
Supported lipid bilayers as dynamic platforms for tethered particles
Kevin L.
Hartman
,
Sungi
Kim
,
Keunsuk
Kim
and
Jwa-Min
Nam
*
Department of Chemistry, Seoul National University, Gwanak-gu, Seoul, 151-747, South Korea. E-mail: jmnam@snu.ac.kr
First published on 23rd October 2014
Abstract
Nanoparticle tethering to lipid bilayers enables the observation of hundreds of diffusing particles that are confined within a single field of view. A wide variety of materials ranging from plasmonic metals to soft matter can be stably tethered to the surface of a fluid bilayer by covalent or non-covalent means. The controlled environment of this experimental platform allows direct control over surface compositions and accurate tracking of nanoparticle interactions. This minireview will cover studies that use bilayer-tethered nanoparticles to investigate physical properties related to lipid mobility, biomolecule sensing, and surface interactions, as well as experiments to reversibly manipulate tethered nanoparticles by electric fields.
 Kevin L. Hartman | Kevin Hartman received his B.S. degree in chemistry from the University of Illinois at Urbana-Champaign in 2007. Under the supervision of Jay T. Groves, he received his Ph.D. in physical chemistry from the University of California, Berkeley in 2013. Kevin's doctoral research involved probing the behavior of E-cadherin mediated cell adhesion using supported lipid bilayers, and a portion of the work was done at the Mechanobiology Institute at the National University of Singapore. Kevin subsequently joined the lab of Jwa-Min Nam at Seoul National University as a postdoctoral researcher to investigate nanoparticle and bilayer biosensing applications. |
 Sungi Kim | Sungi Kim obtained his B.S. degree in chemistry from Pohang University of Science and Technology (POSTECH), South Korea. He has been pursuing his Ph.D. in chemistry in the lab of Jwa-Min Nam at Seoul National University since 2013. His main research subject is bioapplication of plasmonic nanoparticle-tethered supported lipid bilayers. |
 Keunsuk Kim | Keunsuk Kim received his B.S. degree in chemistry from Kyung Hee University, South Korea in 2008. He obtained his M.S. degree in polymer chemistry from Kyung Hee University in 2010. He joined the lab of Jwa-Min Nam at Seoul National University as a Ph.D. student in 2011. His current research focus is the development of an ultra-sensitive detection method using actively controllable supported lipid bilayer platforms for biosensing and plasmonics applications. |
 Jwa-Min Nam | Professor Jwa-Min Nam received his Ph.D. in chemistry from Northwestern University and worked as a postdoctoral fellow at the University of California, Berkeley. He is currently Associate Professor in Chemistry, Seoul National University. He received the Presidential Young Scientist Award from the President of the Republic of Korea, the Young Inorganic Chemist Award from the Korean Chemical Society and the Victor K. LaMer award from the American Chemical Society. His major interests include the synthesis and optics of plasmonic nanostructures, nanocarriers for bio-imaging and delivery, nanoprobe-tethered lipid bilayers, cell-nanostructure interfaces, and biosensors. |
1 Introduction
Nanoparticles, ranging from plasmonic metals to small lipid vesicles, have been employed in a number of biological studies including ultrasensitive detection of biomolecules, cell imaging, and targeted drug delivery.1–4 This wide variety of applications is possible given methods in both surface functionality and particle detection. The nanoparticle surface can be decorated with biological or functional groups for target binding, self-assembly, or other reactions.This mini-review will consider the surface functionalization and applications of nanoparticles (≤200 nm diameter as considered here) tethered to supported lipid bilayers (SLBs). SLBs are synthetic membranes self-assembled on a planar surface, such as a glass slide. These lipid membrane platforms provide 2D fluidity, similar to a live cell membrane, but host a planar geometry and a controllable composition, which allows a rigorous study of subtle lipid and protein interactions using optical microscopy.5 Nanoparticle tethering to a bilayer shares methods with two existing areas of research. The first, single particle tracking (SPT) studies on live cell membranes, has led to an understanding of membrane trafficking, membrane composition, and cytoskeletal influences.6–9 The second, functionalizing SLBs with proteins and other biomolecules, also involves 2D fluidity and studying the interactions and rearrangements of attached molecules.10–14 The tethering techniques and detection methods used in those fields are relevant and essentially modular since they can be interchanged in a variety of ways. With a proper design and a mobile linkage to the bilayer, tethered nanoparticles provide a convenient way to study interparticle reactions, such as dynamic particle clustering or vesicle fusion events. These advantages arise from the planar geometry of the bilayer; in general terms, particles with mobile anchors to a membrane will collide with each other at a higher rate than particles in solution,15 and those collisions can be restricted to the focal plane of the microscope. As several nanoparticles fit into a single field of view, many can be tracked and monitored simultaneously. Nanoparticles separated from each other can be localized to nanometer resolutions if strong signal-to-noise ratios are possible.16 Such signals can be achieved by increasing the incident light intensities, in which case photostable materials like gold particles and quantum dots (QD) hold advantages over faster-bleaching fluorescent dyes.17 Some nanoparticle tethers can persist for weeks,18 and all of these properties enable robust, massive statistical readouts from a single sample of tethered nanoparticles.
This review will discuss published work on tethered nanoparticles, starting with particle composition and tethering techniques for mobile bilayer attachment, followed by the descriptions and results of the experiments. Studies that attempt a more rigorous analysis of tethered nanoparticle tracking will also be considered as well as particle manipulation by electric fields.
2 Nanoparticle-tethering techniques
In the most basic sense, nanoparticle tethering to a bilayer involves a connection between a nanoparticle and the surface of the bilayer. Given the well-known methods for surface functionalization and reliable scattering properties, gold is a common core material for nanoparticle tethering. Part of its popularity also stems from the use of staining transmission electron microscopy (TEM) images with antibody-adsorbed gold colloids, a technique first conceived in 1971.19,20 Following this idea, the first nanoparticle bilayer tethering paper by G. M. Lee et al. involved adsorbing an antibody against fluorescein onto the surface of a 30 nm gold nanoparticle, and allowing it to attach to a bilayer containing 1.7 mol% fluorescein-labeled lipids while maintaining the mobility.21Proteins besides antibodies also adsorb well onto the negatively charged surface of citrate-capped gold nanoparticles, and have been used in creating linkages to the bilayer surface. For example, the tetrameric protein streptavidin can be adsorbed onto gold nanoparticles and linked to a bilayer containing a low percentage (≤1 mol%) of biotinylated lipids.22 The binding interaction between biotin and streptavidin (or its variants avidin and NeutrAvidin) creates the strongest non-covalent bond known and has been used successfully to create fluid displays of a stably-linked protein on bilayers.11,23,24 Hsieh et al. and Spillane et al. also used streptavidin-adsorbed gold nanoparticles, but instead of linking to a biotinylated bilayer, reacted the particles with a limited concentration of biotinylated cholera toxin subunit B (CTB) in solution.17,25 The particles were next added to a bilayer containing ganglioside lipid (GM1), which is the natural ligand of CTB, and thus the tether was formed by the receptor–ligand interaction (Fig. 1a). The native binding to GM1 in a bilayer was used by Kukura et al. to tether a virus-like particle (VLP).26 The biotinylated Simian virus 40 was functionalized with a single streptavidin-modified QD and was attached to a fluid GM1 bilayer. The simultaneous visualization of both the fluorescence signal from the QD and the scattering signal from the VLP revealed its dynamic position and orientation (Fig. 1b). Instead of using a separate linking molecule between a bilayer and a particle, Sagle et al. took a minimalist approach in adsorbing gold nanoparticles directly onto the bilayer.27 Given the strong negative charge of the citrate-capped gold nanoparticle, it could be connected electrostatically to a bilayer containing a positively charged lipid and retain the fluidity. In control experiments the researchers used the peak shift of the gold nanoparticles’ localized surface plasmon resonance to confirm the specificity of the electrostatic tether.
