
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complementary analysis of the hard and soft protein corona: sample preparation critically effects corona composition†
S.
Winzen
a,
S.
Schoettler
a,
G.
Baier
a,
C.
Rosenauer
a,
V.
Mailaender
ab,
K.
Landfester
a and
K.
Mohr
*a
aMax Planck Institute of Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: mohr@mpip-mainz.mpg.de
bDept. of Hematology, Oncology and Pneumology, University Medical Center of the Johannes Gutenberg-University Mainz, Langenbeckstr. 1, 55131 Mainz, Germany
First published on 8th January 2015
Abstract
Here we demonstrate how a complementary analysis of nanocapsule–protein interactions with and without application media allows gaining insights into the so called hard and soft protein corona. We have investigated how both human plasma and individual proteins (human serum albumin (HSA), apolipoprotein A-I (ApoA-I)) adsorb and interact with hydroxyethyl starch (HES) nanocapsules possessing different functionalities. To analyse the hard protein corona we used sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and a protein quantitation assay. No significant differences were observed with regards to the hard protein corona. For analysis of the soft protein corona we characterized the nanocapsule-protein interaction with isothermal titration calorimetry (ITC) and dynamic light scattering (DLS). DLS and ITC measurements revealed that a high amount of plasma proteins were adsorbed onto the capsules’ surface. Although HSA was not detected in the hard protein corona, ITC measurements indicated the adsorption of an HSA amount similar to plasma with a low binding affinity and reaction heat. In contrast, only small amounts of ApoA-I protein adsorb to the capsules with high binding affinities. Through a comparison of these methods we have identified ApoA-I to be a component of the hard protein corona and HSA as a component of the soft corona. We demonstrate a pronounced difference in the protein corona observed depending on the type of characterization technique applied. As the biological identity of a particle is given by the protein corona it is crucial to use complementary characterization techniques to analyse different aspects of the protein corona.
Introduction
Nanomaterials are predominantly administered intravenously where they first come into contact with human blood.1 Blood plasma is the non-cellular component of blood and consists of over 3000 different proteins that are capable of interacting with the nanomaterial surface and form a so called ‘protein corona’.2,3 The corona alters the surface composition and size of the nanomaterial, both of which strongly influence the nanomaterial's biological identity as recognized by cells.4 This biological identity can be rather different from the original chemical identity, as for example targeting structures can eventually lose their functionality after coverage with plasma proteins.5 Also the variation of charge and size can strongly influence the body distribution of the nanomaterials. In addition, the release profiles of biomolecules transported by the nanomaterials could potentially change in the presence of a protein corona, so that the desired effect might not be achieved. It becomes even more difficult when the adsorption leads to changes in the structure of the proteins. Denaturation of those proteins on the nanomaterial surfaces can trigger responses of the immune system and therefore induce inflammatory reactions.6 Thus it is crucial to characterize the protein corona of those nanomaterials before they can be applied in vivo.The newly formed biomolecular interface can be divided into the ‘hard’ and ‘soft’ protein corona, which differ in the binding strength and exchange rates of the proteins associated with the nanomaterial surface. A ‘hard’ protein corona consists of proteins with high binding affinities that are tightly bound and show exchange times greater than the time needed for internalization of a particle.7 This adsorption behaviour is also often referred to as irreversible protein binding. However, proteins which are loosely bound to the nanomaterial surface, or are connected with the hard protein corona via week protein–protein interactions, form the so called ‘soft’ protein corona.3,7–11 These proteins are believed to have high exchange rates and can be replaced easily in a biological environment.7 In practice, the hard corona can be defined as those proteins which are not removed from the nanomaterial's surface during preparation procedures such as washing and centrifugation. These procedures can interrupt the relatively weak protein–protein interactions which would remove the soft protein corona.
However it is still not clear, which form of protein corona exists under physiological conditions that also involve some shear forces in the blood stream. Accordingly it is difficult to determine the biologically relevant entity that for example is read by cellular processing. It has been shown that in some cases the hard protein corona is responsible for the interaction pathways,12 but usually there is no information gained about the role of the soft corona proteins.
The majority of published studies that investigate the protein corona involve isolation of the nanomaterial from the biological media after employing washing steps.8,13–16 As defined above, these investigations tend to explore the hard protein corona. The composition, impact and relevance of the soft protein corona are matters that still require attention.7,17 There are only a few analytical methods available to investigate nanomaterials inside the relevant biological media. Using dynamic light scattering (DLS) one can sensitively detect size changes of particles in undiluted blood plasma.18–20 Additionally, it is possible to monitor the change in heat that results from protein adsorption onto nanomaterials with isothermal titration calorimetry (ITC). As an analytical tool, ITC can provide protein binding affinities and stoichiometry.21,22 An alternative method to investigate nanomaterial–protein interactions in contact media with a high sensitivity is fluorescence correlation spectroscopy (FCS).23–25 The drawback of this technique is that FCS requires either fluorescently labelled nanoparticles or labelled proteins.
Nanomaterials which should be applied as drug delivery devices are often functionalized with poly(ethylene glycol) (PEG) chains to prevent unspecific cell uptake and to suppress protein adsorption to some extent.26–28 Suppressing protein adsorption can increase the circulation time of nanocarriers in the bloodstream, leading to a better bioavailability of transported drugs.26 Since sugar-based polymers like dextranes also exhibit a low protein adsorption,29,30 hydroxyethyl starch (HES) is being discussed as a natural nontoxic alternative for PEG with similar protein repellent characteristics.31–33 The investigated nanocarriers in our study were synthesized from HES to obtain capsules with a shell that decreases protein adsorption.
Here, we have investigated the hard and soft protein corona of HES capsules in human plasma, human serum albumin (HSA) and apolipoprotein A-I (ApoA-I) solution using a combination of sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), a protein quantitation assay, ITC and DLS. In addition to non-functionalized capsules, we also analysed carboxy-functionalized (HES-COOH) and amino-functionalized (HES-NH2) capsules in order to investigate the effect of different charges at neutral pH value.
This approach enables a comparison between the behaviour of the capsules with plasma proteins and with isolated proteins. HSA was chosen for the analysis because it is highly abundant in blood plasma (∼44 g L−1 average)34 and well characterized. Furthermore, HSA was found to be depleted in the hard protein corona of the previously investigated particles with comparison to its concentration in plasma.13 In contrast, ApoA-I was shown to be enriched in the protein corona of the same nanomaterials as well as liposomes,9,13,28,35 and should be considered an interesting protein for adsorption measurements. Our work effectively shows that different analytical investigations are required to provide a full picture of a nanocapsule's hard and soft protein corona.
