
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene-modified electrodes for enhancing the performance of microbial fuel cells
Heyang
Yuan
and
Zhen
He
*
Department of Civil and Environmental Engineering, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA. E-mail: zhenhe@vt.edu
First published on 3rd November 2014
Abstract
Graphene is an emerging material with superior physical and chemical properties, which can benefit the development of microbial fuel cells (MFC) in several aspects. Graphene-based anodes can enhance MFC performance with increased electron transfer efficiency, higher specific surface area and more active microbe-electrode-electrolyte interaction. For cathodic processes, oxygen reduction reaction is effectively catalyzed by graphene-based materials because of a favorable pathway and an increase in active sites and conductivity. Despite challenges, such as complexity in synthesis and property degeneration, graphene-based electrodes will be promising for developing MFCs and other bioelectrochemical systems to achieve sustainable water/wastewater treatment and bioenergy production.
 Heyang Yuan | Mr Heyang Yuan is now a Ph.D. student advised by Dr Zhen He in the Via Department of Civil and Environmental Engineering at Virginia Polytechnic Institute and State University (Virginia Tech). He received his B.S. from Tongji University and M.S. from Technische Universität München. He has conducted research in microbial communities in activated sludge. At Virginia Tech, his research focuses on electrochemistry in microbial fuel cells, the optimization of microbial desalination cells, and nano-structured materials for electrode/catalyst application in bioelectrochemical systems |
 Zhen He | Dr Zhen He is an Associate Professor in the Via Department of Civil and Environmental Engineering at Virginia Tech. Prior to this position, he was an Assistant Professor of Civil Engineering and Mechanics at University of Wisconsin – Milwaukee. He received his BS from Tongji University, MS from Technical University of Denmark, and PhD from Washington University in St. Louis, and all of them were in environmental engineering. His research focuses on the development of biotechnologies for sustainable water and wastewater treatment. He has published more than 70 journal papers and is an associate editor for Water Environment Research. |
Introduction
Proper treatment of wastewater is critical to maintain a sustainable society, and this consumes a large amount of energy. For instance, water and wastewater utilities account for approximately 4% of the electricity consumption in the U.S., namely, one third of the total municipal energy budget.1 It is estimated that the maximal energy that can be extracted from a typical domestic wastewater containing 500 mg L−1 chemical oxygen demand (COD) is 1.9 kWh m−3.2 In contrast, approximately 0.6 kWh m−3 of energy will be consumed to treat this type of domestic wastewater by the prevailing activated sludge method.3 The theoretical net energy benefit using energy-recovering treatment technology compared to activated sludge methods will be 2.5 kWh m−3. Hence, there is an urgent need to develop energy-efficient treatment technologies by reducing energy consumption and increasing energy recovery.Microbial fuel cell (MFC) as a green treatment technology has attracted considerable attention in the past decade.4–9 A typical MFC is composed of two electrodes and an ion exchange membrane, which serves as a separator. Organic compounds (e.g. organic wastes in wastewater) are oxidized by exoelectrogens (electrochemically active microorganisms) growing on the anode electrode, and terminal electron acceptors (e.g. oxygen) are reduced on the cathode electrode (Fig. 1). Because the redox reactions are thermodynamically favorable, electrons released by microorganisms spontaneously flow from the anode electrode to the cathode electrode through an external circuit, and thus electricity is generated.
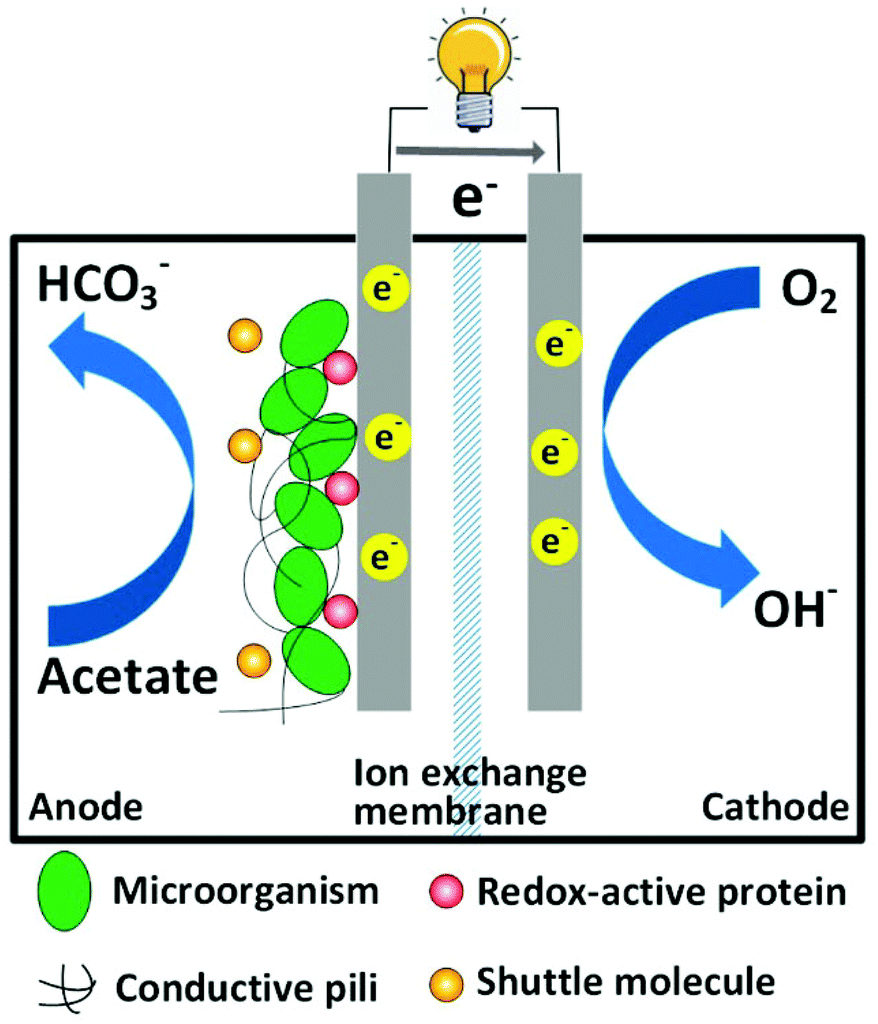 | ||
| Fig. 1 Schematic of a typical two-chamber MFC. | ||
The development of MFC technology has been hindered mainly by the low charge transfer efficiency of electrodes and high cost of catalysts.10,11 To address these issues, a variety of materials and their modifications, including carbon/graphite materials, carbon nanotubes (CNTs), nanostructured materials and non-precious metals, have been studied, as reviewed in detail elsewhere.12,13 While these materials have shown certain promise, their applications are still limited by inherent drawbacks such as low specific surface area, low conductivity, poor biocompatibility, and/or complicated synthesis procedures.
