be used as a sensitivity parameter due to its superior linearity and the large dynamic range of 107. The use of ZnO NRs on the Gr channel significantly increased the VCNP from ∼2.3 mV dec−1 in the Gr FET to ∼8 mV dec−1 in the 3D FET, indicating that the transduction of biomolecular interactions into the electrical conductance change of the Gr channel intensified using the vertical ZnO NRs. The CNP shift is suggested to occur via an electrostatic gating effect, as was previously described in CNT or Si NW bio-FETs.51 If electrostatic gating effects occur, the CNP would be shifted in the positive direction, as the negative charges of bound PSA–ACT Ag molecules increase at pH 7.4. The PSA–ACT complex Ag is negatively charged above its isoelectric point of 6.8. Negative shifts in CNP with the concentration of target Ag molecules (Fig. 3a) suggest electron doping, which contrasts with hole doping by the electrostatic gating effect. The same phenomenon was observed with the Gr FET (Fig. 3b and c) and has also been reported for rGO FET30,31 and CVD Gr FET28,29,52 during protein or DNA interactions. Electron doping in the 3D channel FET is ascribed to the transfer of electrons through the ZnO NRs from biomolecules. Considering the difficulty of direct transfer of biomolecular charges to Gr through ZnO NRs, however, the effects of biomolecular charges on the Fermi level of the n-type ZnO NRs are plausible, because the electron affinities of ZnO NRs (∼−4.4 eV) and Gr (∼−4.3 eV) are nearly equal. Transfer of electrons from ZnO NRs to the Gr channel increases the Fermi level of Gr, which results in a negative CNP shift. A detailed understanding of the sensing mechanism requires further theoretical and experimental studies. Moreover, the 3D channel FET immobilized with PSA–ACT Ab molecules did not show notable tendencies in the shifts of CNP and IDS upon adding a carcinoembryonic antigen (CEA) solution with the wide range of concentrations ranging from 100 fg ml−1 to 100 ng ml−1 during the reaction time of 10 min (Fig. S7†). The results indicate that PSA–ACT Ab–Ag binding in the 3D channel FET was specific.
The changed IDS values were plotted against increasing PSA–ACT complex Ag concentrations for the two types of FETs (Fig. 3d). The IDS change is obtained from the linear region in the transfer curves at VG = 0.6 V. The IDS modulation data also show good linearity in the 3D FET compared with that in the 2D FET. Therefore, modulation of IDS can also be used as a sensitivity factor during immunosensing. The modulation of IDS (IDS/IDS,min) in the 3D FET (∼2.18% dec−1) was larger than that in the 2D FET (∼0.3% dec−1) upon increasing the PSA–ACT complex protein Ag. These results support the potential of our developed 3D FET for immunosensing.
Kinetic measurements were employed to detect PSA–ACT complex Ag by stepwise addition of the Ag molecules. Measurements were carried out at fixed VDS = 0.1 and VG = 0.6 V. There were significant changes in the IDS of the 3D bio-FET upon adding analyte solutions in low concentration ranges (Fig. 4a), but no significant changes were observed in the IDS of the Gr channel FET (Fig. S8†). The IDS in the 3D bio-FET increased in a stepwise fashion when analyte solutions with different concentrations were added. When the same volume of PBS solution without analytes was added into the PDMS well on the 3D FET, the IDS was not modulated (Fig. S9†). Furthermore, the 3D channel FET was applied to detect the target molecules of PSA–ACT Ag in diluted human serum (0.1%) and the measured data for the concentrations of 100 fg ml−1, 100 pg ml−1 and 100 ng ml−1 of the PSA–ACT complex in human serum are shown in Fig. S10a.† The results in Fig. S10a† indicate that the sensor signals in diluted human serum are similar to those of the specific PSA–ACT Ab–Ag binding reaction in diluted PBS solution. However, the sensor responses for the minimum 100 fg ml−1 concentration of the PSA–ACT complex Ag in 1.0 and 10% diluted human serums are negligible (Fig. S10b†), which is attributed to the charge screening of PSA–ACT Ag molecules in the less diluted human serums having larger ionic strength and, in turn, shorter Debye screening length.