Results and discussion
Characterization of prepared hydroxyethyl starch (HES) nanocapsules
For the interaction of proteins with nanocarriers, HES capsules were synthesized in an aqueous dispersion. The obtained nanocapsules were stable colloids and no precipitation or aggregation was observed during six months of storage under constant stirring at room temperature. Scanning electron microscopy (SEM) studies of HES nanocapsules confirmed the formation of a core–shell structure (see Fig. 1). The collapse of the nanocapsule walls is due to drying effects and the electron beam during the SEM measurement.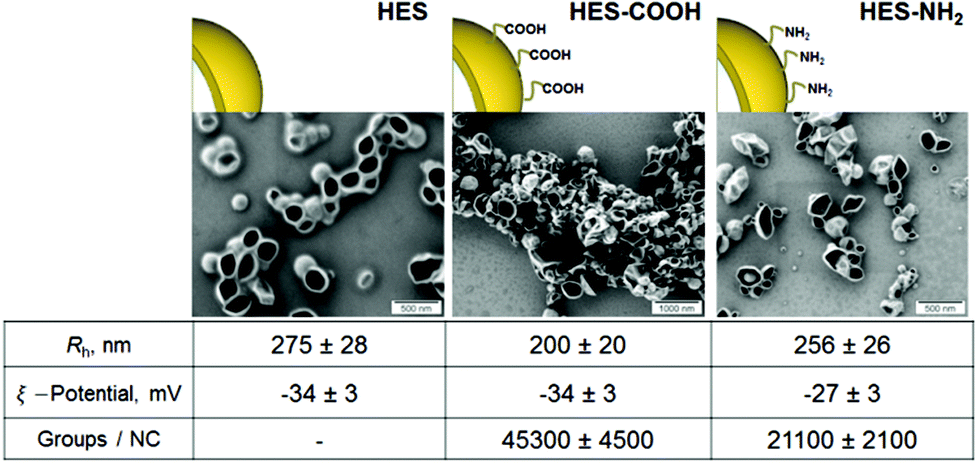 | ||
| Fig. 1 SEM images and characteristics of HES nanocapsules with different surface functionalizations. | ||
All nanocapsules exhibited a negative zeta potential due to a redispersion in water with sodium dodecyl sulphate (SDS). The capsules are purified by extensive dialysis after redispersion to remove most of the SDS. However a small amount is needed on the surface to keep the capsules stable in solution. A functionalization with NH2-groups on the capsule surface leads to a slight increase in zeta potential because of their positive charge at neutral pH. The amount of functional groups per nanocapsule was determined using particle charge detection (PCD, see Methods section).
Protein corona characterization with gel electrophoresis and protein assay
To analyse the proteins strongly associated with the different capsule surfaces, the interaction of the HES capsules with human plasma was investigated with SDS-PAGE and a protein quantitation assay. After incubation with plasma, the capsules were thoroughly washed (three centrifugation steps followed by resuspension in buffer) and the remaining proteins removed from their surface using a mixture of urea, thiourea and buffer (see Materials and methods). Thus the hard protein corona of the HES capsules with varying surface functionalizations was analysed. The protein patterns of the three types of capsules and the pure plasma are shown in the SDS-PAGE gel in Fig. 2. In the pure plasma the most dominant protein bands can be assigned to those proteins that are known to have high plasma concentrations: Albumin (∼44 g L−1, 67 kDa),34 immunoglobulin G (∼10 g L−1, heavy chain 50 kDa, light chain 25 kDa)36 and transferrin (∼2.6 g L−1, 75 kDa).37 The protein patterns recovered from the nanocapsules differ significantly from the pure plasma. It is important to note that the general adsorption pattern is independent of the functionalization of the capsules. The protein bands are very similar in all cases. Interestingly, the most abundant protein in the plasma (albumin) cannot be identified in the hard corona of the capsules.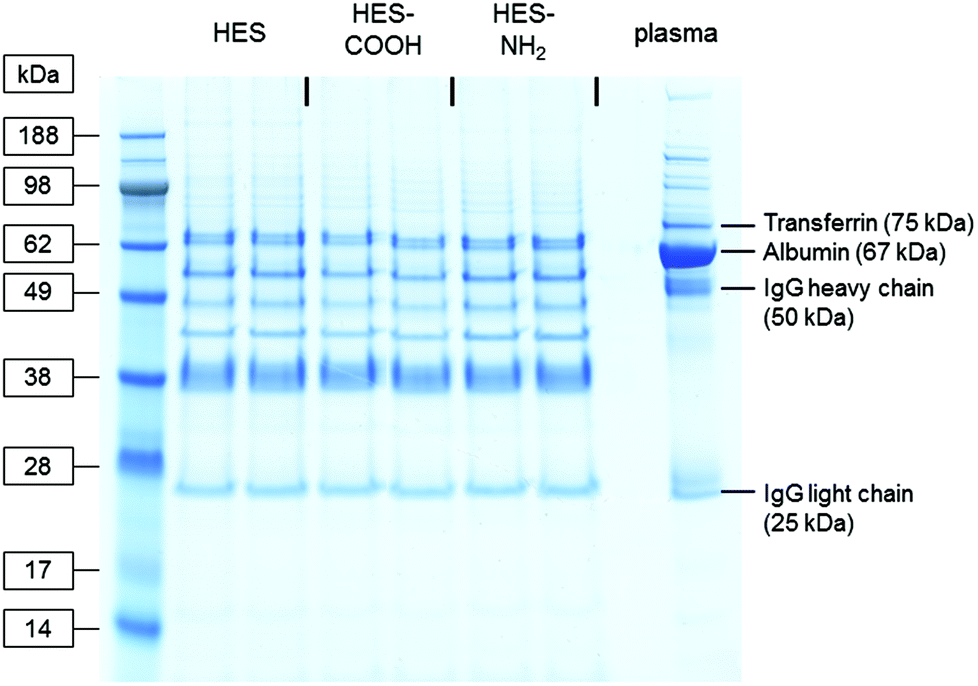 | ||
| Fig. 2 Coomassie-stained gel after SDS-PAGE of plasma proteins recovered from the surface of differently functionalized HES nanocapsules. Pure plasma (right lane) served as a reference. | ||
A quantitative analysis of the adsorbed proteins was performed via a Pierce 660 nm protein assay. Samples were prepared following the SDS-PAGE procedure and analysed photometrically. The obtained protein amounts were normalized with regards to the nanocapsules’ surface area and are shown in Fig. S1.† For the non- and amino-functionalized capsules, values of 0.47 ± 0.11 mg m−2 and 0.46 ± 0.09 mg m−2 respectively were obtained, whereas for the carboxy-functionalized capsule the value decreased to 0.32 ± 0.01 mg m−2. This quantitation suggests that the carboxy-functionalized capsules bind fewer proteins than the other two types of capsules. The described methods require extraction from the interaction medium before analysis. Consequently, proteins that possess a binding affinity below a certain threshold, currently undefined, are likely to be removed during sample preparation. Therefore, we further applied ITC and DLS to investigate the protein–nanocapsule interaction directly in the application medium.