Among these newly developed materials, graphene has been of significant interest since the first demonstration of its facile isolation in 2004.14 This two-dimensional sheet formed of sp2 hybridized carbon atoms exhibits outstanding physical properties, including high mechanical strength, high thermal conductivity and good elasticity.15–17 Its maximal specific surface area is estimated to be 2630 m2 g−1, which is several times higher compared to other carbon-based nanomaterials.18 Furthermore, the superior electrical properties make graphene a promising material for applications in electronics. It has been reported that the charge carrier mobility of suspending single layer graphene can be higher than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1.19 Consequently, electrons travel in graphene at a Fermi velocity of 106 m s−1 without scattering, which is known as ballistic conduction.20 These unique properties, which are tunable towards specific application via different synthetic procedures,21,22 are highly attractive for the development of MFCs. For electrode application, graphene and its modifications may substantially decrease the loss of electrical potential, and thus enhance MFC performance. This review will summarize and discuss graphene-based materials used for MFC application based on their functions, and provide insight into the modification of graphene materials for improving MFC electrodes.
000 cm2 V−1 s−1.19 Consequently, electrons travel in graphene at a Fermi velocity of 106 m s−1 without scattering, which is known as ballistic conduction.20 These unique properties, which are tunable towards specific application via different synthetic procedures,21,22 are highly attractive for the development of MFCs. For electrode application, graphene and its modifications may substantially decrease the loss of electrical potential, and thus enhance MFC performance. This review will summarize and discuss graphene-based materials used for MFC application based on their functions, and provide insight into the modification of graphene materials for improving MFC electrodes.
Enhance the anode performance
Biological oxidation of organics and extracellular electron transfer (EET) are complicated processes affected by several factors. First, electrons are transferred from exoelectrogens to an electrode through three possible mechanisms: (1) soluble electron-shuttling molecules, (2) redox-active proteins on the cell membrane and (3) conductive pili.23 Therefore, an ideal anode electrode is expected to facilitate rapid heterogeneous electron transfer (HET) with microorganisms and electron shuttles.24 Moreover, a bioanode should have biocompatibility and a high specific surface area for accommodating a large quantity of microbes. In this regard, graphene-based materials are promising candidates for highly efficient MFC anodes.25,26 The following sections discuss the study of graphene-based materials in MFC anodes from the aspects of EET efficiency, specific surface area and multi-phase interactions.Improving EET efficiency
It has been determined that graphene-based materials could improve EET efficiency through functional groups. In an early MFC study that involved graphene-based materials, graphene oxide (GO), a common precursor in graphene synthesis, was examined as an anode.27 Cyclic voltammetry (CV) results suggested that the GO nanoribbons yielded a higher response of cytochromes (redox-active proteins), indicating lower activation energy, namely, a lower potential loss. This finding is unexpected because GO is considered as an insulator and only shows graphene-like conductivity after being reduced.28 Thus, researchers attributed the enhanced electrochemical activity to the large amount of functional groups on the material surface. The properties of GO were further studied by forming a polypyrrole/GO composite, which was designed to take advantage of the conductivity of polypyrrole and the electrochemical activity of functional groups from GO.29 As anticipated, the composite achieved a noticeably higher power density compared to reduced GO and pristine polypyrrole. Electrochemical impedance spectroscopy (EIS) also revealed a slight reduction in the ohmic resistance and a significant decrease in the charge transfer resistance.With advances in materials science, graphene is easily obtained by the reduction of GO.30 As a consequence, anode electrodes modified with graphene materials through various methods have been investigated for MFCs. The most straightforward method is to directly coat graphene on anode electrodes, e.g. on stainless steel fiber felts. In comparison to activated carbon and CNTs, graphene-coated anode exhibited remarkably high electrochemical response in CV and the lowest polarization resistance. Although the charge transfer resistance of graphene anode was higher compared to CNT anode, suggesting a relatively inferior HET, it still provided the highest power density among the tested electrodes.31 To enhance HET, Guo et al.32 modified carbon paper with graphene via layer-by-layer assembly and observed increased peak current and decreased peak current separation in CV when the layer number increased (Fig. 2). These results clearly evidenced a promoted electron transfer rate and were further supported by the same trend of charge transfer resistance in EIS tests. Charge transfer resistance was also successfully reduced together with a boosted peak current when tin oxide was anchored on the surface of reduced GO (rGO) using a microwave-assisted method.32
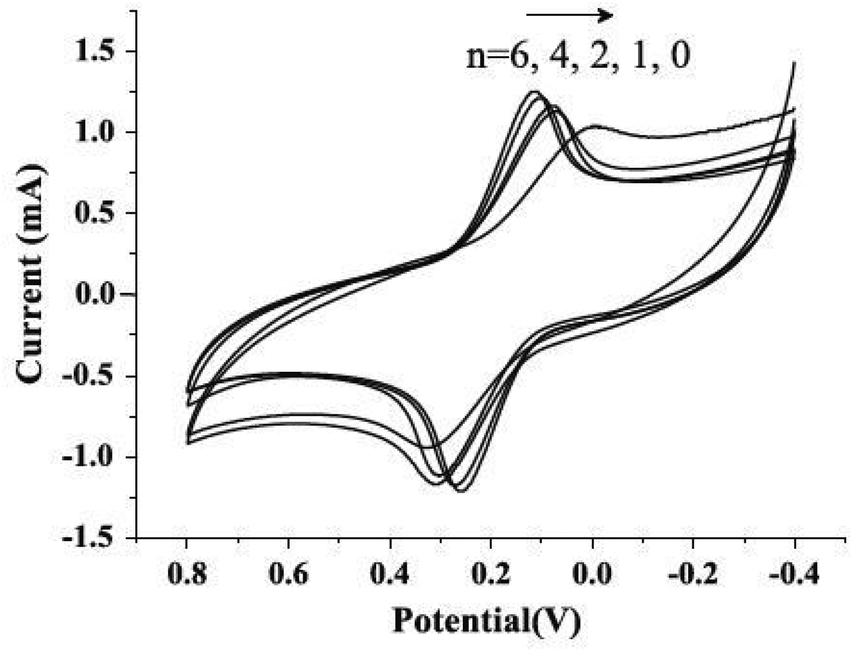 | ||
| Fig. 2 Cyclic voltammograms of n-layer graphene composites (n = 0, 1, 2, 4, 6) in aqueous 0.1 mol L−1 KCl containing 10 mmol L−1 K3[Fe(CN)6] at a scan rate of 20 mV s−1. Reproduction with permission from ref. 32. | ||
