
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Does a nitrogen matter? Synthesis and photoinduced electron transfer of perylenediimide donors covalently linked to C59N and C60 acceptors†
Luis
Martín-Gomis
a,
Georgios
Rotas
b,
Kei
Ohkubo
c,
Fernando
Fernández-Lázaro
a,
Shunichi
Fukuzumi
*cd,
Nikos
Tagmatarchis
*b and
Ángela
Sastre-Santos
*a
aÁrea de Química Orgánica Instituto de Bioingeniería, Universidad Miguel Hernández, Elche, Spain. E-mail: asastre@umh.es; Fax: +34 966658408; Tel: +34 966658351
bTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens 116 35, Greece. E-mail: tagmatar@eie.gr
cDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp
dDepartment of Bioinspired Science, Ewha Womans University, Seoul, 120-750, Korea
First published on 16th March 2015
Abstract
The first perylenediimide (PDI) covalently linked to an azafullerene (C59N) is described. PDI-C59N and PDI-C60 dyads where PDI acts as an electron-donor moiety have been synthesized by connection of the balls to the PDI 1-bay position. Photoexcitation of the PDI unit in both systems results in formation of the charge-separated state by photoinduced electron transfer from the singlet excited state of the PDI moiety to the C59N or to the C60 moiety. The charge-separated state has a lifetime of 400 ps in the case of PDI-C59N and 120 ps for the PDI-C60 dyad in benzonitrile at 298 K. This result has significant implications for the design of organic solar cells based on covalent donor–acceptor systems using C59N as an electron acceptor, indicating that longer-lived charge-separated states can be attained using C59N systems instead of C60 systems.
Introduction
The seeking for efficient molecular systems, based on the covalent combination of suitable and versatile building blocks, capable to mimic the photoinduced electron transfer that naturally occurs in photosynthesis, has been a constant in material science.1 In a simplistic definition, a molecular system which combines an electron-acceptor moiety with an electron-donor counterpart, able to be excited by the action of light, can be considered as a potential photosynthetic system, and therefore useful for photovoltaic applications. [60]Fullerene, due to its extraordinary electron-acceptor character and its fascinating chemical versatility, has been one of the most utilized electron-acceptor building blocks2 for that purpose and, thus, several efficient artificial photosynthetic [60]fullerene-based systems have been described, using different chromophores as photoexcitable electron-donor counterparts.3 Performances of these donor–acceptor materials can be improved by selecting an appropriate electron-donor counterpart, but also by replacing one or more carbon atoms of the fullerene cage by heteroatoms, affording the so-called heterofullerenes.4 The substitution of a tetravalent carbon atom of the three dimensional network by a trivalent nitrogen atom is one of the most interesting alterations. This simple modification leads to azafullerene C59N, a fullerene-based building block with an improved electron-acceptor character, when it is compared to that of pristine C60 fullerene.5 A number of different azafullerene-based donor–acceptor covalent compounds have been previously described, showing interesting photophysical properties that make them ideal candidates to be incorporated in organic photovoltaic devices. Hirsch et al. described the synthesis of an azafullerene-ferrocene dyad that shows strong electronic coupling between the ferrocene and azafullerene moieties in the ground state.6 Additional C59N-based dyads carrying photoactive units such as porphyrin7 and pyrene8 were also prepared. More recently, azafullerene–electron-donor dyads, using phthalocyanine9 and corrole10 units, have been reported to undergo photoinduced electron transfer upon selective irradiation of the electron-donor moiety.On the other hand, perylenediimides (PDI) comprise an outstanding family of perylene derivatives that, due to an extreme synthetic versatility and an easily tunable electronic character, have become one of the most promising classes of molecular materials in organic photovoltaic devices.11 In this context, we can find only a few examples where PDI derivatives have been successfully employed as an electron-donor moiety in fullerene-based systems, eventually undergoing photoinduced electron and/or energy transfer.12