Calorimetric analysis of the adsorption processes
To analyse the soft corona of the HES nanocapsules, the adsorption processes of plasma and single proteins were analysed by ITC measurements. This technique allows the characterization of the capsules directly in physiological media while minimizing alteration of the developed protein corona by additional preparation procedures. Plasma, HSA and ApoA-I solutions were titrated into suspensions of the three types of HES nanocapsules. All titrations were performed at neutral pH, which is above the isoelectric points of both HSA and ApoA-I. The change in heat during every titration step was measured, integrated and corrected for the respective heats of dilution of the titrants (proteins). The heat of dilution from the titration of pure water into the capsule dispersion was negligible. The measured heat changes of the analyzed capsule surfaces and proteins as well as the corresponding adsorption isotherms are shown in Fig. 3. In all monitored adsorption processes, interactions between HES capsules and proteins were observed. In all cases, heat different from the heat of dilution is being released or absorbed. From the integrated heat, the adsorption enthalpy (ΔH), stoichiometry (N) and association constant (Ka) were calculated using a fit according to an independent binding model38,39 (Details on the model are given in the ESI†). The entropy change (ΔS) for each reaction was calculated using the reaction isotherm equation and the Gibbs-Helmholtz equation (see eqn (S2–S4) ESI†). The obtained parameters are summarized in Table 1.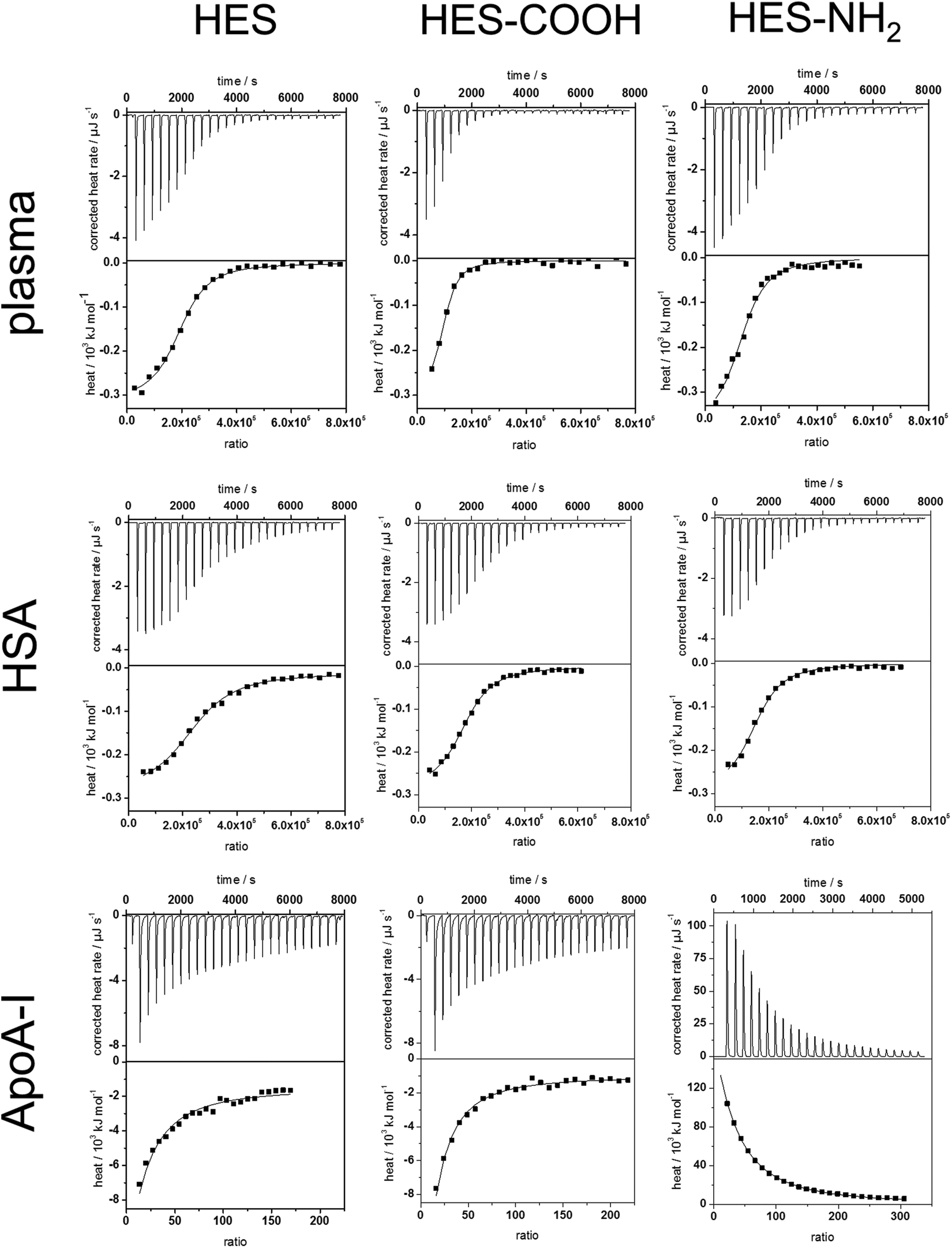 | ||
| Fig. 3 ITC data for the adsorption of plasma, HSA and ApoA-I onto HES nanocapsules. Upper graphs represent the raw data obtained from the titrations (baseline corrected heat rates) and lower graphs the integrated heats of each peak (black squares ■) with a corresponding independent binding fit (straight line –). For plasma measurements the molarity refers to the average HSA concentration of plasma. | ||
| Capsule | Protein | ΔH/kJ mol−1 | K a/106 L mol−1 | ΔS/J K−1 mol−1 | N | Number of proteins per 10 nm2 |
|---|---|---|---|---|---|---|
| HES | Plasma | −285 ± 97 | 1.0 ± 0.4 | −845 ± 329 | 182![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 39000 000 ± 39000 |
2.00 |
| HSA | −277 ± 43 | 0.8 ± 0.3 | −818 ± 147 | 114000 ± 16000 |
1.25 | |
| ApoA-I | −6010 ± 185 | 333 ± 124 | −20000 ± 613 |
10 ± 4 | 1.05 × 10−4 | |
| HES-COOH | Plasma | −306 ± 192 | 1.0 ± 0.8 | −1217 ± 318 | 70000 ± 13500 |
1.40 |
| HSA | −308 ± 30 | 0.4 ± 0.2 | −928 ± 103 | 156000 ± 36000 |
3.00 | |
| ApoA-I | −5150 ± 787 | 188 ± 79 | −17100 ± 2640 |
6 ± 3 | 1.19 × 10−4 | |
| HES-NH2 | Plasma | −281 ± 139 | 1.2 ± 0.6 | −827 ± 468 | 121000 ± 29000 |
1.40 |
| HSA | −277 ± 45 | 0.5 ± 0.1 | −820 ± 152 | 184000 ± 33000 |
2.00 | |
| ApoA-I | 883000 ± 24000 |
5.4 ± 0.1 | 2.4 ± 0.9 × 106 | 8 ± 6 | 0.97 × 10−4 |
These experiments determined that the interactions between human plasma and each type of HES capsule are exothermic and therefore enthalpically favoured. For the analysis of the plasma measurements, the molarity refers to the average concentration of HSA in the plasma. Increasing the concentration consequently leads to a slightly higher stoichiometry N, while the other parameters do not change significantly. The results obtained from the fitting (see Table 1) are in agreement with the analysis of the hard protein corona. The enthalpy change is similar for each different surface functionalization. Furthermore, Ka and ΔS are the same. Given the high number of proteins interacting with one capsule in combination with the small reaction enthalpy, it can be concluded that the proteins are loosely associated with the capsule surface. Additionally, the number of proteins per 10 nm2 of surface area was calculated based on the stoichiometry and the hydrodynamic radius of the capsules. For all functionalities a similar number of around 1–2 proteins was obtained. Thus, the lower protein amount on the carboxy-functionalized particles measured with the protein assay is due to preparation effects. It is important to note that 1–2 proteins per 10 nm2 is not enough space for one protein to obtain a monolayer spread out flat on the surface. Groups of proteins are likely adsorbing at the same time, e.g. protein–protein aggregates formed previously in solution.