An interesting phenomenon, which can be directly related to both EET and GO reduction, is that GO can be in situ reduced via the respiration of bacteria such as Shewanella spp.,33 which is a group of important facultative exoelectrogens in MFCs.34 Electron transfer during the microbial reduction on graphene has been reported to be involved with cytochromes MtrA, MtrB, and MtrC/OmcA, which are all playing pivotal roles in EET.35 This has raised the question if biologically reduced GO interacts better with microbes and shows higher EET efficiency. A further study using microbiologically reduced GO as an anode material demonstrated a 25-fold higher electron transfer rate constant, doubled exchange current and halved ohmic and charge transfer resistance.36 Distinct improvement of these properties is encouraging for the development of highly robust and sustainable anode electrodes via in situ reduction in MFCs.
Increasing specific surface area
A high specific surface area of graphene can provide sufficient sites for the attachment of biofilms, and consequently guarantee a high quantity of biocatalysts for the oxidation of organics.37 For example, the specific surface area of graphene obtained via chemical reduction is 500 times larger compared to woven graphite felt.38,39 However, this specific surface area is only one tenth of the theoretical value, indicating a reduction in area caused by increased layer number or other treatments.21,40 To fully exploit this advantage of graphene materials, modifications were carried out from different aspects. Luo et al.41 introduced an evaporation-induced crumpling process to synthesize compression- and aggregation-resistant graphene balls, which yielded crumpled graphene particles with a specific surface area up to 567 m2 g−1. Electricity generation and power density of the MFC equipped with the crumpled graphene anode (Fig. 3A) was significantly enhanced compared to regular graphene sheets.42 | ||
| Fig. 3 (A) Morphology of crumpled r-GO particles. (B) SEM images of the attachment of bacterial cells on the graphene/PANI anodes. Reproduction with permission from ref. 42, 43. | ||
Polyaniline (PANI) was electro-deposited on graphene nanoribbons to fabricate a composite anode, as this polymer has been reported to easily form porous nanostructures.43 Scanning electron microscopy (SEM) results demonstrated a 3D microporous structure and dense adhesion of microbial cells on the anode surface. As a result of the thickened biofilm (Fig. 3B), the PANI/graphene-anode MFC produced a maximal power density twice as high as that of the control group. Later, a simple synthesis of macroporous 3D graphene sponge was developed by freeze-drying frozen graphene hydrogel.44 A noticeable increase in both specific surface area and pore volume was observed. It could also been seen from SEM images that organisms thrived on a graphene anode, which was in good agreement with the increased power density.
Promoting interaction among microbes, substrates and electrodes
Although the biocompatibility of graphene remains inconclusive,30 graphene anodes have been reported to change microbial metabolism in a manner that favors EET. For example, Liu et al.45 obtained high peak current and lower peak separation when using a graphene-deposited carbon cloth anode. As discussed earlier, these results indicated a facilitated EET. Interestingly, when peak current was plotted versus square root of scan rate or scan rate, which represented mediated and direct electron transfer, respectively, it was observed that the positive effect exerted by graphene anode was more significant to direct EET compared to mediated EET (Fig. 4). This finding, together with a high amount of biomass observed on the anode surface, may be indicative that exoelectrogens integrated graphene materials into their EET pathways. A similar conclusion was drawn in a recent study,46 in which CV response associated with outer membrane c-type cytochrome was detected on a microbiologically reduced graphene/Shewanella biofilm but not on a pristine biofilm, indicating a highly integrated microbe/electrode hybrid. Furthermore, the 3D macroporous structure of the resultant composite ensured good interaction between biocatalyst and substrate.46 It is noteworthy that the enhancement of conductive pili, which is one of the major electron transfer mechanisms, has by far not been reported. Hypothetically, the biocompatibility and porous structure of graphene-based electrode may be favorable for the formation of conductive pili, further facilitating microbe-electrode interaction. Understanding the effects of graphene materials on the pili formation may significantly advance graphene application as bio-electrode materials in MFC. | ||
| Fig. 4 (A) Cyclic voltammograms of MFC in glucose-free medium and (B) in 0.5% glucose medium at a scan rate of 30 mV s−1. (C) Peak current of Peak 1 (−0.6 V vs. SCE) is plotted versus the square root of scan rate. (D) Peak current of Peak 2 (−0.5 V vs. SCE) is plotted versus scan rate. Reproduction with permission from ref. 45. | ||
In addition to creating a porous structure, PANI is positively charged in neutral solutions, and thus shows high affinity towards the negatively charged biomass.47 Moreover, biofilm formation on the hydrophobic graphene is expected to be improved with the hydrophilic PANI.48 In a recent study, CV peak related to a c-type cytochrome (presumably OmcA) was detected using a PANI-hybridized 3D graphene anode, but was absent for carbon cloth anode.49 Furthermore, high performance liquid chromatography did not detect difference in the concentrations of riboflavin, a relevant electron shuttle, between the MFCs equipped with those two anodes. Therefore, it was concluded that electrons were preferentially harvested via direct EET on the graphene/PANI anode due to its 3D integration with biofilm. Hou et al.50 obtained reduced graphene via electrochemical reduction and mixed it with PANI to fabricate MFC anodes. Although the PANI-rGO anode showed slightly higher charge transfer resistance compared to pristine reduced graphene anode, its power density and open circuit voltage outcompeted other materials. It is noteworthy that in the two studies, the diffusion resistance of the PANI-modified graphene was significantly smaller compared to non-modified graphene or other materials, indicating a favorable structure for the interaction between electrolyte and anode electrode.