Here we present the synthesis, characterization and photophysical properties of a new photoexcitable molecular system, consisting of an electron-donor PDI moiety, carrying a pyrrolidinyl substituent in the 1-bay position, which also acts as a linker to the C59N counterpart (Scheme 1). The obtained donor–acceptor PDI-C59N dyad 1, upon selective excitation of the PDI subunit in benzonitrile, shows efficient intramolecular photoinduced electron transfer, generating a 400 ps-lived charge-separated state (CSS). Together with two covalently linked porphyrin-C59N dyads,7 this is the longest CSS lifetime value obtained so far for a C59N-based donor–acceptor dyad in a polar solvent. Moreover, in order to study the effect of the nitrogen atom on the photophysics, the analogous PDI-C60 dyad 2 (Scheme 1) was synthesized, as reference material, obtaining a 3-times shorter CSS lifetime than for the PDI-C59N dyad 1, thus, highlighting the role of nitrogen in the photoinduced electron-transfer processes.
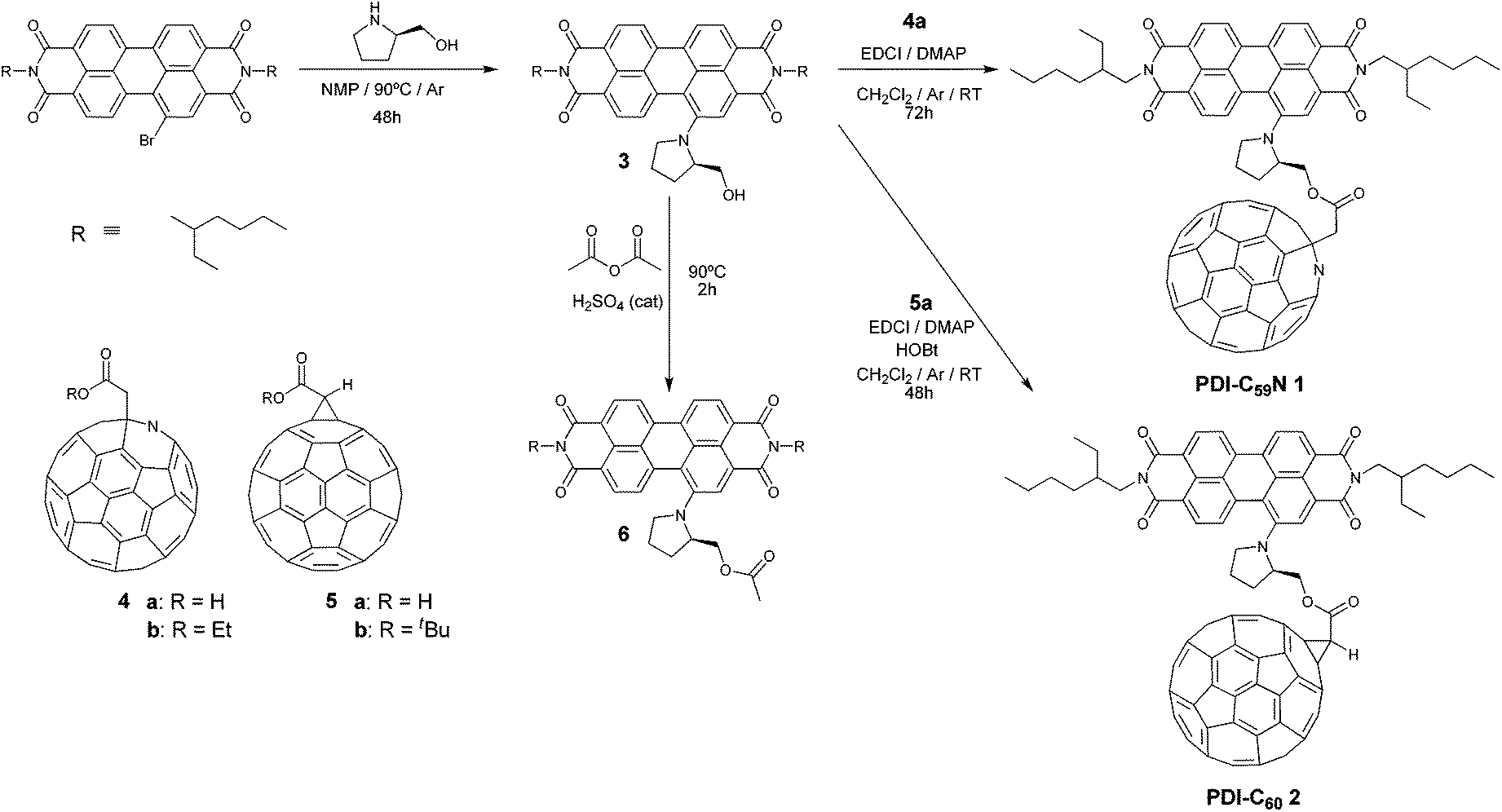 | ||
| Scheme 1 Synthetic routes for preparing PDI-C59N 1 and PDI-C602 dyads as well as PDI-based reference material 6. | ||
Synthesis and characterization
Initially, PDI-C59N and PDI-C60 dyads 1 and 2 were synthesized as described in Scheme 1. Firstly, (R)-1-(2′-hydroxymethylpyrrolidin-N-yl)PDI 3 was obtained upon nucleophilic aromatic substitution on N,N′-di(2-ethylhexyl)-1-bromoperylene-3,4,9,10-tetracarboxy diimide13 by (R)-2-hydroxymethylpyrrolidine in 51% yield. Then, the esterification reaction of PDI-based 3 with azafullerenylacetic acid 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 gave rise to PDI-C59N dyad 1 in 8% yield. A similar esterification process, involving 2,2-[60]fullerenylacetic acid 5a14 and PDI-based 3 was followed, affording in 43% yield PDI-C60 dyad 2. Furthermore, PDI derivative 6 was also synthesized in 75% yield, through an acylation of 3 with acetic anhydride in acidic media, in order to be used as a reliable reference to evaluate optical, electrochemical and photophysical properties.
9 gave rise to PDI-C59N dyad 1 in 8% yield. A similar esterification process, involving 2,2-[60]fullerenylacetic acid 5a14 and PDI-based 3 was followed, affording in 43% yield PDI-C60 dyad 2. Furthermore, PDI derivative 6 was also synthesized in 75% yield, through an acylation of 3 with acetic anhydride in acidic media, in order to be used as a reliable reference to evaluate optical, electrochemical and photophysical properties.
All new compounds were fully characterised using standard analytical techniques such as 1H and 13C NMR, HR-MS, FT-IR, electronic absorption and photoluminescence. Fig. 1 compares the aromatic region of the 1H NMR spectra for 1, 2 and 6 in CDCl3 as solvent. Taking as reference the C1 of the PDI core (the substituted one), and comparing the signals of analogous hydrogens in compounds 1, 2 and 6, it is worth noting that the chemical displacement of the nearby hydrogens (H2, H11 and H12) is significantly shifted in dyad's spectra, while the contrary trend is observed in the displacement of H5, H6, H7 and H8. For example, the H2 signal goes from 8.69 to 9.19 and 9.18 ppm (in 2 and 1 respectively), while H6 goes from 8.45 to 8.40 ppm. Furthermore, the fullerene influence is slightly larger when C59N is present instead of C60, and it can be recognised in the H12 displacement, which changes from 8.11 ppm in 6, to 8.29 and 8.40 ppm in dyads 2 and 1, respectively. Fig. 2 shows the aliphatic regions of the spectra, where some differences can also be found due to the presence of the covalently attached fullerene. For example, the signals corresponding to the hydrogens labeled as HB, HC and HE, which appear as broad signals in the spectrum of PDI-based 6, split into two in the corresponding spectra of PDI-fullerene dyads 1 and 2. All these differences in the 1H NMR spectra of dyads and reference material can be explained by considering the electron-withdrawing character of the fullerene sphere, which is more intense in the case of C59N as compared with C60, and also assuming a pseudo-fixed molecular conformation in PDI-fullerene dyads caused by an electrostatic interaction between the PDI and the fullerene moiety including a concave–convex interaction3l,15 that causes a different influence of the sphere on the chemical shift of the pyrrolidine-linker geminal protons. HR MALDI-TOF (negative mode) experiments clearly confirm the structure of PDI-C59N and PDI-C60 dyads 1 and 2, respectively, showing base peaks due to the molecular ions at 1476.463 amu and 1474.302 amu, with isotopical distributions that exactly match the simulated isotope patterns for C106H52N4O6 and C107H51N3O6, respectively (see ESI, Fig. S1†).
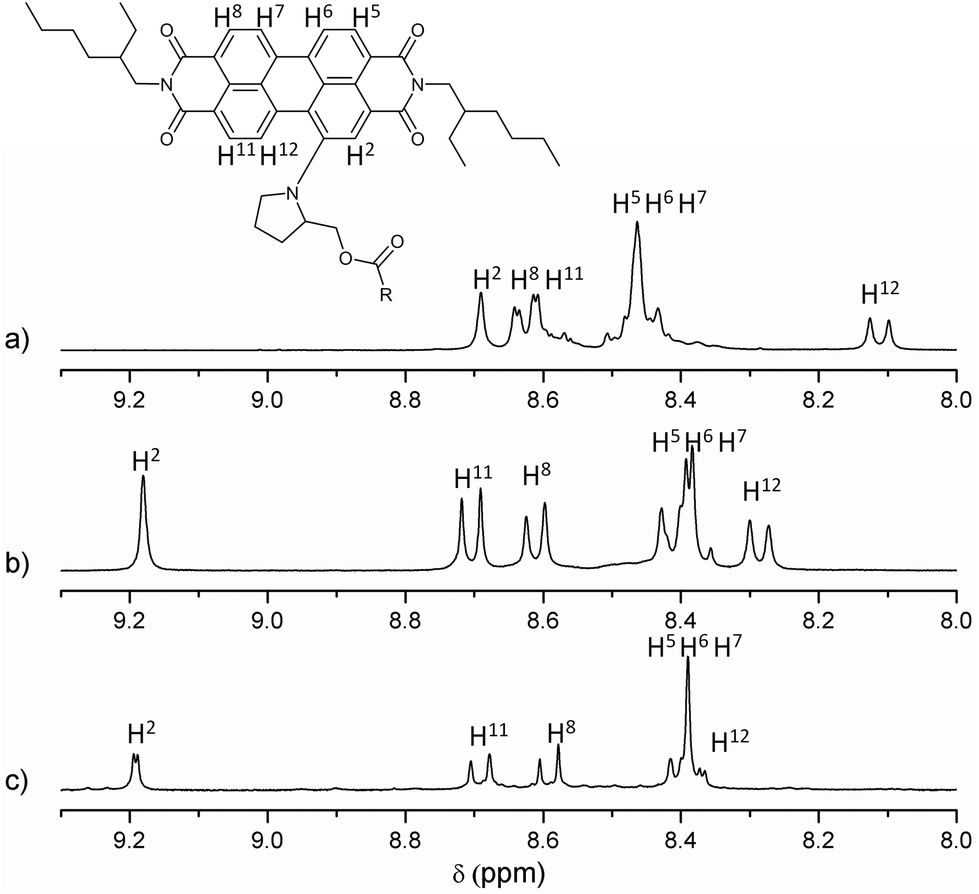 | ||
| Fig. 1 Partial 1H NMR (9.30–8.00 ppm) spectra of (a) PDI-based reference material 6, (b) PDI-C602, and (c) PDI-C59N 1. | ||
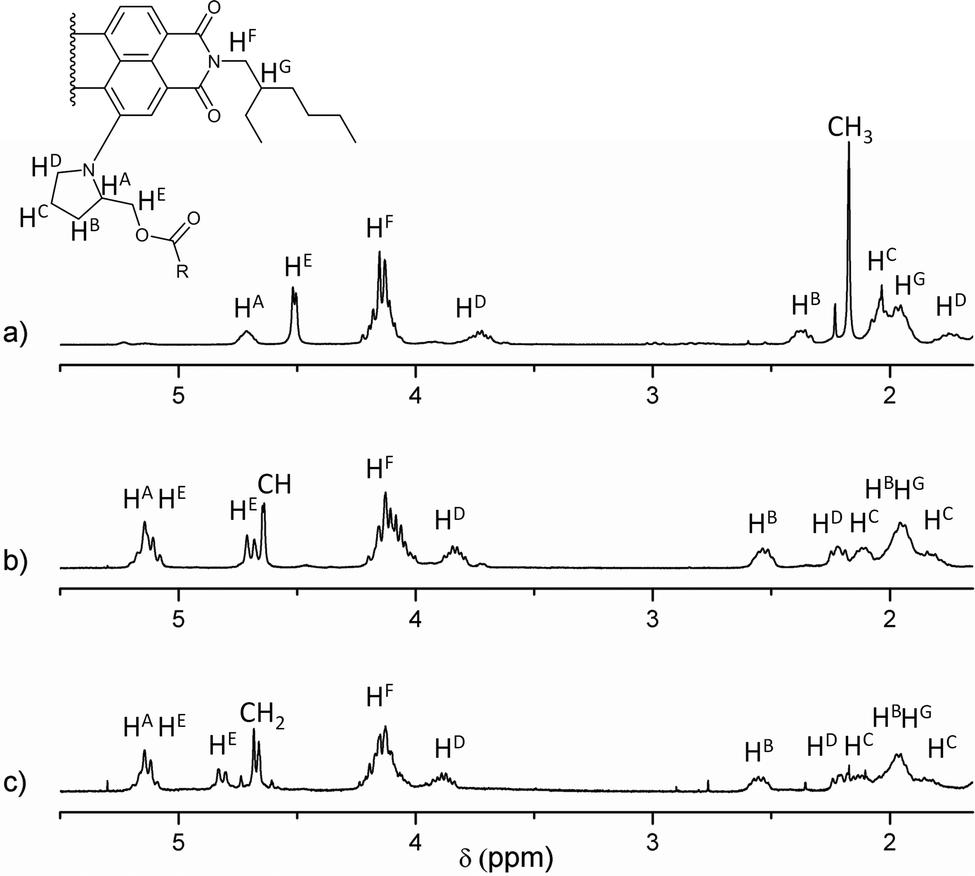 | ||
| Fig. 2 Partial 1H NMR (5.50–1.60 ppm) spectra in CDCl3 as solvent of (a) PDI-based reference material 6, (b) PDI-C602, and (c) PDI-C59N 1. | ||
Photophysical and electrochemical properties
Fig. 3 shows the overlapped UV-vis spectra of dyads 1 (black line) and 2 (blue line) and C605a, C59N 4b and PDI 6 as reference compounds using benzonitrile as solvent. The absorption spectra of PDI-C59N 1 and PDI-C602 dyads are characterised by two sets of absorption maxima around 430 and 638 nm. The band centered at 430 nm can be attributed to transitions 1–0 and 0–0 in the PDI species,16 while the one centred at 638 nm consists of a broad charge transfer (CT) band that usually appears in bay amino-substituted PDI compounds. The latter band appears as a consequence of an electron transfer between the electron-donor pyrrolidine and the electron-acceptor PDI units,17 and therefore is very sensitive to changes in the electronic character of the entire molecule. When those spectra are compared to the absorption profile of reference compound PDI 6, some differences can be found. One of these differences, directly related to the electronic interaction between the electroactive moieties, is a bathochromic displacement of the band centered at 632 nm, which now appears at 638 nm in both PDI-C60 and PDI-C59N. This fact reveals the electronic influence of the fullerene sphere on the perylene subunit in the ground state, as it has been already pointed out in the comparison of 1H NMR spectra (cf. Fig. 1 and 2).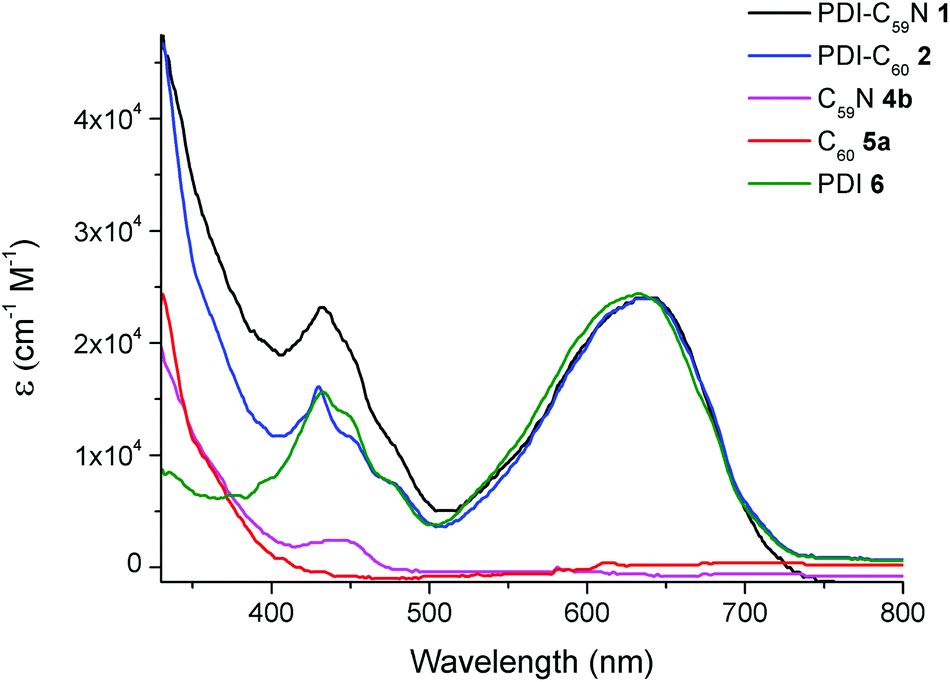 | ||
| Fig. 3 UV/vis spectra of PDI-C59N 1 and PDI-C602, reference compounds 4b and 5a and PDI-based reference 6 registered in benzonitrile as solvent. | ||
On the other hand, Fig. 4 shows the fluorescence emission spectra of PDI-C59N 1 and PDI-C602 dyads, when selectively excited at 429 nm. As can be seen, the fluorescence spectrum of 6 clearly shows three different emission bands, centered at 480, 512 and 691 nm. When the emission spectra of PDI-C59N and PDI-C60 dyads 1 and 2 are compared to that of the reference compound, the first and second bands are partially quenched while the third one is extinguished. The latter is indicative of intra-dyad electronic interactions between the two components of the dyad (i.e., PDI and fullerene) at the excited state. The strong fluorescence quenching of the 691 nm band in both 1 and 2 as compared to 6, supports electron and/or energy transfer as the decay mechanism of the PDI-centred singlet excited state. This effect is slightly more pronounced in the case of PDI-C59N dyad 1 (see inset Fig. 4).
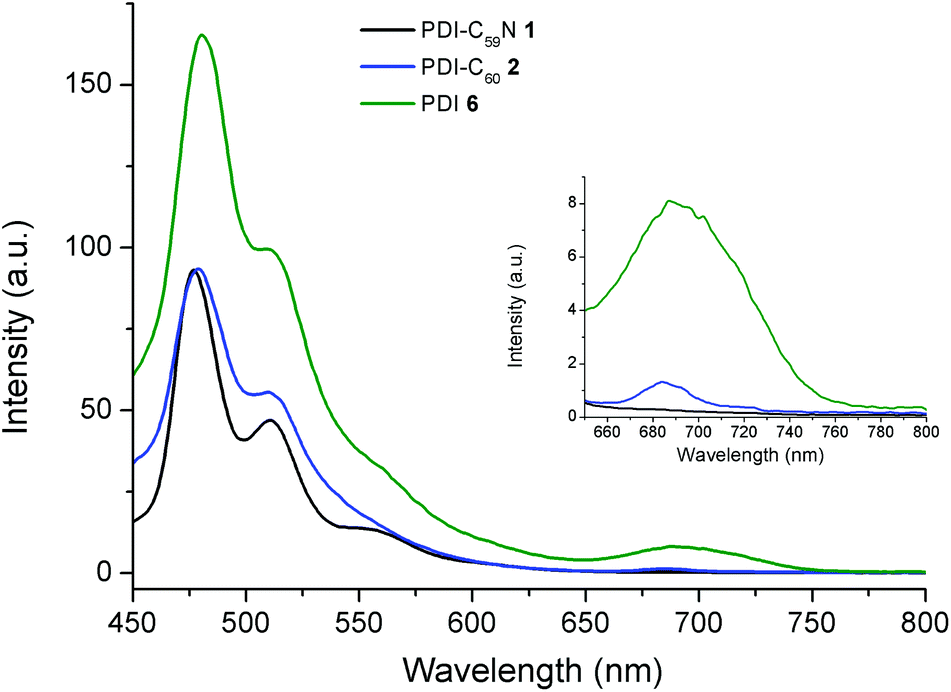 | ||
| Fig. 4 Fluorescence spectra of PDI-C59N 1 and PDI-C602 dyads and PDI-based reference 6, obtained in benzonitrile (4 × 10−6 M; λexc = 429 nm). Fluorescence spectra enlargement of 1, 2 and 6 from 650 to 800 nm. | ||
The electrochemical data of PDI-C59N 1 and PDI-C602 dyads, as well as 6, 4b and 5b as reference materials for PDI-C59N, and C60, respectively, in benzonitrile using Fc/Fc+ as an internal standard are listed in Table 1 (see differential pulse voltammograms in ESI, Fig. S13†). The measurements for PDI 6 reveal a one-electron oxidation and two one-electron reduction processes centred at 0.55, −1.15 and −1.35 V respectively, while the measurements for fullerene reference compounds show two one-electron reversible reduction processes at −1.01 and −1.42 V. The voltammograms of dyads 1 and 2 are very similar, in spite of the nitrogen atom in dyad 1, and can be considered as a combination of the already mentioned electrochemical processes assigned to PDI and fullerene electroactive moieties. A slight influence of the fullerene sphere (either C59N or C60) over the PDI moiety can be recognised, and it is highlighted by a 20–30 mV anodic displacement of the oxidation wave and a 40 mV cathodic displacement of the second reduction process, which is attributed to the PDI counterpart. As contrary to previous results reported by us,9,10 the similar first reduction values obtained for PDI-C60 and PDI-C59N might come from the use of different solvents. Based on the redox data shown in Table 1 and neglecting the Coulombic interactions, the electrochemical band gap for 1 is calculated as 1.59 eV (1.61 eV for 2).
| E 3red (V) | E 2red (V) | E 1red (V) | E ox (V) | |
|---|---|---|---|---|
| PDI-6 | −1.35 | −1.15 | 0.55 | |
| PDI-C602 | −1.39 | −1.19 | −1.03 | 0.58 |
| PDI-C59N 1 | −1.38 | −1.19 | −1.01 | 0.58 |
| C60-ref 5b | −1.42 | −1.01 | ||
| C59N-ref 4b | −1.42 | −1.01 |
Femtosecond laser flash photolysis on a deaerated benzonitrile solution of 1 and 2 indicates the formation of a charge-separated state, providing solid evidence for the electron-transfer deactivation mechanism in 1 and 2, after selective excitation of the perylene moiety. The transient absorption spectrum taken at 1 ps after laser pulse irradiation of the absorption band of the PDI moiety at 490 nm exhibits an absorption maximum at 900 nm, which is assigned to the singlet excited state of C59N of PDI-C59N dyad 1 (Fig. 5a). No indication of the PDI singlet excited state was observed in spite of the selective excitation of PDI, indicating that the rate of energy transfer from the singlet excited state of the PDI moiety to the C59N moiety is too fast to be followed even with the use of femtosecond laser flash photolysis (fwhm = 130 fs). Then, electron transfer from PDI to the singlet-excited state of C59N takes place to form PDI˙+-C59N˙− as the charge-separated state. The transient absorption band decayed with a rate constant of 5.5 × 1010 s−1 giving rise to an absorption band at λmax = 1030 nm due to the C59N˙− radical anion,9,10 which indicates the formation of the charge-separated state. From the decay of the absorption band at 1030 nm, a 400 ps lifetime (k = 2.5 × 109 s−1) was obtained for the charged-separated state (Fig. 5b), being the first example of a charge-separated state in a donor–acceptor dyad based on PDI and azafullerene.
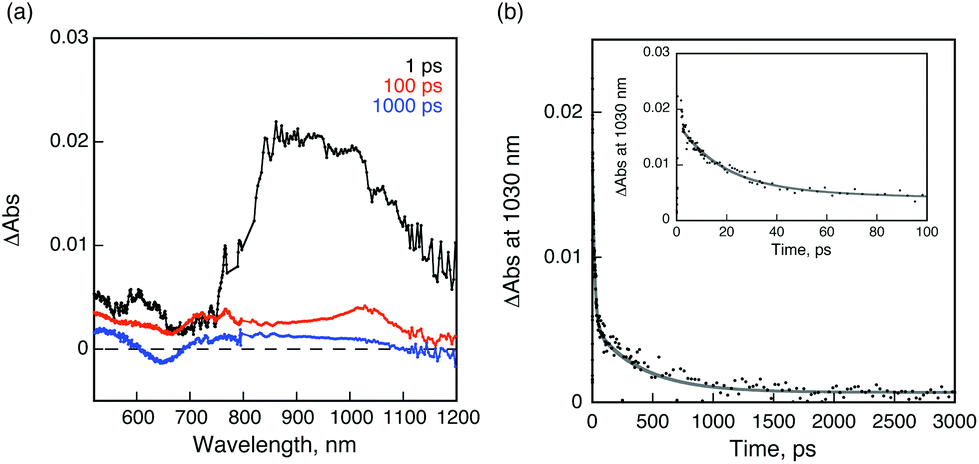 | ||
| Fig. 5 (a) Transient absorption spectra of C59N-PDI (0.5 mM) in deaerated PhCN after femtosecond laser excitation at 510 nm; (b) time profile of the absorbance at 1030 nm. Inset: decay time profile for the short time range. Gray line is drawn by the double exponential curve fitting. | ||
When benzonitrile was replaced by toluene, the rate constant of charge separation became much larger (3.9 × 1011 s−1), whereas the rate constant of charge recombination became smaller (1.3 × 109 s−1) as shown in Fig. S14 (ESI†). The faster charge separation in toluene than in benzonitrile may result from the smaller solvent reorganization energy of toluene than that of the more polar benzonitrile in the Marcus normal region, where the charge separation becomes faster with decreasing the solvent reorganisation energy of electron transfer.1d,18 By the same token, the slower charge recombination in toluene than in benzonitrile results from the smaller solvent reorganisation energy of toluene than that of benzonitrile in the Marcus inverted region, where the charge recombination becomes slower with decreasing the solvent reorganisation energy of electron transfer.1d,18
On the other hand, a similar transient absorption spectral change was observed for PDI-C60 dyad 2 as shown in Fig. 6a. The transient absorption recorded at 1 ps after femtosecond laser pulse irradiation is assigned to the singlet excited state of C60, indicating that ultrafast energy transfer from the singlet excited state of PDI to the C60 moiety also occurs in PDI-C60. Then, electron transfer from PDI to the singlet excited state of C60 takes place to form the charge-separated state. The rate constant for electron transfer is determined to be 8.6 × 1010 s−1 from the rise of the absorbance at 1000 nm (inset of Fig. 6b). This rate is slightly faster than that of PDI-C59N (vide supra). The lifetime of the charge-separated state was determined to be 120 ps from the decay of absorbance at 1000 nm as shown in Fig. 6b. It is worth to mention the much shorter lifetime of the CSS in PDI-C60, less than one third of that in PDI-C59N, pointing to the remarkable influence of the nitrogen atom in C59N, which is not explained only by an increased electron-accepting character as the reduction potentials of 1 and 2 indicate. In both cases, the decay of the charge-separated state by back electron transfer yields the ground state rather than the triplet excited state of PDI as indicated by no triplet transient absorption of 1 and 2 in nanosecond laser flash photolysis measurements (Fig. S14 in ESI†).
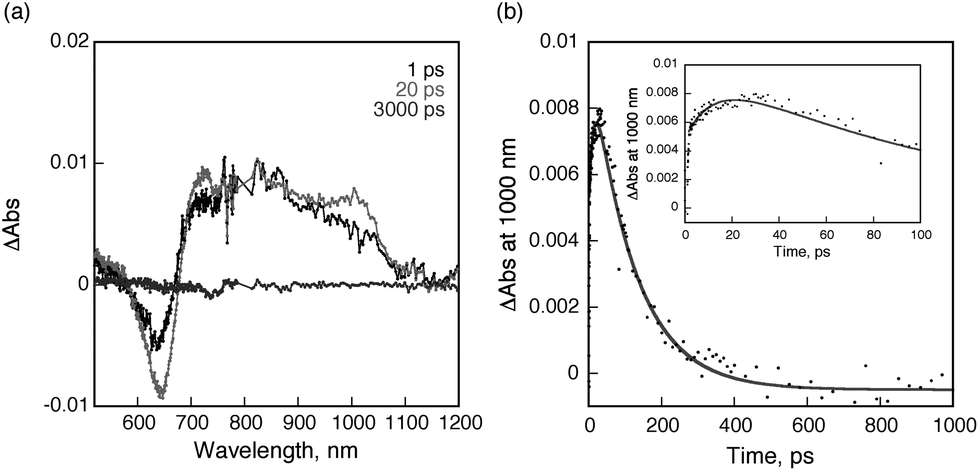 | ||
| Fig. 6 (a) Transient absorption spectra of C60-PDI (0.5 mM) in deaerated PhCN after femtosecond laser excitation at 510 nm; (b) Time profile of the absorbance at 1000 nm. Inset: rise and decay time profile for the short time range. Gray line is drawn by the double exponential curve fitting. | ||
Experimental
General information
All chemicals were reagent grade, purchased from commercial sources, and used as received, unless otherwise specified. Column chromatography: SiO2 (40–63 mm) TLC plates coated with SiO2 60F254 were visualized by UV light. 2 and 6 crude products were finally purified by a Combiflash Rf chromatography system (Teledyne Technologies, Inc., Thousand Oaks, CA), while 1 crude product was finally purified by preparative HPLC (Japan Analytical Industry Co Ltd, utilising a Buckyprep 20 × 250 column with toluene as eluent at 10 mL min−1 flow rate). NMR spectra were measured with a Bruker AC 300. UV/vis spectra were recorded with a Helios Gamma spectrophotometer. Fluorescence spectra were recorded with a PerkinElmer LS 55 Luminescence Spectrometer and IR spectra with a Nicolet Impact 400D spectrophotometer. Mass spectra were obtained from a Bruker Microflex matrix-assisted laser desorption/ionization time of flight (MALDI-TOF).
tert-Butyl (2,2-[60]fullerenyl)acetate 5b14 and ethyl (2-azafullerenyl)acetate 4b, used as C60 and C59N reference compounds, respectively, in electrochemistry measurements, were prepared as reported earlier.
Electrochemistry
Differential pulse voltammetry measurements were performed in a conventional three-electrode cell using a μ-AUTOLAB type III potentiostat/galvanostat at 298 K, over PhCN deaerated sample solutions (∼0.5 mM), containing 0.10 M tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte. A glassy carbon (GC) working electrode, Ag/AgNO3 reference electrode and a platinum wire counter electrode were employed. Ferrocene/ferrocenium was used as an internal standard for all measurements.Synthesis
:1) to obtain 52 mg (51%) of the desired product as a green powder. 1H NMR (300 MHz, 25 °C): 8.46 (1H, s, H-PDI), 8.42–8.35 (2H, m, 3 × H-PDI), 8.18 (1H, d, J = 8.2 Hz, H-PDI), 8.02–7.89 (2H, m, 2 × H-PDI), 7.55–7.47 (1H, m, 2 × H-PDI), 4.51–4.39 (1H, m, CHN-pyrrolidine), 4.31 (1H, t, J = 12.7 Hz, CHH-OH), 4.21–4.03 (4H, m, 2 × CH2-N-alkyl chain), 3.98 (1H, t, J = 12.7 Hz, CHH-OH), 3.57–3.41 (1H, m, CHHN-pyrrolidine), 2.30–2.09 (2H, m, CH2-pyrrolidine), 2.07–1.91 (2H, m, CHH-pyrrolidine + CH-alkyl chain), 1.90–1.76 (1H, m, CH-alkyl chain), 1.71–1.59 (2H, m, CHHN-pyrrolidine + CHH-pyrrolidine) 1.54–1.21 (16H, m, 8 × CH2-alkyl chain) and 1.06–0.78 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C):164.2, 163.9, 163.3, 163.3, 147.2, 134.8, 134.2, 132.0, 131.5, 129.7, 129.7, 128.3, 128.1, 126.6, 126.2, 124.6, 123.2, 123.0, 122.5, 122.3, 120.6, 120.0, 118.0, 116.2, 62.0, 60.2, 56.2, 44.3, 43.9, 38.1, 38.1, 37.8, 30.9, 30.7, 29.6, 28.8, 28.6, 27.9, 25.2, 24.1, 24.0, 23.1, 14.1, 10.7 and 10.6 ppm; IR-FT (KBr) ν/cm−1: 3467, 2956, 2927, 2858, 1691, 1652, 1586, 1427, 1338, 1242, 807, 748; UV/vis (CH2Cl2), λmax/nm (logε): 431 (4.16), 637 (4.36); HR-MS (MALDI-TOF, dithranol): m/z = 713.349, [M]−, calcd for C45H51N3O5: 713.383.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2.11–1.88 (4H, m, CH2-pyrrolidine + 2 × CH-alkyl chain), 1.50–1.24 (16H, m, 8 × CH2-alkyl chain) and 1.01–0.84 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C): 170.9, 164.3, 164.1, 147.8, 135.9, 135.2, 133.2, 131.3, 129.2, 129.1, 127.6, 127.5, 124.6, 124.0, 123.9, 123.3, 122.9, 122.6, 122.1, 121.0, 119.8, 118.9, 65.3, 57.5, 56.4, 44.5, 38.1, 31.0, 29.1, 28.9, 25.4, 24.3, 23.2, 21.0, 14.3, 10.9, 10.8; IR-FT (KBr) 2956, 2927, 2857, 1744, 1693, 1654, 1587, 1419, 1336, 1240, 808, 748 ν/cm−1; UV/vis (CH2Cl2), λmax/nm (logε):332 (3.80), 430 (4.17), 624 (4.36); HR-MS (MALDI-TOF, dithranol): m/z = 755.385, [M]−, calcd for C45H51N3O5: 755.393.
C60), 4.22–3.99 (4H, m, 2 × CH2-N-alkyl chain), 3.90–3.76 (1H, m, CHHN-pyrrolidine), 2.60–2.48 (1H, m, NCH-CHH-pyrrolidine), 2.27–1.76 (6H, m, CHHN-pyrrolidine + NCH-CHH-pyrrolidine + CH2-pyrrolidine + 2 × CH-alkyl chain), 1.50–1.22 (16H, m, 8 × CH2-alkyl chain) and 1.01–0.82 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C): 165.8, 164.3, 164.0, 163.9, 148.8, 148.0, 147.0, 145.5, 145.4, 145.2, 145.1, 145.0, 144.9, 144.8, 144.6, 144.5, 144.4, 144.3, 144.2, 144.1, 143.9, 143.7, 143.6, 143.5, 143.4, 143.3, 143.1, 142.9, 142.8, 142.7, 142.5, 142.2, 142.1, 142.0, 141.9, 141.8, 141.2, 141.0, 140.7, 140.2, 140.0, 139.9, 139.2, 136.3, 136.1, 135.6, 135.1, 133.6, 131.5, 131.1, 129.5, 129.0, 127.8, 127.5, 124.9, 124.5, 124.0, 123.7, 123.1, 122.5, 122.1, 121.1, 120.0, 119.4, 70.2, 70.0, 68.9, 57.1, 44.5, 44.4, 44.3, 38.5, 38.2, 38.0, 30.9, 28.9, 28.8, 28.7, 25.4, 24.2, 23.3, 23.2, 14.3 and 10.9; IR-FT (KBr) 2951, 2923, 2854, 1740, 1693, 1655, 1417, 1333, 1242, 1182, 1152, 807, 747, 527 ν/cm−1; UV/vis (CH2Cl2), λmax/nm (logε): 327 (4.60), 428 (4.18), 628 (4.35); HR-MS (MALDI-TOF, dithranol): m/z = 1473.302, [M]−, calcd for C107H51N3O6: 1473.378.
:1). The first green fraction was collected and further purified in preparative HPLC (buckyprep, toluene, 10 ml min−1. R.t. = 18.6 min) yielding 1 as green dust (3 mg, 8%). 1H NMR (300 MHz, CDCl3, 25 °C): 9.19 (1H, s, H-PDI), 8.69 (1H, d, J = 8.1 Hz, H-PDI), 8.59 (1H, d, J = 8.0 Hz, H-PDI), 8.44–8.35 (4H, m, 4 × H-PDI), 5.21–5.27 (2H, m, CHN-pyrrolidine + CHH-O2C), 4.87–4.77 (1H, m; CHH-O2C), 4.71 (1H, d, J = 16.2 Hz, O2C-CHH-C59N), 4.63 (1H, d, J = 16.2 Hz, O2C-CHH-C59N), 4.24–4.03 (4H, m, 2 × CH2-N-alkyl chain), 3.95–3.83 (1H, m, CHHN-pyrrolidine), 2.62–2.48 (1H, m, NCH-CHH-pyrrolidine), 2.28–1.76 (6H, m, CHHN-pyrrolidine + NCH-CHH-pyrrolidine + CH2-pyrrolidine + 2 × CH-alkyl chain), 1.48–1.22 (16H, m, 8 × CH2-alkyl chain) and 1.02–0.80 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C): 169.5, 164.5, 164.3, 164.1, 164.0, 154.7, 154.4, 148.5, 147.8, 147.6, 147.5, 147.4, 147.1, 147.0, 146.7, 146.6, 146.5, 146.4, 146.3, 145.9, 145.8, 145.7, 145.1, 145.0, 144.8, 144.6, 144.3, 143.8, 143.6, 143.5, 142.7, 142.1, 141.7, 141.6, 141.1, 140.9, 140.8, 140.6, 139.4, 139.2, 137.0, 136.9, 135.8, 135.2, 134.2, 133.7, 133.6, 131.6, 131.1, 129.4, 129.0, 127.9, 127.6, 124.8, 124.3, 124.1, 123.9, 123.8, 123.6, 123.0, 122.4, 122.0, 121.0, 120.0, 119.2, 78.4, 68.4, 57.4, 57.1, 46.9, 44.5, 44.3, 38.2, 37.9, 30.9, 29.8, 29.1, 28.9, 28.8, 28.7, 25.3, 24.2, 23.3, 23.2, 14.3, 10.9 and 10.8 ppm; IR-FT (KBr) ν/cm−1: 3425, 2953, 2925, 2856, 1747, 1694, 1656, 1588, 1421, 1334, 1242, 1185, 808, 748, 525; UV/vis (CH2Cl2), λmax/nm (logε): 321 (4.59), 431 (4.27), 632 (4.37); HR-MS (MALDI-TOF, dithranol): m/z = 1476.463, [M]−, calcd for C106H52N4O6: 1476.389.
O), 2.11–1.88 (4H, m, CH2-pyrrolidine + 2 × CH-alkyl chain), 1.50–1.24 (16H, m, 8 × CH2-alkyl chain) and 1.01–0.84 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C): 170.9, 164.3, 164.1, 147.8, 135.9, 135.2, 133.2, 131.3, 129.2, 129.1, 127.6, 127.5, 124.6, 124.0, 123.9, 123.3, 122.9, 122.6, 122.1, 121.0, 119.8, 118.9, 65.3, 57.5, 56.4, 44.5, 38.1, 31.0, 29.1, 28.9, 25.4, 24.3, 23.2, 21.0, 14.3, 10.9, 10.8; IR-FT (KBr) 2956, 2927, 2857, 1744, 1693, 1654, 1587, 1419, 1336, 1240, 808, 748 ν/cm−1; UV/vis (CH2Cl2), λmax/nm (logε):332 (3.80), 430 (4.17), 624 (4.36); HR-MS (MALDI-TOF, dithranol): m/z = 755.385, [M]−, calcd for C45H51N3O5: 755.393.
C60), 4.22–3.99 (4H, m, 2 × CH2-N-alkyl chain), 3.90–3.76 (1H, m, CHHN-pyrrolidine), 2.60–2.48 (1H, m, NCH-CHH-pyrrolidine), 2.27–1.76 (6H, m, CHHN-pyrrolidine + NCH-CHH-pyrrolidine + CH2-pyrrolidine + 2 × CH-alkyl chain), 1.50–1.22 (16H, m, 8 × CH2-alkyl chain) and 1.01–0.82 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C): 165.8, 164.3, 164.0, 163.9, 148.8, 148.0, 147.0, 145.5, 145.4, 145.2, 145.1, 145.0, 144.9, 144.8, 144.6, 144.5, 144.4, 144.3, 144.2, 144.1, 143.9, 143.7, 143.6, 143.5, 143.4, 143.3, 143.1, 142.9, 142.8, 142.7, 142.5, 142.2, 142.1, 142.0, 141.9, 141.8, 141.2, 141.0, 140.7, 140.2, 140.0, 139.9, 139.2, 136.3, 136.1, 135.6, 135.1, 133.6, 131.5, 131.1, 129.5, 129.0, 127.8, 127.5, 124.9, 124.5, 124.0, 123.7, 123.1, 122.5, 122.1, 121.1, 120.0, 119.4, 70.2, 70.0, 68.9, 57.1, 44.5, 44.4, 44.3, 38.5, 38.2, 38.0, 30.9, 28.9, 28.8, 28.7, 25.4, 24.2, 23.3, 23.2, 14.3 and 10.9; IR-FT (KBr) 2951, 2923, 2854, 1740, 1693, 1655, 1417, 1333, 1242, 1182, 1152, 807, 747, 527 ν/cm−1; UV/vis (CH2Cl2), λmax/nm (logε): 327 (4.60), 428 (4.18), 628 (4.35); HR-MS (MALDI-TOF, dithranol): m/z = 1473.302, [M]−, calcd for C107H51N3O6: 1473.378.
:1). The first green fraction was collected and further purified in preparative HPLC (buckyprep, toluene, 10 ml min−1. R.t. = 18.6 min) yielding 1 as green dust (3 mg, 8%). 1H NMR (300 MHz, CDCl3, 25 °C): 9.19 (1H, s, H-PDI), 8.69 (1H, d, J = 8.1 Hz, H-PDI), 8.59 (1H, d, J = 8.0 Hz, H-PDI), 8.44–8.35 (4H, m, 4 × H-PDI), 5.21–5.27 (2H, m, CHN-pyrrolidine + CHH-O2C), 4.87–4.77 (1H, m; CHH-O2C), 4.71 (1H, d, J = 16.2 Hz, O2C-CHH-C59N), 4.63 (1H, d, J = 16.2 Hz, O2C-CHH-C59N), 4.24–4.03 (4H, m, 2 × CH2-N-alkyl chain), 3.95–3.83 (1H, m, CHHN-pyrrolidine), 2.62–2.48 (1H, m, NCH-CHH-pyrrolidine), 2.28–1.76 (6H, m, CHHN-pyrrolidine + NCH-CHH-pyrrolidine + CH2-pyrrolidine + 2 × CH-alkyl chain), 1.48–1.22 (16H, m, 8 × CH2-alkyl chain) and 1.02–0.80 (12H, m, 4 × CH3-alkyl chain) ppm; 13C NMR (75 MHz, CDCl3, 25 °C): 169.5, 164.5, 164.3, 164.1, 164.0, 154.7, 154.4, 148.5, 147.8, 147.6, 147.5, 147.4, 147.1, 147.0, 146.7, 146.6, 146.5, 146.4, 146.3, 145.9, 145.8, 145.7, 145.1, 145.0, 144.8, 144.6, 144.3, 143.8, 143.6, 143.5, 142.7, 142.1, 141.7, 141.6, 141.1, 140.9, 140.8, 140.6, 139.4, 139.2, 137.0, 136.9, 135.8, 135.2, 134.2, 133.7, 133.6, 131.6, 131.1, 129.4, 129.0, 127.9, 127.6, 124.8, 124.3, 124.1, 123.9, 123.8, 123.6, 123.0, 122.4, 122.0, 121.0, 120.0, 119.2, 78.4, 68.4, 57.4, 57.1, 46.9, 44.5, 44.3, 38.2, 37.9, 30.9, 29.8, 29.1, 28.9, 28.8, 28.7, 25.3, 24.2, 23.3, 23.2, 14.3, 10.9 and 10.8 ppm; IR-FT (KBr) ν/cm−1: 3425, 2953, 2925, 2856, 1747, 1694, 1656, 1588, 1421, 1334, 1242, 1185, 808, 748, 525; UV/vis (CH2Cl2), λmax/nm (logε): 321 (4.59), 431 (4.27), 632 (4.37); HR-MS (MALDI-TOF, dithranol): m/z = 1476.463, [M]−, calcd for C106H52N4O6: 1476.389.
Conclusions
We have synthesized for the first time a dyad where a perylenediimide unit is connected to C59N. Taking advantage of the electron donor character of the mono pyrrolidinyl PDI derivative synthesised, the PDI-C59N dyad undergoes efficient intramolecular photoinduced electron transfer to afford a 400 ps-lived CS state in PhCN. PDI-C60 has also been synthesized, as a reference compound, affording a 3 times shorter CSS lifetime than the PDI-C59N dyad to address the influence of the nitrogen in the photoinduced electron-transfer process. The present results have significant implications for the design of organic solar cells based on covalent donor–acceptor systems using C59N as the acceptor and shows that the longer-lived charge-separated states can be attained using C59N systems instead of C60 systems.Acknowledgements
This work has been supported by the Spanish Ministry of Science and Innovation, Generalitat Valenciana, the European FEDER funds (CTQ2011-26455, PROMETEO 2012/010, ACOMP/2013/024 and ISIC/2012/008), Grants-in-Aid for Scientific Research (no. 26620154 & 26288037) and an Advanced Low Carbon Technology Research and Development (ALCA) and Development of Systems and Technology for Advanced Measurement and Analysis (SENTAN) programs from Japan Science Technology Agency (JST).Notes and references
- Recent and interesting reviews on this topic are (a) A. C. Benniston and A. Harriman, Mater. Today, 2008, 11, 26–34 CrossRef CAS; (b) M. E. El-Khouly, S. Fukuzumi and F. D'Souza, ChemPhysChem, 2014, 15, 30–47 CrossRef CAS PubMed; (c) S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC; (d) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- (a) D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolskia and N. Martín, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC; (b) E. Espildora, J. L. Delgado and N. Martín, Isr. J. Chem., 2014, 54, 429–439 CrossRef CAS; (c) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575–4587 RSC; (d) Y. Kawashima, K. Ohkubo and S. Fukuzumi, Chem. – Asian J., 2015, 10, 44–54 CrossRef CAS PubMed.
- (a) L. Martín-Gomis, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi and Á. Sastre-Santos, Org. Lett., 2007, 9, 3441–3444 CrossRef PubMed; (b) L. Martín-Gomis, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi and Á. Sastre-Santos, Chem. Commun., 2010, 46, 3944–3946 RSC; (c) V. Garg, G. Kodis, M. Chachisvilis, M. Hambourger, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2011, 133, 2944–2954 CrossRef CAS PubMed; (d) T. Fukuda, Y. Kikukawa, S. Takaishi and N. Kobayashi, Chem. – Asian J., 2012, 7, 751–758 CrossRef CAS PubMed; (e) E. Krokos, F. Spänig, M. Ruppert, A. Hirsch and D. M. Guldi, Chem. – Eur. J., 2012, 18, 10427–10435 CrossRef CAS PubMed; (f) M. E. El-Khouly, C. A. Wijesinghe, V. N. Nesterov, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem. – Eur. J., 2012, 18, 13844–13853 CrossRef CAS PubMed; (g) R. F. Enes, J.-J. Cid, A. Hausmann, O. Trukhina, A. Gouloumis, P. Vázquez, J. A. S. Cavaleiro, A. C. Tomé, D. M. Guldi and T. Torres, Chem. – Eur. J., 2012, 18, 1727–1736 CrossRef CAS PubMed; (h) C. B. KC, G. N. Lim, M. E. Zandler and F. D'Souza, Org. Lett., 2013, 15, 4612–4615 CrossRef CAS PubMed; (i) W.-J. Shi, M. E. El-Khouly, K. Ohkubo, S. Fukuzumi and D. K. P. Ng, Chem. – Eur. J., 2013, 19, 11332–11341 CrossRef CAS PubMed; (j) M. E. El-Khouly, J.-H. Kim, K.-Y. Kay and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2013, 17, 1055–1063 CrossRef CAS; (k) R. K. Dubey, M. Niemi, K. Kaunisto, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Chem. – Eur. J., 2013, 19, 6791–6806 CrossRef CAS PubMed; (l) S. Pla, L. Martín-Gomis, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and Á. Sastre-Santos, Asian J. Org. Chem., 2014, 3, 185–197 CrossRef CAS; (m) J. Ranta, K. Kaunisto, M. Niskanen, A. Efimov, T. I. Hukka and H. Lemmetyinen, J. Phys. Chem. C, 2014, 118, 2754–2765 CrossRef CAS.
- O. Vostrowsky and A. Hirsch, Chem. Rev., 2006, 106, 5191–5207 CrossRef CAS PubMed.
- (a) J. C. Hummelen, B. Knight, J. Pavlovich, R. González and F. Wudl, Science, 1995, 269, 1554–1556 CAS; (b) M. Keshavarz-K, R. Gonzalez, R. G. Hicks, G. Srdanov, V. I. Srdanov, T. G. Collins, J. C. Hummelen, C. Bellavia-Lund, J. Pavlovich, F. Wudl and K. Holczer, Nature, 1996, 383, 147–150 CrossRef CAS; (c) M. Keshavarz-K, R. Gonzalez, R. G. Hicks, G. Srdanov, V. I. Srdanov, T. G. Collins, J. C. Hummelen, C. Bellavia-Lund, J. Pavlovich, F. Wudl and K. Holczer, Nature, 1996, 383, 147–150 CrossRef CAS; (d) C. Bellavia-Lund, R. González, J. C. Hummelen, R. G. Hicks, A. Sastre and F. Wudl, J. Am. Chem. Soc., 1997, 119, 2946–2947 CrossRef CAS.
- F. Hauke and A. Hirsch, Chem. Commun., 1999, 2199–2200 RSC.
- (a) F. Hauke, A. Swartz, D. M. Guldi and A. Hirsch, J. Mater. Chem., 2002, 12, 2088–2094 RSC; (b) F. Hauke, S. Atalick, D. M. Guldi and A. Hirsch, Tetrahedron, 2006, 62, 1923–1927 CrossRef CAS PubMed.
- F. Hauke, A. Hirsch, S. Atalick and D. M. Guldi, Eur. J. Org. Chem., 2005, 1741–1751 CrossRef CAS.
- G. Rotas, J. Ranta, A. Efimov, M. Niemi, H. Lemmetyinen, N. Tkachenko and N. Tagmatarchis, ChemPhysChem, 2012, 13, 1246–1254 CrossRef CAS PubMed.
- G. Rotas, G. Charalambidis, L. Glätzl, D. T. Gryko, A. Kahnt, A. G. Coutsolelos and N. Tagmatarchis, Chem. Commun., 2013, 49, 9128–9130 RSC.
- (a) C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed; (b) C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed; (c) E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS PubMed; (d) M. Guide, S. Pla, A. Sharenko, P. Zalar, F. Fernández-Lázaro, Á. Sastre-Santos and T.-Q. Nguyen, Phys. Chem. Chem. Phys., 2013, 15, 18894–18899 RSC; (e) Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389–4394 RSC; (f) X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed; (g) R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435–441 RSC.
- (a) Y. Shibano, T. Umeyama, Y. Matano, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Org. Lett., 2006, 8, 4425–4428 CrossRef CAS PubMed; (b) R. K. Dubey, M. Niemi, K. Kaunisto, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Chem. – Eur. J., 2013, 19, 6791–6806 CrossRef CAS PubMed.
- Y. Liu and J. Zhao, Chem. Commun., 2012, 48, 3751–3753 RSC.
- H. Ito, T. Tada, M. Sudo, Y. Ishida, T. Hino and K. Saigo, Org. Lett., 2003, 5, 2643–2645 CrossRef CAS PubMed.
- E. M. Pérez, M. Sierra, L. Sánchez, M. R. Torres, R. Viruela, P. M. Viruela, E. Ortí and N. Martín, Angew. Chem., Int. Ed., 2007, 46, 1847–1851 CrossRef PubMed.
- (a) S. Prathapan, S. I. Yang, J. Seth, M. A. Miller, D. F. Bocian, D. Holten and J. S. Lindsey, J. Phys. Chem. B, 2001, 105, 8237–8248 CrossRef CAS; (b) W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- Y. Shibano, T. Umeyama, Y. Matano, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Org. Lett., 2006, 8, 4425–4428 CrossRef CAS PubMed.
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization spectra of the new compounds. See DOI: 10.1039/c5nr00308c |
| This journal is © The Royal Society of Chemistry 2015 |