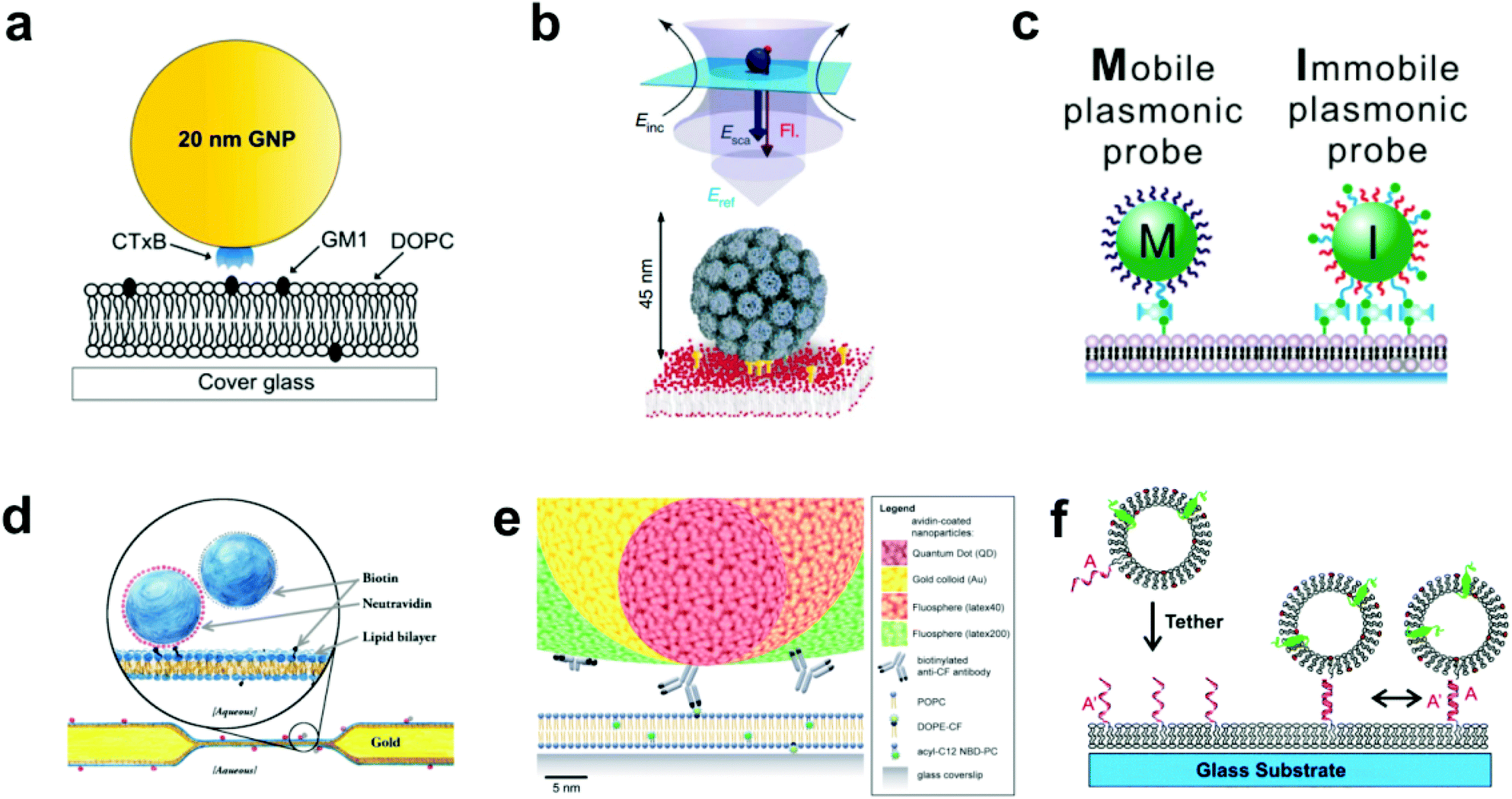 | ||
| Fig. 1 Various techniques to tether nanoparticles onto SLBs. (a) CTB (i.e. CTxB)-modified gold nanoparticle with GM1-lipid.17 The gold nanoparticle is functionalized with streptavidin, and then reacted with a concentration of biotinylated CTB to achieve an average of one CTB pentamer per particle. Copyright 2014, American Chemical Society. (b) Lower portion of illustration shows virus-like particle (VLP) modified with QD (not shown) and tethered to GM1-containing SLB.26 Upper portion shows scattered light and fluorescence detection. Incident laser light (Einc) is scattered (Esca) and reflected (Eref) off the entire VLP + QD to create an interferometric signal, while the single QD emits a fluorescence signal (Fl). Copyright 2009, Macmillan Publishers Ltd. (c) Biotin DNA-modified gold nanoparticles binding to biotinylated-SLB with streptavidin.29 Copyright 2014, American Chemical Society. (d) Tethered particle dimer for rotational and translational diffusion tracking.31 Copyright 2014, The American Physical Society. (e) Scale illustration of different antibody-adsorbed particles for attachment to fluorescent lipids in a SLB.32 Copyright 2012, The Royal Society of Chemistry. (f) DNA-modified vesicle hybridizing and tethering to a DNA-modified SLB.10 Copyright 2005, American Chemical Society. All images in (a) through (f) are reproduced with permission from their respective references. | ||
The gold surface is especially known for its reactivity with thiol functional groups, and this chemistry has been used by Yang et al. and Ota et al. to link gold nanoparticles to bilayers containing thiolated lipids.18,28 This occurs by simply incubating the gold nanoparticles on the SLB. To mediate the reactivity, both research groups first adsorbed BSA onto the nanoparticles to block multivalent thiol–gold linkages. Instead of blocking the surface, Y. K. Lee et al. densely covered gold nanoparticles with thiolated DNA oligomers, some of which were capped with biotin to enable and control binding to a biotin–streptavidin bilayer (Fig. 1c).29 Besides gold, the thiol group is reactive to other species available for tethering methods. For example, Murcia et al. functionalized CdSe/ZnS core–shell quantum dots with hydrophilic groups and maleimide, the latter of which reacts with thiolated lipids to form a covalent bond.30
Creating tethered nanoparticle bilayers is also possible with commercially available nanoparticles, such as latex, polystyrene, quantum dots, or silica particles with functionalized surfaces. Hormel et al. used commercial NeutrAvidin-coated, fluorescent latex particles to link them to a biotin-displaying bilayer.31 They also added similar particles, but with a biotin coating, to form doublets with the tethered NeutrAvidin-coated particles (Fig. 1d). Mascalchi et al. also used streptavidin or NeutrAvidin-coated particles, in this case with latex, quantum dots, or gold cores, but instead of linking them readily to biotin lipids, they used a biotinylated antibody against fluorescein (Fig. 1e).32 This enabled a tether to a fluorescein-containing bilayer, similar to the original tethering technique pioneered by G. M. Lee et al.21
Lipid vesicles on the nanoscale can also be surface-functionalized, tethered to the bilayer, and tracked by fluorophores on the vesicle surface or enclosed within. The first published attempt of this used biotinylated lipids and avidin–streptavidin conjugation for long term, stable observation of single biomolecule fluorescence, but did not allow lateral mobility.33 Mobile tethering of lipids came in 2003 when Yoshina-Ishii and Boxer reported using DNA hybridization for the linking mechanism.34 They achieved this with a disulfide exchange reaction of thiolated 24-mer oligonucleotides to dithiol lipids on the surface of the SLB and on 50 nm lipid vesicles. Upon hybridization of complementary strands, the tethered vesicles were laterally mobile and collided with each other. A subsequent paper by Yoshina-Ishii et al. substituted the disulfide exchange reaction, which could potentially react and denature proteins, with a gentler technique: oligonucleotides were conjugated to lipid headgroups, and these modified lipids were able to spontaneously insert into preformed SLBs and lipid vesicles.10 DNA hybridization following the modified lipid insertion also created a mobile display of tethered lipid vesicles (Fig. 1f). Benkoski and Höök took a similar approach but instead used cholesterol-tagged DNA.35 Following the spontaneous insertion of the cholesterol into preformed vesicles and SLBs, DNA hybridization also produced diffusing, tethered vesicles. For vesicles with a more robust DNA tether, van Lengerich et al. (2010) devised a DNA-templated click reaction to attach the vesicles’ DNA covalently to the SLB.36 Compared to the previous tethering techniques which relied on DNA hybridization alone, this orthogonal covalent linkage has the advantage of stability under different buffer conditions, as low salt concentrations destabilize DNA base pairing.