Enhance the cathode
Oxygen is an ideal electron acceptor for MFC cathode because of its reduction potential and availability. However, the reduction of oxygen is kinetically slow, and hence may contribute to the internal resistance of an MFC more than anode reaction.51 In addition, oxygen reduction reaction (ORR) occurring at the cathode surface involves three phases, i.e., gas (oxygen-electron acceptor), liquid (electrolyte-reaction medium) and solid (electrode-electron donor), making it a complicated process. As such, the cathode process is considered to be challenging and requires efficient catalysts (e.g. simple synthesis and cost effective catalysis) to facilitate ORR.11 Platinum (Pt) is commonly used as a catalyst in bench-scale MFC experiments owing to the small overpotential of ORR, but Pt-based catalysts are not economically viable in large-scale applications, especially for wastewater treatment. A number of carbon-based materials and non-precious metals have been intensively studied as alternative materials for cathode electrodes and catalysts in the past few years, and there is a continuing interest in exploring more efficient catalysts to improve the cathode performance.13 Graphene modification has been demonstrated to be effective to carry out ORR via an efficient pathway (i.e. four-electron transfer), abundant active sites and high conductivity.52Cathode reduction pathways
In principle, oxygen is reduced either through a one-step four-electron pathway (eqn (1)):| O2 + 4H+ + 4e− → 4H2O E° = 1.229 V vs. NHE | (1) |
| O2 + H2O + 2e− → HO2− + OH− E° = −0.065 V vs. NHE | (2) |
| HO2− + H2O + 2e− → 3OH− E° = 0.867 V vs. NHE | (3) |
| 2HO2− → 2OH− + O2 | (4) |
While the 2-e pathway generates highly reactive hydrogen peroxide, which leads to damaged membranes and electrodes, the 4-e pathway is favorable because of the higher reduction potential. It has been determined that four-electron transfer predominates in ORR using nitrogen-doped graphene (NG) as a cathode material.55 The transferred electron number per oxygen molecule was calculated to be 3.69 for a NG cathode synthesized with detonation process, close to that for Pt. In another study, NG obtained via pyrolysis yielded an even higher electron transfer number of 3.87.56 Both studies achieved slightly higher power density with NG cathode compared with Pt in MFCs. The superior ORR catalysis is mainly attributed to the introduction of three nitrogen species by doping: graphitic N adsorbs OOH and reduces O2 to H2O2via 2-e pathway, and pyridinic and pyrrolic N are likely to be responsible for the catalysis of 4-e pathway.56–59
According to Cao et al.,60 ORR can be catalyzed by manganese oxides possibly through the oxidization of MnOOH by adsorbed oxygen. As demonstrated by CV, ORR catalysis by MnO2 coated on graphene was significantly enhanced compared with that on carbon black and multi-wall CNT (MWCNT).61 Moreover, the performance of the MFC equipped with MnO2/graphene cathode was comparable to that with Pt/C in terms of open circuit voltage, power density and coulombic efficiency. A MnO2/GO cathode also provided power density and electricity generation similar to that of a Pt/C cathode.62 Graphene electrodes hybridized with other metals resulted in similar enhancement in the MFC performance.63–66 These studies suggest that graphene can affect ORR pathway either by the modification of its own structure or by the enhancement of catalytic ability with other doped/coated materials.
