
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling “off-target” effects of redox-active polymers and polymer multilayered capsules in prostate cancer cells†
Giovanni L.
Beretta‡
a,
Marco
Folini‡
a,
Francesca
Cavalieri
bc,
Yan
Yan
c,
Enrico
Fresch
b,
Subramanian
Kaliappan
b,
Christoph
Hasenöhrl
d,
Joseph J.
Richardson
c,
Stella
Tinelli
a,
Andreas
Fery
d,
Frank
Caruso
*c and
Nadia
Zaffaroni
*a
aDepartment of Experimental Oncology and Molecular Medicine, Fondazione IRCCS Istituto Nazionale dei Tumori, Via G. Amadeo 42, 20133 Milan, Italy. E-mail: nadia.zaffaroni@istitutotumori.mi.it
bDipartimento di Scienze e Tecnologie Chimiche, Università di Roma Tor Vergata, 00173 Roma, Italy
cARC Centre of Excellence in Convergent Bio-Nano Science and Technology, and the Department of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: fcaruso@unimelb.edu.au
dDepartment of Physical Chemistry II, University of Bayreuth, Bayreuth 95440, Germany
First published on 17th March 2015
Abstract
Redox-active polymers and carriers are oxidizing nanoagents that can potentially trigger intracellular off-target effects. In the present study, we investigated the occurrence of off-target effects in prostate cancer cells following exposure to redox-active polymer and thin multilayer capsules with different chemical properties. We show that, depending on the intracellular antioxidant capacity, thiol-functionalized poly(methacrylic acid), PMASH triggers cell defense responses/perturbations that result in off-target effects (i.e., induction of autophagy and down-regulation of survivin). Importantly, the conversion of the carboxyl groups of PMASH into the neutral amides of poly(hydroxypropylmetacrylamide) (pHPMASH) nullified the off-target effects and cytotoxicity in tested cell lines. This suggests that the simultaneous action of carboxyl and disulfide groups in PMASH polymer or capsules may play a role in mediating the intracellular off-target effects. Our work provides evidence that the rational design of redox-active carriers for therapeutic-related application should be guided by a careful investigation on potential disturbance of the cellular machineries related to the carrier association.
1 Introduction
The assessment of off-target effects and cell survival mechanisms associated with the therapeutic exploitation of nanomaterials represents an active research area in the field of biomedical nanotechnologies. The most widely accepted sources of nanomaterial-mediated side effects deal with the induction of oxidative stress-response and inflammation.1 However, general cell stress-response pathways, including autophagy, have recently emerged as possible nanomaterial-induced mechanisms of either toxicity or cell survival.1,2 Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved mechanism in eukaryotic cells in which cytoplasmic material is trafficked to the lysosomes via intracellular vesicles known as autophagosomes. Autophagy has attracted attention in more recent years because of its critical role in organism homeostasis. Autophagy has pleiotropic functions and promotes cell survival by supplying the cell with nutrients, clearing the intracellular environment from unnecessary or damaged material, and by defending the cell against infection. Despite these beneficial functions of autophagy, constitutive activation of autophagy has been reported to trigger cell death.3 In this context, whether autophagy activation as a response of cancer cells to stimuli represents a pro-survival or a pro-death mechanism still remains to be fully elucidated, although the outcome of autophagy activation has been proposed to be strictly dependent on the cell context.4Several studies suggest that both micro- and nano-particles undergo autophagic sequestration, and that nanomaterial-mediated cell death may occur as a consequence of persistent autophagosome accumulation.5–14 Such an event may be the result of either excessive autophagy induction caused by the accumulation of nondegradable materials, or impairment of the autophagy flux (i.e., nucleation, elongation, maturation and degradation) usually resulting from a blockade in the maturation step (i.e., fusion of autophagosomes with lysosomes) following lysosome dysfunction.3 In this context, it has been reported that micro- and nano-particles may induce damage to the lysosomal compartment, resulting in responses such as lysosomal oxidative stress, alkalization, osmotic swelling, or detergent-like disruption of the lysosomal membrane.1
Redox-responsive micro-nanocarriers bearing disulfide and thiol moieties have been extensively studied for the delivery of therapeutic agents, including small molecules, DNA, RNA and oligopeptides.2,15–22 Redox-active carriers are generally used because of their integrity in the oxidizing bloodstream and in the extracellular environment, thus therapeutic cargo is protected from denaturation or degradation. Upon subsequent cellular internalization, the trafficking of the carrier from the early endosomes to the lysosomes results in the release of cargo due to the intracellular reducing environment.23
In a previous study we designed multilayered redox-active microcapsules (μCs) using disulfide-crosslinked poly(methacrylic acid) (PMASH) for the delivery of a small interfering RNA (siRNA) targeted to the anti-apoptotic gene, survivin, in prostate cancer (PCa) cells.2 To prepare the μCs we used layer-by-layer (LbL) assembly and mesoporous silica templates. LbL assembly is known as a highly versatile technique for the creation of drug delivery vehicles with nanometer thin polymer walls.24 Mesoporous silica particles with a bimodal pore structure (smaller mesopores in the 2–3 nm range and larger mesopores, 10–40 nm) were used as templates.25 The porous template technique provides a method to encapsulate, for example, a positively charged polymer into the redox responsive multilayered μCs and eventually to postload μCs with negatively charged biomacromolecules, such as RNA, by diffusion through the permeable multilayer film and in situ complexation with the polycation. We have shown that the siRNA cargo can be postloaded into PMASH μCs containing poly-L-lysine (PLL) by diffusion through the multilayer shell to form PLL-siRNA polyplexes in situ by electrostatic interactions.2 The μCs protect the PLL-siRNA from enzymatic degradation and it has been hypothesized that PMASH destabilization of the vesicle's membrane may promote the endosomal/lysosomal escape of the cargo.2 Similarly, pH-sensitive carboxylic polymers, such as polyalkylacrylic acid, are known to be membrane-destabilizing agents.26 In the endosome/lysosome, the polymer undergoes a pH-induced coil-to-globule transition, resulting in membrane destabilization and cytoplasmic release. Regardless of the release mechanisms, our previous study shows that the siRNA-PLL complexes embedded into the μCs are at least partially released in the cytosol and are able to significantly inhibit survivin gene expression.2 However, we also observed that the exposure of PCa cells to PMASH μCs resulted in a less pronounced down-regulation of survivin expression levels and induction of autophagy. These effects occurred regardless of the cargo, as both control siRNA-loaded PMASH μCs and empty μCs also elicited a response. The non-dependence on the cargo indicated that the occurrence of off-target effects and the activation of a non-specific response were related to the exposure to the redox-active capsules themselves.
In the present study, we investigate the mechanisms by which redox-active polymers and μCs trigger off-target effects by focusing on the possible correlations between the observed off-target effects and (i) the chemical and functional properties of the μC building blocks, (ii) the physico-chemical properties of the μCs, and (iii) the cellular entry and intracellular trafficking of the μCs. We show that, depending on the physicochemical properties of the μCs and their polymeric components, redox-active materials can elicit a number of cell line-dependent responses. Our findings indicate that redox-active polymers or μCs can potentially interfere with the intracellular milieu and that understanding this behavior has important implications for biomedical applications of biomaterials.
2 Experimental section
2.1 Materials
Roswell Park Memorial Institute medium (RPMI1640), fetal bovine serum (FBS), phosphate buffered saline (PBS) and trypsin were obtained from BioWhittaker (Verviers, Belgium). The human prostate carcinoma cell lines, PC-3 and DU145, were obtained from American Type Culture Collection. Cells were resuscitated soon after arrival, and grown as a monolayer in RPMI 1640 containing 10% FBS. All cultures were maintained at 37 °C in a humidified 5% CO2 incubator. Poly(methacrylic acid, sodium salt), (PMA-Na, Mw = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from Polysciences, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, cystamine hydrochloride, dithiothreitol (DTT), N-hydroxysuccinimide (NHS), and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) 1-amino-2-propanol were purchased from Sigma-Aldrich (St. Louis, MO) and used as received.
000 g mol−1) was purchased from Polysciences, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, cystamine hydrochloride, dithiothreitol (DTT), N-hydroxysuccinimide (NHS), and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) 1-amino-2-propanol were purchased from Sigma-Aldrich (St. Louis, MO) and used as received.
2.2 Preparation of PMASH, pHPMA-co-MA, pHPMASH
PMASH with 15 mol% thiol groups was synthesized and characterized as previously reported.2,27 Briefly, 75 mg of PMA was dissolved in 5 mL of PBS buffer at pH 7.2 and charged with 70 mg of EDC and 40 mg of NHS and stirred for 15 min. Subsequently 7.5 mg of cystamine hydrochloride was added to the reaction mixture. The reaction mixture was kept under stirring for 8 h, purified by dialysis against Milli-Q water and freeze dried. The degree of functionalization was estimated from 1H NMR (D2O): δ (ppm): 0.8 –CH3 from PMA; 1.6 –CH2 from PMA; 2.5–3.4 –CH2 from cystamide moiety. pHPMA-co-MA with 15 mol% MA and 85 mol% pHPMA were prepared as follows: 100 mg of PMA was dissolved in 15 mL of PBS buffer at pH 7.2 and incubated with 302 mg of EDC and 59 mg 1-amino-2-propanol. The reaction mixture was stirred overnight, purified by dialysis against Milli-Q water and freeze dried. The chemical composition of the pHPMA-co-MA copolymer was determined by potentiometric titration of carboxyl groups. The degree of functionalization was also verified from 1H NMR (dmso-d6): δ (ppm): 0.8 –CH3 from MA; 1 –CH2 from MA and HPMA; 2.85 –CH2 from hydroxypropyl amide moiety; 3.65 –CH from hydroxypropyl amide moiety; 4.7 –OH from hydroxypropyl amide moiety. pHPMASH with 15 mol% thiol groups was prepared as follows: 100 mg of PMASH was dissolved in 10 mL PBS buffer at pH 7.2 and incubated with 440 mg of EDC and 185 mg of 1-amino-2-propanol. The reaction mixture was stirred overnight, purified by dialysis against Milli-Q water and freeze dried. The complete neutralization of PMASH was assessed by potentiometric titration.2.3 Preparation of PMASH and pHPMASH μCs
Mesoporous silica particles were prepared as previously reported.28 100 μL of PLL (2 mg mL−1 in sodium acetate buffer at pH 4) was infiltrated into 1 mg of mesoporous silica particles. After infiltration of PLL into the template, the polycation was labeled with 2.3 μL of AF488 (1 mg mL−1). Next, the particles were washed three times with acetate buffer to remove the excess AF488 and the free unbound polymer. Loading of PLL in the nanopores was confirmed by fluorescence microscopy. The PLL-infiltrated particles were incubated alternately with PMASH (1 g L−1) and PVP (1 g L−1) for 15 min, as previously described.2 Three centrifugation/wash cycles (900g for 30 s) with sodium acetate buffer (50 mM, pH 4) were conducted after deposition of each polymer layer. Polymers were added sequentially until 20 layers were deposited. The core–shell particles were treated with chloramine T (2 mM) in MES buffer (10 mM, pH 6) for 2 min. The particles were then suspended in sodium acetate buffer, to which HF (2 M) in NH4F (8 M, pH 5) was added. Caution! Hydrofluoric acid and ammonium fluoride are highly toxic. Extreme care should be taken when handling HF solution and only small quantities should be prepared. The resulting μCs were washed via centrifugation (4500g for 5 min) four times. pHPMASH μCs were prepared as follows: PMASH microparticles were washed with 100 μL sodium acetate buffer (50 mM) and centrifuged. The supernatant was removed, the particles were resuspended with 20 μL of MES buffer pH 6 and then incubated with 340 μL of EDC (20 mg mL−1 in MES pH 6) and 270 μL of 1-amino 2-propanol (20 mg mL−1 in MES pH 6) for 2 h. Subsequently, the microparticles were washed with PBS buffer three times. Finally the templates were removed by treatment with 5 M HF. ζ-potential measurements of PMASH and pHPMASH microparticles were carried out on a Malvern Zetasizer. The ζ-potential values in Milli-Q water were −29 ± 3 mV and +2 ± 3 mV for PMASH and pHPMASH microparticles, respectively. The μCs were imaged on an Olympus IX71 digital wide-field fluorescence microscope equipped with a fluorescein isothiocyanate (FITC) and tetramethyl rhodamine isocyanate (TRITC) filter cube, a differential interference contrast (DIC) slider (U-DICT, Olympus), and a 60× objective lens. A CCD Camera (CoolSNAP fx, Photometrics, Tucson, AZ) was mounted on the left-hand port of the microscope. The diameters of the PMASH and pHPMASH μCs were measured by optical microscopy for at least 200 μCs.2.4 AFM characterization
The mechanics of PMASH and pHPMASH μCs were investigated using a combination of a Nanowizard I AFM (JPK Instruments AG, Berlin, Germany) and an optical inverted microscope obtained from Zeiss, (Zeiss Axiovert 200, Germany), following the protocol reported by Dubreuil et al.29 Briefly, prior to determining the force-deformation relationship of the nanocarriers, the glass support was dip-coated in an aqueous solution of 1 mg mL−1 polyethylene imine (PEI, Mw = 30000, Sigma-Aldrich) for 20 min and rinsed in distilled water afterwards. Microcapsules were subsequently adsorbed from the sample solution onto the polymer film for 3 min, before the solution was washed twice with PBS. Force measurements were carried out in a filtered PBS solution to mimic biological conditions. A glass bead with a diameter of ∼40 μm (Polysciences Inc., USA) was glued using a two component epoxy glue (UHU Plus endfest 300, UHU GmbH & Co. KG, Germany) onto the tip-less cantilever (spring constant 16.9–46.6 mN m−1, NSC12 or CSC12, MicroMash, Estonia). At least 35 capsules were measured for each set of substrates, and for each individual capsule six measurements were performed to monitor changes in the deformation behavior. The maximum applied force ranged from 8–80 nN with a typical value of 10 nN. The approaching and retracting velocity were set at 5 μm per second.
AFM Imaging was performed as follows: prior to adsorption of the capsules, bare Si-wafers were cleaned by ultrasonication in a 1:1 solution of water and propan-2-ol for 15 min. Further, the wafers were rinsed with water and cleaned in a boiling solution of a 5:1:1 mixture of water, hydrogen peroxide and ammonia for 10 min. The wafers were repeatedly rinsed with water and dried in a nitrogen flow afterwards. To promote the adhesion of the capsules, the Si-wafers were coated with polyethylene imine (PEI) following the protocol reported above. After extensively rinsing the PEI-coated wafers, they were placed into diluted solutions of the nanocarriers (5 μL mL−1 or 15 μL mL−1) for 30 min. The samples were imaged in the dried state using a Dimension 3100 equipped with a Nanoscope V controller (Veeco Instruments Inc., USA) operating in Tapping Mode. The Si3N4 cantilevers (OMCL-AC160TS, Olympus) had a typical spring constant of 42 N m−1, a generic resonance frequency of ∼300 kHz and a tip radius less than 7 nm. The obtained images were processed with NanoScope Analysis v. 1.40 (Build R2Sr1.83411) using Plane Fit in the XY-Mode and Flatten of the 1st order.
2.5 Cytotoxicity assay
The cytotoxic activity of polymers and capsules was assessed by the MTS assay (Promega, Milano, Italy), according to the manufacturer's protocol. Briefly, cells were seeded at the appropriate density into 96-well plates. The following day, the cells were treated with polymer or μCs for the indicated time points in a final volume of 100 μL of RPMI1640. Twenty μL of a MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt) solution was then added to each well. The absorbance was measured using a FLUOstar OPTIMA plate reader (BMG Labtech GmbH, Offenburg, Germany) at 492 nm after 4 h incubation at 37 °C in 5% CO2. The cytotoxic activities of GSH and GSSG were assessed by a growth inhibition assay. Briefly, 24 h after seeding in 6-well plates, cells were exposed for 72 h to the compounds, collected and counted in a particle counter (Coulter Counter, Coulter Electronics, Luton, UK). Each experimental sample was run in triplicate. The concentration of compounds causing 50% cell growth inhibition (IC50) was determined by dose response curves generated by testing at least 5 different drug concentrations.2.6 Evaluation of the “redox potential” of PC-3 and DU145 cells
The amounts of total glutathione in PC-3 and DU145 cells were estimated using the GSH/GSSG-Glo™ Assay Kit (Promega), according the manufacturer's instructions. Such a luminescence-based system allows the detection and quantification of total GSH and GSSG as well as the GSH/GSSG ratios in cultured cells by comparison with a GSH standard curve.The Total Antioxidant Capacity (TAC) of PC-3 and DU145 cells was measured by a TAC Assay Kit (Cell Biolabs Inc., San Diego, CA), according to the manufacturer's protocol. Briefly, 24 h after seeding the cells were collected, washed and lysed by sonication in PBS. Cell lysates were then diluted with a reaction reagent and, upon addition of Cu2+ solution, the reaction was stopped and read with a spectrophotometric microplate reader at 490 nm. Antioxidant capacity, expressed as copper reducing equivalent (CRE), was determined by using a standard curve generated by using fixed concentrations of uric acid.