The above studies represent a range of bilayer tethering techniques for nanoparticles that can be easily adapted or combined into new strategies. But more important than the tether's composition or physical behavior is the number of tethers per particle. This number exists on a continuum where a nanoparticle with several attachment points to a bilayer will exhibit limited or no mobility, even on a fluid bilayer. At the other extreme, a nanoparticle with very few, or just one attachment, will diffuse freely on a fluid bilayer. Thus, a successful bilayer platform of tethered nanoparticles involves controlling this degree of attachment. Part of this requires an optimal concentration of attaching lipids, which are mixed into the bilayer constituent lipids at concentrations ranging from 0.1 to 4 mol% depending on the chemistry involved.18,27 For some experiments, it is more important to regulate the surface chemistry to reduce the number of surface anchoring sites. This was key for the original tethering by G. M. Lee et al. where mobile attachment was not achieved until blocking most of the fluorescein antibodies on the nanoparticle with a secondary antibody.21 Also limiting multiple tether formation, Y. K. Lee et al. used 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :799 molar ratio of biotinylated (tethering) DNA to non-biotinylated (target capture) DNA on their nanoparticles to produce a highly mobile attachment with an assumed single anchor on a bilayer with 0.1 mol% biotin–streptavidin.29
:799 molar ratio of biotinylated (tethering) DNA to non-biotinylated (target capture) DNA on their nanoparticles to produce a highly mobile attachment with an assumed single anchor on a bilayer with 0.1 mol% biotin–streptavidin.29
In some cases a nanoparticle's lack of curvature will generate multiple binding sites: larger nanoparticles with less curvature have more exposed surface area for multiple anchors. Mascalchi et al. cited this tendency to explain the slow diffusion of their 40 nm gold nanoparticles.32 Considering this disadvantage, Lin et al. and Hsieh et al. instead used 20 nm gold nanoparticles to limit the number of tethers formed per particle.17,22 However, there are bounds to reducing the size as smaller nanoparticles produce weaker scattering, with a signal contrast pattern that can be confused with lipid vesicles on the surface.17,25 Despite the disadvantage of lower mobility from multiple surface anchors, Hormel et al. attached larger particles (200 nm diameter NeutrAvidin-coated latex) to a bilayer displaying a relatively high concentration (1 mol%) of biotin lipids.31 Their goal was to create lipid footprint areas underneath the particles to better respond to the rotational and translational effects of the bilayer viscosity, and this approach would otherwise be limited with singly-anchored nanoparticles. Recently, Johnson-Buck et al. tethered rectangular DNA origami tiles to a fluid bilayer using the technique of cholesterol-DNA hybridization similar to Benkoski and Höök.35,37 Each 60 × 90 × 2 nm tile was designed with 187 ssDNA of the identical nucleotide sequence exposed on one side. Of the hybridizing oligomers added, only 25% contained the bilayer-anchoring cholesterol. Though the exact number of bilayer connections per tile was not calculated and probably less than maximal due to charge repulsion, each particle was likely anchored by multiple strands. However, the modest 0.71 μm2 s−1 diffusion of the tiles on the bilayer suggests that the spacing of the tethers, or perhaps the complete lack of particle curvature, may nullify the conditions leading to immobile tethering.
Luckily, a percentage of immobile particles does not spell disaster for a tethering experiment. An advantage of bilayer tethering is that the nanoparticles at the extremes of immobilization or no attachment can easily be identified: immobile particles stand out as defects among mobile particles. Non-attached particles can be spotted by their fast diffusion in and out of the focal plane, and can furthermore be washed away. This purification ability of the bilayer attachment is advantageous for producing mobile, singly anchored particles. This is because the surface chemistry of the nanoparticles can err on the side of being fixed with a low average number of anchoring groups or anchoring sites. For instance, the 1:799 molar ratio of anchoring to target capture DNA by Y. K. Lee et al. generated an average of only 0.57 anchoring DNA strands per particle.29 If a nanoparticle surface composition results in a statistical population with no anchoring sites, or those that otherwise have no chance to bind, those non-attached nanoparticles can be rinsed out, leaving behind only the tethered mobile particles.
3 Interactions of nanoparticles with the immediate lipid environment
A common subject of nanoparticle tethering is the effect of CTB binding to GM1. This is a convenient model of protein binding to a membrane surface: both components are commercially available, many cells express GM1 on their membranes,38,39 and CTB–GM1 binding has high affinity and stability.40 Interest in studying GM1–CTB binding on SLBs also exists given the receptor–ligand complex's formation of nanodomains that can limit bilayer fluidity. When using 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) as the major component lipid near its bilayer gel phase transition temperature of 24 °C, this decrease in fluidity is significant. For example, under these conditions, CTB binding to just 0.25 mol% GM1 in the bilayer substantially reduces fluorescence recovery after photobleaching.41 Closer inspection by infrared spectroscopy and fluorescence correlation spectroscopy revealed that the binding of CTB creates an ordered arrangement in the alkane chains of the GM1 complex, which induces a gel phase in the neighboring DMPC lipids.42Yang et al. took advantage of this gel phase ordering in their SLB experiment.18 Gold nanoparticles were tethered to a bilayer containing DMPC, thiolated lipids for particle attachment, and GM1. The nanoparticles were tracked by dark-field scattering, and the CTB binding decreased the particle diffusion rate (Fig. 2a). This decrease in diffusion rate was proportional to the solution concentration of CTB and could be used to sense the protein in solution at concentrations as low as 100 pM. This experiment was a significant demonstration of tethered nanoparticles for sensitive and quantitative protein detection. Hsieh et al. also examined CTB–GM1 complexes, but in the environment of high lipid fluidity.17 Their nanoparticles, being tethered to the CTB, could track the diffusion of the entire CTB–GM1 complex across the membrane. With high-speed, high-resolution particle tracking (1.9 nm spatial precision and 1 ms temporal resolution by interferometric scattering), the researchers found that as the GM1 concentration in the bilayer was increased from 0.01 to 10 mol%, the number of particle trajectories showing single component diffusion decreased from 25.7 to 7.4%, as two-component diffusion became more predominant. Spillane et al. used the same CTB–GM1 interferometric scattering experiment but achieved a higher temporal resolution (20 μs).25 The data from the shorter time scales showed that interactions of GM1 with the substrate created sub-diffusion nanodomains.
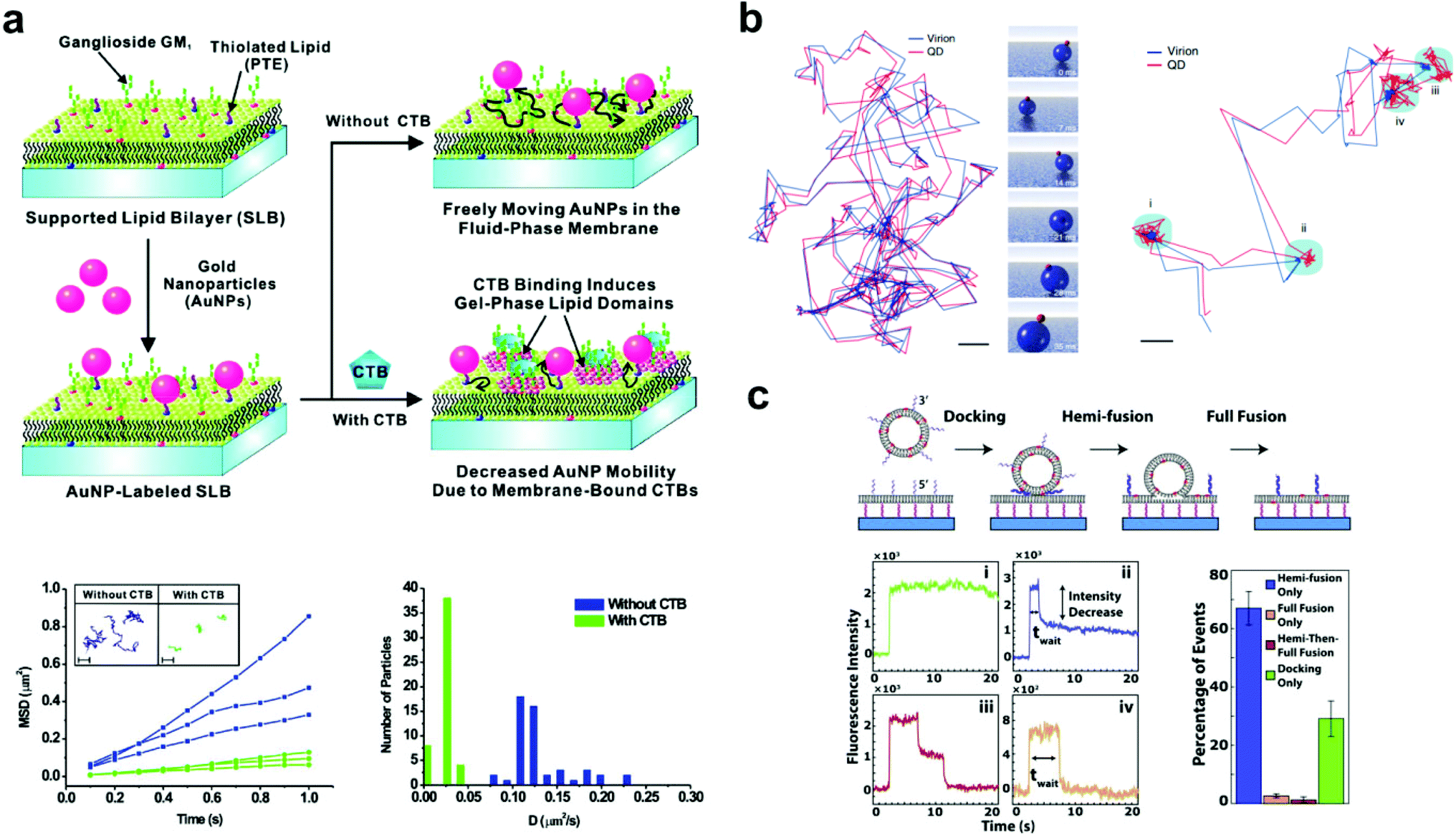 | ||