Increasing active sites
As discussed above, a high content of pyridinic and pyrrolic nitrogen in NG is desirable because they might facilitate the more efficient four-electron transfer in ORR. To increase the proportion of these nitrogen active sites, Feng et al.67 implanted mesoporous graphitic carbon nitride (C3N4) into NG nanosheet. This novel approach resulted in a high nitrogen/carbon ratio of 19.7% and an increased content of pyridinic N, which is distinctly higher than that synthesized using pyrrole, aniline and ammonia as precursors.57 The remarkable ORR catalysis of the implanted NG was later testified by electrochemical analysis, in which CV showed a higher peak current, whereas linear sweep voltammetry provided an earlier onset potential compared to Pt/C. Furthermore, the transferred electron number was calculated to be 4, indicating a complete four-electron transfer pathway.67 When tested in MFCs, the modified NG yielded 20% higher power density (Fig. 5A) and noticeably higher stability compared to Pt/C (Fig. 5B). The increase in nitrogen activity can also be implemented from other aspects. For example, iron was added to stabilize nitrogen incorporation on graphene.68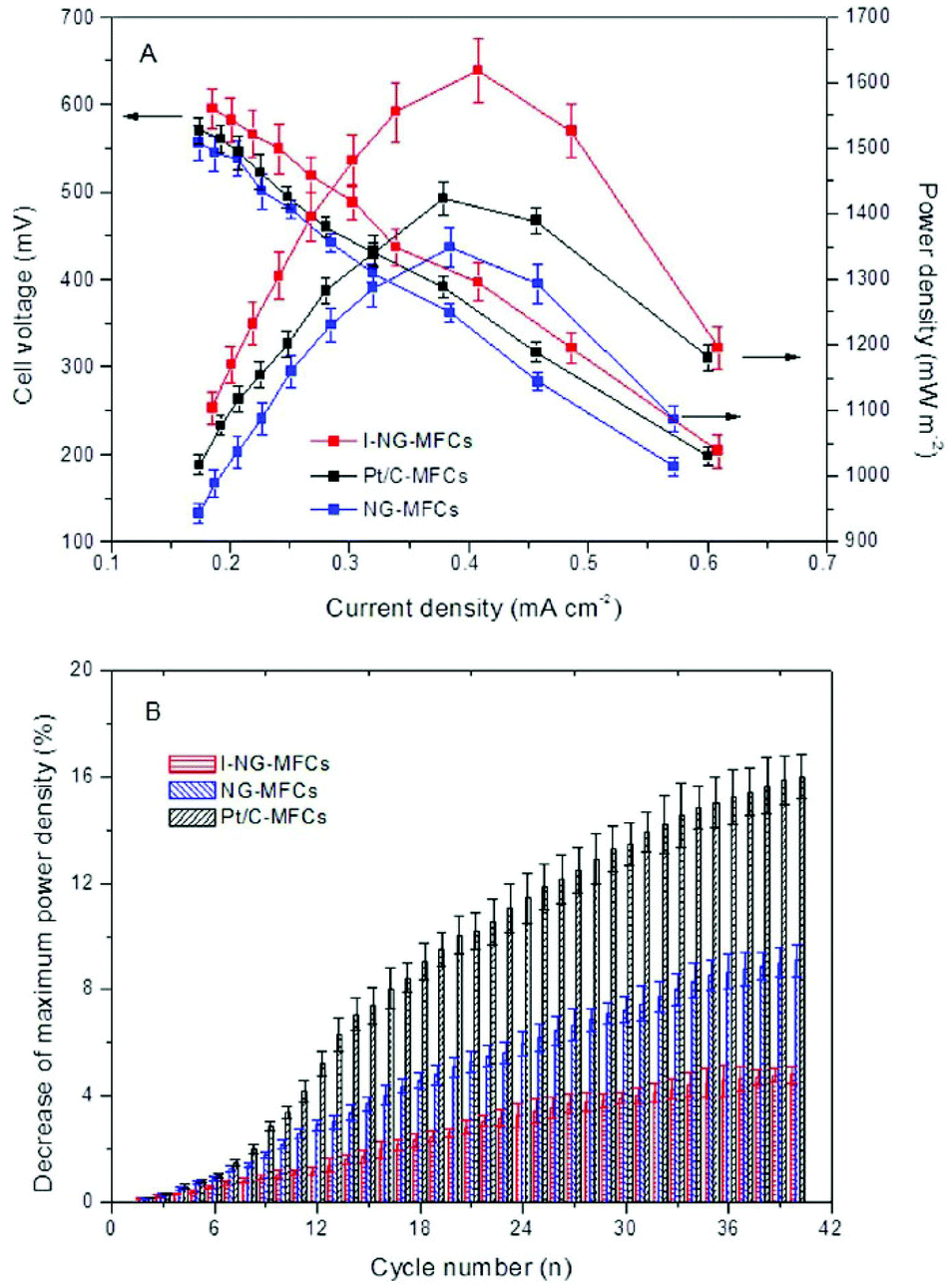 | ||
| Fig. 5 (A) Power densities and cell voltages of mesoporous graphitic C3N4-NG MFCs, regular NG-MFCs and Pt/C-MFCs. (B) Reduction of maximum power density with cycle number at an external resistance of 1000 Ω. Reproduction with permission from ref. 67. | ||
In addition to the amount of nitrogen sites, sufficient exposure of active sites to reactants using a large specific surface area could effectively promote ORR. PANI was hybridized with GO to produce porous nitrogen-doped carbon nanosheet on graphene with a specific surface area of 1398 m2 g−1, leading to a prominent CV peak and power density.69 In the same study, oxygen content in the modified material reached a high value of 9.13%, which could be another explanation for the efficient catalysis. Oxygen species were also observed in C3N4-NG, which was speculated to be advantageous to ORR because of the high adsorption of O2.70 Liu et al.56 has proposed that the presence of O species not only decreases the active energy carrier of ORR, but also protects the catalytic C-N groups from being attacked by protons (Fig. 6). Apart from oxygen, other polar functional groups on the graphene surface have been reported to encourage the formation of small silver particles, whose size is inversely correlated to ORR catalysis, resulting in an electron transfer number of 3.8 and comparable power density and electricity generation to Pt/C.71
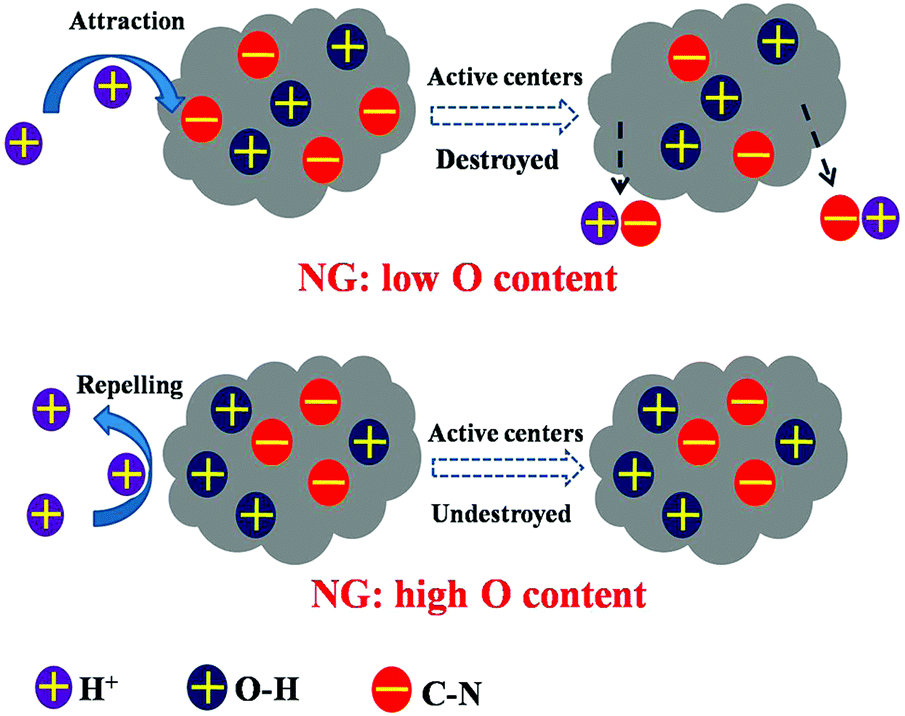 | ||
| Fig. 6 NG is protected by O–H groups from the attack of protons. Reproduction with permission from ref. 56. | ||
Enhancing conductivity
Although in theory graphene possesses high conductivity, non-ballistic conduction was observed in bulk samples,72 possibly resulted from impurity and topological lattice defects.21,73,74 This increases the internal resistance of the cathode electrode, and thus hinders ORR. Therefore, it is of crucial importance to maximize the conductivity of graphene cathodes in practical applications. To reduce resistance, graphene is doped with nitrogen. The conductivity of regular NG was measured to be 35 S m−1, which is 10 S m−1 lower compared to Pt/C,55 whereas that of C3N4-implanted NG increased to 66 S m−1.67 Accordingly, the MFC with an implanted NG cathode, which showed a 31% decrease of internal resistance, produced a 20% higher power density compared to the one with a regular NG cathode. Similar to graphene-based anode electrodes, the ohmic and charge transfer resistance of cathode electrodes can be reduced with graphene/biofilm composites or crumpled graphene particles.42,75 The versatile polymer PANI has also been doped with graphene to fabricate cathode electrodes.76 EIS results revealed the diminished ohmic and charge transfer resistance for the PANI-graphene cathodes compared to the pristine PANI cathode, with the PANI/graphene ratio of 1:9 being the most conductive electrode. The PANI/graphene composites doped with para-toluenesulfonic acid exhibited high electrical conductivity at high temperature due to the high mobility of the charge carriers.77
Perspectives
It is worth noting that in most cases, the enhanced performance of anode electrodes and/or cathode electrodes, conferred by graphene materials, is a synergistic effect.78–81 For example, a chitosan/vacuum-stripped graphene scaffold composite with hierarchically porous 3D structure had a specific surface area of 248 m2 g−1 owing to the large specific areas of graphene.82 Moreover, its charge transfer resistance was reduced with increased graphene loading, with the lowest value of 150 Ω for 50 wt% among a series of loading rates. Together with the good biofilm adhesion and high production of endogenous mediator stimulated by the biocompatible chitosan, current and power density of the MFC containing this composite as its anode electrode were 16 and 78 times higher, respectively, compared to the control with the carbon cloth anode. On the other hand, multi-directional improvement was also observed in cathode modifications, for example implantation of mesoporous graphitic C3N4 simultaneously facilitated favorable ORR pathway and increased active sites and conductivity.67 Therefore, synergistic effects are desirable as electrode performance benefited from multiplication instead of addition of the enhancement of individual aspects. This may be helpful for the preparation and modification of graphene-based electrode and catalysts in future.Despite their great promise in MFCs, graphene materials suffer from relatively complicated synthesis procedures. In general, graphene electrodes are prepared through three steps: GO synthesis, GO reduction to graphene, and modification.83 This approach, and other methods, including chemical vapor deposition,84 unzipping of MWCNTs and detonation are either troublesome, energy/time-consuming or involved with hazardous chemicals,27,49,55 thereby increasing capital cost in large-scale applications. The in situ reduction of GO to graphene by bacteria or phytoextracts could be an effective option for anode fabrication as it meets the requirement of sustainability and green chemistry.85 For cathode, NG is a competitive material because of its outstanding ORR catalysis, but does not seem to prevail before economic synthesis methods with optimized nitrogen types and contents are developed.59
MFCs are a major type of bioelectrochemical systems (BES), which also include various ramifications with different functions such as the hydrogen-producing microbial electrolysis cells (MECs), the chemical-synthesizing microbial synthesis cells (MSCs) and the salt-removing microbial desalination cells.4 To date, there is very limited study using graphene to enhance performance in BES other than MFCs.86 In a study of MECs, 3D MoS2/NG nanosheet aerogels were synthesized through a facile hydrothermal approach and used to catalyze hydrogen evolution reaction (HER). Although the loading rate of MoS2/NG aerogels on the cathode electrode was only 25% that of Pt/C, its current density in MFC test was comparable to that of Pt/C. In addition, the MoS2/NG aerogels outperformed pristine MoS2 nanosheets and NG aerogels with respect to electrochemistry and catalysis. It can be expected that with advances in graphene research, there will be more research using graphene materials as electrode and catalyst for improving BES performance in a broad range of applications, and in return this will be beneficial for identifying proper application niches for graphene materials and stimulating graphene research in future.
Acknowledgements
The authors acknowledge the financial support from a faculty startup fund of Virginia Polytechnic Institute and State University.Notes and references
- R. Goldstein and W. Smith, Water & Sustainability (Volume 4): US Electricity Consumption for Water Supply & Treatment-The Next Half Century, Electric Power Research Institute, 2002 Search PubMed.
- P. L. McCarty, J. Bae and J. Kim, Environ. Sci. Technol., 2011, 45, 7100–7106 CrossRef PubMed.
- T. P. Curtis, Environ. Microbiol., 2010, 2, 301–318 Search PubMed.
- H. Wang and Z. J. Ren, Biotechnol. Adv., 2013, 31, 1796–1807 CrossRef CAS PubMed.
- W.-W. Li, H.-. Yu and Z. He, Energy Environ. Sci., 2013, 7, 911–924 Search PubMed.
- V. B. Wang, N. Yantara, T. M. Koh, S. Kjelleberg, Q. Zhang, G. C. Bazan, S. C. J. Loo and N. Mathews, Chem. Commun., 2014, 50, 8223–8226 RSC.
- V. B. Wang, S.-L. Chua, Z. Cai, K. Sivakumar, Q. Zhang, S. Kjelleberg, B. Cao, S. C. J. Loo and L. Yang, Bioresour. Technol., 2014, 155, 71–76 CrossRef PubMed.
- V. B. Wang, N. D. Kirchhofer, X. Chen, M. Y. L. Tan, K. Sivakumar, B. Cao, Q. Zhang, S. Kjelleberg, G. C. Bazan, S. C. J. Loo and E. Marsili, Electrochem. Commun., 2014, 41, 55–58 CrossRef PubMed.
- V. B. Wang, J. Du, X. Chen, A. W. Thomas, N. D. Kirchhofer, L. E. Garner, M. T. Maw, W. H. Poh, J. Hinks, S. Wuertz, S. Kjelleberg, Q. Zhang, J. S. C. Loo and G. C. Bazan, Phys. Chem. Chem. Phys., 2013, 15, 5867–5872 RSC.
- R. A. Rozendal, H. V. Hamelers, K. Rabaey, J. Keller and C. J. Buisman, Trends Biotechnol., 2008, 26, 450–459 CrossRef PubMed.
- B. E. Logan, Appl. Microbial. Biotechnol., 2010, 85, 1665–1671 CrossRef PubMed.
- Y. Qiao, S.-J. Bao and C. M. Li, Energy Environ. Sci., 2010, 3, 544–553 Search PubMed.
- M. Zhou, M. Chi, J. Luo, H. He and T. Jin, J. Power Sources, 2011, 196, 4427–4435 CrossRef PubMed.
- K. S. Novoselov, A. K. Geim, S. Morozov, D. Jiang, Y. Zhang, S. Dubonos, I. Grigorieva and A. Firsov, Science, 2004, 306, 666–669 CrossRef PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef PubMed.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef PubMed.
- R. S. Edwards and K. S. Coleman, Nanoscale, 2013, 5, 38–51 RSC.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef PubMed.
- D. R. Lovley, Annu. Rev. Microbiol., 2012, 66, 391–409 CrossRef PubMed.
- F. Zhao, R. C. Slade and J. R. Varcoe, Chem. Soc. Rev., 2009, 38, 1926–1939 RSC.
- C. Zhao, Y. Wang, F. Shi, J. Zhang and J.-J. Zhu, Chem. Commun., 2013, 49, 6668–6670 RSC.
- Y. Zhu, J. Ji, J. Ren, C. Yao and L. Ge, Colloids Surf., A, 2014, 455, 92–96 CrossRef PubMed.
- Y.-X. Huang, X.-W. Liu, J.-F. Xie, G.-P. Sheng, G.-Y. Wang, Y.-Y. Zhang, A.-W. Xu and H.-Q. Yu, Chem. Commun., 2011, 47, 5795–5797 RSC.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef PubMed.
- Z. Lv, Y. Chen, H. Wei, F. Li, Y. Hu, C. Wei and C. Feng, Electrochim. Acta, 2013, 111, 366–373 CrossRef PubMed.
- J. Filip and J. Tkac, Electrochim. Acta, 2014, 136, 340–354 CrossRef PubMed.
- J. Hou, Z. Liu, S. Yang and Y. Zhou, J. Power Sources, 2014, 258, 204–209 CrossRef PubMed.
- W. Guo, Y. Cui, H. Song and J. Sun, Bioprocess Biosyst. Eng., 2014, 37, 1749–1758 CrossRef PubMed.
- G. Wang, F. Qian, C. Saltikov, Y. Jiao and Y. Li, Nano Res., 2011, 4, 563–570 CrossRef PubMed.
- E. C. Salas, Z. Sun, A. Lüttge and J. M. Tour, ACS Nano, 2010, 4, 4852–4856 CrossRef PubMed.
- S. Patil, C. Hägerhäll and L. Gorton, Bioanal. Rev., 2012, 4, 159–192 CrossRef.
- Y. Yuan, S. Zhou, B. Zhao, L. Zhuang and Y. Wang, Bioresour. Technol., 2012, 116, 453–458 CrossRef PubMed.
- A. Mehdinia, E. Ziaei and A. Jabbari, Int. J. Hydrogen Energy, 2014, 39, 10724–10730 CrossRef CAS PubMed.
- D. H. Park and J. G. Zeikus, Biotechnol. Bioeng., 2003, 81, 348–355 CrossRef CAS PubMed.
- Y. Zhang, G. Mo, X. Li, W. Zhang, J. Zhang, J. Ye, X. Huang and C. Yu, J. Power Sources, 2011, 196, 5402–5407 CrossRef CAS PubMed.
- H. Skulason, P. Gaskell and T. Szkopek, Nanotechnology, 2010, 21, 295709 CrossRef CAS PubMed.
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943–8949 CrossRef CAS PubMed.
- L. Xiao, J. Damien, J. Luo, H. D. Jang, J. Huang and Z. He, J. Power Sources, 2012, 208, 187–192 CrossRef CAS PubMed.
- C. Zhao, P. Gai, C. Liu, X. Wang, H. Xu, J. Zhang and J.-J. Zhu, J. Mater. Chem. A, 2013, 1, 12587–12594 CAS.
- W. Chen, Y.-X. Huang, D.-B. Li, H.-Q. Yu and L. Yan, RSC Adv., 2014, 4, 21619–21624 RSC.
- J. Liu, Y. Qiao, C. X. Guo, S. Lim, H. Song and C. M. Li, Bioresour. Technol., 2012, 114, 275–280 CrossRef CAS PubMed.
- Y.-C. Yong, Y.-Y. Yu, X. Zhang and H. Song, Angew. Chem., Int. Ed., 2014, 53, 4480–4483 CrossRef CAS PubMed.
- B. Lai, X. Tang, H. Li, Z. Du, X. Liu and Q. Zhang, Biosens. Bioelectron., 2011, 28, 373–377 CrossRef CAS PubMed.
- P. Avouris and C. Dimitrakopoulos, Mater. Today, 2012, 15, 86–97 CrossRef CAS.
- Y.-C. Yong, X.-C. Dong, M. B. Chan-Park, H. Song and P. Chen, ACS Nano, 2012, 6, 2394–2400 CrossRef CAS PubMed.
- J. Hou, Z. Liu and P. Zhang, J. Power Sources, 2013, 224, 139–144 CrossRef CAS PubMed.
- Y. Fan, E. Sharbrough and H. Liu, Environ. Sci. Technol., 2008, 42, 8101–8107 CrossRef CAS.
- Y. Zhang, G. Mo, X. Li and J. Ye, J. Power Sources, 2012, 197, 93–96 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 1980 Search PubMed.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707–4716 CrossRef CAS PubMed.
- L. Feng, Y. Chen and L. Chen, ACS Nano, 2011, 5, 9611–9618 CrossRef CAS PubMed.
- Y. Liu, H. Liu, C. Wang, S.-X. Hou and N. Yang, Environ. Sci. Technol., 2013, 47, 13889–13895 CrossRef CAS PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- Y. L. Cao, H. X. Yang, X. P. Ai and L. F. Xiao, J. Electroanal. Chem., 2003, 557, 127–134 CrossRef CAS.
- S. Khilari, S. Pandit, M. M. Ghangrekar, D. Das and D. Pradhan, RSC Adv., 2013, 3, 7902–7911 RSC.
- G. Gnana kumar, Z. Awan, K. Suk Nahm and J. Stanley Xavier, Biosens. Bioelectron., 2014, 53, 528–534 CrossRef CAS PubMed.
- G. H. Qi, X. Q. Li and J. Cao, Adv. Mater. Res., 2014, 881, 310–314 CrossRef.
- Q. Wen, S. Wang, J. Yan, L. Cong, Z. Pan, Y. Ren and Z. Fan, J. Power Sources, 2012, 216, 187–191 CrossRef CAS PubMed.
- Z. Yan, M. Wang, B. Huang, R. Liu and J. Zhao, Int. J. Electrochem. Sci., 2013, 8, 149–158 CAS.
- Y. Su, Y. Zhu, X. Yang, J. Shen, J. Lu, X. Zhang, J. Chen and C. Li, Ind. Eng. Chem. Res., 2013, 52, 6076–6082 CrossRef CAS.
- L. Feng, L. Yang, Z. Huang, J. Luo, M. Li, D. Wang and Y. Chen, Sci. Rep., 2013, 3 Search PubMed.
- S. Li, Y. Hu, Q. Xu, J. Sun, B. Hou and Y. Zhang, J. Power Sources, 2012, 213, 265–269 CrossRef CAS PubMed.
- Q. Wen, S. Wang, J. Yan, L. Cong, Y. Chen and H. Xi, Bioelectrochemistry, 2014, 95, 23–28 CrossRef CAS PubMed.
- P. G. Collins, K. Bradley, M. Ishigami and A. Zettl, Science, 2000, 287, 1801–1804 CrossRef CAS.
- S. Hongliang, X. Kongliang, L. Guolong, L. Hongbin and L. Zhenning, IEEE Trans. Nanotechnol., 2014, 13, 789–794 CrossRef.
- F. Miao, S. Wijeratne, Y. Zhang, U. C. Coskun, W. Bao and C. N. Lau, Science, 2007, 317, 1530–1533 CrossRef CAS PubMed.
- A. M. Ilyin, E. A. Daineko and G. W. Beall, Physica E, 2009, 42, 67–69 CrossRef CAS PubMed.
- S. Adam, E. H. Hwang, E. Rossi and S. Das Sarma, Solid State Commun., 2009, 149, 1072–1079 CrossRef CAS PubMed.
- L. Zhuang, Y. Yuan, G. Yang and S. Zhou, Electrochem. Commun., 2012, 21, 69–72 CrossRef CAS PubMed.
- Y. Ren, D. Pan, X. Li, F. Fu, Y. Zhao and X. Wang, J. Chem. Technol. Biotechnol., 2013, 88, 1946–1950 CrossRef CAS.
- M. O. Ansari, M. M. Khan, S. A. Ansari, I. Amal, J. Lee and M. H. Cho, Chem. Eng. J., 2014, 242, 155–161 CrossRef CAS PubMed.
- X. Xie, G. Yu, N. Liu, Z. Bao, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2012, 5, 6862–6866 CAS.
- H. Wang, G. Wang, Y. Ling, F. Qian, Y. Song, X. Lu, S. Chen, Y. Tong and Y. Li, Nanoscale, 2013, 5, 10283–10290 RSC.
- Y. Qiao, X.-S. Wu, C.-X. Ma, H. He and C. M. Li, RSC Adv., 2014, 4, 21788–21793 RSC.
- C.-. Zhao, W.-J. Wang, D. Sun, X. Wang, J.-R. Zhang and J.-J. Zhu, Chem. – Euro. J., 2014, 20, 7091–7097 CrossRef CAS PubMed.
- Z. He, J. Liu, Y. Qiao, C. M. Li and T. T. Y. Tan, Nano Lett., 2012, 12, 4738–4741 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- M. Agharkar, S. Kochrekar, S. Hidouri and M. A. Azeez, Mater. Res. Bull., 2014, 59, 323–328 CrossRef CAS PubMed.
- Y. Hou, B. Zhang, Z. Wen, S. Cui, X. Guo, Z. He and J. Chen, J. Mater. Chem. A, 2014, 2, 13795–13800 CAS.
| This journal is © The Royal Society of Chemistry 2015 |