The adsorption of pure HSA onto the HES capsules was investigated. In general, the titration isotherms and fit parameters obtained from plasma and HSA are quite similar. This can be explained by the high HSA content in human plasma. Still, slight deviations of Ka, N and the protein per surface area values can be observed. For all capsule types, Ka of HSA was reduced compared to the Ka of plasma, which is the result of additional proteins in the plasma that have a higher binding affinity than HSA. The number of proteins per surface area changes only slightly in comparison to plasma. Those changes can be attributed to different protein compositions of the soft coronas formed in plasma. Moreover, the affinity of HSA towards the HES capsules is roughly 4 times smaller than the protein's affinity towards polystyrene particles, which has been previously investigated.40 This finding can be explained by taking into account the more hydrophilic nature of HES compared to polystyrene, which results in less hydrophobic interactions between the capsule surface and the proteins. However, the number of proteins per 10 nm2 is approximately 10–20 times higher than the values reported for the polystyrene particles.40 From the protein amounts adsorbed it can be concluded that the soft corona formed for HES nanocapsules contains much more proteins than the soft corona of polystyrene particles. Therefore, the difference between the hard and soft corona is more significant for the HES capsules.
The low affinity of HSA to the HES capsules, given by the parameter Ka, in combination with the stoichiometry and the depletion of HSA in the hard protein corona (observed in SDS-PAGE experiments) leads to the identification of HSA as a soft corona protein. These results match the findings reported in literature.41,42
In contrast to the HSA adsorption, a significant amount of heat is generated when ApoA-I is titrated into pure HES capsules (see Fig. 3). Also, Ka differs by about an order of magnitude, which implies that the capsules have a greater affinity towards ApoA-I than to HSA. Meanwhile, N and the protein number per surface area are several orders of magnitude smaller than the values for the HSA molecules. These findings identify ApoA-I as a hard corona protein for the investigated system. This is in good agreement with a previous study of Cedervall et al., who found a high binding affinity of ApoA-I to N-isopropylacrylamide (NIPAM) particles.43 Our results also support the theory that the high binding affinity of ApoA-I is due to the surface characteristics of the nanomaterial rather than the adsorption of fats from the plasma before interaction with the protein.
While the non-functionalized and the carboxy-functionalized capsules display similar behaviour, the amino-functionalized capsules maintain an endothermic interaction with ApoA-I. The large amount of heat absorbed during the titration suggests an entropy-driven interaction process. The independent binding fit (Table 1) confirms these findings, revealing an entropy gain contrasting the entropy loss in the other interactions studied. This entropy gain could partially be attributed to protein structural changes and unfolding during adsorption. An unfolded protein occupies a higher surface area on the capsule and, therefore, more water molecules of the hydration shell are released. However, the amount of heat generally needed for an unfolding of proteins44,45 is lower than the heat absorbed in this reaction. This suggests that there is another interaction process going on, which could involve the surfactant SDS. To clarify this, we are continuing to investigate when denaturation occurs and to which extent surfactants influence the adsorption processes. At least the surfactant does not seem to screen the functional groups of the capsule surfaces, since the endothermic reaction was only observed for the NH2-functionalised capsule.
Dynamic light scattering (DLS) and zeta-potential analysis of capsules after protein adsorption
The adsorption of certain proteins can significantly influence the aggregation behaviour of the nanocapsules. The functionalized HES capsules were further studied with regards to their aggregation behaviour in human plasma, HSA and ApoA-I solutions via DLS. The analysis was performed by adapting the method after Rausch et al.18 In contrast to the previous study, the analysis was performed in human plasma instead of human serum to stay as close as possible to the final in vivo application. Human plasma contains fibrinogen and other coagulation factors, which are removed during serum preparation.46 As fibrinogen is one of the high abundance proteins in blood (∼2.8 g L−1),47 human plasma is used for all analyses instead of serum. The autocorrelation function (ACF) of human plasma could be perfectly described by a sum of three exponentials (eqn (1)), like human serum.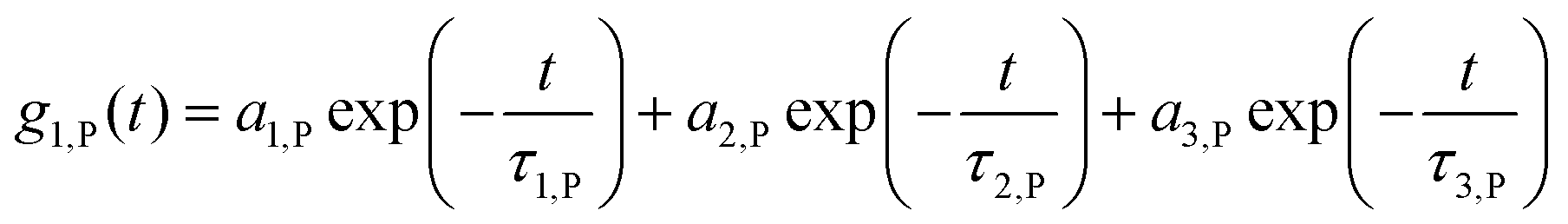 | (1) |
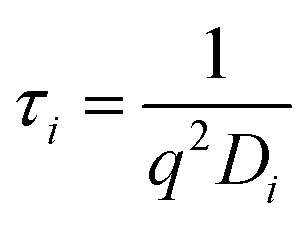 while q is the absolute scattering vector
while q is the absolute scattering vector 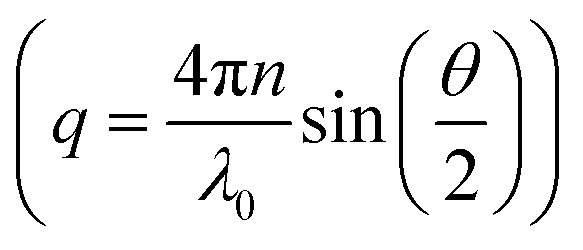 and Di the Brownian diffusion coefficient of component i. Data obtained from light scattering analysis of human plasma are provided in the ESI (Fig. S2†). The ACFs for the HES capsules alone can successfully be fitted by a sum of two exponentials (eqn (2)).
and Di the Brownian diffusion coefficient of component i. Data obtained from light scattering analysis of human plasma are provided in the ESI (Fig. S2†). The ACFs for the HES capsules alone can successfully be fitted by a sum of two exponentials (eqn (2)).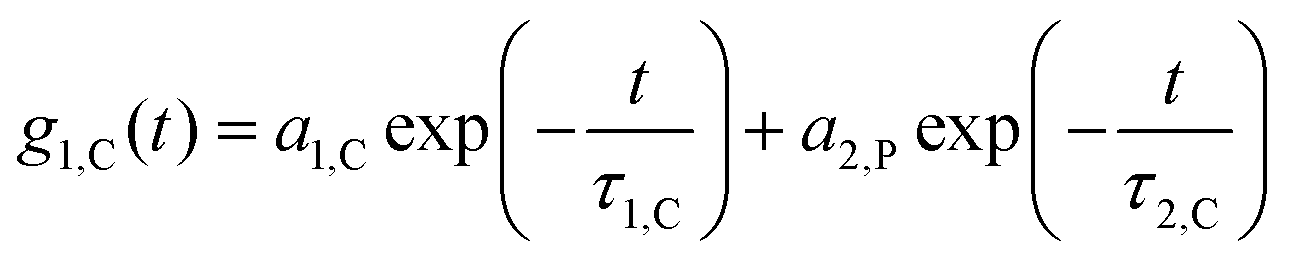 | (2) |
Knowing the ACF of human plasma and the respective HES capsule, the correlation function of the plasma capsule mixtures could be analysed. If no aggregation were to occur, the resulting ACF of the plasma capsule mixture would correlate to the so-called force fit. In the force fit, the sum of the individual correlation functions with the known parameters of the two compounds (plasma/capsule) is kept fixed and the intensity contributions for plasma fP and capsule fC are the only fit parameters (eqn (3)).
| g1,m(t) = fPg1,P(t) + fCg1,C(t) | (3) |
However, interactions of HES capsules with plasma components resulted in larger sizes than the plasma components and HES capsules themselves; consequently, the ACFs could not be described by the force fit.
The fit needed to be modified by an additional, longer ACF relaxation time related to the size of the formed structures (eqn (4)).
| g1,m(t) = fPg1,P(t) + fCg1,C(t) + faggg1,agg(t) | (4) |
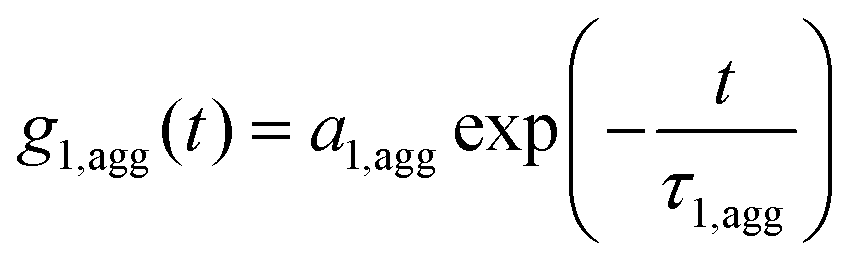 | (5) |
The multicomponent analyses of the HSA and ApoA-I capsule mixtures were performed accordingly. The exemplary analysis of the ACF (g1,m(t)) of the mixture of non-functionalized HES capsules with human plasma is shown in Fig. 4A. The multicomponent analyses of the other mixtures are provided in the ESI.† The force fit (sum of individual components) did not describe the data correctly. In contrast, the fit including an additional aggregate component was suitable for the capsule/plasma mixture. The fitting procedure for each angle produced the corresponding hydrodynamic radius Rh shown in Fig. 4B. From the extrapolated diffusion coefficients the z-averaged hydrodynamic radius <1/Rh>z−1 was calculated by the application of Stokes law and can be found in Table 2. Fig. 4 demonstrates the increase of Rh, which occurs after interaction with plasma. The size increase of around 100 nm for non-functionalized capsules is too small to be caused by aggregation between several capsules, so it is attributed to coating with plasma proteins. Additionally, the intensity contribution of the aggregates (see Table 2) was calculated and is also shown in Fig. 4B. The fraction of the new species (protein-coated capsules) being formed is significant in comparison to the fraction of pure capsules. The remaining fraction of the capsules in the mixture can be attributed to the natural distribution of the thickness of the protein corona and the polydispersity of capsules and aggregates. As the applied method is highly sensitive for the detection of aggregates with sizes larger than the largest size present in the pure components solutions, changes within the size distribution of plasma and capsules are only detectable if the amplitudes (i.e. the intensity fractions) of the newly formed sizes are sufficiently large (and thus detectable by DLS). Typically, intensity fractions between 3% (for sizes larger than the largest component in the mixture) and 20% (for sizes in the same size range of the mixture components) of newly formed particles are necessary in order to become detectable by the described fitting procedure.48 Consequently, small capsules with a thin protein corona are not recognized as aggregates. The DLS analysis has been carried out for all capsule types and proteins and the corresponding sizes and intensity fractions are displayed in Table 2. The DLS experiments were repeated after 24 h of incubation time to check for any changes related to the protein adsorption kinetics. However there was no significant difference found in the aggregation behaviour. According to reports in the literature, the composition of the protein corona changes quantitatively but not qualitatively. Therefore it is likely that slight changes in the protein corona compositions of the mixtures with plasma cannot be detected with this type of analysis.
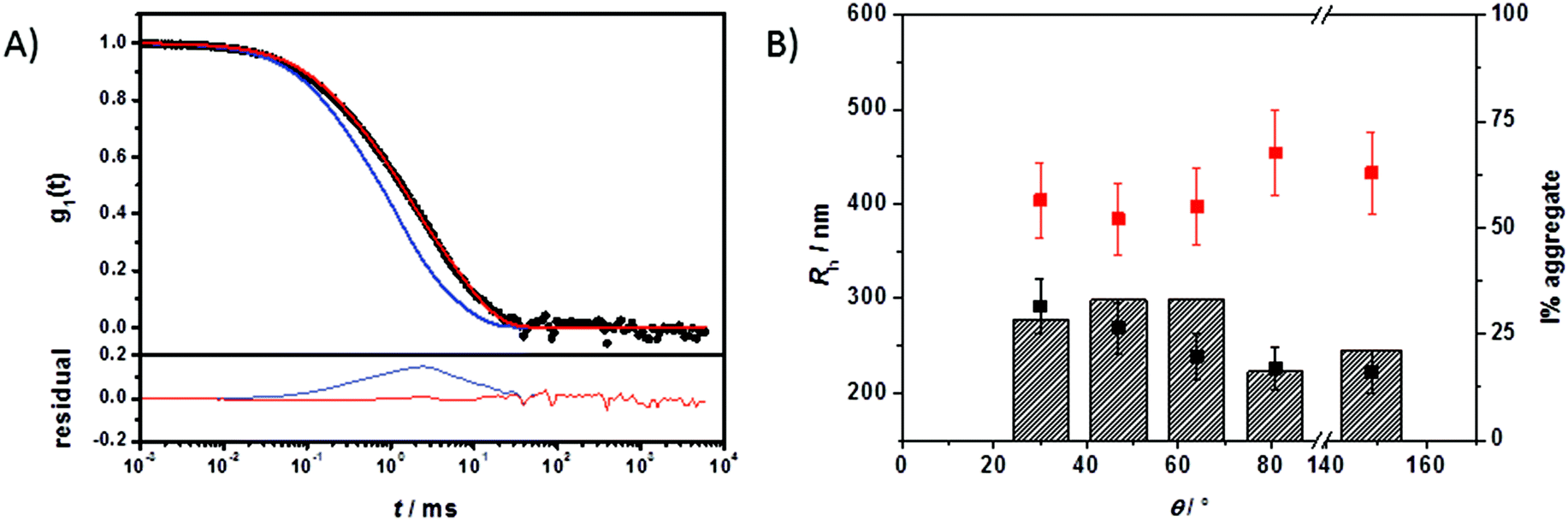 | ||
Fig. 4 A) Upper graph: Autocorrelation function g1(t) (black circles ●) of non-functionalized HES capsules mixed with plasma at θ = 64°. The blue line ( ) represents the forced fit composed of the sum of the individual components whereas the red line ( ) represents the forced fit composed of the sum of the individual components whereas the red line ( ) represents the fit with an additional aggregation function. Lower graph: Corresponding residuals resulting from the difference between the data and the two fits. B) Hydrodynamic radii of pure non-functionalized HES capsules (black squares ■) and of the aggregate formed in plasma (red squares ) represents the fit with an additional aggregation function. Lower graph: Corresponding residuals resulting from the difference between the data and the two fits. B) Hydrodynamic radii of pure non-functionalized HES capsules (black squares ■) and of the aggregate formed in plasma (red squares  ). Striped columns represent the intensity fraction of aggregates in the mixture. ). Striped columns represent the intensity fraction of aggregates in the mixture. | ||
| Capsule | No protein | Plasma | HSA | ApoA-I | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R h/nm | ξ-potentiala/mV | R h,Agg/nm | I%Aggb | ξ-potentiala/mV | R h,Agg/nm | I%Aggb | ξ-potentiala/mV | R h,Agg/nm | ξ-potentiala/mV | |
| a ξ-potential at pH 7 in 0.001 M KCl solution. b Intensity fractions of the aggregates (I%Agg) are exemplarily given for a scattering angle of 64°. For the intensity fractions of other scattering angles see Fig. S3, S5 and S6. | ||||||||||
| HES | 275 ± 26 | −34 ± 3 | 390 ± 39 | 33 | −22 ± 9 | 347 ± 35 | 28 | −18 ± 1 | — | −7 ± 6 |
| HES-COOH | 200 ± 20 | −34 ± 3 | 319 ± 32 | 25 | −13 ± 5 | 352 ± 35 | 47 | −15 ± 4 | — | −8 ± 7 |
| HES-NH2 | 256 ± 26 | −27 ± 3 | — | — | −11 ± 4 | — | — | −10 ± 1 | — | −18 ± 8 |
Additionally, the zeta-potential was measured for all samples before and after the mixture with proteins (see Table 2) to monitor the change of the capsules’ surface charge after protein adsorption.
The average size increase of around 120 nm for the non- and carboxy-functionalized capsules coated with plasma (Fig. S5†) and the significant intensity fraction of the resulting larger structures can be attributed to the formation of a protein corona. For the amino-functionalized capsules no component larger than the components of the mixture was found via DLS (Fig. S8†), even though the ITC measurements suggested adsorption processes similar to the other capsules. The DLS results are in agreement with previous studies of amino-functionalized nanoparticles that did not show any aggregate formation.20 It has to be noted that the aggregation behaviour cannot be attributed to a positive overall particle charge due to the amino functionalization as the amino-functionalized particles observed a negative zeta potential because of their redispersion in water with SDS. Concerning the difference between the findings from ITC and DLS, it is important to note that the samples taken from ITC measurements were diluted for the DLS experiments (see Methods section). While a high concentration is needed to obtain a sufficient signal in ITC, it has to be decreased for light scattering due to the high turbidity of the capsules above a certain concentration. Light scattering measurements would then be complicated by multiple and back scattering artifacts.49,50 Therefore, it is possible that a formed protein corona changes upon dilution due to concentration dependencies. The soft protein corona is especially affected because of the high protein exchange rates reported in literature.7 Nevertheless, the zeta-potential for all three surface functionalizations was increased after protein adsorption. This suggests coverage with proteins, as the negative charges from SDS are shielded, or even replaced, by proteins with a lower negative charge compared to their volume or positive charge.
The same analysis was performed with HSA and yielded results similar to the plasma measurements (see Table 2, Fig. S3 and S6†). This reaffirms the conclusion drawn from the ITC experiments that HSA is adsorbed to the capsule surface with a high stoichiometry and thus leads to the formation of a thick soft protein corona. The combination of these results with the depletion of HSA in the SDS-PAGE identifies HSA as a soft corona protein. Again, no additional aggregates were found in the sample containing amino-functionalized capsules (Fig. S9†). Also, the zeta-potential increase suggests the coverage with proteins.
In contrast to plasma and HSA, no additional aggregates were formed from the interaction of all capsule types with ApoA-I (see Fig. S4, S7 and S10†). This agrees with the ITC results, given that few molecules (around 10 ApoA-I molecules per capsule) adsorbed to the capsule surface. The formation of aggregates was not expected because the size increase due to formation of an ApoA-I monolayer would not be greater than 8 nm in radius. The size of the protein (Rh ≈ 3.8 nm)51 is only around 1.5% of the capsule size, so the size change is in the experimental error of the light scattering experiment. These results in combination with the high binding affinity for ApoA-I (Table 1) classify ApoA-I as a hard corona protein.
Experimental
Materials
Human Serum Albumin (HSA) was purchased from Sigma Aldrich (St. Louis, USA; Product No. A3782) and Apolipoprotein A-I (ApoA-I) was purchased from Biopur AG (Reinach, Switzerland; Product no. BP 10-61-1101). The proteins were used without further purification. All protein solutions were freshly prepared with water (Millipore, Milli-Q water with a conductivity <18.2 MΩ cm).Blood was taken at the Transfusion Centre of the University Medical Centre of the Johannes Gutenberg-University Mainz from 10 healthy donors after obtaining informed consent. The study was approved by the local ethics committee. To prevent blood clotting Li-Heparin was added. The blood was centrifuged to pellet red and white blood cells and the plasma supernatant was pooled. Aliquots were stored at −80 °C. After thawing, the plasma was centrifuged at 20000g for 1 h at 4 °C to remove any residual protein precipitates. A protein concentration of 66 g L−1 was determined for the plasma.
Capsule preparation
HES nanocapsules were synthesized by a polyaddition reaction performed at the miniemulsion droplet's interface similar to the previously published procedure.52 Afterwards, the HES nanocapsules were functionalized to create positively and negatively charged HES nanocapsules. With the carboxymethylation procedure, the HES nanocapsules were covered with carboxylic groups.52 For the NH2-functionalization of HES nanocapsules, 2.0 g of HES nanocapsules dispersion (in cyclohexane as continuous phase, solid content 3.0 wt%) were mixed with 20 mg 2,4-Toluene diisocyanate (TDI, Sigma Aldrich) and stirred for 24 h at 25 °C. The nanocapsules were then transferred into the aqueous phase using the following procedure: 1 g of the nanocapsules dispersion in cyclohexane (polymer solid content 3 wt%) was mixed with 5 g sodium dodecyl sulphate (SDS; Sigma Aldrich) aqueous solution (0.1 wt%) and kept under mechanical stirring conditions for 24 h at 25 °C. Next, the samples were redispersed for 15 min at 50 °C in a sonication bath (power 50%, 25 kHz). Finally, the nanocapsule dispersion was centrifuged (Sigma 3k-30, RCF 1467, 20 min). The supernatant was removed, the nanocapsules were redispersed in demineralized water and dialyzed for 24 h (MWCO: 12000 g mol−1) in order to remove residues of SDS.
Capsule characterization
The amount of surface charged groups was calculated from the results of the titration experiments performed on a Mütek particle charge detector (BTG, Herrsching, Germany) in combination with a Titrino Automatic Titrator (Metrohm AG, Herisau, Switzerland). The carboxylic groups were titrated against the positively charged polycation poly(diallyl dimethyl ammonium chloride) (poly-DADMAC). The amine groups on the nanocapsules surface were titrated against the negatively charged polyelectrolyte poly(ethylene sulphonate) (PES-Na). The titrations were performed on 10 mL of the nanocapsules dispersion with a solid content of 1 g L−1. The amount of groups per gram of polymer was calculated from the consumed volume of the polyelectrolyte solution. Morphological studies were performed with scanning electron microscopy (SEM). The images were recorded by using a field emission microscope (LEO (Zeiss) 1530 Gemini, Oberkochen, Germany) operated at an accelerating voltage of 170 V. The samples were prepared by diluting the nanocapsule dispersion to about 0.01% solid content and by placing a droplet onto silica wafers and drying under ambient conditions.Preparation of samples for gel electrophoresis and protein quantification
The nanocapsule dispersions were diluted with ultrapure water to a constant particle surface concentration (0.1 m2 in 150 μL) and incubated with 500 μL human blood plasma for 1 h at 37 °C under constant agitation. The particles were separated from the supernatant by centrifugation at 15000g for 1 h. The nanoparticles were washed with phosphate-buffered saline (PBS; Thermo Fisher Scientific, Waltham, USA) in three centrifugation steps at 15000g for 1 h. To elute the adsorbed proteins, the particle pellet was resuspended in 7 M urea, 2 M thiourea and 4% CHAPS, and the nanoparticles were again pelleted. The supernatant was then used for protein quantitation and SDS-PAGE.
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE)
16.25 μL of each protein sample was loaded onto a NuPAGE® Novex® 10% Bis-Tris Gel (Thermo Fisher Scientific) and subjected to SDS-PAGE according to standard procedures. As a molecular marker SeeBlue Plus2 Pre-Stained Standard (Thermo Fisher Scientific) was run in parallel. Proteins were fixed in 10% acetic acid for 1 h and subsequently visualized by staining with 0.1% Coomassie brilliant blue G-250 in 10% ammonium sulphate, 2% phosphoric acid and 25% methanol for 24 h.Protein assay
Protein concentrations were determined using Pierce 660 nm protein Assay (Thermo Scientific, Rockford, USA) according to manufacturer's instructions with BSA as a standard. Each sample was measured in triplicate.Isothermal titration calorimetry (ITC)
The calorimetric measurements were performed using a NanoITC Low Volume (TA Instruments, Eschborn, Germany) with an effective cell volume of 170 μL. In an experiment 50 μL of an aqueous protein solution (human plasma 15 g L−1, HSA 10 g L−1 or ApoA-I 0.084 g L−1) were titrated to 300 μL of a suspension of HES nanocapsules (0.1 g L−1 in water for titration with human plasma and HSA; 4 g L−1 for titration with ApoA-I). The experimental temperature was kept constant at 25 °C. Additionally, the same amount of each protein solution was titrated into pure water to determine the heat of dilution for reference. The number and injected volume of the titration steps were the same for all measurements (25 × 2 μL). The spacing between injections was set to 300 s. The integrated reference heats were then subtracted from the integrated heats of the adsorption experiments. The normalized heats were then analyzed with a fitting procedure according to an independent binding model (see ESI†) to obtain the association constant (Ka), the reaction enthalpy (ΔH) and the reaction stoichiometry (N) as the fitting parameters.Dynamic light scattering (DLS)
All light scattering experiments were performed with an ALV-CGS 8F SLS/DLS 5022F goniometer equipped with eight simultaneously working ALV 7004 correlators and eight QEAPD Avalanche photodiode detectors (ALV, Langen, Germany). A HeNe laser (632.8 nm, 25 mW output power) was utilized as the light source.For measurements of nanocapsules–protein mixtures the samples were prepared according to the ITC titration procedure. In every case, 300 μL of a capsule suspension was mixed with 50 μL of protein solution or water for the reference measurements (concentrations were the same as in ITC experiments). Plasma and individual proteins were prepared by adding the solutions into 300 μL of water to maintain the same dilutions. All plasma and HSA containing samples were then diluted with water up to 2 mL sample volume. ApoA-I containing samples were diluted up to 5 mL total volume. Capsule concentrations of 0.015 g L−1 and 0.24 g L−1 respectively were achieved. The solutions were then filtered through Millex SV filters with a pore size of 5 μm (Merck Millipore, Billerica, USA) into dust-free quartz light scattering cuvettes (inner diameter 18 mm, Hellma, Müllheim), which were cleaned before with acetone in a Thurmont-apparatus.
Zeta-potential measurements
For zeta-potential measurements, 20 μL of each sample mixture obtained after ITC were diluted with 1 mL of 0.001 M KCl solution. The samples were then analyzed with a Zetasizer Nano Z (Malvern Instruments GmbH, Herrenberg, Germany).Conclusions
There is currently a major gap in knowledge with regards to a defined physicochemical characterization of the protein corona that forms on a nanomaterial's surface once it enters the blood stream. Effects of the preparation procedure required for different techniques are unknown and with that the understanding of the true biological identity is still challenging. In our study we have applied different techniques to obtain complementary information about the protein corona of HES nanocapsules. For the first time we have compared data concerning the hard protein corona derived from SDS-PAGE and protein quantitation with information from ITC and DLS about the soft corona. We have demonstrated that ITC and DLS are valuable methods to investigate the soft protein corona as they allow us to characterize particles in the incubation medium. This is in stark contrast to SDS-PAGE and protein assays, which require particle extraction prior to characterization. With a combination of these techniques we have been able to compare the characteristics of the hard and soft corona and at the same time detect differences in the adsorption behaviours of single proteins. For future investigations it still remains crucial to obtain a better understanding of the soft corona. The true biologically relevant corona composition is still not clear and most probably not only involves the hard protein corona in the case of low-affinity nanocapsules.Acknowledgements
We thank B. Mueller for scientific support with nanocapsule characterization and J. Schwabacher for helpful discussions. We acknowledge the DFG SFB 1066 as a funding agency for funding the project Q1 and Q2. We are also very grateful to Professor M. Schmidt for providing us admission to his equipment.Notes and references
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Nanomed. Nanotechnol., 2009, 5, 106–117 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggård, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed.
- W. Liu, J. Rose, S. Plantevin, M. Auffan, J.-Y. Bottero and C. Vidaud, Nanoscale, 2013, 5, 1658–1668 RSC.
- F. Wang, L. Yu, M. P. Monopoli, P. Sandin, E. Mahon, A. Salvati and K. A. Dawson, Nanomed. Nanotechnol., 2013, 9, 1159–1168 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharm., 2008, 5, 487–495 CrossRef CAS PubMed.
- N. P. Mortensen, G. B. Hurst, W. Wang, C. M. Foster, P. D. Nallathamby and S. T. Retterer, Nanoscale, 2013, 5, 6372–6380 RSC.
- M. Lundqvist, Nat. Nanotechnol., 2013, 8, 701–702 CrossRef CAS PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef CAS PubMed.
- L. Nuhn, S. Gietzen, K. Mohr, K. Fischer, K. Toh, K. Miyata, Y. Matsumoto, K. Kataoka, M. Schmidt and R. Zentel, Biomacromolecules, 2014, 15, 1526–1533 CrossRef CAS PubMed.
- K. Mohr, M. Sommer, G. Baier, S. Schöttler, P. Okwieka, S. Tenzer, K. Landfester, V. Mailänder, M. Schmidt and R. G. Meyer, J. Nanomed. Nanotechnol., 2014, 5, 193–203 Search PubMed.
- K. Bouchemal, Drug Discovery Today, 2008, 13, 960–972 CrossRef CAS PubMed.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- C. Rocker, M. Potzl, F. Zhang, W. J. Parak and G. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577–580 CrossRef PubMed.
- P. Maffre, K. Nienhaus, F. Amin, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2011, 2, 374 CrossRef CAS PubMed.
- P. d. Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 RSC.
- V. Torchilin, in Long Acting Injections and Implants, ed. J. C. Wright and D. J. Burgess, Springer US, 2012, pp. 263–293 Search PubMed.
- M. D. Howard, M. Jay, T. D. Dziubla and X. Lu, J. Biomed. Nanotechnol., 2008, 4, 133–148 CrossRef CAS PubMed.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Lagana, Nanoscale, 2014, 6, 2782–2792 RSC.
- R. E. Marchant, S. Yuan and G. Szakalas-Gratzl, J. Biomater. Sci., Polym. Ed., 1995, 6, 549–564 CrossRef PubMed.
- E. Österberg, K. Bergström, K. Holmberg, T. P. Schuman, J. A. Riggs, N. L. Burns, J. M. Van Alstine and J. M. Harris, J. Biomed. Mater. Res., 1995, 29, 741–747 CrossRef PubMed.
- M. Orlando, Ph.D. Thesis, University of Giessen, 2003.
- A. Besheer, J. Vogel, D. Glanz, J. Kressler, T. Groth and K. Mäder, Mol. Pharm., 2009, 6, 407–415 CrossRef CAS PubMed.
- C. Lemarchand, R. Gref and P. Couvreur, Eur. J. Pharm. Biopharm., 2004, 58, 327–341 CrossRef CAS PubMed.
- T. Le, V. Bhushan and D. A. Rao, First Aid for the USMLE Step 1: 2008, McGraw Hill Professional, 2007 Search PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- A. Gonzalez-Quintela, R. Alende, F. Gude, J. Campos, J. Rey, L. Meijide, C. Fernandez-Merino and C. Vidal, Clin. Exp. Immunol., 2008, 151, 42–50 CrossRef CAS PubMed.
- A. Helander, Clin. Chem., 1999, 45, 131–135 CAS.
- E. Lewis and K. Murphy, in Protein-Ligand Interactions, ed. G. U. Nienhaus, Humana Press, 2005, vol. 305, pp. 1–15 Search PubMed.
- E. Freire, O. L. Mayorga and M. Straume, Anal. Chem., 1990, 62, 950A–959A CrossRef CAS.
- G. Baier, C. Costa, A. Zeller, D. Baumann, C. Sayer, P. H. H. Araujo, V. Mailänder, A. Musyanovych and K. Landfester, Macromol. Biosci., 2011, 11, 628–638 CrossRef CAS PubMed.
- D. Dell'Orco, M. Lundqvist, C. Oslakovic, T. Cedervall and S. Linse, PLoS One, 2010, 5, e10949 Search PubMed.
- A. Cifuentes-Rius, H. de Puig, J. C. Y. Kah, S. Borros and K. Hamad-Schifferli, ACS Nano, 2013, 7, 10066–10074 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggård, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- A. Michnik, K. Michalik, A. Kluczewska and Z. Drzazga, J. Therm. Anal. Calorim., 2006, 84, 113–117 CrossRef CAS PubMed.
- M. J. Pikal, D. Rigsbee and M. L. Roy, J. Pharm. Sci., 2008, 97, 5122–5131 CrossRef CAS PubMed.
- E. A. Martin, in Concise medical dictionary, Oxford University Press, 2010 Search PubMed.
- Fibrinogen Studies Collaboration, JAMA, 2005, 294, 1799–1809 Search PubMed.
- M. Hemmelmann, K. Mohr, K. Fischer, R. Zentel and M. Schmidt, Mol. Pharm., 2013, 10, 3769–3775 CAS.
- P. Štěpánek, J. Chem. Phys., 1993, 99, 6384–6393 CrossRef PubMed.
- S. Bantle, M. Schmidt and W. Burchard, Macromolecules, 1982, 15, 1604–1609 CrossRef CAS.
- G. V. Annathur, S. Kawas, T. K. Das and S. V. Ho, J. Membr. Sci., 2010, 353, 41–50 CrossRef CAS PubMed.
- G. Baier, D. Baumann, J. M. Siebert, A. Musyanovych, V. Mailänder and K. Landfester, Biomacromolecules, 2012, 13, 2704–2715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Pierce 660 nm protein assay, ITC data evaluation, DLS data evaluation, autocorrelation functions of protein – HES capsule mixtures. See DOI: 10.1039/c4nr05982d |
| This journal is © The Royal Society of Chemistry 2015 |