| Fig. 2 Examples of bilayer-tethered nanoparticles interacting with the lipid environment. (a) The binding of CTB to a SLB displaying GM1 and tethered nanoparticles decreases the nanoparticle mobility.18 The lower left graph shows MSD vs. t analysis of single particle trajectories with and without 100 nM CTB while the lower right graph shows the distributions of D obtained from tracking. Copyright 2009, American Chemical Society. (b) Motion and orientation of a single SV40 virion at low GM1 concentrations (left, scale bar 100 nm) and at high GM1 concentrations (right, scale bar 50 nm).26 The rendered figure in middle panels illustrates sliding and tumbling motion of the VLP + QD. Copyright 2009, Macmillan Publishers Ltd. (c) Top: DNA-mediated vesicle fusion process to a tethered bilayer patch. Bottom left: examples of fluorescence intensity profiles of a single vesicle from each fusion mode: (i) docking-only, (ii) hemi-fusion only, (iii) hemi-then-full-fusion, (iv) full-fusion-only. Bottom right: probability of each mode.43 Copyright 2013, Elsevier. All images (a) through (c) are reproduced with permission from their respective references. | ||
Also using a fluid bilayer with GM1, Kukura et al. examined the dynamic connection and diffusion of single virus particles that natively attach to the ganglioside lipid from several surface binding sites.26 As mentioned earlier, the QD on the VLP allowed the combination of both fluorescence and interferometric light scattering tracking data to analyze the position and orientation with nanometer precision. The trajectory of the VLP + QD on a bilayer with 0.05 mol% GM1 showed sliding and tumbling motions, while at 1% GM1 showed alteration between diffusion and confined motion (Fig. 2b). In the confined state, the particle showed a back-and-forth rocking motion that suggested an exchange of binding sites with the GM1 on the bilayer. This dynamic behavior likely occurs when the virus encounters a real cell membrane.
Without CTB, a bilayer with ample GM1 can create nanodomains, hindering the diffusion of other lipids; high densities of GM1 can even perturb CTB binding.44 Sagle et al. tethered gold nanoparticles to fluid lipid bilayers containing GM1 to study the effect of the nanodomains on fluidity.27 Particle tracking by dark-field scattering revealed the nanoparticles to become confined with higher GM1 concentrations. By graphing the percentage of confined nanoparticles at different GM1 densities, extrapolation revealed a percolation threshold of 22 mol% GM1, which corresponds to the minimum level of GM1 required to restrict all the tethered nanoparticles to diffusion nanodomains, which measured about 50 nm in diameter. This is relevant to interactions on a live cell membrane, where the lateral movement of transmembrane proteins can be restricted by immobile components on the bilayer or by interactions with the cytoskeleton.45,46 The high-resolution tracking of CTB–GM1 by Hsieh et al. also revealed diffusion nanodomains, but these were believed to result from imperfections on the glass substrate.17 However, in comparing different substrates, and a form of GM1 with an unsaturated lipid tail, Spillane et al. concluded that the sub-diffusion nanodomains of ≤40 nm diameter resulted from interactions of GM1 with the substrate, which were transmitted to the surface of the bilayer through interleaflet coupling.25
Without CTB and GM1, both Benkoski & Höök, and Yoshina-Ishii et al. (2006) studied the diffusion of bilayer-tethered vesicles by SPT analysis and fluorescence recovery after photobleaching (FRAP).35,47 Despite the differences between their DNA tethering methods and experimental conditions, overall trends regarding vesicle motion were similar. The tethered vesicles of both studies showed random movement with a diffusion coefficient (D) one order of magnitude lower than that of bilayer lipids or DNA without vesicles. Both studies also ruled out the effect of vesicle dimensions, as increasing the diameter up to double the size showed no effect on its diffusion coefficient. Benkoski and Höök explained that multiple tethers form between the vesicle and SLB, leading to slower diffusion.35 However, Yoshina-Ishii et al. also experienced a decrease in diffusion for vesicles containing an average of one DNA linker or less.47 They hypothesized that the slower diffusion might arise from transient interactions and frictional coupling of the vesicle with the SLB surface. The researchers promoted some such interactions with the addition of polymers or Ca2+. When they added polyethylene glycol (PEG 8000), the vesicles were reversibly immobilized, likely by the polymer's effect of displacing water within the vesicle–SLB interface. When both SLB and vesicles contained negatively charged lipids, the addition of Ca2+ increased vesicle–SLB interactions, reducing diffusion. Both results of polymers and Ca2+ on vesicle mobility indicated that the DNA tether does not prevent the vesicle from interacting with the SLB surface in ways that limit mobility.
However, promoting such interactions between vesicles and a planar bilayer leads to the complete fusion and vesicle content transfer, which are important biologically in processes such as protein transfer within cells, synaptic vesicle fusion, and virus entry through a host cell membrane.48 Addressing this, the next studies used mobile, DNA tethered vesicles to study the conditions that lead to fusion and the kinetics of the event. Simonsson et al. first published results of DNA-induced membrane fusion reactions by single vesicle monitoring on SLBs, noting the importance of the DNA hybridization geometry.49 Simple DNA hybridization modes, such as those caused by complementary oligomers both tethered from the 3′ or the 5′ ends, as the earlier Yoshina-Ishii DNA tethers,10,34 generally maintain space between the vesicle and bilayer surfaces and are less likely to cause fusion. However, the “zippering” hybridization mode, where one DNA strand is attached at the 5′ end and the complementary strand at the 3′ end, forces the lipid surfaces into contact, inducing fusion. This zippering configuration is similar to the geometry of the SNARE family proteins, which form a complex to bring together apposing membrane surfaces, promoting fusion. Simonsson et al. used cholesterol-tagged DNA to tether vesicles to fluid bilayers to observe the two hybridization modes and test the conditions promoting vesicle fusion. It was found that with Ca2+, most zippering DNA hybridization modes lead to vesicle fusion, and that in this close contact, a moderate amount of DNA oligomers is required to bring the membranes together without causing electronic repulsion or steric hindrance.
Two vesicle fusion studies following from the Boxer Lab, Rawle et al. and van Lengerich et al. (2013), used DNA to tether a planar bilayer patch 8 nm above a glass coverslip to observe not only vesicle fusion but also lipid and content transfer.43,50 In the first, Rawle et al. observed zippering DNA-mediated vesicle fusion to the bilayer surface by imaging the transfer of an aqueous fluorescent dye from the vesicle.50 The fluorescence decay following the fusion event fits better to a 2D rather than a 3D diffusion model, indicating content transfer into the gap between the tethered membrane and the coverslip in a way that could be more easily tracked and studied. van Lengerich et al. recently used this approach with the same experimental platform to study the kinetics of different zippering DNA-mediated vesicle fusion events in the perspective of lipid movement.43 In this experiment the waiting time from initial binding to a lipid dye transfer event was recorded for each vesicle. Based on the decrease of fluorescence intensity, the lipid fusion events were categorized into four binding modes ranging from no fluorescence decrease (vesicle docking without fusion) to a complete decrease (total vesicle fusion) (Fig. 2c). The cumulative distribution functions of the waiting times in each mode showed exponential behavior, suggesting that a vesicle's transition from docking to fusion with a bilayer occurs as a stochastic process.