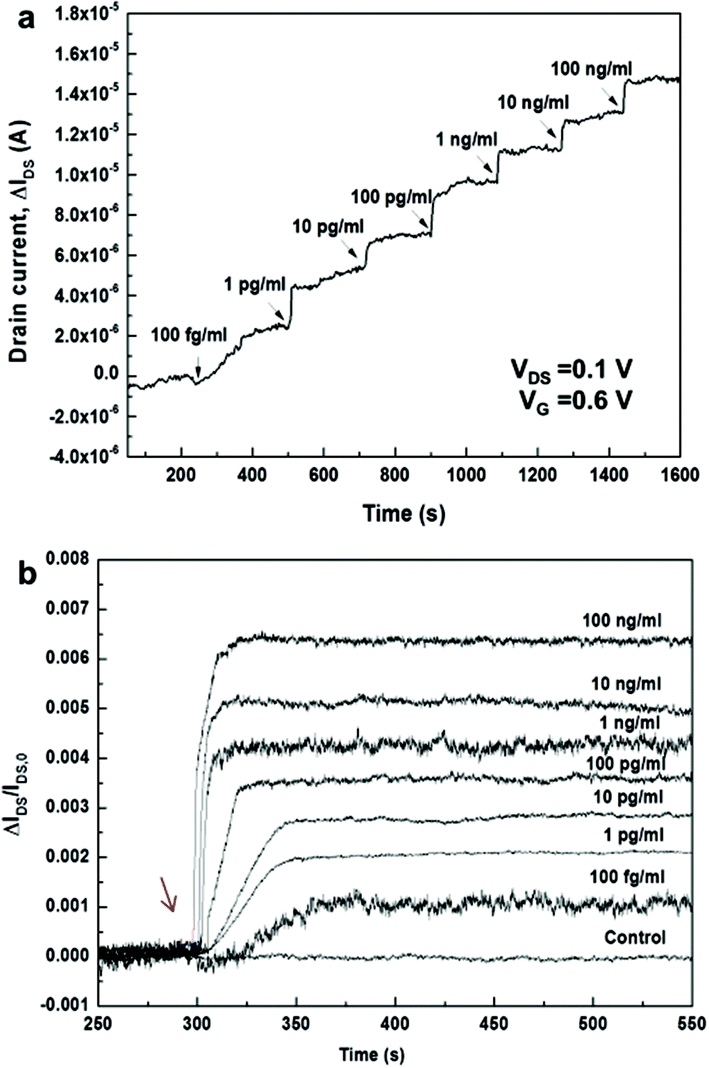 | ||
| Fig. 4 (a) Responses of the 3D channel FET upon adding the analyte solutions in different concentrations with a step-wise increase. (b) Time-dependent kinetic responses of 3D channel FETs upon adding the analyte solutions at different concentrations. Each response curve was obtained from different devices. The arrow indicates the time when the antigen solutions are added. | ||
Time-dependent responses of bio-FETs upon adding each target solution with different Ag concentrations from 100 fg ml−1 to 100 ng ml−1 were measured and plotted by normalizing the responses as ΔIDS/IDS,o for each concentration (Fig. 4b). As the Ag target concentration increased, saturation occurred more rapidly; the increase in the response time matched well with the results of the static measurements at VG = 0.6 V. At concentrations of 100 fg ml−1 and 100 pg ml−1, for example, the normalized signal was saturated after 64 and 44 s, respectively. Based on the Langmuir model,53,54 an association constant, KA, of ∼16 pM−1 and a kon of ∼106 M−1 s−1 were obtained (see Fig. S11†). This value is different from the reported values of, for example, ∼0.9 nM−1 for immunoreactions of PSA mAb and anti-PSA from the surface plasmon resonance biosensor55 and 0.4 pM−1 for immunoreactions of PSA mAb and anti-PSA in the SiNW bio-FET.56 The KA value in this work was also smaller than that in the 2D rGO bio-FET (∼4.2 nM−1) reported previously by our group.30 Even though the reason for this discrepancy for planar 2D and vertically nanostructured 3D surfaces is not clearly understood, it is presumed that the observed discrepancy is possibly attributed to differences in the density, orientation or binding states of immobilized Ab biomolecules on planar 2D and vertical 1D surfaces, resulting in the different binding reactions at equilibrium. A better understanding of the discrepancy may require a further study of the quantitative kinetic analysis in microfluidic systems similarly to the approach by Duan et al.20
In the well containing bulk target solutions with a relatively large volume (400 μl), the mass transport of target molecules occurs by diffusion. While the diffusivity D of proteins in solution is of the order of 1–10 μm2 s−1,1 the bulk diffusion time of the target proteins towards the ZnO NRs is of the order of 101–102 min. Indeed, the diffusion time on the 2D rGO channel bio-FET was of the order of tens of minutes.30 On the other hand, the observed ultrafast response time in the 3D channel FET is presumably attributed to fast binding of the Ag molecules in between ZnO NRs as well as in the vicinity of vertical 1D ZnO NRs. Since the time scale of mass transport of target molecules in between ZnO NRs towards the NR surface is of the order of 10−5 s considering an average distance between NRs of 50–100 nm, the diffusion of target molecules from the bulk solution only near the top surface of ZnO NRs vertically towards the region of ZnO NRs can be considered. The time scale of vertical mass transport to the bottom of the ZnO NRs was approximated to be of the order of a few seconds after considering the height of the ZnO NRs, H, of ∼2.5 μm. In this situation, the collection rate of target molecules towards the ZnO NRs was approximated by JD = AcDCo/H, where the target concentrations at the top and bottom of the ZnO NRs were assumed to be Co and zero by neglecting the depletion of target molecules above ZnO NRs. Then, the Damköhler number is given by Da = konbm(As/Ac)/(D/H). Due to the difficulty of direct measurement of bm for the PSA Ab–Ag interactions on the ZnO NRs, the surface density of biotin-Au NPs from streptavidin–biotin interactions, ∼103 μm−2, was used as the bm value on the ZnO sensing surface. This value was lower than the surface density of the PSA Ab probes on rGO, which was indirectly estimated at ∼2 × 105 μm−2.30 The Da value was estimated as ∼11, which indicates a mass-transport limited regime during Ab–Ag interactions in the 3D channel. We presume that, while the mass transport from the bulk solution towards the ZnO NRs determines the overall binding kinetics, added Ag molecules would be easily captured because of the large number of probe molecules (a large bmAs value) and the short distance in between vertical ZnO NRs and, as a result, faster binding kinetics would lead to a faster electrical response. The range of detection time observed in the present immunosensing method, close to 1 min at the fM level, is much shorter than the several tens of minutes to hours typically observed for 2D rGO30 or 1D NW20 biosensors.
As an alternative reason for the ultrafast response, some charged species near the ZnO NR surface in proximity under the electrical operation of the device might be related to the fast binding kinetics of Ag molecules because negatively charged Ag molecules or other charged particles can be drifted. To investigate the effect of gate electric field on binding kinetics, detection of immunoreactions was measured in the 3D channel FET under no gate biasing (VG = 0 V). The reaction time of the PSA–ACT complex at 100 fg ml−1 concentration under no gate biasing was ∼70 s (Fig. S12†) similarly to that of the 3D FET under positive gate biasing, which presumably indicates that the drift of charged particles by gate biasing negligibly affects the binding kinetics. The feature of fast response time in the 3D bio-FET could be very important for applications of miniaturized nanobiosensors in diagnostic technology utilizing microfluidic channels where the binding kinetics are limited due to the depletion of target molecules near the 2D sensing surface.1 Further verification of binding kinetics in microfluidics with mass transport of simultaneous convection and diffusion is needed to elucidate the optimal sensor mechanism and design. The measurement of immunoreactions in the 3D bio-FET indicates the clear advantages of ultrahigh sensitivity, large dynamic range, and ultrafast response time in the field of immunosensing.
Conclusions
Here, we presented a new sensing platform for a 3D channel bio-FET based on nanostructured materials. We successfully developed a bio-FET with a 3D channel composed of TiO2-encapped ZnO NRs vertically grown on CVD Gr for a label-free, ultrasensitive, and ultrarapid electrical biosensor for immunosensing. An ultrathin layer of TiO2 solved the problem of chemical instability of ZnO NRs in the electrolyte. The use of a 3D channel dramatically increased the number of probe molecules immobilized on the dense, vertical TiO2-encapped ZnO NRs in a given channel area compared to that on the 2D Gr channel. Increased probe molecules in a given channel area resulted in a significant improvement in static measurement sensitivity. The results from the 3D channel bio-FET also indicated an extremely large dynamic range of 107 and a sensitivity of 8 mV dec−1 from CNP shifts. The large sensing surface area and shorter distance between vertically grown ZnO NRs facilitated the binding kinetics of analyte-target protein molecules, resulting in a fast response time of ∼64 s at an analyte concentration of 100 fg ml−1 (∼1 fM). More research including reusability of the 3D bio-FET sensor and a detailed kinetic analysis need to be performed for further advancement of 3D channel bio-FETs in the near future. The proposed 3D biosensing platform can be easily extended to the detection of various biomolecules other than proteins and various electrochemical detection schemes including voltammetry, amperometry or impedimetry. Therefore, the newly developed 3D electrochemical 3D bio-FET platform based on nanostructured materials has great potential to be applied to a variety of biosensing systems including disease diagnostics, environmental monitoring, and food safety.Experimental
Graphene synthesis
SLG was synthesized using the CVD method on a copper foil (0.025 mm, Alfa Aesar). The copper foil was cleaned with acetic acid, placed in the middle of a quartz tube furnace, and heated at 1000 °C under a H2 gas flow of 40 sccm for 20 min. Subsequently, 20 sccm of CH4 gas was flowed into the tube in order to grow Gr followed by a mixture of H2 and CH4 gases at a 2:1 ratio and a pressure of 300 mTorr for 20 min. The heated copper foil was cooled to room temperature under an H2 atmosphere.
Graphene transfer
Synthesized SLG was transferred using a gold layer method that can reduce defects and give a very clean Gr surface. A 25 nm gold layer was deposited on Gr grown on copper foils at a deposition rate of 0.1 Å s−1 using a thermal evaporator. Gold/graphene was separated from the copper foil with a FeCl3 copper etchant for 2 h (Alfa Aesar) and then subsequently transferred to an acetone/ethanol-cleaned SiO2 (300 nm)/Si substrate. The sample was cleaned with three rinses of DI water over a 4 h period. After cleaning, the gold/graphene/SiO2 was naturally dried and annealed to remove water molecules from the interface graphene between SiO2 at 180 °C for 1 h. The gold layer was then wet etched using a gold etchant (Alfa Aesar) consisting of KI/I2 and DI water.Fabrication process of a 3D channel FET
A schematic illustration of the device is shown in Fig. S1.† Source and drain electrodes of Cr (5 nm) and Au (45 nm) were deposited onto patterned Gr using a shadow mask via an e-beam evaporator (Fig. S1a and S1b†). The entire device was covered with an Al2O3 layer of 10 nm as a buffer via ALD (Fig. S1c†), and the active area was patterned for active area opening through positive PR photolithography. Using phosphoric acid (∼85 wt%, Sigma-Aldrich), the Al2O3 on the Gr surface was etched at 45 °C for 90 s and immediately washed with DI water (Fig. S1d†). The Al2O3 layer was used as a buffer layer to prevent direct contact with various polymers during the photolithographic process and to reduce the leakage current of the device in an aqueous environment. Acetone at 40 °C for 30 min was used to remove positive PR (Fig. S1e†).ZnO NR growth and capping by TiO2
The solution growth method has been widely used to grow ZnO NRs because of its low temperature growth, size control, and low cost. A dispersion solution of 10 wt% amine-modified ZnO (NPs) in water was dropped onto the graphene surface as a seed and allowed to dry for 10 min (Fig. S1f†). Subsequently, ZnO NPs were washed using DI water and were immersed in a mixture of hexamethylenetetramine (25 mM, HMTA, C6H12N4) and zinc nitrate hexahydrate (25 mM, Zn(NO3)2·6H2O) in water at 90 °C for 3 h. Grown ZnO NRs were rinsed with DI water and heated at 200 °C for 1 h (Fig. S1g†). The ZnO NRs grown on Gr as an active layer were capped with an ultrathin 1 nm TiO2 layer using ALD.FET encapsulation
All areas except for the channel and electrode pad (contact region for probing) of the 3D channel and Gr FETs were encapsulated using SU-8 negative PR as follows: (1) the device was coated with SU-8 using a spin coater at an optimized spinning speed and time, (2) the fully coated SU-8 device was soft-baked at 95 °C for 3 min, (3) SU-8 was exposed to UV light to make the encapsulation pattern, (4) the exposed device was baked once more at 95 °C for 2 min (post exposure bake), and (5) a development process was carried out in an SU-8 developer for 90 s (Fig. S1h†). To contain the physiological solution, a PDMS well (diameter of 8 mm) was attached onto the device (Fig. S1i†).Functionalization of probe biomolecules
(1) The 3D channel FET: To produce hydroxyl groups on the surface of the 3D channel, oxygen plasma treatment was carried out in a microwave plasma reactor. ATPES of 2.5% in ethanol was poured on the channel region at room temperature (RT) for 2 h; it was subsequently rinsed with ethanol and dried using nitrogen gas. To activate amine group-terminated APTES, the device was heated at 150 °C for 20 min. Activated amine groups were reacted with 2.5% GA in a PBS solution at RT for 2 h, and the device was washed using PBS and DI water in sequence. To immobilize streptavidin (Sigma-Aldrich) or PSA–ACT complex Ab molecules (Fitzgerald) onto the surface of the 3D channel, the device was incubated with 100 μl of probe (100 μl ml−1) in PBS overnight. After incubation, the channel region was rinsed with PBS.(2) Gr channel FET: The Gr surface was treated with 1-pyrenebutanoic acid and 10 mM succinimidyl ester (PBSE) in dimethylformamide (DMF) at RT for 2 h in a dark environment. After treatment, the sample was washed to remove unstacked PBSE molecules using DMF and DI water. Immobilization of probe molecules utilized the same process as the 3D channel FET.
Measurements
Functionalized 3D and Gr channel FETs were used to detect PSA–ACT complex Ag molecules (Fitzgerald). The binding of functionalized streptavidin with biotin-grafted gold nanoparticles (biotin-AuNPs) (Cytodiagnostics) was confirmed by addition of PBS containing 100 nM biotin-AuNPs. For electrical detection of immunoreactions, the 3D and Gr channel FETs with PDMS were placed on the sample holder with the probe tips. During measurements, PBS solutions with different concentrations of PSA–ACT complex Ag molecules (Fitzgerald) were added into the PDMS well, and transfer characteristics were obtained using a semiconductor parameter analyzer (HP4145B).Acknowledgements
This research was supported by the Basic Science Research Program (2013R1A2A1A01015232) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT, & Future Planning, South Korea.Notes and references
- T. M. Squires, R. J. Messinger and S. R. Manalis, Nat. Biotechnol., 2008, 26, 417 CrossRef CAS PubMed.
- P. Yager, G. J. Domingo and J. Gerdes, Annu. Rev. Biomed. Eng., 2008, 10, 107 CrossRef CAS PubMed.
- D. Mark, S. Haeberle, G. Roth, F. V. Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153 RSC.
- V. Gubala, L. F. Harris, A. J. Ricco, M. X. Tan and D. E. Williams, Anal. Chem., 2012, 84, 487 CrossRef CAS PubMed.
- Y. Wan, Y. Su, X. Zhu, G. Liu and C. Fan, Biosens. Bioelectron., 2013, 47, 1 CrossRef CAS PubMed.
- F. Patolsky, B. P. Timko, G. Zheng and C. M. Lieber, MRS Bull., 2007, 32, 142 CrossRef CAS.
- J. F. Rusling, G. Sotzing and F. Papadimitrakopoulosa, Bioelectrochemistry, 2009, 76, 189 CrossRef CAS PubMed.
- D. Wei, M. J. A. Bailey, P. Andrew and T. Ryhänen, Lab Chip, 2009, 9, 2123 RSC.
- W. Yang, K. R. Ratinac, S. P. Ringer, P. Thordarson, J. J. Gooding and F. Braet, Angew. Chem., Int. Ed., 2010, 49, 2114 CrossRef CAS PubMed.
- I. M. Feigel, H. Vedala and A. Star, J. Mater. Chem., 2011, 21, 8940 RSC.
- R. Stine, S. P. Mulvaney, J. T. Robinson, C. R. Tamanaha and P. E. Sheehan, Anal. Chem., 2012, 85, 509 CrossRef PubMed.
- S. Liu and X. Guo, NPG Asia Mater., 2012, 4, e23 CrossRef PubMed.
- S. Wu, Q. He, C. Tan, Y. Wang and H. Zhang, Small, 2013, 9, 1160 CrossRef CAS PubMed.
- B.-R. Li, C.-C. Chen, U. R. Kumar and Y.-T. Chen, Analyst, 2014, 139, 1589 RSC.
- F. Yan, M. Zhang and J. Li, Adv. Healthcare Mater., 2014, 3, 313 CrossRef CAS PubMed.
- B. Zhan, C. Li, J. Yang, G. Jenkins, W. Huang and X. Dong, Small, 2014, 10, 4042 CrossRef CAS PubMed.
- J. L. Arlett, E. B. Myers and M. L. Roukes, Nat. Nanotechnol., 2011, 6, 203 CrossRef CAS PubMed.
- F. Patolsky, B. P. Timko, G. Yu, Y. Fang, A. B. Greytak, G. Zheng and C. M. Lieber, Science, 2006, 313, 1100 CrossRef CAS PubMed.
- K.-I. Chen, B.-R. Li and Y.-T. Chen, Nano Today, 2011, 6, 131 CrossRef CAS PubMed.
- X. Duan, Y. Li, N. K. Rajan, D. A. Routenberg, Y. Modis and M. A. Reed, Nat. Nanotechnol., 2012, 7, 401 CrossRef CAS PubMed.
- P. R. Nair and M. A. Alam, Appl. Phys. Lett., 2006, 88, 233120 CrossRef PubMed.
- P. E. Sheehan and L. J. Whitman, Nano Lett., 2005, 5, 803 CrossRef CAS PubMed.
- G. Zheng, F. Patolsky, Y. Cui, W. U. Wang and C. M. Lieber, Nat. Biotechnol., 2005, 23, 1294 CrossRef CAS PubMed.
- Y. Cui, Q. Wei, H. Park and C. M. Lieber, Science, 2001, 293, 1289 CrossRef CAS PubMed.
- R. J. Chen, S. Bangsaruntip, K. A. Drouvalakis, N. W. S. Kam, M. Shim, Y. Li, W. Kim, P. J. Utz and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2013, 100, 4984 CrossRef PubMed.
- Y. L. Bunimovich, Y. S. Shin, W.-S. Yeo, M. Amori, G. Kwong and J. R. Heath, J. Am. Chem. Soc., 2006, 128, 16323 CrossRef CAS PubMed.
- G. Zheng, X. P. A. Gao and C. M. Lieber, Nano Lett., 2010, 10, 3179 CrossRef CAS PubMed.
- X. Dong, Y. Shi, W. Huang, P. Chen and L.-J. Li, Adv. Mater., 2010, 22, 1649 CrossRef CAS PubMed.
- J. H. Jung, I. Y. Sohn, D. J. Kim, B. Y. Kim, M. Jang and N. E. Lee, Carbon, 2013, 62, 312 CrossRef CAS PubMed.
- D.-J. Kim, I. Y. Sohn, J.-H. Jung, O. J. Yoon, N.-E. Lee and J.-S. Park, Biosens. Bioelectron., 2013, 41, 621 CrossRef CAS PubMed.
- D.-J. Kim, H.-C. Park, I. Y. Sohn, J.-H. Jung, O. J. Yoon, J.-S. Park, M.-Y. Yoon and N.-E. Lee, Small, 2013, 9, 3352 CAS.
- D. Sarkar, W. Liu, X. Xie, A. C. Anselmo, S. Mitragotri and K. Banerjee, ACS Nano, 2014, 8, 3992 CrossRef CAS PubMed.
- L. Wang, Y. Wang, J. I. Wong, T. Palacios, J. Kong and H. Y. Yang, Small, 2014, 10, 1101 CrossRef CAS PubMed.
- V. Q. Dang, D.-I. Kim, T. D. Le, B.-Y. Kim, B.-U. Hwang, M. Jang, K.-S. Shin, S.-W. Kim and N.-E. Lee, Nanoscale, 2014, 6, 15144 RSC.
- M. Jang, T. Q. Trung, J.-H. Jung, B.-Y. Kim and N.-E. Lee, Phys. Chem. Chem. Phys., 2014, 16, 4098 RSC.
- W. Choi, K.-S. Shin, H. Lee, D. Choi, K. Kim, H.-J. Shin, S.-M. Yoon, J.-Y. Choi and S.-W. Kim, Nano Res., 2011, 4, 440 CrossRef CAS PubMed.
- M. Guo, P. Diao and S. Cai, J. Solid State Chem., 2005, 178, 1864–1873 CrossRef CAS PubMed.
- J. O. Hwang, D. H. Lee, J. Y. Kim, T. H. Han, B. H. Kim, M. Park, K. No and S. O. Kim, J. Mater. Chem., 2011, 21, 3432 RSC.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 4430 CrossRef CAS PubMed.
- D. Polsongkram, P. Chamninok, S. Pukird, L. Chow, O. Lupan, G. Chai, H. Khallaf, S. Park and A. Schulte, Phys. B (Amsterdam, Neth.), 2008, 403, 3713 CrossRef CAS PubMed.
- J. Zhou, N. Xu and Z. L. Wang, Adv. Mater., 2006, 18, 2432 CrossRef CAS PubMed.
- M. Liu, C.-Y. Nam, C. T. Black, J. Kamcev and L. Zhang, J. Phys. Chem. C, 2013, 117, 13396 CAS.
- I. Heller, S. Chatoor, J. Männik, M. A. G. Zevenbergen, C. Dekker and S. G. Lemay, J. Am. Chem. Soc., 2010, 132, 17149 CrossRef CAS PubMed.
- E. Uesugi, H. Goto, R. Eguchi, A. Fujiwara and Y. Kubozono, Sci. Rep., 2013, 3, 1595 Search PubMed.
- F. Chen, J. Xia, D. K. Ferry and N. Tao, Nano Lett., 2009, 9, 2571 CrossRef CAS PubMed.
- F. Chen, Q. Qing, J. Xia, J. Li and N. Tao, J. Am. Chem. Soc., 2009, 131, 9908 CrossRef CAS PubMed.
- A. B. Artyukhin, M. Stadermann, R. W. Friddle, P. Stroeve, O. Bakajin and A. Noy, Nano Lett., 2006, 6, 2080 CrossRef CAS PubMed.
- W.-J. Kim, S. Kim, B. S. Lee, A. Kim, C. S. Ah, C. Huh, G. Y. Sung and W. S. Yun, Langmuir, 2009, 25, 11692 CrossRef CAS PubMed.
- Y.-E. Choi, J.-W. Kwak and J. W. Park, Sensors, 2010, 10, 428 CrossRef CAS PubMed.
- R. B. M. Schasfoort, P. Bergveld, R. P. H. Kooyman and J. Greve, Anal. Chim. Acta, 1990, 238, 323 CrossRef CAS.
- I. Heller, A. M. Janssens, J. Männik, E. D. Minot, S. G. Lemay and C. Dekker, Nano Lett., 2007, 8, 591 CrossRef PubMed.
- T. Y. Chen, P. T. K. Loan, C. L. Hsu, Y. H. Lee, J. T.-W. Wang, K. H. Wei, C. T. Lin and L. J. Li, Biosens. Bioelectron., 2013, 41, 103 CrossRef CAS PubMed.
- A. Halperin, A. Buhot and E. B. Zhulina, J. Phys.: Condens. Matter, 2006, 18, S463 CrossRef CAS.
- A. Lin, A. S.-Y. Lee, C.-C. Lin and C.-K. Lee, Curr. Proteomics, 2006, 3, 271 CrossRef.
- P. S. Katsamba, I. Navratilova, M. Calderon-Cacia, L. Fan, K. Thornton, M. Zhu, T. V. Bos, C. Forte, D. Friend, I. Laird-Offringa, G. Tavares, J. Whatley, E. Shi, A. Widom, K. C. Lindquist, S. Klakamp, A. Drake, D. Bohmann, M. Roell, L. Rose, J. Dorocke, B. Roth, B. Luginbuhl and D. G. Myszka, Anal. Biochem., 2006, 352, 208 CrossRef CAS PubMed.
- Y. Cui, W. U. Wang, L. Huynh and C. M. Lieber, https://web.stanford.edu/group/cui_group/papers/PSA.pdf.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr00909j |
| This journal is © The Royal Society of Chemistry 2015 |