2.7 Evaluation of intracellular internalization of capsules
Cells were seeded in 6-well plates and the day after treated with AF647-labeled μCs at a particle:cell ratio of 72:1 at 37 °C and 5% CO2. At specific intervals (0, 3, 6 and 24 h) cells were washed (×3) with PBS and harvested by trypsinization followed by centrifugation at 400g for 5 min. The cell pellet was resuspended in PBS and analyzed by flow cytometry (FACS Canto, BD Biosciences, San Jose, CA). Cells that had internalized capsules were identified in the TRITC fluorescence channel. The percentage of positive cells within the overall cell population (20000 per analysis) was calculated by FACSDiva™ software (BD Biosciences).
For deconvolution microscopy experiments, PC-3 cells were plated into 8-well Lab-Tek I chambered coverglass slides (Thermo Fisher Scientific, Rochester, NY) and allowed to adhere overnight. Cells were then incubated with capsules (125 or 500 capsules:cell) for 24 or 96 h (37 °C, 5% CO2), followed by three washes with PBS. Cells were then fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.1% Triton X-100 in PBS for 1 min, and blocked with 1% BSA in PBS for 5 min. Samples were then incubated with mouse anti-human LAMP1 monoclonal antibody (2.5 μg mL−1 in PBS containing 0.25% BSA) (BD Pharmingen, San Diego, CA) and anti-human LC3 antibody (2 μg mL−1 in PBS containing 0.25% BSA) (Sigma-Aldrich, Sydney, Australia), followed by detection with AF647-labeled goat anti-mouse IgG (2 μg mL−1 in PBS containing 0.25% BSA) (Invitrogen, San Giuliano Milanese, Italy) or AF647-labeled goat anti-mouse antibody IgG (2 μg mL−1 in PBS containing 0.25% BSA) (Invitrogen) for 1 h at 23 °C. Optical sections were collected with a TCS SP2 laser scanning confocal unit (Leica, Solms, Germany). Colocalization analysis was performed with Imaris software (Bitplane AG, Zürich, Switzerland).
2.8 Western immunoblotting
Total protein extracts were prepared according to standard methods. Twenty μg of protein extracts was fractioned by denaturating polyacrylamide gel electrophoresis and transferred to nitrocellulose using standard protocols. The filters were blocked with PBS containing 5% (w/v) skim milk and incubated overnight with primary antibodies specific for survivin (Sigma-Aldrich), LC3 (Cell Signaling) and topoisomerase I (BD Pharmingen). The filters were then incubated with secondary peroxidase-linked whole antibodies (Life Technologies Europe BV, Monza, Italy). Bound antibodies were detected using the Novex ECL, HRP Chemiluminescent substrate Reagent Kit (Life Technologies). Filters were autoradiographed and images were acquired by using a BioSpectrum Imaging System (Ultra-Violet Products Ltd, Cambridge, UK). An anti-β-actin antibody (Sigma-Aldrich) was used on each blot to ensure equal loading of proteins.2.9 Evaluation of survivin concentration with enzyme-linked immunosorbent assay (ELISA)
Survivin expression levels were assessed in PC-3 cells treated with building block polymers using the Surveyor IC Immunoassay (R&D Systems, Abingdon, UK), according to the manufacturer's protocol. Briefly, 24 h after seeding, cells were treated with polymers (1.25 ng per cell) for 96 h. Cells were then collected and processed. The kit employs a two-site sandwich ELISA to quantitate survivin in cell lysates. An antibody specific for survivin was pre-coated onto a microplate. Standards and samples were added and any survivin present was bound by the immobilized antibody. After washing away unbound material, a biotinylated detection antibody recognizing survivin was used for detection utilizing a standard streptavidin-HRP format. Color developed in proportion to the amount of survivin content. The color development was stopped and the intensity measured. A survivin standard curve was used to determine the concentration of survivin in samples.2.10 Human proteome array
The expression profiles of cell stress-related factors were analyzed using the Human Apoptosis Array kit (R&D Systems), according to the manufacturer's protocol. Briefly, 24 h after seeding, cells were treated with polymers (1.25 ng per cell) for 96 h. Cells were then collected, lysed and incubated overnight onto the array. After washing, the array was incubated with a cocktail of biotinylated detection antibodies. Streptavidin-HRP and chemiluminescent detection reagents were applied. The signal produced at each capture spot corresponded to the amount of bound protein.3 Results and discussion
3.1 Preparation of redox-active polymers and μCs
To ascertain the role of the chemical composition of PMASH μCs in inducing off-target effects, a number of polymers and μCs were prepared, namely PMASH (Fig. 1A), pHPMASH (Fig. 1B), PMASH μCs and pHPMASH μCs (Fig. 1C). The carboxyl groups (85% mol) of PMA (15 kDa) were converted to neutral amide groups to obtain pHPMA-co-MA. Similarly, to obtain a redox-active neutral polymer, pHPMASH, all carboxyl groups of PMASH were converted to neutral amide groups. The degree of functionalization of polymers was estimated from 1H NMR and potentiometric titration, as described in the experimental section.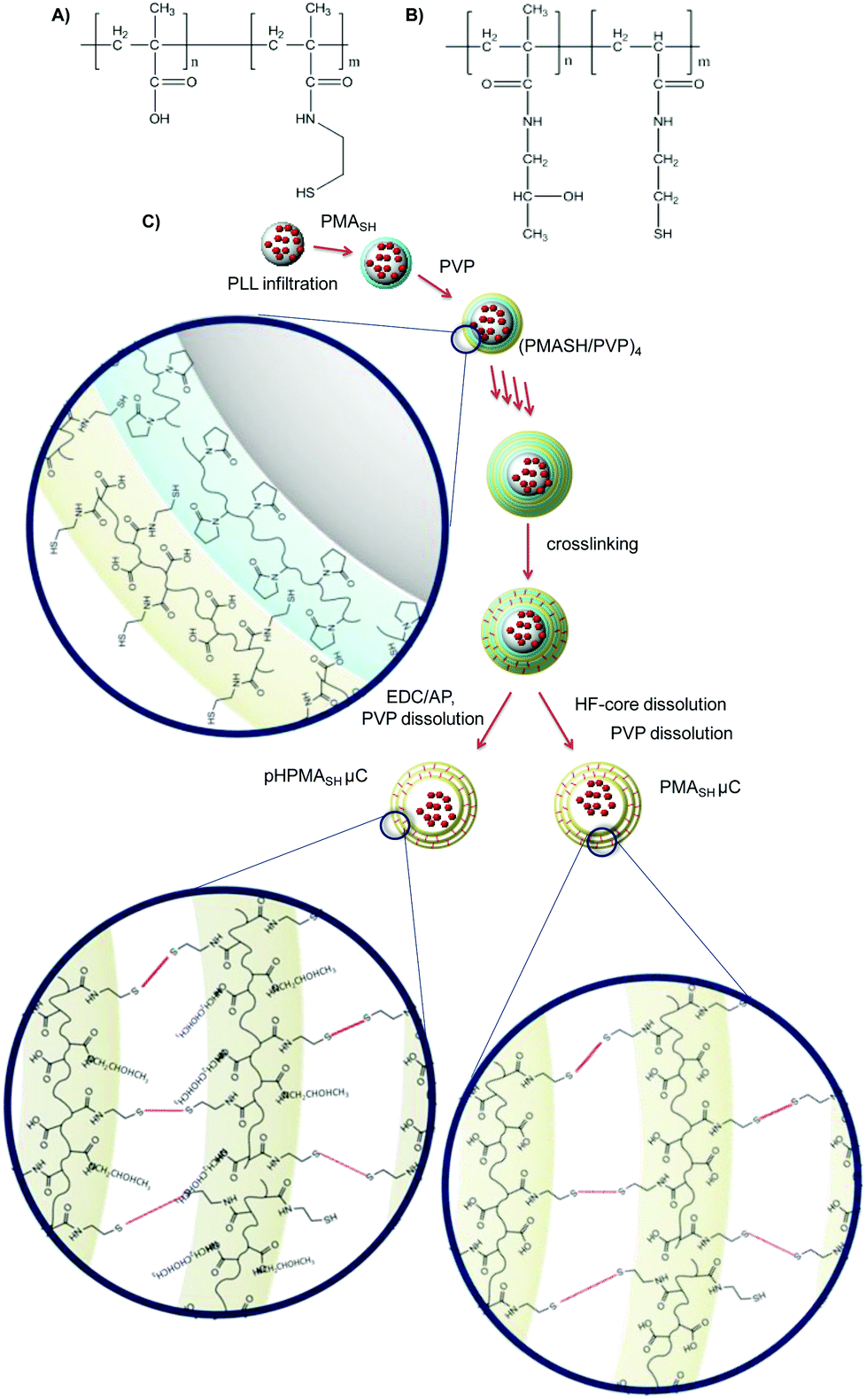 | ||
| Fig. 1 Schematic representation of (A) PMASH polymer, (B) pHPMASH, polymer and (C) PMASH and pHPMASH μCs preparation routes. | ||
For the preparation of multilayered PMASH μCs loaded with PLL (Fig. 1), PLL was infiltrated into the mesoporous silica templates prior to PMASH/PVP multilayer assembly. LbL assembly was performed at pH 4 via the alternate deposition of PMASH and PVP until the nanometer thick film could retain PLL (five bilayers). Disulfide crosslinks were formed by the oxidation of thiols and after core removal, PVP was washed out from the μCs at pH 7.17 The carboxyl groups of PMASH μCs were converted to neutral amide groups to obtain redox active pHPMASH μCs by treatment with EDC (20 mg mL−1 in MES pH 6) and 1-amino 2-propanol (20 mg mL−1 in MES pH 6). The reaction was performed before silica template dissolution.