4 Interactions between nanoparticles
For some signaling proteins, the recruitment to, or attachment on, cell membranes leads to significant increases in local concentration, which can enhance signal transduction several-fold.51,52 The local concentration of nanoparticles is increased in a similar way by SLB tethering, giving the particles a higher probability of interaction with each other.15 Thus for bilayer tethering, the nanoparticle surface chemistries can be engineered for meaningful interactions that would otherwise be too difficult or rare to observe in bulk solution.The nanoparticles used by Y. K. Lee et al. were designed to interact on the basis of DNA hybridization.29 The bilayer consisted of two types of tethered particles: highly mobile particles and purposefully immobile particles. The target capture DNA sequence displayed on the nanoparticle surface allowed the mobile particles to link to the immobile particles in the presence of the DNA target, while remaining tethered to the bilayer. Using dark-field scattering, many immobile particles could be simultaneously observed as interaction hubs, and the step-wise increase in their scattering intensity indicated their oligomerization with the mobile particles (Fig. 3). This increase in scattering intensity could be interpreted in terms of cluster size, allowing the dynamics of particle growth to be rigorously described by a 3-step reaction kinetics model. Furthermore, it was found that by counting the number of clusters formed, the tethered nanoparticle platform could be used to reliably detect the target DNA strand at concentrations as low as 30 fM, with outstanding single-base mismatch sensitivity.
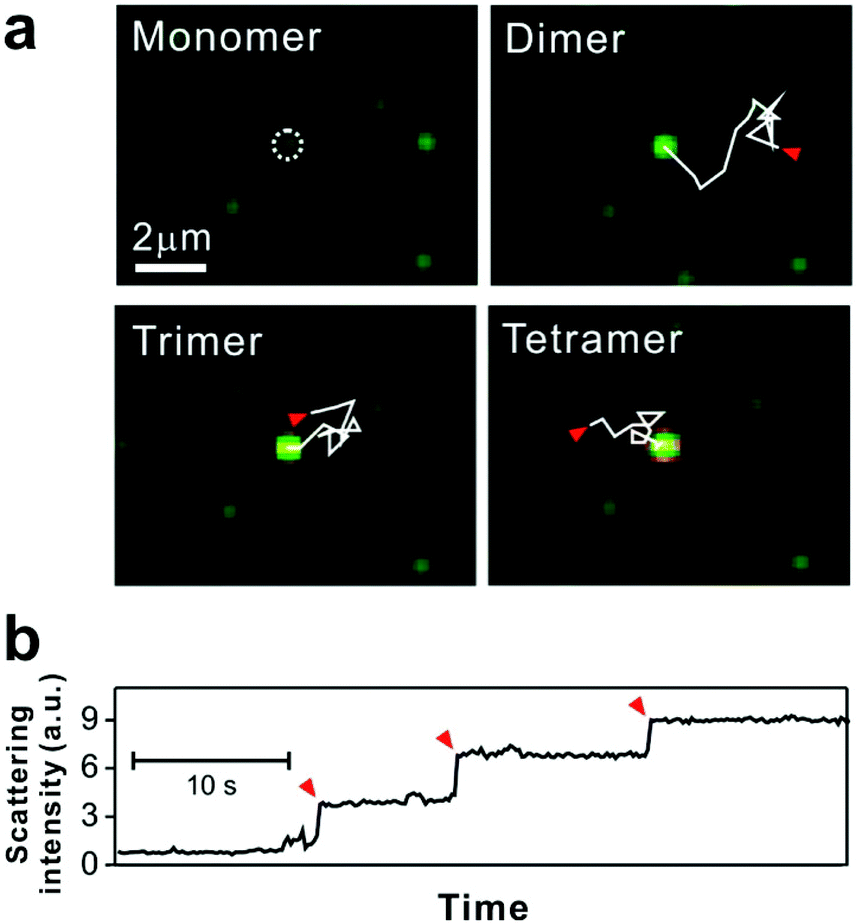 | ||
| Fig. 3 Tethered 50 nm plasmonic gold nanoparticle interactions on SLB. (a) Dark-field microscopic images of target DNA hybridization-induced nanoparticle cluster growth. The trajectories, each 2.82 seconds long and highlighted with solid while lines, show the diffusion of mobile particles from starting points (red arrows) to an immobile particle binding site (white dashed circle). (b) Stepwise increase of scattering intensity by nanoparticle clustering. Red arrows show single nanoparticle addition. Reproduced with permission from ref. 29. Copyright 2014, American Chemical Society. | ||
Chan et al. (2007) also used DNA to study particle–particle interactions.53 In this case, lipid vesicles displaying oligonucleotides were tethered to fluid bilayers to observe docking events between vesicles, demonstrating how tethered vesicles could be used to explore interactions between membrane proteins displayed on the surfaces. Vesicles of 50 nm diameter were tethered on the sides of a bilayer within a microfluidic channel using previously described oligonucleotide-modified lipids.10 The vesicles on each of the two sides of the channel displayed an additional DNA sequence complementary to the other side. With laminar flow, diffusive mixing occurred, leading to vesicle collisions and DNA-mediated docking. Vesicles that docked together diffused in tandem without fusion or lipid mixing as the DNA position blocked membrane–membrane contact. Kinetic analysis of the particle events showed that the probability of docking after collision was affected by the length, sequence, and number of oligonucleotides on the vesicles. These results implied that docking depends on the DNA hybridization efficiency, which was also predicted by simulations of the experiment.
5 Rigorous methods for bilayer-tethered particle tracking analysis
The lipid bilayer translational diffusion coefficient, D, is a standard measure of membrane component mobility in two dimensions, which is dependent on the surrounding lipid environment. Fluid bilayers usually have a D in the range of 1–10 μm2 s−1, which varies between experiments depending on the lipid composition, substrate cleaning method, and temperature.32,54 Diffusion at length scales relevant to cell membrane receptor translocation (∼10−7–10−6 m) can be measured by fluorescence correlation spectroscopy (FCS),42 fluorescence recovery after photobleaching (FRAP) analysis,55 or single particle tracking (SPT).56 SPT combines a spatial resolution on the order of nanometers—better than the diffraction-limited FCS—with the ability to measure up to micron length scales. Using particle tracks, traditional methods in SPT calculate the diffusion coefficient from the two-dimensional diffusion equation: D = <r2>/4t.57 This relationship is typically represented as the slope of a trajectory's <r2> (mean squared displacement, MSD) vs. t, which is linear for ideal Brownian motion. By graphing the particle trajectories from a single sample, this plot allows one to easily monitor large variations in the diffusion coefficient, as shown in a study by Yang et al. (Fig. 2a).18 In spite of its convenience, the MSD vs. t analysis averages out information on individual particle tracks (just as FRAP and FCS).58 Simulations of 2D diffusing particles show that the divergence in the MSD trajectories produces a wide spread in the uncertainty of D (Fig. 4) and masks multi-component diffusion.32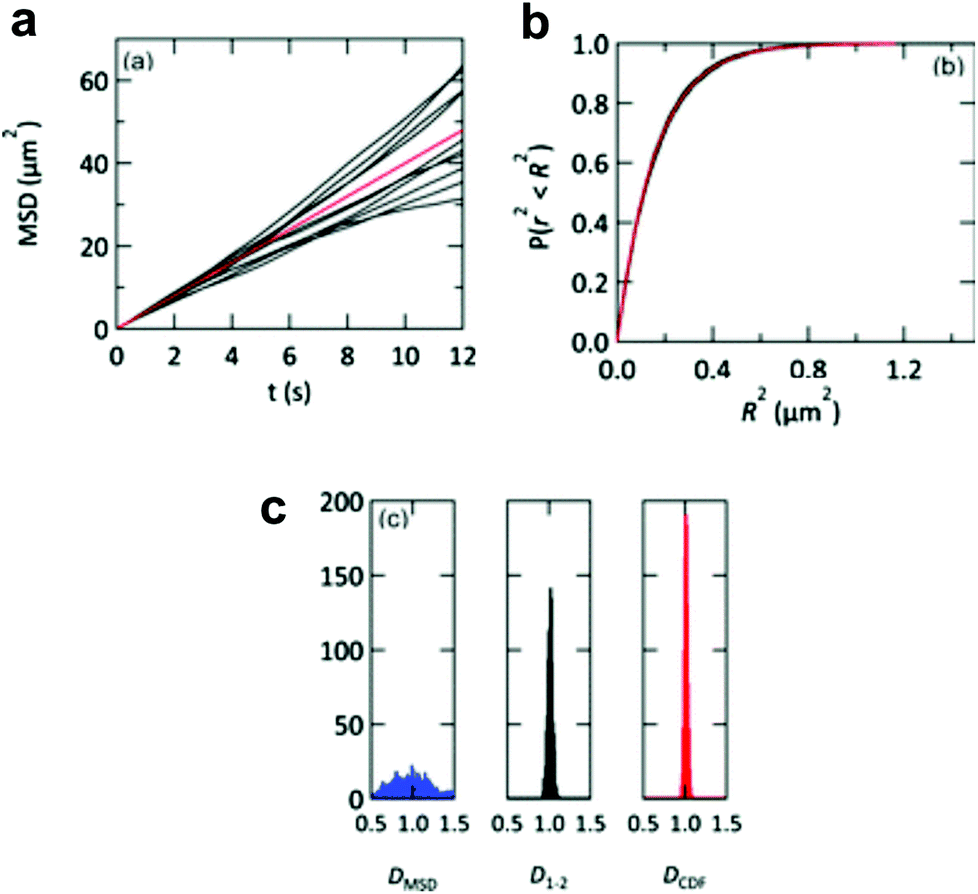 | ||
| Fig. 4 Representations of D from simulated particle diffusion. Particle trajectories underwent 3000 steps each of 2D-Brownian motion with an average D = 1 μm2 s−1. (a) Ten simulated MSD vs. t plots (black profiles). These diverge from the red profile, which is the theoretical MSD vs. t plot. (b) The same 10 trajectories (black profiles) and theoretical prediction (red profile) converge when plotted as cumulative distribution functions P(r2, t). (c) Histograms of the diffusion coefficients calculated from 1000 simulated particle trajectories. The calculated diffusion coefficient from each trajectory is derived from either a linear regression of the MSD vs. t plot (DMSD, blue histogram), a linear fit to only the first two points of the MSD vs. t plot (D1–2, black histogram), or an exponential fit to the cumulative distribution function (DCDF, red histogram). Reproduced with permission from ref. 32. Copyright 2012, The Royal Society of Chemistry. | ||
In Hsieh et al.'s tracking of CTB–GM1 diffusion, the researchers argued that it is more accurate to measure the diffusion coefficient using only two early time points of the MSD vs. t relationship to obtain a diffusion coefficient D1–2.17,59 The authors also considered representing particle trajectories in terms of the cumulative probability distribution of the square of displacement, P(r2, t), which represents the probability of finding the particle within a radius r given a time t after starting at the origin.60 This analysis has the property that multiple particle trajectories converge with time, unlike MSD vs. t.32 However, Hsieh et al. decided that the D1–2 analysis was sufficiently accurate for their purposes, and using that approach with short trajectories of less than 10 ms, the researchers found that a two-component diffusion model fits the probability distribution of measured diffusion coefficients more accurately than a one-component fit.
If nanoparticles are tethered directly to lipids in a bilayer, is the diffusion coefficient of the nanoparticles close to that of the bilayer lipids? The answer depends more on the size of the movable cluster of molecules embedded in the bilayer, rather than the size of the particle itself. This cluster consists not only of the tethering molecules but also of any ligands, hydrophobic chains, or associating lipid groups that move with the particle. The effect of this cluster on mobility exists because of differences in fluidity between the bulk solution and the membrane. Saffman and Delbrück looked at the theoretical effects of a membrane's viscosity on the rotational and translational diffusion of an embedded cylinder.61 They pointed out that such a structure would exhibit substantially slower diffusion than it would in solution, as the viscosity of the membrane is two orders of magnitude larger than that of the surrounding solution. This discrepancy in viscosities means that the diffusion of a tethered particle is dictated by the lipid attachment—more specifically, the size of the cluster and the interaction with neighboring lipids, assuming no frictional contact with the substrate.62,63 This property has been observed and noted in several of the tethered nanoparticle papers reviewed thus far: that the number of lipid anchoring points or the area of the lipid footprint in the membrane has a stronger influence on the particle mobility than the size of the particle35,47 or changes in the solution's viscosity.21 However, on a large enough scale, the particle size eventually becomes a determining factor of mobility. For instance, Mascalchi et al. examined the diffusion of differently sized particles linked to a bilayer by antibodies.32 They found that 200 nm latex particles showed slower diffusion than the 40 nm latex particles or the 20 nm quantum dots, the latter of which showed diffusion values (D = 0.936 and 1.115 μm2 s−1, respectively) close to what they obtained from FRAP analysis of a fluorescent lipid dye (D = 1.05 μm2 s−1). Thus, D of the mobile lipids could be determined from SPT of the quantum dots or the smaller latex beads in their specific experiment. The study showed an unexpected result of the 40 nm gold nanoparticles diffusing slower, with a diffusion coefficient about one-third that of the 40 nm latex beads. The authors did not attempt to refute the original theories of Saffman and Delbrück, but explained that the gold nanoparticle must be non-specifically binding to the bilayer and forming multiple tethers.
Particle tracking measurements are not limited to translational diffusion: the particle dimers created by Hormel et al. furthermore enabled tracking of rotational diffusion, due to the dimers’ elongated shapes.31 The viscosity of the bilayer (in this case, a bilayer spanning a 100 μm pore) was calculated from theories of Saffman and Delbrück and from later studies, the more recent of which takes membrane deformations into account.65,66 Hormel et al. calculated the rotational and translational mobilities from individual particles, which they then used to calculate the membrane viscosity and the theoretical lipid footprint areas. The calculations showed that particles with larger lipid footprints had slower rotational and translational diffusion. This footprint averaged 170 nm diameter and ranged up to twice the diameter of the 200 nm attached particle. The three different models of membrane viscosity produced values in the range of 13.1–15.9 Pa s m. The binding of a trafficking protein Sar1p to the bilayer led to more than a magnitude increase in viscosity, showing that the tethered particles can be used to monitor changes in membrane fluidity beyond that of translational diffusion.
6 Manipulation of tethered nanoparticles by electric fields
Tethered nanoparticles are not only available to study as passively diffusing objects but also can be collectively manipulated on a single platform, as two studies illustrate. In the first, Yoshina-Ishii and Boxer (2006) showed that the movement of tethered vesicles can be actively controlled with electric fields.64 Two main forces, electrophoresis and electro-osmotic flow, describe the movement of charged vesicles on the SLB. Through electrophoresis, the negatively charged lipid vesicles experience constant attraction towards the positive electrode. In the opposite direction, the positive shielding charges above the negatively charged supported bilayer cause electro-osmotic flow of the bulk solution towards the negative electrode (Fig. 5a). In the experiment, the electro-osmotic effect was found to predominate at first: the lipid vesicles moved towards the negative electrode, with a velocity dependent on the amount of negatively charged lipids in the vesicle and SLB. However, within patterned bilayers the electro-osmotic flow became a spatial gradient due to the uneven distribution of charged lipids, and the vesicles moved to an equilibrium point where electro-osmosis and electrophoresis were balanced. Furthermore, the migration of the vesicles was found to be reversible, with free diffusion without an electric field, and movement in the opposite direction when the electrodes were switched. This type of electrophoretic particle manipulation within patterned bilayers could easily be applied to the detection of membrane-binding proteins, based on the extent of particle migration. This was demonstrated by Lee and Nam using charged lipids in the bilayer (without particles),67 but could be performed with any charged tethered particle, besides vesicles. This detection method would likely yield greater sensitivity and reproducibility than the previously discussed methods that used diffusion tracking to detect protein binding.17,18,27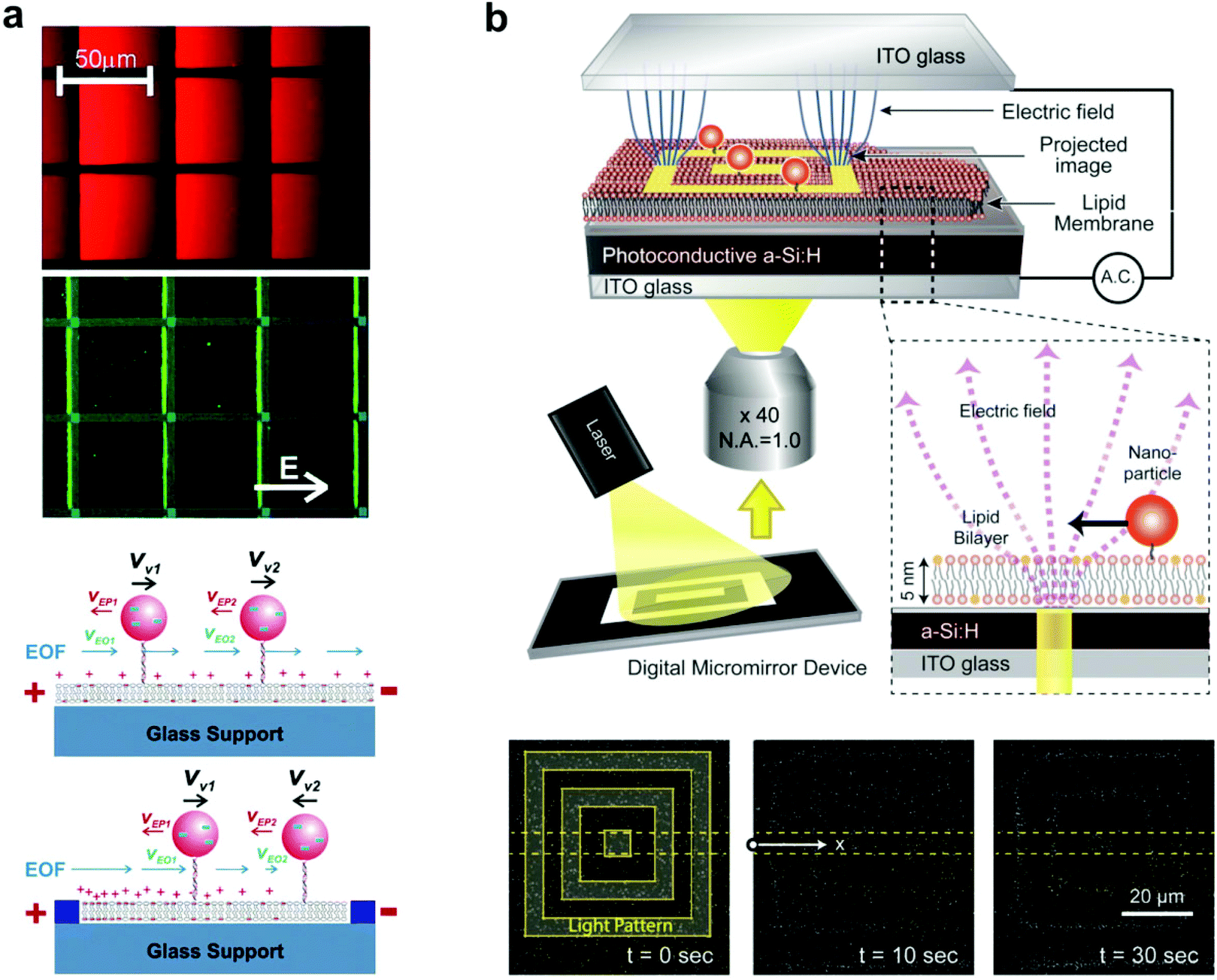 | ||
| Fig. 5 Manipulation of tethered nanoparticles with electric fields. (a) Top: steady-state fluorescence images of SLBs containing negatively charged 1 mol% Texas Red DHPE and tethered vesicles containing Oregon Green content dye under the influence of a 10 V cm−1 electric field. Immobile fibronectin patterns are highlighted with blue lines. Bottom: schematic illustration of the effect of electro-osmotic flow (EOF) (blue arrows) and electrophoretic force (red arrows) under an electric field, with and without patterns. With patterns, the heterogeneous distribution of charged lipids in SLBs generates a spatial gradient of EOF.64 Copyright 2006, American Chemical Society. (b) Schematic of the experimental setup (top) and interferometric scattering images (bottom) of SLB-tethered gold nanoparticle manipulation using optoelectronic tweezers.28 An image sequence shows particle localization with 10 and 30 seconds of laser light application. Copyright 2013, American Chemical Society. Both images (a) and (b) are reproduced with permission from their respective references. | ||
The second experiment of tethered nanoparticle manipulation was performed more recently by Ota et al. using optoelectronic tweezers, which rely on force generated by the dielectric effect.28 60 nm gold nanoparticles were tethered to a bilayer supported by a planar photoconductive electrode, which was held within an alternating electric field. A laser light pattern reflected onto the photoconductive electrode created local variations in the electric field, which interacted with the induced dipole moments of the nanoparticles to create a dielectrophoretic force. When the light pattern was applied, the nanoparticles localized to the 5 μm wide lines of the pattern, and removing the light pattern restored free diffusion of the particles (Fig. 5b). Bilayer-tethering was advantageous for restricting the nanoparticles to the photoconductive substrate where they could experience the maximum dielectrophoretic force, and allowed for the reversible manipulation of hundreds of gold nanoparticles.
7 Conclusion
We briefly reviewed various bilayer-tethered nanoparticle experiments that present a wide spectrum of materials and applications, ranging from simulations of biological phenomena to reversible manipulation by electric fields. All of these studies exploited the advantages of the SLB system's geometric and compositional control, which allowed complex systems to be restricted within a single focal plane of a microscope and at fixed densities. This regularity made it possible to adapt the bilayer platform to applications that rely on a massive readout of particle interactions, such as that used for biosensing of DNA,29 which would otherwise be too difficult in bulk solution.On the bilayer, we showed how nanoparticles with gold, fluorescent lipid vesicles, quantum dots, and fluorophore-modified latex cores could be individually tracked to nanometer precision. Gold remains a popular material due to its excellent light scattering properties and high photostability. Still, there is much room for creativity: two of the studies we reviewed had created asymmetric particles in order to detect changes in rotation.26,31 Another study combined mobile particles with purposely immobile particles that acted as reaction and observation hubs.29 For bilayer attachment schemes, we reviewed diverse tethering methods adapted from existing techniques, such as biotin–streptavidin conjugation,22 or developed for the first time in the experiment, such as vesicle-DNA tethering.10 Despite different chemistries, these tethers all illustrated that the highest mobility attachment is formed when particles have the lowest number of contact points with the bilayer.
In establishing a bilayer-tethered nanoparticle experiment, one obviously knows the composition of the lipids used and the makeup of the particles. However, many of the experiments reviewed here used tethered nanoparticles to study unknown physical characteristics of lipid membranes, including the effects of protein binding, nanodomains, and interleaflet coupling.25,27 In other experiments, the emphasis was to uncover properties of the particles themselves, for example the virus-like particle's dynamic binding to GM126 or the conditions for vesicle fusion.43,49,50 With expanding research in nanomaterials and detection techniques, we believe that the combination of nanoparticles with bilayers will continue to produce unique results relating to soft matter, biophysics, and biosensing applications.
Acknowledgements
J.-M. N. was supported by the National Research Foundation (NRF) of Korea (2011-0018198) and the BioNano Health-Guard Research Center funded by the Ministry of Science, ICT & Future Planning (MSIP) of Korea as the Global Frontier Project (H-GUARD_2013M3A6B2078947). We also acknowledge the support from the Pioneer Research Center Program (2012-0009586) through the NRF of Korea funded by the Ministry of Science, ICT, and Future Planning.References
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS PubMed.
- A. E. Prigodich, P. S. Randeria, W. E. Briley, N. J. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2012, 84, 2062–2066 CrossRef CAS PubMed.
- S. A. Jensen, E. S. Day, C. H. Ko, L. A. Hurley, J. P. Luciano, F. M. Kouri, T. J. Merkel, A. J. Luthi, P. C. Patel, J. I. Cutler, W. L. Daniel, A. W. Scott, M. W. Rotz, T. J. Meade, D. A. Giljohann, C. A. Mirkin and A. H. Stegh, Sci. Transl. Med., 2013, 5, 209ra152 Search PubMed.
- K. T. Thurn, E. Brown, A. Wu, S. Vogt, B. Lai, J. Maser, T. Paunesku and G. E. Woloschak, Nanoscale Res. Lett., 2007, 2, 430–441 CrossRef CAS PubMed.
- W.-C. Lin, C.-H. Yu, S. Triffo and J. T. Groves, Curr. Protoc. Chem. Biol., 2010, 2, 235–269 Search PubMed.
- A. Kusumi, Y. Sako and M. Yamamoto, Biophys. J., 1993, 65, 2021–2040 CrossRef CAS.
- A. Kusumi, C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo and T. Fujiwara, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 351–378 CrossRef CAS PubMed.
- H. M. van der Schaar, M. J. Rust, C. Chen, H. van der Ende-Metselaar, J. Wilschut, X. Zhuang and J. M. Smit, PLoS Pathog., 2008, 4, e1000244 Search PubMed.
- H. Ba, J. Rodríguez-Fernández, F. D. Stefani and J. Feldmann, Nano Lett., 2010, 10, 3006–3012 CrossRef CAS PubMed.
- C. Yoshina-Ishii, G. P. Miller, M. L. Kraft, E. T. Kool and S. G. Boxer, J. Am. Chem. Soc., 2005, 127, 1356–1357 CrossRef CAS PubMed.
- K. Salaita, P. M. Nair, R. S. Petit, R. M. Neve, D. Das, J. W. Gray and J. T. Groves, Science, 2010, 327, 1380–1385 CrossRef CAS PubMed.
- M. P. Coyle, Q. Xu, S. Chiang, M. B. Francis and J. T. Groves, J. Am. Chem. Soc., 2013, 135, 5012–5016 CrossRef CAS PubMed.
- C.-H. Yu, N. B. M. Rafiq, A. Krishnasamy, K. L. Hartman, G. E. Jones, A. D. Bershadsky and M. P. Sheetz, Cell Rep., 2013, 5, 1456–1468 CrossRef CAS PubMed.
- L. Iversen, H.-L. Tu, W.-C. Lin, S. M. Christensen, S. M. Abel, J. Iwig, H.-J. Wu, J. Gureasko, C. Rhodes, R. S. Petit, S. D. Hansen, P. Thill, C.-H. Yu, D. Stamou, A. K. Chakraborty, J. Kuriyan and J. T. Groves, Science, 2014, 345, 50–54 CrossRef CAS PubMed.
- J. T. Groves and J. Kuriyan, Nat. Struct. Mol. Biol., 2010, 17, 659–665 CAS.
- J. C. Crocker and B. D. Hoffman, Methods Cell Biol., 2007, 83, 141–178 CAS.
- C.-L. Hsieh, S. Spindler, J. Ehrig and V. Sandoghdar, J. Phys. Chem. B, 2014, 118, 1545–1554 CrossRef CAS PubMed.
- Y.-H. Yang and J.-M. Nam, Anal. Chem., 2009, 81, 2564–2568 CrossRef CAS PubMed.
- W. P. Faulk and G. M. Taylor, Immunochemistry, 1971, 8, 1081–1083 CrossRef CAS.
- J. F. Hainfeld and R. D. Powell, J. Histochem. Cytochem., 2000, 48, 471–480 CrossRef CAS PubMed.
- G. M. Lee, A. Ishihara and K. A. Jacobson, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6274–6278 CrossRef CAS.
- Y.-H. Lin, W.-L. Chang and C.-L. Hsieh, Opt. Express, 2014, 22, 9159–9170 CrossRef CAS PubMed.
- O. H. Laitinen, V. P. Hytönen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- J.-M. Nam, P. M. Nair, R. M. Neve, J. W. Gray and J. T. Groves, ChemBioChem, 2006, 7, 436–440 CrossRef CAS PubMed.
- K. M. Spillane, J. Ortega-Arroyo, G. de Wit, C. Eggeling, H. Ewers, M. I. Wallace and P. Kukura, Nano Lett., 2014, 14, 5390–5397 CrossRef CAS PubMed.
- P. Kukura, H. Ewers, C. Müller, A. Renn, A. Helenius and V. Sandoghdar, Nat. Methods, 2009, 6, 923–927 CrossRef CAS PubMed.
- L. B. Sagle, L. K. Ruvuna, J. M. Bingham, C. Liu, P. S. Cremer and R. P. Van Duyne, J. Am. Chem. Soc., 2012, 134, 15832–15839 CrossRef CAS PubMed.
- S. Ota, S. Wang, Y. Wang, X. Yin and X. Zhang, Nano Lett., 2013, 13, 2766–2770 CrossRef CAS PubMed.
- Y. K. Lee, S. Kim, J.-W. Oh and J.-M. Nam, J. Am. Chem. Soc., 2014, 136, 4081–4088 CrossRef CAS PubMed.
- M. J. Murcia, D. E. Minner, G.-M. Mustata, K. Ritchie and C. A. Naumann, J. Am. Chem. Soc., 2008, 130, 15054–15062 CrossRef CAS PubMed.
- T. T. Hormel, S. Q. Kurihara, M. K. Brennan, M. C. Wozniak and R. Parthasarathy, Phys. Rev. Lett., 2014, 112, 188101 CrossRef.
- P. Mascalchi, E. Haanappel, K. Carayon, S. Mazères and L. Salomé, Soft Matter, 2012, 8, 4462–4470 RSC.
- E. Boukobza, A. Sonnenfeld and G. Haran, J. Phys. Chem. B, 2001, 105, 12165–12170 CrossRef CAS.
- C. Yoshina-Ishii and S. G. Boxer, J. Am. Chem. Soc., 2003, 125, 3696–3697 CrossRef CAS PubMed.
- J. J. Benkoski and F. Höök, J. Phys. Chem. B, 2005, 109, 9773–9779 CrossRef CAS PubMed.
- B. van Lengerich, R. J. Rawle and S. G. Boxer, Langmuir, 2010, 26, 8666–8672 CrossRef CAS PubMed.
- A. Johnson-Buck, S. Jiang, H. Yan and N. G. Walter, ACS Nano, 2014, 8, 5641–5649 CrossRef CAS PubMed.
- E. A. Merritt, S. Sarfaty, F. Van Den Akker, C. L'Hoir, J. A. Martial and W. G. J. Hol, Protein Sci., 1994, 3, 166–175 CrossRef CAS PubMed.
- H. Wu, A. E. Oliver, V. N. Ngassam, C. K. Yee, A. N. Parikh and Y. Yeh, Integr. Biol., 2012, 4, 685–692 RSC.
- J. M. Moran-Mirabal, J. B. Edel, G. D. Meyer, D. Throckmorton, A. K. Singh and H. G. Craighead, Biophys. J., 2005, 89, 296–305 CrossRef CAS PubMed.
- V. Yamazaki, O. Sirenko, R. J. Schafer and J. T. Groves, J. Am. Chem. Soc., 2005, 127, 2826–2827 CrossRef CAS PubMed.
- M. B. Forstner, C. K. Yee, A. N. Parikh and J. T. Groves, J. Am. Chem. Soc., 2006, 128, 15221–15227 CrossRef CAS PubMed.
- B. van Lengerich, R. J. Rawle, P. M. Bendix and S. G. Boxer, Biophys. J., 2013, 105, 409–419 CrossRef CAS PubMed.
- J. Shi, T. Yang, S. Kataoka, Y. Zhang, A. J. Diaz and P. S. Cremer, J. Am. Chem. Soc., 2007, 129, 5954–5961 CrossRef CAS PubMed.
- D. E. Golan and W. Veatch, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 2537–2541 CrossRef CAS.
- M. J. Saxton and K. Jacobson, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 373–399 CrossRef CAS PubMed.
- C. Yoshina-Ishii, Y.-H. M. Chan, J. M. Johnson, L. A. Kung, P. Lenz and S. G. Boxer, Langmuir, 2006, 22, 5682–5689 CrossRef CAS PubMed.
- T. H. Söllner, Curr. Opin. Cell Biol., 2004, 16, 429–435 CrossRef PubMed.
- L. Simonsson, P. Jönsson, G. Stengel and F. Höök, ChemPhysChem, 2010, 11, 1011–1017 CrossRef CAS PubMed.
- R. J. Rawle, B. van Lengerich, M. Chung, P. M. Bendix and S. G. Boxer, Biophys. J., 2011, 101, L37–L39 CrossRef CAS PubMed.
- B. N. Kholodenko, J. B. Hoek and H. V. Westerhoff, Trends Cell Biol., 2000, 10, 173–178 CrossRef CAS.
- J. Gureasko, W. J. Galush, S. Boykevisch, H. Sondermann, D. Bar-Sagi, J. T. Groves and J. Kuriyan, Nat. Struct. Mol. Biol., 2008, 15, 452–461 CAS.
- Y.-H. M. Chan, P. Lenz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18913–18918 CrossRef CAS PubMed.
- K. J. Seu, A. P. Pandey, F. Haque, E. A. Proctor, A. E. Ribbe and J. S. Hovis, Biophys. J., 2007, 92, 2445–2450 CrossRef CAS PubMed.
- D. M. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef CAS.
- H. Qian, M. P. Sheetz and E. L. Elson, Biophys. J., 1991, 60, 910–921 CrossRef CAS.
- S. Chandrasekhar, Rev. Mod. Phys., 1943, 15, 1–89 CrossRef.
- D. Ernst and J. Köhler, Phys. Chem. Chem. Phys., 2012, 15, 845–849 RSC.
- X. Michalet, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2010, 82, 041914 CrossRef.
- G. J. Schütz, H. Schindler and T. Schmidt, Biophys. J., 1997, 73, 1073–1080 CrossRef.
- P. G. Saffman and M. Delbrück, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3111–3113 CrossRef CAS.
- M. P. Clausen and B. C. Lagerholm, Curr. Protein Pept. Sci., 2011, 12, 699–713 CrossRef CAS.
- E. Sackmann, Science, 1996, 271, 43–48 CAS.
- C. Yoshina-Ishii and S. G. Boxer, Langmuir, 2006, 22, 2384–2391 CrossRef CAS PubMed.
- B. D. Huges, B. A. Pailthorpe and L. R. White, J. Fluid Mech., 1981, 110, 349–372 CrossRef.
- A. Naji, A. J. Levine and P. A. Pincus, Biophys. J., 2007, 93, L49–L51 CrossRef CAS PubMed.
- Y. K. Lee and J.-M. Nam, Small, 2012, 8, 832–837 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |