
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diameter-dependent release of a cisplatin pro-drug from small and large functionalized carbon nanotubes†
Laura
Muzi
ab,
Cécilia
Ménard-Moyon
a,
Julie
Russier
a,
Jian
Li
c,
Chee Fei
Chin
d,
Wee Han
Ang
 de,
Giorgia
Pastorin
*ce,
Gianfranco
Risuleo
*b and
Alberto
Bianco
*a
de,
Giorgia
Pastorin
*ce,
Gianfranco
Risuleo
*b and
Alberto
Bianco
*a
aCNRS, Institut de Biologie Moléculaire et Cellulaire, Laboratoire d'Immunopathologie et Chimie Thérapeutique, Strasbourg, France. E-mail: a.bianco@ibmc-cnrs.unistra.fr
bDipartimento di Biologia e Biotecnologie, Sapienza Università di Roma, Italy. E-mail: gianfranco.risuleo@uniroma1.it
cDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore. E-mail: phapg@nus.edu.sg
dDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore
eNUS Graduate School of Integrative Sciences and Engineering, National University of Singapore, Singapore
First published on 16th February 2015
Abstract
The use of platinum-based chemotherapeutic drugs in cancer therapy still suffers from severe disadvantages, such as lack of appropriate selectivity for tumor tissues and insurgence of multi-drug resistance. Moreover, drug efficacy can be attenuated by several mechanisms such as premature drug inactivation, reduced drug uptake inside cells and increased drug efflux once internalized. The use of functionalized carbon nanotubes (CNTs) as chemotherapeutic drug delivery systems is a promising strategy to overcome such limitations due to their ability to enhance cellular internalization of poorly permeable drugs and thus increase the drug bioavailability at the diseased site, compared to the free drug. Furthermore, the possibility to encapsulate agents in the nanotubes’ inner cavity can protect the drug from early inactivation and their external functionalizable surface is useful for selective targeting. In this study, a hydrophobic platinum(IV) complex was encapsulated within the inner space of two different diameter functionalized multi-walled CNTs (Pt(IV)@CNTs). The behavior of the complexes, compared to the free drug, was investigated on both HeLa human cancer cells and RAW 264.7 murine macrophages. Both CNT samples efficiently induced cell death in HeLa cancer cells 72 hours after the end of exposure to CNTs. Although the larger diameter CNTs were more cytotoxic on HeLa cells compared to both the free drug and the smaller diameter nanotubes, the latter allowed a prolonged release of the encapsulated drug, thus increasing its anticancer efficacy. In contrast, both Pt(IV)@CNT constructs were poorly cytotoxic on macrophages and induced negligible cell activation and no pro-inflammatory cytokine production. Both CNT samples were efficiently internalized by the two types of cells, as demonstrated by transmission electron microscopy observations and flow cytometry analysis. Finally, the platinum levels found in the cells after Pt(IV)@CNT exposure demonstrate that they can promote drug accumulation inside cells in comparison with treatment with the free complex. To conclude, our study shows that CNTs are promising nanocarriers to improve the accumulation of a chemotherapeutic drug and its slow release inside tumor cells, by tuning the CNT diameter, without inducing a high inflammatory response.
1. Introduction
Chemotherapy, either alone or in combination with surgery and radiation therapy, is widely used to treat different types of cancer. However, limited drug solubility, rapid elimination and lack of appropriate selectivity for tumor tissues, causing severe side effects, are some of the drawbacks faced by current chemotherapeutics. Moreover, various cancers display both intrinsic and acquired multidrug-resistance. This phenomenon can be ascribed to P-glycoprotein, an efflux pump able to recognize and transport the drug out from the cell once internalized, therefore attenuating the drug-induced cytotoxic effect.1 Among all, Pt(II) cisplatin analogues, together with carboplatin or oxaliplatin, have a central role in cancer chemotherapy, including testicular, cervical, head and neck, ovarian and non-small-cell lung cancer.2 Pt(IV) complexes have also been synthesized as inactive cisplatin prodrugs, which can be converted to their active Pt(II) form upon cleavage of the axial ligands.3 On the other hand, the poor specificity of these drugs induces numerous side effects, such as nephrotoxicity, emetogenesis, ototoxicity and neurotoxicity, thus limiting their applications.4 Cisplatin's mechanism of action involves the formation of DNA interstrand cross-links. Severe cellular DNA damage disrupts the normal replication and transcription processes activating the cell cycle checkpoint leading to apoptosis.5 To make this possible, once internalized, cisplatin undergoes a series of aquation reactions. The active mono-aquated form of cisplatin is a highly reactive species which can be recognized by several endogenous nucleophiles, thus making the drug potentially vulnerable and leading to premature cytoplasmic inactivation.6 Moreover, in some cancer cells, reduction in drug accumulation is the main mechanism of cisplatin resistance. In addition to an increased expression of efflux pumps, the mechanism is also associated with the downregulation of the plasma membrane copper transporter CTR1 involved in cisplatin uptake. An increased DNA damage tolerance and the loss of propagation of the DNA damage signals to the apoptotic machinery are other downstream cellular resistance mechanisms to cisplatin.6–8Nanomaterials can help in overcoming the limitations described above, offering great advantages to the delivery of therapeutic molecules to the tumors. They can help the delivery of drugs with poor solubility and stability, reduce their non-specific accumulation and toxicity, improve the pharmacokinetics and bioavailability and increase local drug concentrations. As a whole effect, they can enhance both drug permeability and retention at the target tissue. Several nanocarriers have been used for the delivery of platinum-based anticancer drugs including liposomes, micelles, dendrimers, polymeric nanoparticles and carbon nanotubes.9–11
CNTs are among the most attractive nanovectors under investigation.12 Functionalized CNTs (f-CNTs) have shown great promise as tools for biomedical applications. This is due to their well demonstrated ability to cross biological barriers entering and accumulating inside cells.13,14 In addition, their high surface area provides multiple attachment sites for molecules and are in turn useful for selective targeting.15 In fact, CNTs have been used as biosensors, as scaffolds for regenerative medicine and as delivery systems of many bioactive molecules, including various anti-cancer agents.16–19 Therapeutic molecules can be either attached on the external CNT wall or encapsulated within their inner cavity, thus protecting the drug from premature inactivation and enabling the possibility of controlled drug release.20 On the other hand, different approaches have been developed to make this material biocompatible and recently, it has been demonstrated that f-CNTs can be degraded by oxidative enzymes.21
In this study, a hydrophobic Pt(IV) prodrug of cisplatin was entrapped within the inner cavity of two different diameter fluorescently labeled multi-walled CNTs (MWCNTs) and the complexes were tested for their activity on two different cellular models. A number of studies on the efficacy of cisplatin-filled carbon-based nanomaterials, in particular single-walled CNTs (SWCNTs), have been published.22–27 However, the smaller diameter of SWCNTs allows encapsulating a lower amount of drug molecules compared to MWCNTs. Furthermore, another advantage of our complexes is the use of a prodrug, designed to achieve intracellular controlled release of the cytotoxic Pt(II) species. In fact, upon chemical reduction by endogenous reducing agents such as glutathione, the prodrug can be released from the CNT cavity being converted into its active hydrophilic Pt(II) form able to react with DNA.28 This avoids a fast and premature drug release (unlike what happens with cisplatin), preventing also its possible inactivation. Our groups previously demonstrated that Pt(IV)@CNTs can significantly enhance drug accumulation in some tissues after being intravenously injected in mice without inducing any abnormal inflammation. Moreover, both kidney and liver accumulation were reduced thus decreasing eventual nephrotoxicity, a typical side effect of cisplatin.29 For a better understanding of Pt(IV)@CNT complex activity, in the current study the cytotoxicity, the possible induction of a pro-inflammatory response and the cellular uptake were investigated on human cancer cells HeLa and murine macrophages RAW 264.7. The intracellular platinum accumulation after exposure to different complexes was also explored. In addition, the effect of the MWCNT diameter on the release and activity of the Pt(IV) prodrug was examined for the first time.
2. Results and discussion
2.1. Preparation of Pt(IV)@CNTs
Two types of MWCNTs with different diameters and length distributions were used in this study. The CNT diameter distributions are shown in Fig. 1A. MWCNTs (S-CNTs) from Nanocyl company have an average external diameter and length of 10 nm (n = 178, σ = 2.8) and 1.5 μm, respectively. We also used non-commercial MWCNTs (L-CNTs) with a bigger external diameter (38 nm in average, n = 144, σ = 13.6) and a length ranging from 200 nm to several microns. The average inner diameter of S- and L-CNTs is 4 nm30 and 10 nm,29 respectively. To reduce their length, both types of MWCNTs were treated under acid conditions using a mixture of sulfuric and nitric acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under sonication (Scheme 1). The length distribution of both types of MWCNTs after oxidation is shown in Fig. 1B: the average length of shortened S- and L-CNTs is 274 nm (n = 222, σ = 227) and 855 nm (n = 100, σ = 537), respectively. Both types of MWCNTs have a similar aspect ratio in the range of 23–27.
:1) under sonication (Scheme 1). The length distribution of both types of MWCNTs after oxidation is shown in Fig. 1B: the average length of shortened S- and L-CNTs is 274 nm (n = 222, σ = 227) and 855 nm (n = 100, σ = 537), respectively. Both types of MWCNTs have a similar aspect ratio in the range of 23–27.
 | ||
| Fig. 1 Histograms of the diameter of S- and L-CNTs (A) and length distribution of functionalized S- and L-CNTs (B). The diameter and length were measured on hundreds of CNTs in TEM images. | ||
 | ||
| Scheme 1 Functionalization of S-CNTs with cyanine 5. | ||
To allow monitoring of the trafficking of MWCNTs upon interactions with cells, we labeled the nanotubes with the fluorescent probe cyanine 5 (Cy5). The synthetic strategy for the derivatization of S-CNTs is shown in Scheme 1. We first derivatized the oxidized S-CNTs 1 by amidation with Boc-monoprotected diamine derivative 2, after activation of the carboxylic groups using oxalyl chloride. The Boc protecting group was cleaved using HCl in 1,4-dioxane. The Kaiser test was used to assess the amount of amino groups: 400 μmol per gram of S-CNTs 4.31 The ammonium functions were derivatized with Cy5 derivative 5 in the presence of O-(N-succinimidyl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TSTU) as the coupling agent.32 According to the Kaiser test, the amine loading drastically decreased (165 μmol g−1), which confirmed the efficiency of the amidation reaction. We deduced that the level of functionalization was 235 μmol of Cy5 per gram of S-CNTs 6. We followed a similar strategy to label the L-CNTs with Cy5. Amino-L-CNTs S1 were prepared according to a protocol previously reported by our group.29 The amines were derivatized with Cy5 derivative 5 using TSTU (Scheme S1†). After the reaction, the Kaiser test decreased from 670 to 105 μmol g−1. Thus, the Cy5 loading was 565 μmol per gram of L-CNTs S2.
2.2. Characterization of functionalized MWCNTs
The functionalized MWCNTs were characterized by Transmission electron microscopy (TEM) and thermogravimetric analysis (TGA). The TEM images of S-CNTs 4 and L-CNTs S1 are shown in Fig. 2A and 2B. The shortening of both types of CNTs is clearly visible in comparison with pristine purified S-CNTs and L-CNTs (Fig. S1†). | ||
| Fig. 2 TEM images of S-CNTs 4 (A) and L-CNTs S1 (B). TGA of pristine S-CNTs, oxidized S-CNTs 1, S-CNTs 4 and 6 under N2 (C). | ||
The TGA profiles of pristine purified S-CNTs, oxidized S-CNTs 1, and S-CNTs 4 and 6 are displayed in Fig. 2C. As the analyses were performed under an inert atmosphere, the weight loss observed corresponds to the amount of functional groups present on the nanotube surface. As expected, the pristine purified S-CNTs show a negligible weight loss under these conditions, while the weight loss gradually increases after each step of derivatization, which shows successful functionalization. The TGA of L-CNTs S2 in comparison with the precursor L-CNTs is shown in Fig. S2.†
The Cy5-CNTs 6 and S3 were filled with platinum(IV) prodrug S3 (Fig. S3†). The entrapment of the Pt(IV) complex was performed according to the nano-extraction procedure we recently reported for the encapsulation of S3 in amino-CNTs S1.29 The platinum content was determined using inductively coupled plasma atomic emission spectrometry (ICP/AES). High Pt(IV) levels were measured inside both types of CNTs: 37.3% and 35.6% w/w of Pt(IV) for S-CNTs 6 and L-CNTs S2, respectively. The Pt(IV)@CNTs 6 and S2 were characterized by high-resolution TEM (HR-TEM) coupled to EDX (Fig. 3 and S4†). The energy-dispersive X-ray spectroscopy (EDX) analyses were performed at different locations, either in the inner core of the nanotubes or on their sidewall. We could observe the appearance of the Pt M peak at 2.05 keV (Fig. 3A) when analyzing the cavity of Pt(IV)@L-CNTs S2, while no significant amount of Pt was detected in the sidewall (Fig. 3B). Similar results were also obtained for Pt(IV)@S-CNTs 6, confirming that the majority of Pt detected by EDX was found in the inner core of the nanotubes, which is indicative of a high entrapment efficiency.
 | ||
| Fig. 3 HRTEM images and EDX spectra of Pt(IV)@L-CNTs S2 taken at two different locations (see red circles): in the cavity (A) and in the sidewall (B). Scale bars correspond to 10 nm (A) and 5 nm (B). | ||
2.3. Cellular viability
As a first step in our investigation, the cytotoxicity of the two different Pt(IV)@CNTs compared to the free drug was evaluated in cells with different characteristics and origin. For this purpose, human cervix cancer cells HeLa (non-phagocytic) and murine macrophages RAW 264.7 (phagocytic) were chosen. As previously described, S- and L-CNTs differ only in their diameter and length, whereas they have similar aspect ratio and drug loading. In an initial experiment, cells were incubated with Pt(IV)@CNT complexes at Pt(IV) concentrations ranging from 1 to 15 μM (HeLa) or from 0.5 to 10 μM (RAW 264.7) for 24 h. The corresponding amount of MWCNTs used was back-calculated from the loading of the prodrug inside the tubes (Table 1). As Pt(IV) loading inside S- and L-CNTs was rather similar (37.3% and 35.6%, respectively), almost equal concentrations of the two CNT types were used. At the end of the incubation, cellular viability was determined by flow cytometry. Results are shown in Fig. 4 (where only live cells are reported) and S5 (where also dead and pre-apoptotic cells are reported). In the case of HeLa cells (Fig. 4A and S5A†), a concentration-dependent cytotoxicity was observed with both CNT samples and also the free drug. Furthermore, there was no significant difference when comparing the free drug with Pt(IV)@L-CNTs in the whole concentration range. In contrast, both the free drug and the larger CNTs were more cytotoxic than the smaller ones.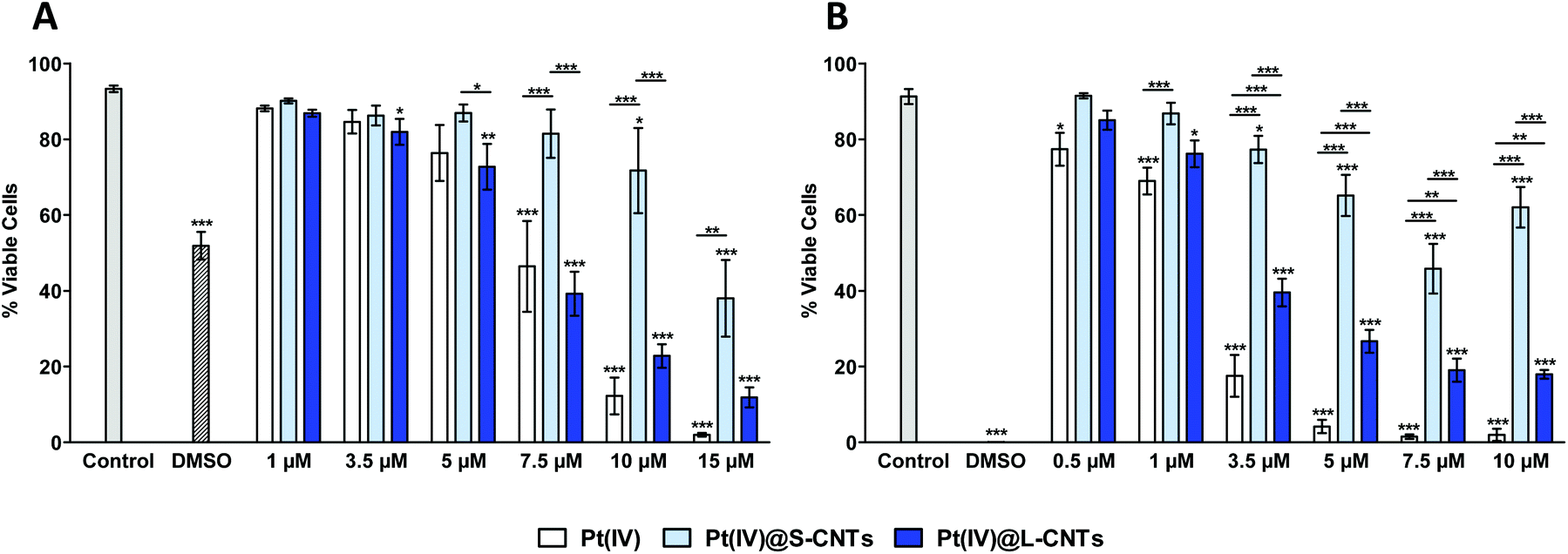 | ||
| Fig. 4 Flow cytometry analysis of cellular viability in HeLa cells (A) and RAW 264.7 cells (B) exposed to different concentrations of Pt(IV)@CNTs for 24 h. AnnV−/PI− live cells are shown in this graph. Two-way ANOVA followed by Bonferroni's post-test was performed to determine the statistical differences versus control cells and to compare the two CNT samples with each other and with Pt(IV) (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
| Pt(IV) (μM) | Pt(IV) (μg ml−1) | Pt(IV)@S-CNTs (μg ml−1) | Pt(IV)@L-CNTs (μg ml−1) |
|---|---|---|---|
| 0.1 | 0.05 | 0.15 | 0.15 |
| 0.2 | 0.11 | 0.29 | 0.30 |
| 0.5 | 0.27 | 0.72 | 0.76 |
| 1 | 0.54 | 1.45 | 1.52 |
| 2 | 1.08 | 2.90 | 3.00 |
| 3.5 | 1.89 | 5.07 | 5.31 |
| 5 | 2.70 | 7.24 | 7.58 |
| 7.5 | 4.05 | 10.86 | 11.38 |
| 10 | 5.40 | 14.48 | 15.17 |
| 15 | 8.10 | 21.75 | 22.80 |
This first experiment allowed us to demonstrate the role of MWCNT diameter in the release of the embedded drug. As previously described, the hydrophobic Pt(IV) prodrug is selectively released by chemical reduction and hydrophobicity reversal from the inner cavity of CNTs by reducing agents, being converted into its active Pt(II) species.28 In view of this, one hypothesis to explain the different efficacy of the two CNT samples could be a reduced accessibility to the intracellular reducing agents, such as glutathione, in the case of MWCNTs with smaller diameter compared to the larger tubes. Consequently, the active form of the drug could be released more slowly by S-CNTs compared to L-CNTs, thus explaining their lower cytotoxicity after 24 h.
On the other hand, RAW 264.7 cells displayed a general higher sensitivity to the samples compared to HeLa cells. In fact, the same dose-dependent cytotoxicity but at lower concentrations was observed in RAW 264.7 cells (Fig. 4B and S5B†). Moreover, the free drug was significantly more effective than both Pt(IV)@L-CNTs and Pt(IV)@S-CNTs. This is consistent with the results obtained after exposure to 20% DMSO used as the positive control. In fact, the residual cellular viability was about 50% in the case of HeLa and 0% in RAW 264.7, suggesting an overall higher sensitivity of the murine macrophages compared to the other cell line. Despite this, as observed in HeLa cells, Pt(IV)@S-CNTs were again less cytotoxic compared to Pt(IV)@L-CNTs, a result consistent with the previous hypothesis. It has to be noticed that the observed toxicity on both cell types was only due to the Pt(IV)@CNT hybrid since the empty CNTs caused no alteration of HeLa and RAW 264.7 cell viability at the same corresponding concentrations (shown in μg ml−1 in Fig. S5A and B†). This observation is important, since CNT's intrinsic cytotoxicity is always an important issue to be investigated to avoid unexpected toxicological effects in normal cell populations that are absolutely unwanted in the case of an anticancer therapy.33,34
Similar results were obtained when a 48 h treatment (Fig. S6A and B†) and a time course experiment (Fig. S7A and B†) were performed on the same cell types.
As a slower release of the encapsulated drug was hypothesized in the case of S-CNTs compared to L-CNTs, long-term cell viability experiments were performed. For this purpose, both HeLa and RAW 264.7 cells were incubated with the CNTs at concentrations ranging from 0.1 to 2 μM Pt(IV) for 6 hours before the medium was renewed with fresh medium and cells were kept in culture for further 72 h. In this experiment, HeLa cells (Fig. 5A and S8A†) began to show a significant mortality compared to the control already at 0.2 μM Pt(IV) concentration for both CNT samples. In this case, Pt(IV)@S-CNTs’ cytotoxicity was comparable to the free drug although they were poorly effective after 24 h even at higher concentrations. The gain of function of Pt(IV)@S-CNTs after a relatively long period of time is fully consistent with our hypothesis. In fact, the intracellular reducing agents could have had more time to induce the release of the drug from the small diameter CNTs, making the Pt(IV)@S-CNTs more cytotoxic. On the other hand, the larger CNTs induced higher cellular mortality in comparison with both the free drug (from 0.5 to 1 μM) and the smaller ones (from 0.5 to 2 μM) even though they displayed the same cytotoxicity as the free drug in the previous experiment. This again reinforces our explanation. In contrast to the first experiment, the two CNT samples but also the free drug displayed a negligible cytotoxicity on RAW 264.7 cells in comparison with the HeLa response (Fig. 5B and S8B†). In fact, the residual viability after the treatment with all the conjugates was not less than 75% up to a drug concentration of 1 μM for both the free drug and CNTs. In all the concentration ranges, the free drug was more cytotoxic than the Pt(IV)@CNTs and there was no difference in the effect between the two CNT types. This result could be explained by a recovery of the murine macrophages after relatively short exposure to low concentrations of the conjugates, contrarily to HeLa cells, while continuous exposure to high doses of the complexes for 24 h induced severe cytotoxicity (Fig. 4B and S5B†). Similar to the first cell viability experiments, no reduction in cell viability was observed in both cell lines after exposure to the empty CNTs (Fig. S8A and B†).
 | ||
| Fig. 5 Flow cytometry analysis of cellular viability in HeLa cells (A) and RAW 264.7 cells (B) exposed to different concentrations of Pt(IV)@CNTs for 6 h and allowed to grow for further 72 h. AnnV−/PI− live cells are shown in this graph. Two-way ANOVA followed by Bonferroni's post-test was performed to determine the statistical differences versus control cells and to compare the two CNT samples with each other and with Pt(IV) (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
The results obtained in the cell viability experiments demonstrate the efficacy of both Pt(IV)@CNT complexes and suggest their possible use as nanovectors for anticancer drugs. Moreover, as already mentioned, one of the advantages of our complexes is the use of a platinum(IV) prodrug that can be reduced to release the cytotoxic compound cisplatin upon entry into the cells. The advantages of Pt(IV)@CNT complexes compared to Pt(II)@CNT, leveraging on the greater aqueous stability of Pt(IV) prodrugs, has already been demonstrated by the group of Lippard.23,24 In their previous reports, the Pt(IV) prodrug was non-covalently linked on the external surface of CNTs while in our work, the drug was entrapped within the inner cavity of CNTs. This strategy of hydrophobic entrapment shields the prodrug from premature inactivation6 and enables controlled drug release by reduction, contrary to Pt(II) complexes that are rapidly extruded from the cavity.28 In this report, we demonstrated that the diameter of CNT plays an important role in determining the rate of drug release and directly affects the efficacy of the Pt(IV)@CNT construct. Therefore, the CNT diameter should be taken into account in the design and fine-tuning of these delivery vectors.
2.4. Cellular activation and cytokine determination
Avoiding inflammation is important, especially in cancer therapy, where it can also play decisive roles at different stages of tumors and affect responses to therapy.35,36 Since the induction of a pro-inflammatory response is an important issue of investigation in the case of CNTs,37,38 cellular activation and cytokine production by RAW 264.7 macrophages were explored after exposure to all CNT samples. Macrophages were chosen since they can be considered the primary sentinels of our body not only to infectious organisms but also to exogenous materials.39 For the first purpose, CD86 expression was chosen as readout since this molecule is expressed by this cell population upon activation.40 RAW 264.7 cells were treated with different concentrations of CNTs as previously described for the cellular viability experiments (from 0.5 to 10 μM for 24 h or from 0.1 to 2 μM for 6 h followed by a further 72 h cell growth). CD86 expression on live cell gated population was then measured by flow cytometry (Fig. 6A and B). After 24 h, a significant increase in CD86 expression compared to both the control and free drug was observed for the two types of CNTs from a Pt(IV) concentration of 3.5 μM. Moreover, at this concentration, a major activation in the case of Pt(IV)@L-CNTs was observed compared to Pt(IV)@S-CNTs. This behavior was not measured at higher concentrations. In fact, while in the case of S-CNTs CD86 expression followed a concentration-dependent trend, CD86 levels decreased as the concentration of the L-CNTs increased. This can be explained by the fact that L-CNTs promoted higher cell mortality at a drug concentration higher than 3.5 μM compared to the smaller tubes (Fig. 4B). Therefore, the observed reduction of CD86 expression could be due to the death of highly activated cells that were consequently not detected. On the other hand, while the cytotoxicity experiments showed an increased responsiveness of HeLa cells to low Pt(IV)@CNT concentrations 72 h after 6 h exposure (Fig. 5A), only a slight RAW 264.7 activation (*p < 0.05) at the highest Pt(IV)@CNT doses was observed (Fig. 6B). Furthermore, there was no difference between the two CNT types. | ||
| Fig. 6 Flow cytometry analysis of CD86 expression in RAW 264.7 exposed to different concentrations of Pt(IV)@CNTs for 24 h (A) or 6 h and allowed to grow for further 72 h (B). Two-way ANOVA followed by Bonferroni's post-test was performed to determine the statistical differences versus control cells and to compare the two Pt(IV)@CNT samples with each other and with Pt(IV) (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
In order to further investigate the macrophage activation profile in response to the conjugates, the cell culture supernatants were collected from the previous experiments on RAW 264.7 macrophages and the levels of two pro-inflammatory cytokines (IL6 and TNF α) were determined by ELISA. After a 24 hour treatment, no IL6 production was detected in cell supernatants (Fig. 7A). In contrast, the TNF α secretion started to arise significantly compared to the control from a 3.5 μM Pt(IV) concentration in the case of L-CNTs (Fig. 7B). A minor effect on TNF α production was detected after treatment with Pt(IV)@S-CNTs. In fact a significant difference between the two types of nanotubes, starting from 5 μM Pt(IV), could be observed. This is consistent with the previous result obtained for RAW 264.7 cell activation. Before the cells start to display a high mortality rate, we could observe a more important increase in CD86 levels concerning the largest CNTs in comparison with the smaller tubes (Fig. 6A). On the other hand, neither IL6 nor TNF α production was detected 72 hours after the 6 hour cellular exposure to the different complexes at concentrations ranging from 0.1 to 2 μM Pt(IV) (Fig. 7C and D). This experimental observation is again consistent with the results obtained previously (Fig. 6B), where only a slight macrophage activation was observed. We can also exclude an intrinsic MWCNT pro-inflammatory action41 because no effect on cellular activation or cytokine production was observed after exposure to both the empty CNTs (Fig. S9†).
 | ||
| Fig. 7 Cytokine production by RAW 264.7. IL6 (A) and TNF α (B) levels were determined after incubation with different concentrations of Pt(IV)@CNTs for 24 h or after the cells were grown for further 72 h after a 6 h treatment (C and D for IL6 and TNF α, respectively). Two-way ANOVA followed by Bonferroni's post-test was performed to determine the statistical differences versus control cells and to compare the two Pt(IV)@CNT samples with each other and with Pt(IV) (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
Taken together the results on cellular activation and cytokine production encourage their use as nanovectors for cancer therapy. Obviously, as is the case of any other type of drug, safety is dependent on Pt(IV)@CNT concentrations.
2.5. Cellular uptake
The results obtained with the cell viability experiments evidenced that the Pt(IV)@CNTs can preserve the cytotoxic activity of the encapsulated Pt(IV) prodrug. In fact, upon cellular exposure to the complexes, a reduction in cancer cell viability was observed and it was comparable to the free drug, or even increased in the case of Pt(IV)@L-CNTs (Fig. 5A). These results suggested efficient drug transport inside the cells and thus the cellular internalization of both CNT complexes. To confirm and visualize the CNT cellular uptake and possibly identify its mechanisms, HeLa or RAW 264.7 cells were exposed to 10 μg ml−1 of S- or L-CNTs for 6 h and the samples were processed for TEM observations. Both CNT types were found inside the human and murine cells, thus confirming their cellular uptake (Fig. 8). In more detail, aggregation of CNTs inside vesicles can be observed in all cases. This suggests an endocytic (HeLa) or phagocytic (RAW 264.7) internalization pathway.42,43 It was also possible to identify some CNTs free into the cytoplasm. The latter observation is consistent with the previously reported ability of CNTs to penetrate the cells by passive diffusion as nanoneedles.42 In fact, upon inhibition of energy-dependent internalization pathways, CNTs can be still internalized by different types of cells, as has been reported in previous work performed in our laboratories.43 On the other hand, the free CNTs could be the result of endosome rupture. In Fig. 8, a single CNT outside the endosome is evidenced by a red arrow. Such behavior is particularly relevant, as nanomaterials’ ability to escape from the endolysosomal network could facilitate the drug access to its target.44 No CNTs were found in the nucleus of the two cell types. Despite this, the drug was able to reach the nucleus and exert its function blocking DNA replication5 as evidenced by the cellular viability experiments. Besides the nuclear DNA, the free drug could have triggered apoptosis also by affecting the mitochondrial DNA or by other mechanisms, such as damage of cytoplasmic proteins or of the cytoskeleton.45 A model for the Pt(IV)@CNT activity mechanism could first consist of their internalization by both passive diffusion or endocytosis/phagocytosis. The internalized CNTs could be then transported throughout the endolysosomal pathway, subsequently escaping from these compartments and slowly releasing the Pt(II) active form of the drug within the cytoplasm.28 Finally, the free drug translocates into the nucleus or to the mitochondrion exerting its cytotoxic function. Incubating the cells with the same concentration of Pt(IV)@CNTs for 6 h, similar results were obtained (data not shown). Finally, the same TEM observations were carried out after an incubation of 24 hours with Pt(IV)@CNTs. In this case, a strong impairment of the general cellular structure due to the transported drug effect could be observed. Some examples are reported in Fig. S10,† in which the cytoplasm of both cell lines shows a wide presence of vacuoles and lysis residues. The cellular uptake of Pt(IV)@CNTs was also followed by flow cytometry thanks to the functionalization with the Cy5 fluorophore (Fig. S11†). A fluorescence shift of HeLa (Fig. S11A†) or RAW 264.7 (Fig. S11B†) cell populations (corresponding to the Cy5 on the CNT surface) in comparison with the control was observed, thus confirming their intracellular uptake. We can assert that only a negligible amount of fluorescence could be derived from the CNTs interacting with the cellular surface as the samples were extensively washed before the analysis. | ||
| Fig. 8 TEM images of HeLa and RAW 264.7 incubated with the different S- and L-CNTs (10 μg ml−1) for 6 h. The dotted areas in the top images are enlarged in the respective bottom pictures. | ||
2.6. Analysis of the cellular platinum content
Flow cytometry analysis did not allow for a precise quantification of Pt(IV)@CNTs inside the cells as the occurrence of fluorescent quenching phenomena by CNTs could not be excluded.46For this reason we used ICP/AES to quantitatively analyze the cellular content of platinum in HeLa and RAW 264.7 macrophages after 6 h exposure to 1 μM of both free Pt(IV) and Pt(IV)@CNT samples. It is important to mention that almost equal concentrations of the two CNT types were utilized, as Pt(IV) loading inside S- and L-CNTs was rather similar (Table 1). As shown in Fig. 9A and B, an increase in the cellular platinum content (both entrapped or released) compared to the free drug could be observed for both CNT conjugates in the two cell lines. In particular, the amount of platinum inside both HeLa and the macrophages was about two times higher compared to the free drug when the cells were exposed to Pt(IV)@S-CNTs. Concerning Pt(IV)@L-CNTs, the increase in the intracellular drug accumulation compared to free Pt(IV) was again more evident. In fact the platinum amount found inside HeLa and RAW 264.7 cells was about three and four times higher, respectively, than the free drug. Such results allow also quantifying the CNT cell internalization. Interestingly, not a great difference in the ability to internalize the complexes between HeLa and RAW 264.7 cells was observed. In fact, as macrophages are phagocytic cells, a major CNT uptake compared to HeLa cells was expected. This was not the case, demonstrating the ability of non-phagocytic cells to internalize a considerable amount of Pt(IV)@CNTs by endocytosis or by passive diffusion.43 On the other hand, L-CNTs were able to enter both cell types more efficiently as compared to S-CNTs; this result is consistent with their higher cytotoxicity after 24 hours (Fig. 4A). However, although less internalized, Pt(IV)@S-CNTs showed a cytotoxicity comparable to Pt(IV)@L-CNTs in HeLa cells during the long-term cell viability experiment (Fig. 5A). The latter observation evidences how the slower Pt(IV) release by CNTs having a smaller diameter could be advantageous to achieve an enhanced long-lasting cytotoxicity against tumor cells, compared to CNTs with a larger diameter.
 | ||
| Fig. 9 ICP/AES analysis of the cellular platinum content in HeLa (A) and RAW 264.7 cells (B) exposed to Pt(IV) or to Pt(IV)@CNTs (1 μM) for 6 h. One-way ANOVA followed by Bonferroni's post-test was performed to determine the statistical differences versus control cells and to compare the two Pt(IV)@CNT samples with each other and with Pt(IV) (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
The results obtained by the cell uptake experiments are encouraging, given the well-documented problems in cisplatin cancer therapy. In fact, despite being one of the most potent antitumor agents known, cisplatin's decreased intracellular accumulation is one of the main mechanisms of resistance to the drug.2,6 The results show that our Pt(IV)@CNT complexes could promote drug accumulation inside the cells. This, together with the slow drug release achieved by using S-CNTs as Pt(IV) carriers, could result in an enhanced drug cytotoxicity and retention in cancer cells compared to the free drug.
3. Conclusion
In this study, a hydrophobic Pt(IV) prodrug of cisplatin was entrapped within the cavity of two functionalized MWCNTs having different inner diameters. The complexes were tested for their activity on two different cellular models. The activity of the entrapped drug was preserved in both conjugates and the role of the CNT diameter in the release of the encapsulated drug was demonstrated. Both Pt(IV)@CNT complexes induced negligible cell activation and no pro-inflammatory cytokine production on RAW 264.7 macrophages 72 h after the end of the treatment. Both samples were efficiently internalized by the two types of cells and high platinum levels were found in the cells after Pt(IV)@CNT exposure. Our study shows that f-MWCNTs are promising nanocarriers to improve the accumulation of a chemotherapeutic drug inside the cells and demonstrates that the CNT diameter is an important parameter to consider in the preparation of the complexes. In particular, a smaller CNT diameter could be advantageous, allowing a slower drug release inside tumor cells compared to CNTs with a larger diameter. This behavior could allow the drug to be retained in tumor cells and exert its function over a longer period of time. Future studies will include evaluation of the Pt(IV)@CNT constructs in both sensitive and Pt-resistant cell lines, to assess whether these nanocarriers are not only efficient transporters and delivery systems for chemotherapeutics, but they are also able to overcome the cisplatin-associated resistance due to P-glycoprotein efflux pump mechanisms. Additional in vivo studies in mice will also be necessary to assess whether S- and L-CNT samples show a different biodistribution in tissues and organs. This would be of fundamental importance in view of a possible application of Pt(IV)@CNTs in cancer therapy. In fact, besides having different diameters, the two types of nanotubes possess different lengths and such a characteristic is likely responsible for different accumulation in the body.474. Experimental section
4.1. Materials and characterization
The large diameter MWCNTs (abbreviated hereafter as L-CNT) were synthesized by Prof. Ramaprabhu (Indian Institute of Technology, Chennai, India) with a purity >95% and a length in the range of 200 nm to several microns. The small diameter MWCNTs (abbreviated hereafter as S-CNT) were provided by Nanocyl (thin MWCNT 95+ % C purity, Nanocyl 3100, batch no. 071119, average length of 1.5 μm). TEM was performed either on a Hitachi H7500 microscope with an accelerating voltage of 80 kV or on a Hitachi 600 microscope with an accelerating voltage of 75 kV. The CNTs were dispersed in a mixture of deionized water and ethanol (1:1) by water bath sonication, 10 μL of the resulting dispersion were deposited onto carbon-coated copper TEM grids (Formvar/Carbon 300 Mesh; Cu from Delta Microscopies) and dried at room temperature. The ImageJ software was used to measure the length and diameter of CNTs in TEM images. HR-TEM analyses were performed on a JEOL 2100F TEM/STEM electron microscope operating at 200 kV coupled to EDX. TGA was performed using a TGA Q500 TA instrument with a ramp of 10 °C min−1 under nitrogen using a flow rate of 60 mL min−1. ICP/AES analyses were performed on Agilent 7500ce apparatus. For this purpose, HeLa or RAW 264.7 were seeded in quadruplicate into 6-well plates at a density of 6 × 105 cells per well and allowed to adhere overnight prior to exposure to the free drug or to Pt(IV)@CNTs at a platinum concentration of 1 μM for 6 h. Cells were then detached as previously described and the cells in the 4 wells were collected together in a glass vial.
4.2. Cellular cultures
To compare the behavior of the conjugates in non-phagocytic and phagocytic cells, human cervix cancer cells HeLa and Abelson murine leukemia virus transformed macrophages RAW 264.7 were selected, respectively, as common cancer models to study the anticancer efficacy of Pt(IV)@CNTs and for the evaluation of their pro-inflammatory effect. Both cell lines were purchased from ATCC (VA, USA) and were maintained in RPMI 1640 medium supplemented with 10% heat inactivated fetal bovine serum (FBS) and 100 U ml−1 gentamycin (with the addition of 50 μM β-mercaptoethanol and 20 mM HEPES for RAW 264.7 macrophages) and incubated at 37 °C in humidified air containing 5% CO2. Media and solutions were purchased from Lonza. When the confluence reached 70–80%, HeLa or RAW 264.7 cells were washed with phosphate-buffered saline (PBS), detached with trypsin or with SE buffer (PBS containing 2 mM EDTA and 2% FBS), respectively, and subcultured every 2–3 days. Prior to CNT exposure, cells were detached, counted and reseeded in appropriate well size and density (see each experiment for details) and allowed to adhere overnight.4.3. Flow cytometry analysis
000 events using a Gallios flow cytometer (Beckman Coulter, Villepinte, France) and analyzing the data with FlowJo software.
000 events using the Gallios flow cytometer and analyzing the data on live cell gated population (AnnV−/PI−) with FlowJo software.
000 events was measured using the Gallios flow cytometer and the data on live cell gated population were analyzed with FlowJo software.
4.4. Cytokine determination
RAW 264.7 macrophage supernatants were collected after treatment for the cellular viability experiment and the levels of murine pro-inflammatory cytokine tumor necrosis factor alpha (TNF α) and interleukin-6 (IL6) were detected by a double sandwich ELISA. LPS (1 μg ml−1) in combination with interferon-γ (IFN γ, 1 ng ml−1) was used as the pro-inflammatory cytokine production positive control. Polyvinyl microtiter plates (Falcon) were coated overnight at 4 °C with 50 μl per well of purified hamster anti-mouse/rat TNF-α (BD Pharmingen 557516) or purified rat anti-mouse IL6 (BD Pharmingen 554400) diluted in 0.05 M carbonate buffer, pH 9.6. After washing with PBS containing 0.05% Tween (PBS-T), a saturation step was performed by adding 100 μl per well of PBS containing 10% FBS for 1 h at 37 °C. After washing with PBS-T, 50 μl per well of culture supernatants or recombinant mouse TNF (BD Pharmingen 554589) or recombinant mouse IL6 (BD Pharmingen 554582), diluted in PBS-10% FBS, were added for 2 h at 37 °C. Plates were then washed with PBS-T and 50 μl of secondary biotin rabbit anti-rat/mouse TNF (BD Pharmingen 557432) or biotin rat anti-mouse IL6 (BD Pharmingen 554402), diluted in PBS-10% FBS, were added per well and incubated for 1 h at 37 °C. Plates were washed with PBS-T, and 50 μl of streptavidin conjugated to horseradish peroxidase diluted in PBS-10% FBS were added per well. After 30 min incubation at 37 °C, the plates were washed extensively and the enzymatic reaction, revealing the presence of cytokines in the tested supernatants, was visualized by adding 75 μl per well of 3,3′,5,5′-tetramethylbenzidine diluted in 0.1 M citrate buffer, pH 5, in the presence of H2O2. The resulting absorbance was measured at 450 nm after the reaction was stopped with 25 μl per well of 1 N HCl.4.5. Transmission electron microscopy
For TEM observations, the cells were seeded into 24-well plates on glass coverslips at a density of 5 × 105 cells per well and allowed to adhere overnight prior to exposure to empty or Pt(IV)@CNTs at a concentration of 10 μg ml−1 (corresponding to a drug concentration of about 7.5 μM) for 6 or 24 h. The cells were then washed with PBS and fixed overnight with 2.5% glutaraldehyde diluted in phosphate buffer (PB 0.1 M, pH 7.2). The day after, cells were washed with distilled water and fixed again with 0.5% aqueous osmium tetraoxide for 1 h at room temperature. Cells were rinsed extensively with distilled water and a series of dehydration baths was performed: twice with 50% ethanol (10 min), once with 70% ethanol (20 min), once with ethanol 95% (10 min), twice with absolute ethanol (10 min) and finally twice with propylene oxide (for 10 min each). Samples were then embedded in epoxy resin Epon (LX 112 Embedding Kit, LADD). Mixtures of propylene oxide and Epon resin in 2:1 (1 h) and 1:2 (1 h) ratios and finally with pure resin (1 h) were added to the samples. On the last day, the resin was replaced with the fresh one and further incubated for 3 h. Polymerized blocks with embedded cells were then prepared by filling gelatin capsules with fresh resin and placing them upon the glass coverslips on which cells were grown. The capsules were then polymerized at 60 °C for 48 h, afterwards glass coverslips were removed from the polymerized block surface and ultrathin sections (70 nm thick) were obtained using an ultramicrotome (Leica) with a diamond knife (DiATOME). The ultrathin sections were then collected on butvar-coated single-slot copper grids and examined by TEM (Hitachi H600 or Hitachi H7500).
4.6. Statistical analysis
We present a summary of the data from at least three separate experiments run in triplicate for each cell line. Data are presented as ±SEM (Standard Error of the Mean). Statistical analyses were performed using a two-way ANOVA test or a one-way ANOVA test in the case of ICP-AES analysis, followed by Bonferroni's post-test. All p values < 0.05 were considered significant.Acknowledgements
This research was supported by the CNRS (Centre National pour la Recherche Scientifique), the National University of Singapore, Department of Pharmacy [(AcRF) Tier 1-FRC Grant R-148-000-164-112; NUSAGE grant N-148-000-009-001] and by MOE of Singapore (grantMOE T2, R-144-000-306-112). The authors acknowledge Dris Ihiawakrim for HR-TEM/EDX analysis and Professor Ramaprabhu, from the Department of Physics, Indian Institute of Technology Madras, Chennai (India), for providing the L-MWCNTs. G.P. and A.B. acknowledge the support from the 2011 Merlion Programme (project number: R-148-000-162-133). TEM was performed at the RIO Microscopy Facility Plateform of Esplanade Campus and at the Plateforme Imagerie in Vitro at the Center of Neurochemistry (Strasbourg, France). Alessia Battigelli is gratefully acknowledged for help with TGA.References
- F. Thiebaut, T. Tsuruo, H. Hamada, M. M. Gottesman, I. Pastan and M. C. Willingham, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7735–7738 CrossRef CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- Y. Shi, S. A. Liu, D. J. Kerwood, J. Goodisman and J. C. Dabrowiak, J. Inorg. Biochem., 2012, 107, 6–14 CrossRef CAS PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- V. Cepeda, M. A. Fuertes, J. Castilla, C. Alonso, C. Quevedo and J. M. Pérez, Anticancer Agents Med. Chem., 2007, 7, 3–18 CrossRef CAS.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef CAS PubMed.
- M. Rebucci and C. Michiels, Biochem. Pharmacol., 2013, 85, 1219–1226 CrossRef CAS PubMed.
- N. R. Patel, B. S. Pattni, A. H. Abouzeid and V. P. Torchilin, Adv. Drug Deliver. Rev., 2013, 65, 1748–1762 CrossRef CAS PubMed.
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Deliver. Rev., 2013, 65, 1667–1685 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- K. Kostarelos, A. Bianco and M. Prato, Nat. Nanotechnol., 2009, 4, 627–633 CrossRef CAS PubMed.
- D. Pantarotto, J. P. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16–17 RSC.
- N. W. Kam, Z. Liu and H. Dai, Angew. Chem., Int. Ed., 2006, 45, 577–581 CrossRef CAS PubMed.
- C. Fabbro, H. Ali-Boucetta, T. Da Ros, K. Kostarelos, A. Bianco and M. Prato, Chem. Commun., 2012, 3911–3926 RSC.
- M. Prato, K. Kostarelos and A. Bianco, Acc. Chem. Res., 2008, 41, 60–68 CrossRef CAS PubMed.
- E. Heister, E. V. Brunner, G. R. Dieckmann, I. Jurewicz and A. B. Dalton, ACS Appl. Mater. Interfaces, 2013, 5, 1870–1891 CAS.
- R. G. Mendes, A. Bachmatiuk, B. Büchner, G. Cunibertib and M. H. Rümmeli, J. Mater. Chem. B, 2013, 1, 401–428 RSC.
- B. S. Wong, S. L. Yoong, A. Jagusiak, T. Panczyk, H. K. Ho, W. H. Ang and G. Pastorin, Adv. Drug Deliver. Rev., 2013, 65, 1964–2015 CrossRef CAS PubMed.
- S. Boncel, P. Zając and K. K. Koziol, J. Control. Release, 2013, 169, 126–140 CrossRef CAS PubMed.
- A. Bianco, K. Kostarelos and M. Prato, Chem. Commun., 2011, 10182–10188 RSC.
- K. Ajima, M. Yudasaka, T. Murakami, A. Maigné, K. Shiba and S. Iijima, Mol. Pharm., 2005, 2, 475–480 CrossRef CAS PubMed.
- R. P. Feazell, N. Nakayama-Ratchford, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 8438–8439 CrossRef CAS PubMed.
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS PubMed.
- K. Ajima, T. Murakami, Y. Mizoguchi, K. Tsuchida, T. Ichihashi, S. Iijima and M. Yudasaka, ACS Nano, 2008, 2, 2057–2064 CrossRef CAS PubMed.
- A. A. Bhirde, V. Patel, J. Gavard, G. Zhang, A. A. Sousa, A. Masedunskas, R. D. Leapman, R. Weigert, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 307–316 CrossRef CAS PubMed.
- A. Guven, I. A. Rusakova, M. T. Lewis and L. J. Wilson, Biomaterials, 2012, 33, 1455–1461 CrossRef CAS PubMed.
- J. Li, S. Q. Yap, C. F. Chin, Q. Tian, S. L. Yoong, G. Pastorin and W. H. Ang, Chem. Sci., 2012, 2083–2087 RSC.
- J. Li, A. Pant, C. F. Chin, W. H. Ang, C. Ménard-Moyon, T. R. Nayak, D. Gibson, S. Ramaprabhu, T. Panczyk, A. Bianco and G. Pastorin, Nanomedicine, 2014, 10, 1465–1475 CrossRef CAS PubMed.
- J.-P. Tessonnier, D. Rosenthal, T. W. Hansen, C. Hess, M. E. Schuster, R. Blume, F. Girgsdies, N. Pfänder, O. Timpe, D. S. Su and R. Schlögl, Carbon, 2009, 47, 1779–1798 CrossRef CAS PubMed.
- (a) E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS; (b) C. Samorì, R. Sainz, C. Ménard-Moyon, F. M. Toma, E. Venturelli, P. Singh, M. Ballestri, M. Prato and A. Bianco, Carbon, 2010, 48, 2447–2454 CrossRef PubMed.
- M. Lapeyre, J. Leprince, M. Massonneau, H. Oulyadi, P. Y. Renard, A. Romieu, G. Turcatti and H. Vaudry, Chem. Eur. J., 2006, 12, 3655–3671 CrossRef CAS PubMed.
- L. Lacerda, A. Bianco, M. Prato and K. Kostarelos, Adv. Drug Deliver. Rev., 2006, 58, 1460–1470 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, B. Sun and C. Chen, Acc. Chem. Res., 2013, 46, 702–713 CrossRef CAS PubMed.
- L. M. Coussens and Z. Werb, Nature, 2002, 420, 860–867 CrossRef CAS PubMed.
- S. I. Grivennikov, F. R. Greten and M. Karin, Cell, 2010, 140, 883–899 CrossRef CAS PubMed.
- C. Qu, L. Wang, J. He, J. Tan, W. Liu, S. Zhang, C. Zhang, Z. Wang, S. Jiao, S. Liu and G. Jiang, Gene, 2012, 493, 9–12 CrossRef CAS PubMed.
- P. Wang, X. Nie, Y. Wang, Y. Li, C. Ge, L. Zhang, L. Wang, R. Bai, Z. Chen, Y. Zhao and C. Chen, Small, 2013, 9, 3799–3811 CrossRef CAS PubMed.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS PubMed.
- A. Nolan, M. Weiden, A. Kelly, Y. Hoshino, S. Hoshino, N. Mehta and J. A. Gold, Am. J. Respir. Crit. Care Med., 2008, 177, 301–308 CrossRef CAS PubMed.
- M. Pescatori, D. Bedognetti, E. Venturelli, C. Ménard-Moyon, C. Bernardini, E. Muresu, A. Piana, G. Maida, R. Manetti, F. Sgarrella, A. Bianco and L. G. Delogu, Biomaterials, 2013, 34, 4395–4403 CrossRef CAS PubMed.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J. P. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- L. Lacerda, J. Russier, G. Pastorin, M. A. Herrero, E. Venturelli, H. Dumortier, K. T. Al-Jamal, M. Prato, K. Kostarelos and A. Bianco, Biomaterials, 2012, 33, 3334–3343 CrossRef CAS PubMed.
- L. Y. Chou, K. Ming and W. C. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC.
- V. M. Gonzalez, M. A. Fuertes, C. Alonso and J. M. Perez, Mol. Pharmacol., 2001, 59, 657–663 CAS.
- C. F. Chiu, N. Dementev and E. Borguet, J. Phys. Chem. A, 2011, 115, 9579–9584 CrossRef CAS PubMed.
- H. Ali-Boucetta and K. Kostarelos, Adv. Drug Deliver. Rev., 2013, 65, 2111–2119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr00220f |
| This journal is © The Royal Society of Chemistry 2015 |