The stoichiometric neutralization of carboxyl groups in pHPMASH μCs was verified by the potentiometric titration of carboxyl groups and microelectrophoresis measurements. No residual carboxyl groups were detected. The ζ-potentials of PMASH and pHPMASH μCs were −28 mV and 2 mV respectively, indicating the amidation reaction was efficiently performed. The diameters of PMASH μCs (1.7 ± 0.2 μm) and pHPMASH μCs (1.3 ± 0.2 μm) were measured by optical microscopy over a set of 200 μCs (Fig. S1†). The deconstruction of both PMASH2 and pHPMASH μCs was verified by optical microscopy after treatment with a reducing agent (DTT 0.5 M, Fig. S1†). This feature confirmed that the multilayered μCs are held together by redox-responsive disulfide bonds (S–S).
Moreover, using AFM we investigated how the chemical modification of PMASH μCs into pHPMASH μCs affects the thickness and stiffness (Table S1†). This provided insights into the effects of μC architecture on mechanical resistances at deformations on the nanoscale, as previously reported.30 AFM imaging analysis (Fig. S2†) showed that the shell thicknesses for the dried PMASH μCs and pHPMASH μCs were 10 ± 6 and 31 ± 3 nm, respectively. Since the number of polymer layers was the same, the increase in thickness in the pHPMASH μCs indicated neutralization of carboxyl groups, leading to a less compact and more amorphous nanostructured film. AFM force spectroscopy measurements (Fig. S2†) were then performed to quantify the mechanical properties of swollen PMASH μCs and pHPMASH μCs.31 Microcapsule stiffness was determined from the slope of force–distance plots (Fig. S2†). Both PMASH μCs and pHPMASH μCs show a similar stiffness (Table S1†), corresponding to 7 ± 3 and 6 ± 3 mN m−1, respectively. Previous work reported that the stiffness plays an important role in microcapsule cellular uptake,32 PMASH and pHPMASH μCs show comparable deformability in spite of differences in wall thickness, suggesting that no mechanical influence of the stiffness on capsule-cell interactions should be expected.
We next studied the cellular response of these redox-active polymers and μCs and attempted to correlate their structure to cellular responses.
3.2 Cytotoxicity of redox-active polymers and μCs on DU145 and PC-3 PCa cell lines
We previously reported that PMASH μCs have cytotoxic effect on PC-3 PCa cells (300:1 particles per cell).2 As methacrylic copolymers are widely used as excipients (Eudragit®) in a large number of pharmaceutical preparations and have shown little or no toxicity,33 we hypothesized that the observed toxicity could be correlated directly to the redox properties of PMASH and would be dependent on the selected cell line. Hence, the cytotoxic activity of PMASH polymer and PMASH μCs was assessed on DU145 cells, another well-established cell model of human castration-resistant PCa.
We found that, unlike PC-3 cells, the survival of DU145 cells was negligibly affected by either polymer building block or μC, even at dosages up to 2.5 ng per cell and 1000 particles per cells, respectively (Fig. S3† and 2A), thus suggesting the occurrence of a cell line-dependent cytotoxic response.
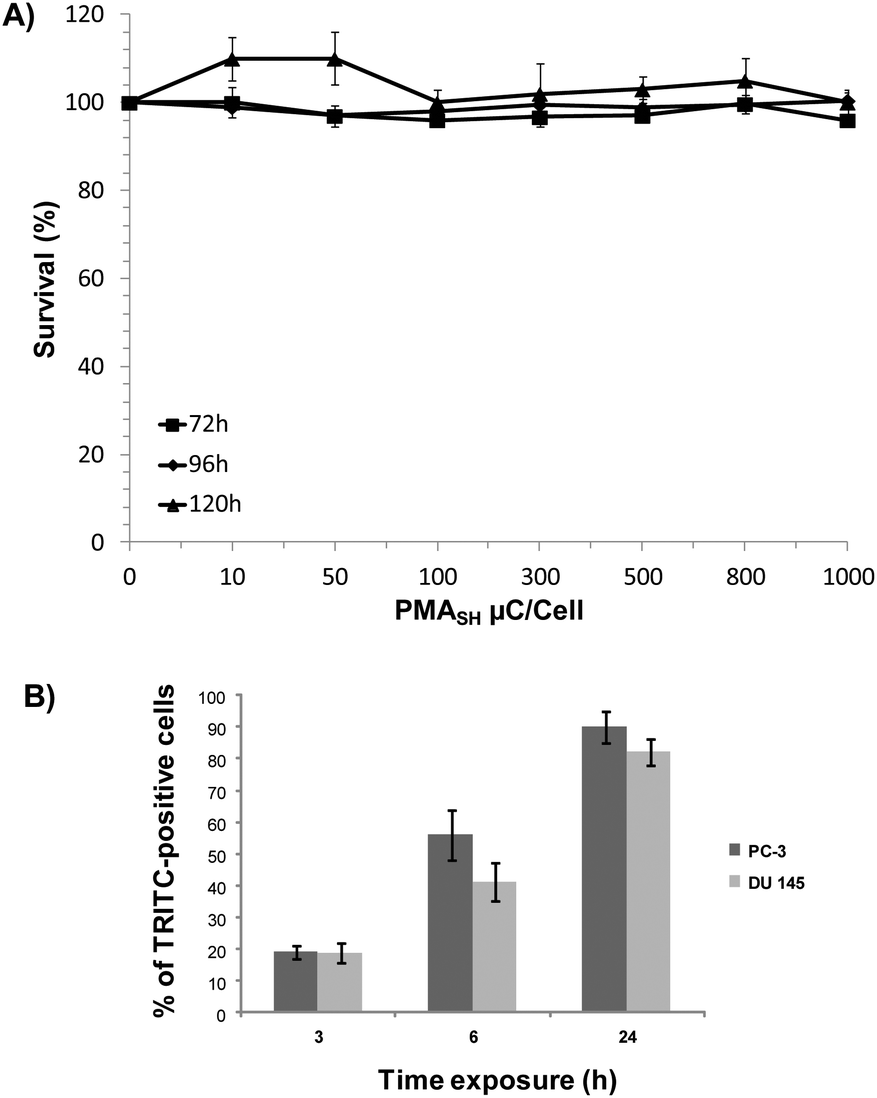 | ||
| Fig. 2 (A) DU145 cell survival curves after incubation for 72, 96 and 120 h with PMASH μCs. Data are reported as a percentage of viable cells exposed to μCs compared with untreated cells. All data represent mean values ± SD. (B) Time dependent evaluation of PMASH μC internalization in DU145 and PC-3 cells. Cells were exposed to AF647-labeled capsules at a capsule to cell ratio of 72:1 for 3, 6 and 24 h. Cells were analyzed using flow cytometry. | ||
Since we cannot exclude that the absence of toxicity of PMASH μCs in DU145 cells may be a consequence of the reduced or inefficient internalization of μCs, their uptake was investigated in both cell lines by flow cytometry. Results showed a time-dependent increase in the percentage of TRITC-positive cells in both cell lines after the exposure to AlexaFluor647-labeled μCs at a particle per cell ratio of 72:1 (Fig. 2B). Specifically, after 24 h exposure to labeled capsules, 82% and 90% of positive cells were observed in DU145 and PC-3 cells, respectively. This finding ruled out that a modest uptake capability of DU145 cells could account for their lower susceptibility to μCs compared with PC-3 cells.
To gain further insight into the cell line-dependent cytotoxic response, the basal redox properties of PC-3 cells were compared with those of DU145 cells. The analysis of both total intracellular reduced (GSH) and oxidized (GSSG) glutathione content revealed that PC-3 cells produced significantly (P < 0.05) lower amounts of GSH (116 ± 4 fmol per cell) compared with DU145 cells (156 ± 2 fmol per cell), whereas no difference in total GSSG content was observed between the two cell lines (1.13 ± 0.06 and 1.2 ± 0.1 fmol per cell in PC-3 and DU145, respectively). Moreover, in comparison to DU145, PC-3 cells showed a two- to three-fold reduction (P < 0.05) in sensitivity to both GSH (27.95 ± 0.08 mM versus 12.8 ± 1.2 mM) and GSSG (34 ± 7 mM versus 12.5 ± 0.8 mM), as revealed by a cytotoxicity assay. In addition, evaluation of the total reducing capability (e.g., CRE per μg of protein) showed that PC-3 cells were characterized by a higher total antioxidant performance (16.8 ± 1.9 CRE per μg protein) compared to DU145 cells (12.7 ± 0.14 CRE per μg protein).
Taken together, these results suggest that the cytotoxic effect of PMASH μCs on PC-3 cells could be the result of the μC-mediated oxidative stress produced by a more efficient cellular reduction of μCs by PC-3 cells.
To ascertain whether disulfide groups of PMASH μCs were responsible for the cytotoxic effect observed in PCa cells, the cellular interactions of pHPMASH polymer and pHPMASH μCs were assessed. The intracellular uptake and trafficking of pHPMASH μCs were investigated in PC-3 cells. Flow cytometry analyses showed a time-dependent increase in the percentage of AlexaFLuor647-positive cells that reached 90% of the overall cell population after 24 h exposure to 72:1 particles per cell (Fig. S4A†). Fluorescence deconvolution microscopy analyses showed that after 24 h exposure to AlexaFLuor488-labeled μCs (500:1 particles per cell) the majority of internalized μCs were localized close to the late endosomes/lysosomes, as revealed by the overlap of the fluorescent signals originating from both capsules and the lysosome associated membrane protein 1 (LAMP1), detected by an AlexaFluor647 secondary antibody (Fig. S4B†). This confirms that, like PMASH μCs,23,34 pHPMASH μCs travel through the endocytic route, and then converge into the lysosomes, for their intracellular accumulation.35–37
However, no cytotoxic effects were observed in both cell lines exposed for 96 h to up to 2.5 ng per cell of polymers (pHPMA-co-MA, pHPMASH, Fig. S5†) and 1000 particles per cell of pHPMASH μCs (Fig. 3).
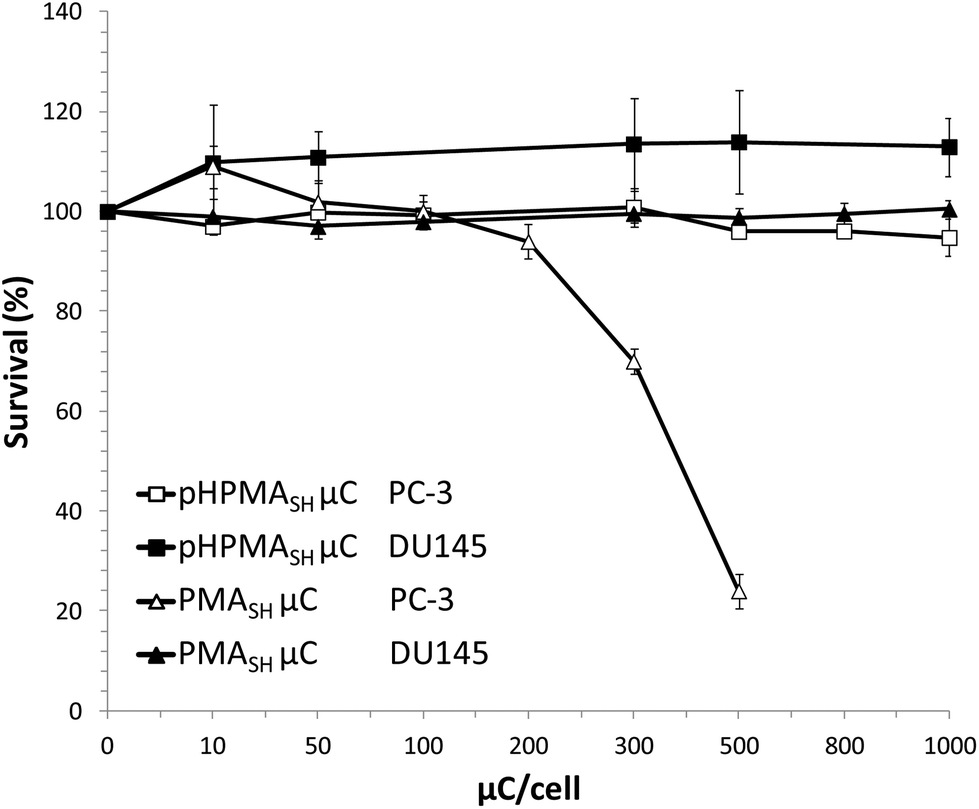 | ||
| Fig. 3 Cell survival curves for PC-3 and DU145 PCa cells after incubation for 96 h with pHPMASH and PMASH μCs. Data are reported as a percentage of viable cells exposed to μCs compared with untreated cells. All data represent mean values ± SD. | ||
Overall, these data indicate that the cytotoxicity of redox-active μCs on PC-3 cells depends on the chemical composition (e.g., combination of carboxyl and disulfide moieties) of the polymeric building blocks and that the redox properties of PC-3 cells appear to play a pivotal role on cell viability after exposure to PMASH μCs.
3.3 Assessment of autophagy in PC-3 and DU145 cells exposed to redox-active polymers and μCs
It has been reported that internalized μCs or derived fragments may undergo ubiquitination or colocalize with polyubiquitin complexes that are then targeted to the autophagosome via a p62-LC3-II-dependent pathway.38,39 In this context, we previously showed that PC-3 cells exposed to PMASH μCs were able to activate the autophagic pathway, regardless of the cargo, likely as a consequence of their attempt to degrade what they perceive as a foreign material and, consequently, to cope with μC-induced detrimental effects (e.g., oxidative stress).2To gain further insight into the autophagosome formation in response to PMASH μC exposure, the localization of LC3-II (a marker of autophagosome membrane) in the cytosol of PC-3 cells was investigated by deconvolution fluorescence microscopy (Fig. 4).
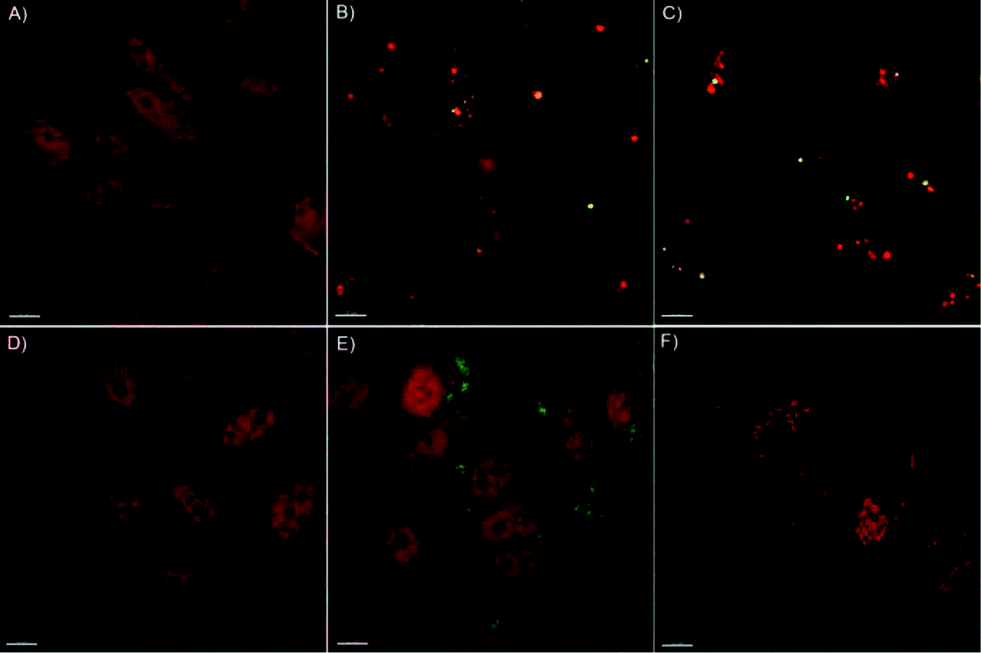 | ||
| Fig. 4 Representative deconvolution microscopy images of PC-3 cells: (A) Control, treated for 96 h with (B) PMASH μCs (125 μCs per cell), (C) PMASH μCs (500 μCs per cell), (D) PMASH polymer (1.25 ng per cell), (E) pHPMASH μCs (500 μCs per cells), and (F) chloroquine (100 μM). Chloroquine was used as a positive control for autophagosome accumulation. μCs were labeled with AF488 dye (green) and the immunostaining of LC3 was performed using an antibody raised against the LC3 labeled with AF647 dye (red). Scale bars: 10 μm. | ||
As expected, a dose-dependent formation of autophagosomes was observed in PC-3 cells treated for 96 h with PMASH μCs (Fig. 4B, C). Strikingly, complete co-localization of PMASH μCs and autophagosomes (stained for LC3 protein) was observed, indicating that PMASH μCs were recognized by the autophagy machinery and accumulated into the autophagosomes.
Conversely, when incubated for 96 h with PMASH polymer building blocks, PC-3 cells did not show any autophagosome formation (Fig. 4D). This indicates that, contrary to what was observed with PMASH μCs, the soluble form of PMASH fails to induce autophagy and toxicity (Fig. S5†), suggesting that the polymer per se does not represent a threat for PC-3 cells. In addition, fluorescence microscopy analysis did not show autophagosome formation in PC-3 cells incubated with 500 particles per cell of pHPMASH μCs (Fig. 4E), whereas it revealed the capability of PC-3 cells to engulf a large number of pHPMASH μCs per cell (Fig. 4E) with no detrimental effects on their vitality (Fig. 3).
These data gain further support by the biochemical analysis of the conversion of the LC3 protein from the cytosolic (LC3-I) to the autophagosome-associated (LC3-II) form assessed by Western immunoblotting. Specifically, no changes in LC3-II expression levels were observed in either PCa cell lines exposed to pHPMASH μCs (125:1 μCs per cell, Fig. 5A) or to soluble polymer building blocks (pHPMASH, pHPMA-co-MA and PMASH, Fig. 5B).
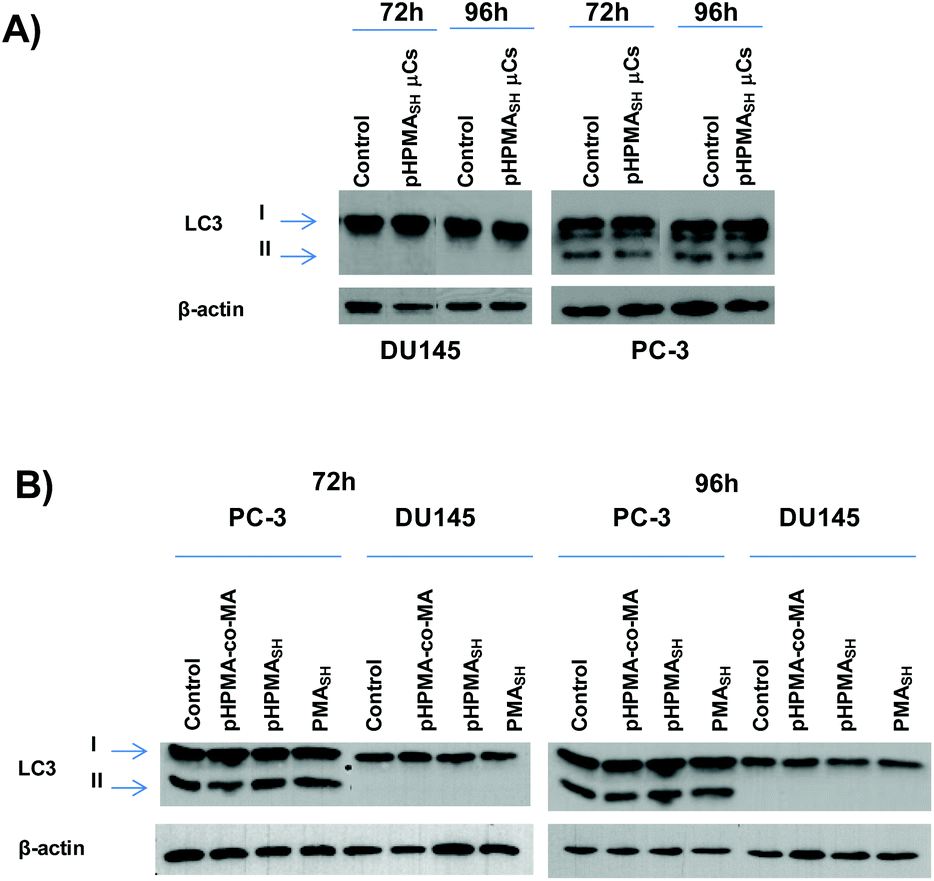 | ||
| Fig. 5 Representative Western immunoblotting showing LC3-I and -II expression levels in PC-3 and DU145 cells exposed for 72 h and 96 h to (A) pHPMASH μCs (125 μCs per cell) and (B) building block polymers (1.25 ng per cell). β-actin was used as control for equal protein loading. | ||
Of note, no conversion of LC3-I to LC3-II was observed in DU145 cells treated for 72 and 96 h with 125 PMASH μCs per cell (Fig. S6†). Overall, these findings indicate that the lack of any sign of autophagy induction may lead to an improved biocompatibility profile compared to PMASH μC (Fig. 3 and S5†).
3.4 Assessment of the occurrence of off-target effects in PCa cells exposed to redox active polymers and μCs
To further corroborate our previous study2 in which we observed a non-specific down regulation of survivin expression levels in PC-3 cells exposed to PMASH μCs, regardless of the cargo (e.g., siRNA against the target gene), we evaluated the expression levels of this protein in DU145 cells treated with PMASH polymer (1.25 ng per cell) or μCs (125:1 particles per cell).
A decrease of survivin expression levels was observed in PC-3 cells exposed for up to 96 h to soluble PMASH polymer (Fig. 6A and Table 1), as assessed by both Western immunoblotting and enzyme-linked immunosorbent assay (ELISA). On the contrary, no significant changes in survivin expression were seen in DU145 cells after up to 96 h exposure to either soluble PMASH polymer or μCs (Fig. 6A and S7†). Moreover, no perturbations in the expression levels of topoisomerase I (a protein that does not share any domain or sequence homology with survivin) were observed in PMASH-treated DU145 cells (Fig. 6A), whereas a slight decrease of the protein amount was detected in PMASH-treated PC-3 cells (Fig. 6A).
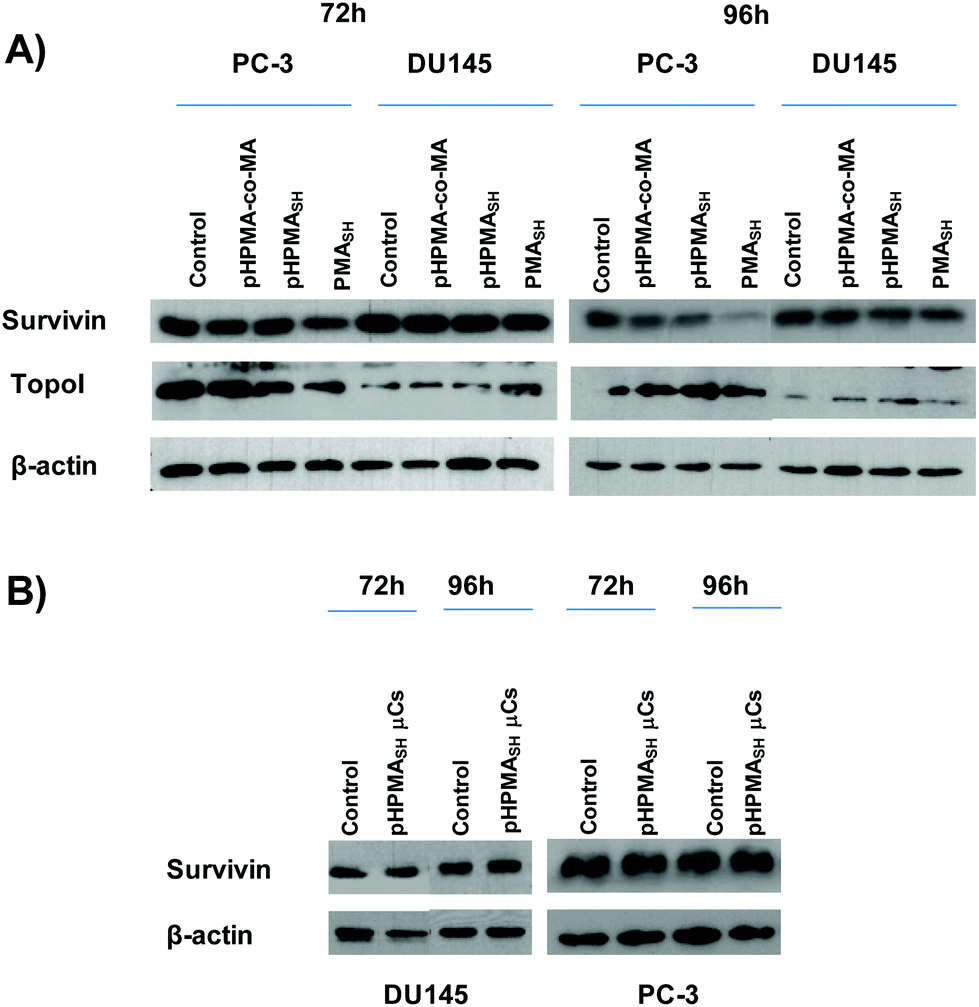 | ||
| Fig. 6 (A) Representative Western immunoblotting showing survivin and topoisomerase I expression levels in PC-3 and DU145 cells exposed for 72 h and 96 h to the indicated building block polymers (1.25 ng per cell). (B) Representative Western immunoblotting showing survivin expression in PC-3 and DU145 cells exposed for 72 h and 96 h to pHPMASH μCs (125 particles per cell). β-actin was used as a control for loading. | ||
| pg Survivin/μg total proteina | |
|---|---|
| a Survivin expression level was assessed by enzyme-linked immunosorbent assay performed on PC-3 cells exposed for 96 h to 1.25 ng per cell of the indicated building block polymers. *P < 0.05 (ANOVA). | |
| Control | 11.57 ± 0.40 |
| PMASH | 9.56 ± 0.42* |
| pHPMA-co-MA | 12.47 ± 0.45 |
| pHPMASH | 13.17 ± 1.04 |
Overall our data indicate that both PMASH polymer and PMASH μCs promote off-targets effects, in terms of protein expression, in PC-3 cells compared to DU145 cells (Fig. 6A and S7†).
Conversely, no evidence of significant perturbations in the expression levels of survivin and topoisomerase I was detected in PC-3 and DU145 cells exposed for up to 96 h to (1.25 ng per cell) pHPMASH polymer (Fig. 6A and Table 1). Similarly, no changes in survivin expression levels were found in both PCa cell lines (Fig. 6B) after the exposure for up to 96 h to pHPMASH μC, (125:1 particles per cells).
These findings are also supported by the observation, obtained through a protein array system, that the exposure of PC-3 cells to PMASH polymer impaired the expression levels of factors involved in the response of cells to different insults (Fig. 7).
 | ||
| Fig. 7 Analysis of protein expression levels by proteome array carried out in PC-3 cells treated with polymer building blocks. PC-3 cells were treated for 96 h with 1.25 ng per cell of PMASH, pHPMA-co-MA, and pHPMASH polymers. Cells were then collected and analyzed. | ||
Specifically, PMASH-treated cells were characterized by a marked up-regulation of the pro-apoptotic cell membrane death receptors TRAIL-R1/DR4 and TRAIL-R2/DR5, which are important players in the apoptotic extrinsic pathway, and a slight up-modulation of two proteins belonging to the Bcl-2 family of proteins, Bax and Bcl-2. Bax redistributes to the mitochondria from the cytosol during apoptosis and its increased expression can accelerate cell death.40,41 Conversely, Bcl-2 acts as an anti-apoptotic factor by repressing the induction of programmed cell death triggered by diverse stimuli.41 PMASH-treated PC-3 cells were also characterized by an increase in the expression levels of HSP60 and a down-regulation of HTRA2/Omi, both involved in the control of protein folding.42,43 Specifically, HSP60 plays a role in assisting nascent polypeptides to reach a native conformation, whereas HTRA2/Omi is a protease that degrades mis-folded and non-assembled polypeptides. In addition, a slight increase in the amounts of p21CIP1/WAF1, a negative regulator of the cell cycle progression during cell stress,44 and of PON2, a membrane-bound enzyme that protects cells from oxidative stress,45 was also observed in PC-3 cells exposed to free PMASH polymer. Of note, neither pHPMA-co-MA nor pHPMASH elicited such a global perturbation in the expression levels of cell stress-related factors, except for an evident increase in the amount of HSP60 in pHPMASH-treated PC-3 cells, probably as a result of an oxidative insult produced by disulfide moieties on the polymer.
Taken together, these data indicate that: (i) the physico-chemical properties of redox active PMASH polymer and capsules play a pivotal role in eliciting cell-dependent off-target responses, which can be avoided by using the negatively charged polymer pHPMA-co-MA or the neutral disulfide polymer pHPMASH; and (ii) depending on the cell context, the simultaneous action of carboxyl and disulfide groups in PMASH polymer or μCs may play a role in mediating the off-target effects.
4 Conclusion
In the present study, we investigated the occurrence of off-target effects in cancer cells following exposure to redox-active biomaterials with different chemical properties. Specifically, we showed that depending on the cell context, PMASH polymer triggers cell defense responses/perturbations that restrict its exploitation to selected tumor cell models. Cell-dependent response to redox active PMASH based material correlates with intracellular antioxidant capacity. This highlights that the chemical composition of a given polymer that forms the μCs represents an important parameter for the biological properties of the μCs themselves. Conversion of the carboxylic acids in PMASH to neutral amides (pHPMASH) resulted in a more inert redox-active system, which resulted in the reduction of the off-target effects, as suggested by both the relevant absence of cytotoxic activity and the unaltered expression levels of stress-related factors when administered to cancer cells. Our results provide evidence that the rational design of micro-nanovectors for therapeutic-related applications should be guided by an investigation on potential disturbances of the cellular machineries related to μC association.Acknowledgements
This work was supported in part by grants from Marie Curie Actions–International Research Staff Exchange Scheme (grant #247542, Nanosirna), Fondazione Italo Monzino, by the Endeavour Research Fellowship Scheme 2014 (F. Cavalieri), and was conducted and funded by the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036), and the Australian Research Council under the Australian Laureate Fellowship (F. Caruso, FL120100030) and Discovery Early Career Researcher Award (Y.Y., DE130100488) schemes. The authors are thankful to J. Cui (The University of Melbourne) for kindly providing the mesoporous silica particles.Notes and references
- S. T. Stern, P. P. Adiseshaiah and R. M. Crist, Part. Fibre Toxicol., 2012, 14, 9–20 Search PubMed.
- A. L. Becker, N. I. Orlotti, M. Folini, F. Cavalieri, A. N. Zelikin, A. P. R. Johnston, N. Zaffaroni and F. Caruso, ACS Nano, 2011, 5, 1335–1344 CrossRef CAS PubMed.
- A. Eisenberg-Lerner and A. Kimchi, Apoptosis, 2009, 14, 376–391 CrossRef PubMed.
- N. I. Orlotti, G. Cimino-Reale, E. Borghini, M. Pennati, C. Sissi, F. Perrone, M. Palumbo, M. G. Daidone, M. Folini and N. Zaffaroni, Autophagy, 2012, 8, 1185–1196 CrossRef CAS PubMed.
- X. Ma, Y. Wu, S. Jin, Y. Tian, X. Zhang, Y. Zhao, L. Yu and X. J. Liang, ACS Nano, 2011, 5, 8629–8639 CrossRef CAS PubMed.
- M. I. Khan, A. Mohammad, G. Patil, S. A. Naqvi, L. K. Chauhan and I. Ahmad, Biomaterials, 2012, 33, 1477–1488 CrossRef CAS PubMed.
- B. Halamoda Kenzaoui, C. Chapuis Bernasconi, S. Guney-Ayra and L. Juillerat-Jeanneret, Biochem. J., 2012, 44, 813–821 CrossRef PubMed.
- L. Yu, Y. Lu, N. Man, S. H. Yu and L. P. Wen, Small, 2009, 5, 2784–2787 CrossRef CAS.
- H. Li, Y. Li, J. Jiao and H. M. Hu, Nat. Nanotechnol., 2011, 6, 645–650 CrossRef CAS PubMed.
- H. Eidi, O. Joubert, C. Nemos, S. Grandemange, B. Mograbi, B. Foliguet, J. Tournebize, P. Maincent, A. Le Faou, I. Aboukhamis and B. H. Rihn, Int. J. Pharm., 2012, 422, 495–503 CrossRef CAS PubMed.
- C. Li, H. Liu, Y. Sun, H. Wang, F. Guo, S. Rao, J. Deng, Y. Zhang, Y. Miao, C. Guo, J. Meng, X. Chen, L. Li, D. Li, H. Xu, H. Wang, B. Li and C. J. Jiang, Mol. Cell Biol., 2009, 1, 37–45 CrossRef CAS PubMed.
- H. L. Liu, Y. L. Zhang, N. Yang, Y. X. Zhang, X. Q. Liu, C. G. Li, Y. Zhao, Y. G. Wang, G. G. Zhang, P. Yang, F. Guo, Y. Sun and C. Y. Jiang, Cell Death Dis., 2011, 2, e159 CrossRef PubMed.
- L. Harhaji, A. Isakovic, N. Raicevic, Z. Markovic, B. Todorovic-Markovic, N. Nikolic, S. Vranjes-Djuric, I. Markovic and V. Trajkovic, Eur. J. Pharmacol., 2007, 568, 89–98 CrossRef CAS PubMed.
- O. Seleverstov, O. Zabirnyk, M. Zscharnack, L. Bulavina, M. Nowicki, J. M. Heinrich, M. Yezhelyev, F. Emmrich, R. O'Regan and A. Bader, Nano Lett., 2006, 6, 2826–2832 CrossRef CAS PubMed.
- A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010, 6, 1836–1852 CAS.
- A. N. Zelikin, A. D. Price and B. Städler, Small, 2010, 6, 2201–2207 CrossRef CAS PubMed.
- A. N. Zelikin, Q. Li and F. Caruso, Angew. Chem., Int. Ed., 2006, 45, 7743–7745 CrossRef CAS PubMed.
- A. N. Zelikin, A. L. Becker, A. P. R. Johnston, K. L. Wark, F. Turatti and F. Caruso, ACS Nano, 2007, 1, 63–69 CrossRef CAS PubMed.
- R. De Rose, A. N. Zelikin, A. P. R. Johnston, A. Sexton, S.-F. Chong, C. Cortez, W. Mulholland, F. Caruso and S. J. Kent, Adv. Mater., 2008, 20, 4698–4703 CrossRef CAS.
- A. Sexton, P. G. Whitney, S.-F. Chong, A. N. Zelikin, A. P. R. Johnston, R. De Rose, A. G. Brooks, F. Caruso and S. J. Kent, ACS Nano, 2009, 3, 3391–3400 CrossRef CAS PubMed.
- S. F. Chong, A. Sexton, R. De Rose, S. J. Kent, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5178–5186 CrossRef CAS PubMed.
- S. Sivakumar, V. Bansal, C. Cortez, S.-F. Chong, A. N. Zelikin and F. Caruso, Adv. Mater., 2009, 21, 1820–1824 CrossRef CAS.
- Y. Yan, A. P. R. Johnston, S. J. Dodds, M. M. Kamphuis, C. Ferguson, R. G. Parton, E. C. Nice, J. K. Heath and F. Caruso, ACS Nano, 2010, 4, 2928–2936 CrossRef CAS PubMed.
- K. Ariga, Y. Yamauchi, G. Rydzek, Q. Ji, Y. Yonamine, K. C. W. Wu and J. P. Hill, Chem. Lett., 2014, 43, 36–68 CrossRef CAS.
- Y. Yan, M. Björnmalm and F. Caruso, Chem. Mater., 2014, 26, 452–460 CrossRef CAS.
- C. Y. Cheung, N. Murthy, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 906–910 CrossRef CAS PubMed.
- F. Cavalieri, M. Zhou, F. Caruso and M. Ashokkumar, Chem. Commun., 2011, 47, 4096–4098 RSC.
- Y. Wang, V. Bansal, A. N. Zelikin and F. Caruso, Nano Lett., 2008, 8, 1741–1745 CrossRef CAS PubMed.
- F. Dubreuil, N. Elsner and A. Fery, Eur. Phys. J., 2003, 12, 215–221 CAS.
- M. P. Neubauer, M. Poehlmann and A. Fery, Adv. Colloid Interface Sci., 2014, 207, 65–80 CrossRef CAS PubMed.
- J. P. Best, M. P. Neubauer, S. Javed, H. H. Dam, A. Fery and F. Caruso, Langmuir, 2013, 29, 9814–9823 CrossRef CAS PubMed.
- R. Hartmann, M. Weidenbach, M. Neubauer, A. Fery and W. J. Parak, Angew. Chem., Int. Ed., 2015, 54, 1365–1368 CrossRef CAS PubMed.
- A. Vollrath, S. Schubert, N. Windhab, C. Biskup and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 2053–2058 CrossRef CAS PubMed.
- Y. Yan, Y. Wang, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2011, 23, 3916–3921 CrossRef CAS PubMed.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- N. M. Zaki and N. Tirelli, Expert Opin. Drug Deliv., 2010, 7, 895–913 CrossRef CAS PubMed.
- Y. Ichimura and M. Komatsu, Semin. Immunopathol., 2010, 32, 431–436 CrossRef PubMed.
- H. M. Ni, A. Bockus, A. L. Wozniak, K. Jones, S. Weinman, X. M. Yin and W. X. Ding, Autophagy, 2011, 7, 188–204 CrossRef CAS.
- M. Suzuki, R. J. Youle and N. Tjandra, Cell, 2000, 103, 645–654 CrossRef CAS.
- S. Zinkel, A. Gross and E. Yang, Cell Death Differ., 2006, 13, 1351–1359 CrossRef CAS PubMed.
- H. Saibil, Nat. Rev. Cancer, 2013, 13, 630–642 CrossRef PubMed.
- D. L. Vaux and J. Silke, Cell, 2003, 115, 251–253 CrossRef CAS.
- T. Abbas and A. Dutta, Nat. Rev. Cancer, 2009, 9, 400–414 CrossRef CAS PubMed.
- S. Altenhöfer, I. Witte, J. F. Teiber, P. Wilgenbus, A. Pautz, H. Li, A. Daiber, H. Witan, A. M. Clement, U. Förstermann and S. J. Horke, J. Biol. Chem., 2010, 285, 24398–24403 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr07240e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |