
Sunlight-assisted route to antimicrobial plasmonic aminoclay catalysts†
Sudhir
Ravula‡
a,
Jeremy B.
Essner‡
a,
Wendy A.
La
ab,
Luis
Polo-Parada
c,
Roli
Kargupta
d,
Garret J.
Hull
d,
Shramik
Sengupta
d and
Gary A.
Baker
*a
aDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA. E-mail: bakergar@missouri.edu
bDepartment of Chemistry, Truman State University, Kirksville, MO 63501, USA
cDepartment of Medical Pharmacology and Physiology and Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO 65211, USA
dBiological Engineering, University of Missouri, Columbia, MO 65211, USA
First published on 23rd October 2014
Abstract
We present a straightforward, environmentally-benign, one-pot photochemical route to generate alloyed AgAu bimetallic nanoparticle decorated aminoclays in water at room temperature. The protocol uses no reducing agent (e.g., NaBH4) nor is photocatalyst required. These hybrid materials show excellent promise as dual catalysts/antibacterial agents.
In recent years, metal nanoparticles (MNPs), especially those derived from gold and silver, have received great attention due to their attractive optical, thermal, electronic, and chemical properties. These properties are all strongly influenced by the size, shape, and composition of the MNPs, as well as through hybridization with other materials to form nanohybrids or composites. Due to their tailorable features, MNP hybrids have been widely utilized as (bio)catalytic, surface plasmonic, sensory, or antimicrobial agents, as well as platforms for surface-enhanced Raman spectroscopy (SERS).1–6 As one area of chief interest, bimetallic MNPs (BMNPs) frequently show enhanced performance over their monometallic counterparts.7,8 For instance, Yen and co-workers reported that Au and Ag BMNPs supported on an acidic mesoporous aluminosilicate showed enhanced catalytic activity towards low-temperature CO oxidation over their single MNP analogues.9
Historically, a variety of approaches involving laser ablation,10 phase-transfer,11 microwave irradiation12 or chemical reductants (e.g., NaBH4,13 hydrazine14) have been followed to generate MNPs. For practical application, these MNPs are generally supported or stabilized through the use of various surfactants,15 polymers,12 or clays.16,17 A serious downside to many of these classical approaches, however, is that they often require lengthy, multi-step, and complex procedures, or utilize hazardous chemicals to accomplish the metal reduction. Therefore, simple, environmentally-responsible, and cost-effective approaches to generate supported MNPs are highly sought, particularly when they can be facilely extended to the preparation of supported and highly-active BMNPs. For example, Saha et al. recently reported the green photochemical synthesis of Ag and Au NP embedded calcium alginate gel beads which were applied for the catalytic reduction of 4-nitrophenol (4-NP).4 More recently, Murugadoss et al. reported that bimetallic AuAg nanoclusters could be produced on a large scale through a sequential reduction of the metallic precursors by simple mortar grinding.18
The further exploration and development of greener synthetic approaches to supported BMNPs is vital to propel these materials forward within the field of nanoscience. In light of this, the use of organic nanoclays as MNP stabilizing agents has already shown promise due to the nanoclay's unique properties (mechanical strength, thermal stability, antimicrobial activity, permselectivity, ion exchange capacity, etc.).19–22 The organic nanoclay comprising 3-aminopropyl-functionalized magnesium phyllosilicate (commonly referred to simply as aminoclay, AC) has excellent prospects for MNP stabilization since it can be delaminated and stably dispersed in aqueous media due to repulsive electrostatic forces between pendant quaternary ammonium groups.23,24 Eswaramoorthy and co-workers recently reported that MNPs (M = Au, Ag, Pd, Pt) formed through NaBH4 reduction effectively decorated the exfoliated AC surfaces.16 Upon addition of hexadecanethiol, the water-soluble AC@Au composites were quantitatively transferred to the aqueous/organic interface, demonstrating the tenacity of binding between the AC surface and the AuNP as well as the potential for use of these nanocomposite materials in interfacial biphasic catalytic reactions. The same group also reported that the AC matrix provided an oxygen-impermeable barrier, stabilizing air-sensitive CuNPs against oxidation for extended periods (weeks).17 Finally, unmodified AC nanosheets showed antimicrobial activity against Escherichia coli, Staphylococcus aureus, and Candida albicans with the assumption that the inhibition of the microorganisms arose from charge interactions with the aminopropyl group by a membrane disruption mechanism.20
Herein, we present a green, facile, and expedient aqueous photo-assisted route to produce tailored BMNP-decorated AC nanosheets (denoted AC@BMNP) that requires neither heating nor external reducing agent (e.g., NaBH4). The essence of our strategy is captured in Scheme 1. Our approach entails the initial deposition of AgCl NPs onto the surfaces of AC lamellae as a result of chloride residues remaining from the Mg source employed. AgCl acts as an in situ photocatalyst for the light-assisted reduction of AgCl colloids to generate Ag NPs directly on the water-dispersed AC nanosheets within a few minutes. Significantly, both artificial light and natural sunlight proved equally effective in the preparation of AC@Ag.
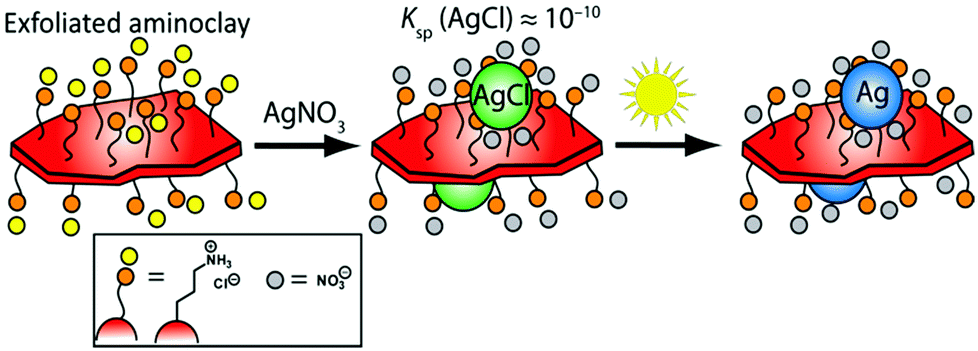 | ||
| Scheme 1 Pathway illustrating the formation of AC@Ag hybrids. | ||
Detailed experimental procedures for preparing the exfoliated AC nanosheets and their AC@Ag and AC@AgxAuy nanohybrid counterparts are provided in the ESI.† Direct ambient temperature treatment of the transparent AC suspensions with appropriate water-soluble metal salts (AgNO3 or HAuCl4) afforded AC@MNP, however, the synthesis required several weeks and suffered from spontaneous aggregation upon standing, a problem especially evident for Au (Fig. S1 in the ESI†). The AC@MNP formation could be successfully shortened to several days by treating at various temperatures ranging from 40 to 90 °C. Nonetheless, this approach was still accompanied by agglomeration and marked precipitation, again particularly in the case of Au. Photochemical reduction proved to be a remarkably facile, expedient, and less energy-intensive route to stable AC@MNP hybrids. It is noteworthy that exposing the Ag and Au reaction mixtures to natural (unfocused) sunlight provided sufficient light energy for generating these hybrids, as illustrated in the central image series of Fig. S1.†
The room temperature formation of AC@Ag using Xe arc lamp irradiation was easily monitored via the progression in the intensity of the localized surface plasmon resonance (LSPR) band for Ag at 421 nm, as shown in Fig. 1A. Under arc lamp irradiation, the solution color changed from a cloudy white to yellowish-orange within a few minutes, eventually becoming dark orange within 15 min. As shown in the inset of Fig. 1A, the formation of AC@Ag was essentially complete within 30 min. Interestingly, placement of the initial solution outside in direct sunlight for various times gave results indistinguishable from those obtained using a Xe arc lamp. In contrast, a control experiment conducted in the absence of AC showed no colour development after 1 h of continuous arc lamp irradiation, demonstrating the important role played by the dispersed AC. The effect of photochemical reaction time on AC@Au creation was also monitored in a similar way. In this case, the solution changed from a cloudy yellow to lavender after a 2 h period, becoming light red after 3.5 h of constant illumination. Upon storage of the same sample in the dark, further Au reduction took place post-irradiation and the samples continually darkened over the course of 2 days to become a wine red colour, signifying the maturation of AC@Au. Remarkably, addition of silver salt to the gold salt-containing reaction mixture prior to irradiation resulted in rapid simultaneous reduction of both metals, permitting the preparation of AC@BMNP composites.
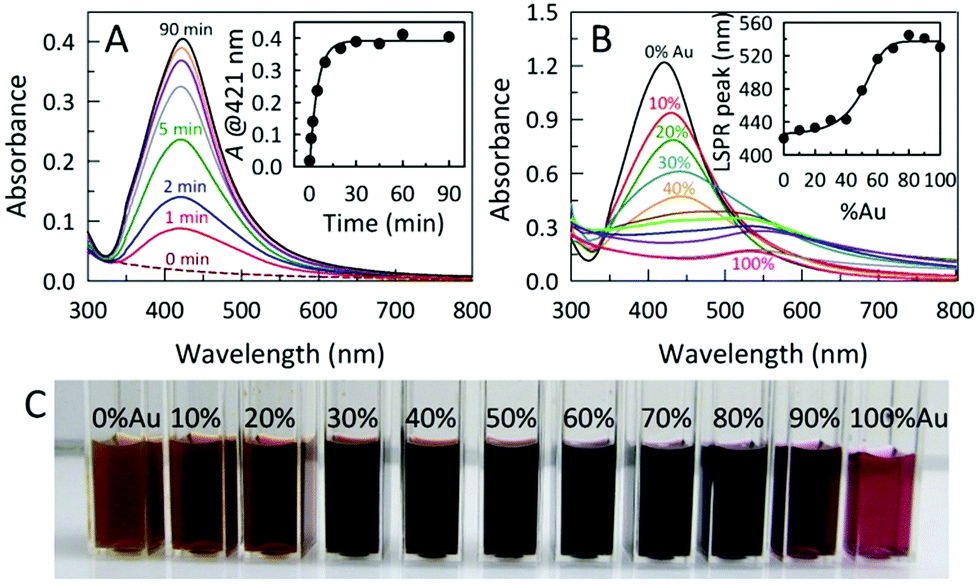 | ||
Fig. 1 (A) Time-resolved UV-Vis spectra following the photo-assisted evolution of AC@Ag hybrids. Inset: time dependence of the AgNP LSPR absorbance at 421 nm. (B) UV-Vis spectra for AC@AgxAuy made via photoirradiation for 60 min employing varied Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratios at a fixed noble metal precursor (Ag + Au) concentration. Inset: the plasmon-“tuning” made possible simply by varying the Ag:Au ratio used during photoreduction. (C) Photograph of the AC@AgxAuy hybrids showing a visually-distinct color transition from left to right as the initial fraction of the Au precursor was increased in 10% increments. :Au ratios at a fixed noble metal precursor (Ag + Au) concentration. Inset: the plasmon-“tuning” made possible simply by varying the Ag:Au ratio used during photoreduction. (C) Photograph of the AC@AgxAuy hybrids showing a visually-distinct color transition from left to right as the initial fraction of the Au precursor was increased in 10% increments. | ||
In this fashion, varying the initial Ag:Au ratio in the reaction mixture and irradiating for 60 min afforded a palette of AC@AgxAuy hybrids displaying continuously tunable LSPR band maxima in the range from 420–540 nm (Fig. 1B, C and S2†). As the initial Au content was increased, the intensity of the absorption bands for the AC@AgxAuy hybrids decreased and shifted to the longer wavelength region, consistent with an increased Au content in the alloy. Formation of alloyed AgxAuy BMNPs is evidenced by the appearance of a single band with a LSPR maximum located between those of pure Au and Ag NPs. Moreover, the absorption band width of the alloyed BMNPs is broader than that of the pure Au or Ag NPs.
Fig. 2 displays representative transmission electron microscopy (TEM) images of the exfoliated AC and AC@Ag. The AC exfoliation process generally yielded AC nanoplatelets in the size range from 50 to 150 nm (Fig. 2A). The inset in Fig. 2A displays an example of a fairly large isolated AC particle, which appears to be a single exfoliated sheet, while many of the larger “sheets” seen in Fig. 2A actually appear to comprise an assembly of smaller sheets. The overall average particle size of the exfoliated AC was 156 ± 137 nm, based on counting 400 individual sheets. For the AC@Ag composites (Fig. 2B), Ag NPs in the range of 5–30 nm were generally produced, with a mean NP size of 18.4 ± 10.3 nm. It should be noted that, due to the differential z-contrast, it is difficult to clearly visualize the AC sheets and the Ag NPs simultaneously under the TEM, although the outlines of the AC itself are evident upon close inspection.
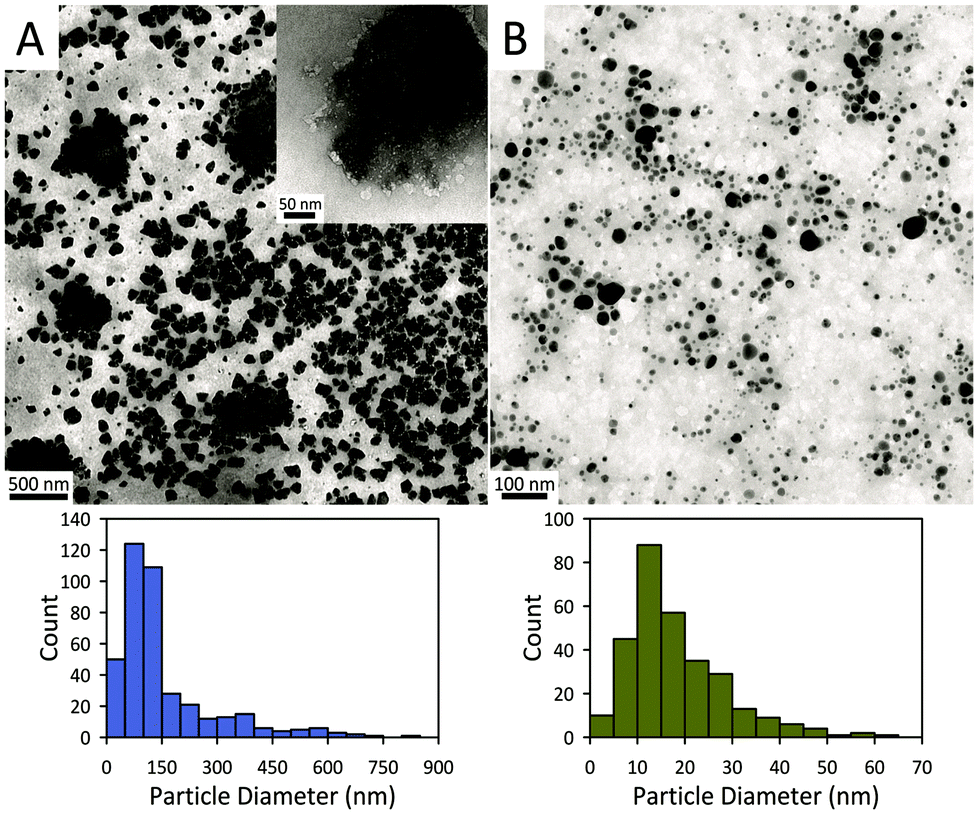 | ||
| Fig. 2 TEM images of (A) AC platelets and (B) AC@Ag hybrids and corresponding histograms of their sizes. | ||
In the AC@AgxAuy hybrids, the BMNPs possessed varying particle geometries which included quasi-spherical particles, hexagonal or triangular plates, and donut-like particles (Fig. S3†). The average sizes of the BMNPs within the AC@Ag0.3Au0.7, AC@Ag0.5Au0.5 and AC@Ag0.1Au0.9 hybrids were 8.9 ± 4.1 nm, 12.1 ± 6.3 nm and 8.0 ± 6.1 nm, respectively. Fascinatingly, at the highest Au content employed (AC@Ag0.1Au0.9), the TEM image provides clear evidence for hollow nanostructures suggestive of a galvanic replacement reaction (Fig. S3C†).25 This result provides some mechanistic insight into the co-reduction process that presumably occurs during AC@AgxAuy formation. That is, under these conditions, Ag NPs are at least partially formed initially and then hollowed out during Au deposition. Notably, however, hollow NPs are not observed in the cases of AC@Ag0.3Au0.7 and AC@Ag0.5Au0.5 (Fig. S3A,B†). Furthermore, the normalized absorbance profile for AC@Ag0.5Au0.5 (Fig. S2†) is fully consistent with an alloy AgAu NP, not a hollowed Au NP. In this light, we posit that nascent AgCl NPs are initially photoreduced to form Ag “seeds” which are autocatalytic for the rapid growth of Ag or AgAu NPs. This accounts for the ability to produce AC@AgxAuy in under 1 h while AC@Au ultimately requires several hours of photoillumination followed by aging for a couple days. More broadly, this outcome suggests that galvanic replacement, coupled with other processes like the Kirkendall effect and cold welding, might have future potential to engineer the properties of metal nanostructures within next-generation AC@MNP hybrids in terms of size, composition, structure, shape, and morphology, leading to further tailored optical and catalytic properties.
Fig. 3 displays powder X-ray diffraction (XRD) patterns of dried AC, AC@Ag, and AC@Ag0.5Au0.5 hybrids. The diffraction pattern of the as-synthesized AC shows broad in-plane peaks at higher angles that correspond to the (020,110), (130,200) and (060,330) planes. The presence of the weak low-angle reflection at 4.9° (d001 = 1.80 nm) corresponds to a bilayer arrangement of propylamine groups, indicating efficient exfoliation with the possible coexistence of stacks made up of a few AC layers. The intralayer reflection (d060,330 = 1.56 Å), denoted by #, is characteristic of the 2:1 trioctahedral Mg-phyllosilicate clay. These indices match well with values reported in the literature for AC.16,24 The interlayer (001) reflection disappears for the AC@Ag and AC@Ag0.5Au0.5 hybrids, signifying that exfoliation of AC to individual sheets has occurred. The XRD peaks at 37.92, 44.08, 64.36, and 77.28°, corresponding to the (111), (200), (220) and (311) planes (denoted by asterisks), respectively, arose from metallic Ag (or Ag0.5Au0.5) NPs. This indicates that the Ag and AgAu NPs were of a face centred cubic structure.9,14,16
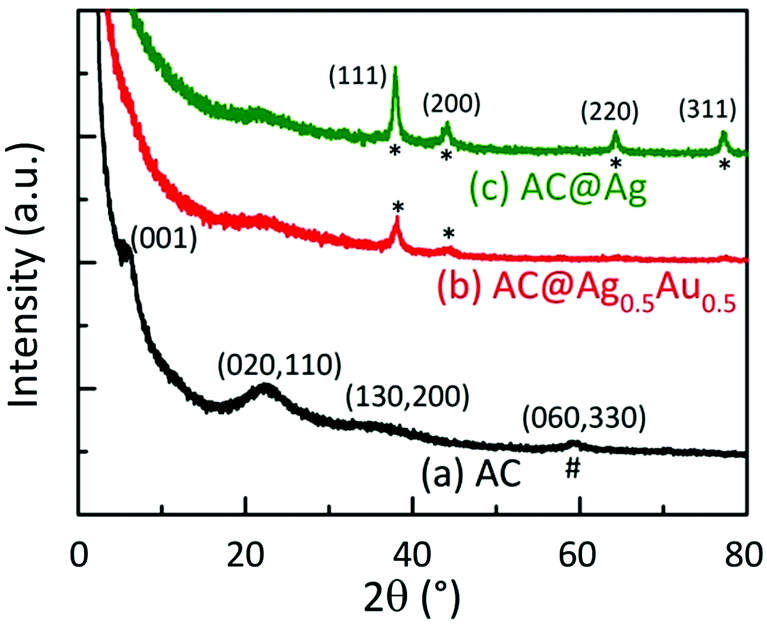 | ||
| Fig. 3 XRD patterns of (a) as-synthesized AC, (b) AC@Ag0.5Au0.5, and (c) AC@Ag. The broad in-plane reflections at higher angles and the intralayer reflection (d060,330 = 1.56 Å), denoted by #, are characteristic of the 2:1 trioctahedral magnesium phyllosilicate clay with a talc-like structure. The peaks marked with asterisks (*) arise from metallic Ag or AgAu NPs. | ||
The catalytic activities of these AC hybrids were evaluated by using the widely-studied conversion of the toxic pollutant 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) using NaBH4 as the reducing agent.26 The reaction kinetics of this catalytic conversion are readily monitored by UV-Vis spectroscopy. The solution of neat 4-NP displays little colour (pale yellow), with an absorbance band at 320 nm. Upon addition of NaBH4, the colour immediately turned dark yellow showing a bathochromic shift to 401 nm, corresponding to the formation of the 4-nitrophenolate ion (4-NPO) under alkaline conditions (Fig. S4A†). In order to assess the catalytic activity of the AC hybrids, UV-Vis spectra were collected every 15 s, monitoring the decrease in absorbance at 401 nm (loss of 4-NP) which is concomitant with the appearance of new peaks for 4-AP at 230 and 300 nm. Addition of the metal NP-decorated AC hybrids to the 4-NPO solution resulted in a rapid drop in absorbance at 401 nm, indicating 4-NP conversion to 4-AP. The complete reduction of 4-NP to 4-AP (bleaching of the initial yellow solution) was accomplished within 675 s, 195 s, and 480 s for AC@Ag, AC@Ag0.5Au0.5, and AC@Au hybrids, respectively. The four isosbestic points (indicated by vertical bars in Fig. 4A) clearly indicate the clean conversion of 4-NP to 4-AP, without the formation of by-products.27 In the presence of just NaBH4 or NaBH4 plus plain AC, no conversion was observed after 1 h (Fig. S4B,C†), validating that the (bi)metallic NPs were indeed responsible for the catalytic activity.
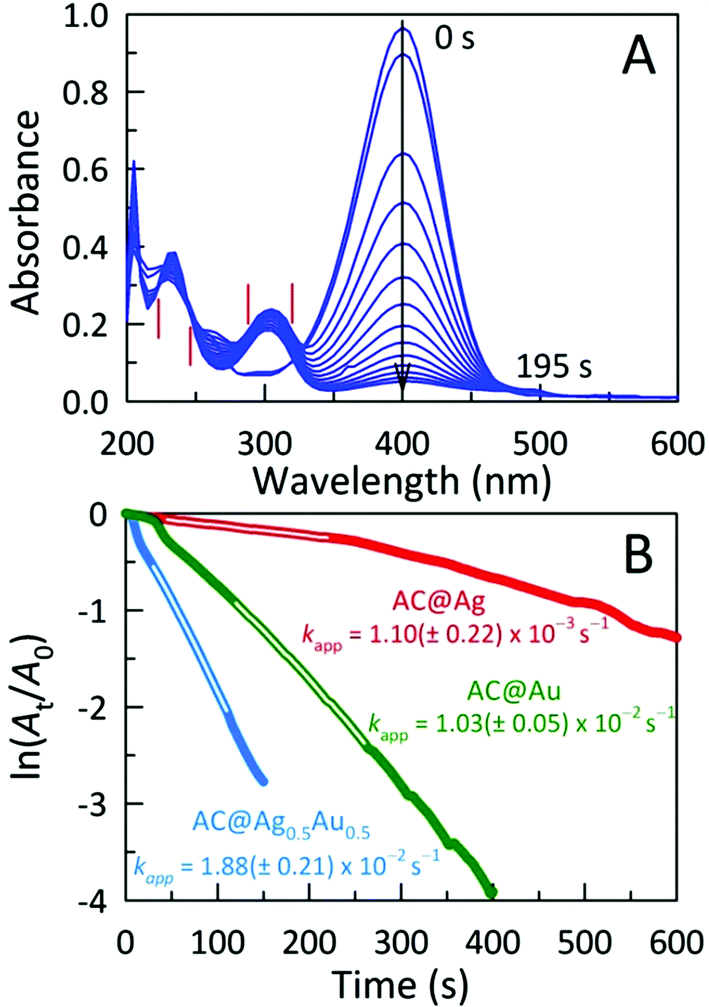 | ||
| Fig. 4 (A) Time-dependent electronic absorption spectra for NaBH4-assisted 4-NP reduction catalyzed by AC@Ag0.5Au0.5. Spectra were acquired every 15 s. (B) Plots of ln(At/A0) of 4-NP against time for various AC@MNP catalysts, monitored at 401 nm. Each catalyst was tested six weeks after preparation. | ||
The reduction mechanism can be understood through the Langmuir-Hinshelwood mechanism in which the analyte of interest adsorbs onto the metallic surface, which also reacts with the BH4− to form metal hydrides. The metal hydrides combine with the sorbed reactant (in this case, 4-NPO), facilitating its reduction. The transfer of electrons from the metal hydride to the 4-NPO is a slow process and is considered the rate determining step of this reaction.28 This lag phase or induction time (t0) was minimized by first degassing the 4-NP solution for 20 min to remove dissolved oxygen. In these studies, the concentration of NaBH4 employed far exceeds that of 4-NP (by a factor of ∼200), allowing us to safely assume that conversion follows pseudo-first-order kinetics. In this fashion, the apparent rate constants (kapp) can be estimated from the slopes of the linear correlations between ln(At/A0) and time (t), where A0 and At denote the initial absorbance for 4-NPO (measured at 401 nm) and the time-dependent absorbance at 401 nm, respectively. The kapp values determined for the AC@Ag, AC@Au, and AC@Ag0.5Au0.5 catalysts are 1.10 ± 0.22 × 10−3 s−1, 1.03 ± 0.05 × 10−2 s−1, and 1.88 ± 0.21 × 10−2 s−1, respectively (Fig. 4B). The activities of these catalysts are excellent, especially considering the very low catalyst dosage used with respect to the 4-NP (3.0 mol%). Indeed, examinations of catalytic efficiency using this model reaction are typically performed at catalyst dosages of 20–50 mol%.29 As might be expected, the AC@Au proved to be a more effective catalyst (in this case, by an order of magnitude) over AC@Ag. Much more surprising is the observation that the AC@Ag0.5Au0.5 hybrid shows nearly twice the catalytic activity of AC@Au, despite containing essentially half the amount of Au. We tentatively attribute this enhanced catalytic behaviour to the presence of miniscule Au domains distributed randomly across the alloyed AgAu NP surface. Essentially, these putative discrete Au clusters or islands are functionally akin to imparting a multiplicity of smaller Au NPs, achieving a greater effective surface-to-volume ratio and a concomitantly higher catalytic activity.
In order to evaluate the robustness and stability of the highest-performing AC@Ag0.5Au0.5 catalyst for 4-NP reduction, we measured the activity of a freshly-made catalyst alongside one aged for 6 months for several consecutive reactive cycles. The activity of the freshly-prepared catalyst remained very high even after four cycles, retaining essentially half of the initial activity (Fig. 5A). Such a decrement in 4-NP reduction rate for iterative cycles is not without precedent, however, we note that a significant portion of this loss in apparent activity stems from the fact that only 4-NP is added to trigger each subsequent cycle and thus a significant population of the initial NaBH4 is either consumed by reaction or is hydrolysed to NaB(OH)4. Noteworthy is the fact that, even following storage for 6 months in water, the aged AC@Ag0.5Au0.5 still shows half of the activity of the just-prepared material (Fig. 5B). This result bodes well for the long-term shelf life of these materials, particularly if they are stored in the dry state.
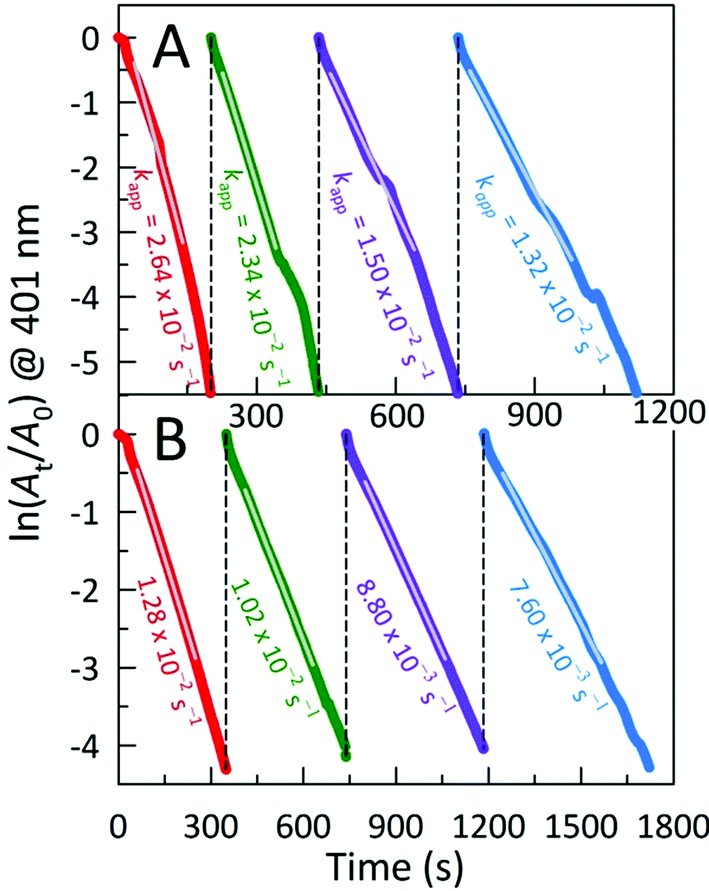 | ||
| Fig. 5 Influence of the number of repeated uses of AC@Ag0.5Au0.5 as catalyst for 4-NP reduction on the apparent reaction rate for (A) freshly-prepared (5-day old) catalyst versus (B) material that had been aged for 6 months in a laboratory drawer. | ||
In order to place the performance of these catalysts in context with existing 4-NP reduction catalysts, we determined turnover frequency (TOF) values, calculated from the time required to reach 50% conversion of 4-NP to 4-AP. Quite high TOFs of 245 h−1 and 436 h−1 were calculated for the AC@Ag and AC@Au hybrids, respectively. Remarkably, however, the TOF for AC@Ag0.5Au0.5 (Ag and Au are each present at 1.5 mol% of the concentration of 4-NP) reaches 1121 h−1, making it amongst the fastest catalysts for 4-NP reduction in water ever reported. For example, our AC@Ag0.5Au0.5 catalyst performs as well as, or indeed better than, the well-dispersed AuNPs supported on two-dimensional graphene oxide/SiO2 composite nanosheets reported by the Dong group,30 and even the “click” synthesis of nona-PEG-branched arene-cored triazole dendrimers for the stabilization/encapsulation of AuNPs published earlier this year by Astruc and co-workers,31 two noteworthy nanoscale materials from the recent literature that efficiently catalyze 4-NP reduction.
Energy-dispersive X-ray (EDX) microanalysis of AC@AgxAuy was performed to reveal the composition of the bimetallic colloids and to provide evidence for the presence of nanodomains within the BMNPs. As shown in Fig. 6A, the actual composition is nominally the same as the starting stoichiometry of Ag:Au used for photoreduction, although for a low fraction of Ag initially mixed (AC@Ag0.1Au0.9), an enrichment in Au arises, consistent with the notion of galvanic replacement posited earlier. The EDX mapping results provided in the lower panel of Fig. 6 reveal that, in general, Au is somewhat more evenly dispersed throughout the BMNPs while both metals occasionally form small clusters or islands (see also Fig. S5†). Fig. S6 of the ESI† provides representative EDX spectra for the AC@AgxAuy samples. As expected, as the Ag:Au ratio decreases, the Ag peaks become less prominent whereas the Au peaks become dominant. X-ray lines for Mg and Si arising from the clay itself are also present.
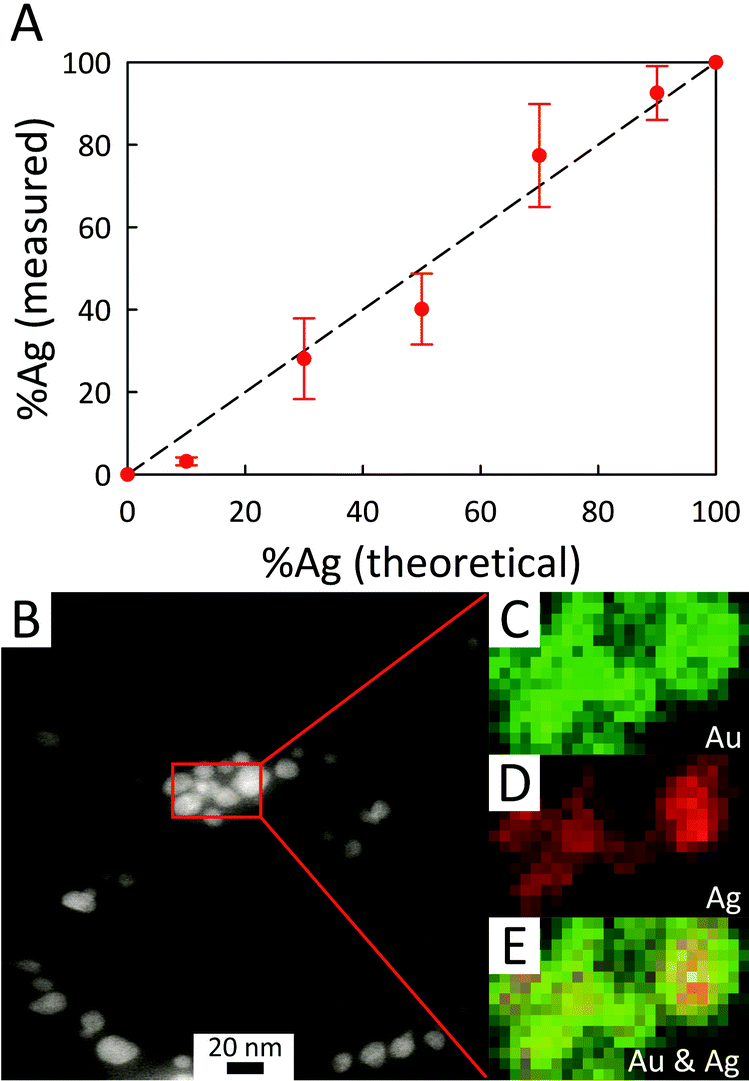 | ||
| Fig. 6 (A) Plot showing the actual %Ag (determined via EDX microanalysis) within BMNPs decorating the AC sheets versus the content predicted from the initial Ag:Au stoichiometry. The broken line represents the ideal case where the AgxAuy composition reflects the reagent metallic ratio. (B) Representative STEM image of AC@Ag0.5Au0.5 and (C–E) corresponding elemental EDX mapping images performed on the area within the red box of (B). Maps of (C) Au and (D) Ag are denoted in green and red, respectively. Panel (E) provides a composite mapping pattern revealing the co-distribution of the elements Au and Ag. The EDX maps shown have a 2 nm resolution with a 5 s dwell. | ||
The antibacterial activities of the AC hybrids were tested against gram-negative E. coli (ATCC 25922), a standard strain of the commonly occurring human pathogen. Fig. 7 summarizes the bacterial inhibition resulting from exposure of the bacterial cultures to AC@AgxAuy hybrids for fixed periods of 0, 2, and 5 h. The initial bacterial counts in the suspension, as measured using plating, were ∼5 × 105 colony forming units (CFU) mL−1 in all cases. In the absence of any candidate antimicrobial agent, the number of bacteria in the control reached a saturation value (>109 CFU mL−1) after 5 h of incubation. Similarly, after 5 h, the sample containing native AC absent of any metal NPs reached ∼4 × 108 CFU mL−1, indicating that this material possesses essentially zero antimicrobial activity at this concentration (1.43 mg mL−1). We note that, in previous work, unmodified AC was shown to inhibit the growth of E. coli, S. aureus, and C. albicans. However, the minimal inhibitory concentration (MIC) against E. coli reported for AC in phosphate buffer and phosphate buffered saline was 3.12 mg mL−1 and 6.25 mg mL−1, respectively,20 partially accounting for this discrepancy.
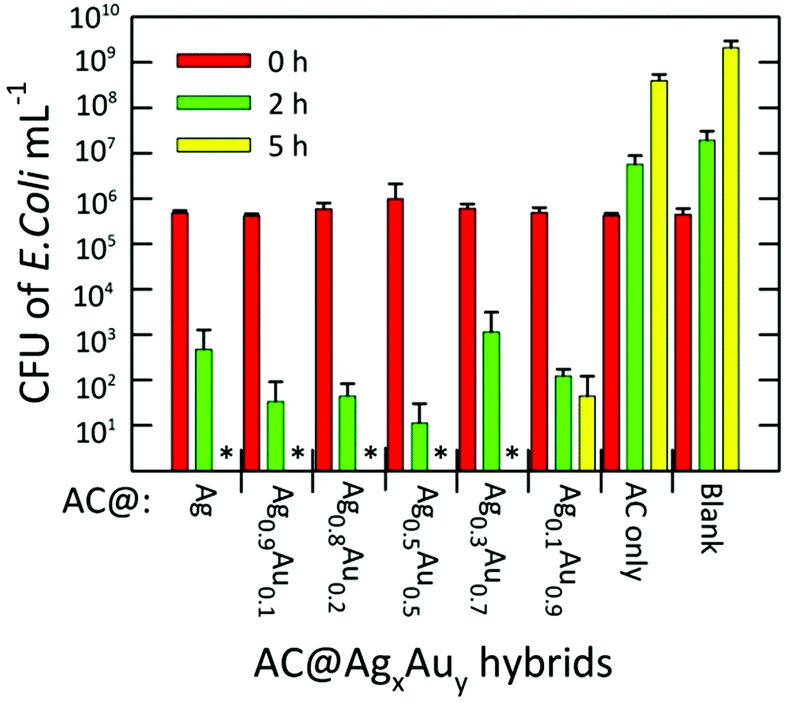 | ||
| Fig. 7 Inactivation efficiency of various AC@AgxAuy hybrids against E. coli expressed in colony forming units (CFU) mL−1 counted after various periods of exposure. The symbol * indicates that no live bacteria were detected after 5 h. The clay concentration used in each assay was held constant at 1.43 mg mL−1. The error bars are standard deviations for triplicate measurements. | ||
Nevertheless, a complete inhibition of the bacteria was observed for the AC@Ag hybrid after 5 h. In fact, for all other Ag-containing samples besides AC@Ag0.1Au0.9, no live bacteria were detected at 5 h. For the AC@Ag0.1Au0.9 agent, the number of living bacteria decreased to ∼102 CFU mL−1 after 5 h. Extraordinarily, if one compares the number of viable E. coli colonies present after 2 h, it is apparent that as the precursor Ag content decreases upon going from AC@Ag to AC@Ag0.5Au0.5, the inhibition in the growth of E. coli actually improves. Indeed, after 2 h, an additional 1.62 log10 units of inactivation of E. coli are found for AC@Ag0.5Au0.5 beyond the inactivation induced by AC@Ag. One can evoke a similar rationale to explain this result as the one proposed earlier to discuss the improved catalytic performance of supported AgAu alloy NPs over monometallic NPs. In this case, of course, it is the nanodispersed Ag phase that derives the benefit. In this manner, these results highlight the capacity of alloyed bimetallic NPs to concurrently derive benefits for widely divergent applications, suggesting potential in other avenues such as SERS and metal-enhanced fluorescence.
In summary, we have developed a green, facile, single-pot photochemical route for preparing aminoclays decorated by tailored bimetallic AgAu nanoparticles within aqueous media. This method requires neither heating nor the use of an external reductant. These AC@AgxAuy hybrids showed excellent catalytic activity towards the reduction of a model nitroaromatic compound and are potent bactericidal agents against E. coli. Intriguingly, the intermediate bimetallic hybrid showed superior activity in both of these applications compared with the pure Ag or Au analogues. The approach followed here has broad generality and may open up additional avenues in (inter alia) drug delivery, catalysis, membranes, bio-imaging, theranostics, (bio)chemical sensing, groundwater purification, and surface-enhanced spectroscopies.
Acknowledgements
GAB thanks the donors of the Petroleum Research Fund, administered by the American Chemical Society, for support of this research. WAL thanks the Stevens’ Summer Research Fellowship program in the Department of Chemistry at the University of Missouri-Columbia for sponsorship. We thank Dr Tommi White and the MU Electron Microscopy Core Facility for providing access to imaging resources and Dr Thomas Lam in particular for kind help with the EDX microanalysis.References
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2009, 26, 2885–2893 CrossRef PubMed.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- C. Yu and J. Irudayaraj, Anal. Chem., 2006, 79, 572–579 CrossRef PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- C.-W. Yen, M.-L. Lin, A. Wang, S.-A. Chen, J.-M. Chen and C.-Y. Mou, J. Phys. Chem. C, 2009, 113, 17831–17839 CAS.
- I. Lee, S. W. Han and K. Kim, Chem. Commun., 2001, 1782–1783 RSC.
- M. J. Hostetler, C.-J. Zhong, B. K. H. Yen, J. Anderegg, S. M. Gross, N. D. Evans, M. Porter and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 9396–9397 CrossRef CAS.
- A. Pal, S. Shah and S. Devi, Colloids Surf., A, 2007, 302, 51–57 CrossRef CAS PubMed.
- L. M. Liz-Marzan and A. P. Philipse, J. Phys. Chem., 1995, 99, 15120–15128 CrossRef CAS.
- D.-H. Chen and C.-J. Chen, J. Mater. Chem., 2002, 12, 1557–1562 RSC.
- L. Gao, L. Fan and J. Zhang, Langmuir, 2009, 25, 11844–11848 CrossRef CAS PubMed.
- K. K. R. Datta, M. Eswaramoorthy and C. N. R. Rao, J. Mater. Chem., 2007, 17, 613–615 RSC.
- K. K. R. Datta, C. Kulkarni and M. Eswaramoorthy, Chem. Commun., 2010, 46, 616–618 RSC.
- A. Murugadoss, N. Kai and H. Sakurai, Nanoscale, 2012, 4, 1280–1282 RSC.
- L. J. Bonderer, A. R. Studart and L. J. Gauckler, Science, 2008, 319, 1069–1073 CrossRef CAS PubMed.
- G. Chandrasekaran, H.-K. Han, G.-J. Kim and H.-J. Shin, Appl. Clay Sci., 2011, 53, 729–736 CrossRef CAS PubMed.
- K. K. R. Datta, A. Achari and M. Eswaramoorthy, J. Mater. Chem. A, 2013, 1, 6707–6718 CAS.
- A. J. Patil, M. Li, E. Dujardin and S. Mann, Nano Lett., 2007, 7, 2660–2665 CrossRef CAS PubMed.
- A. J. Patil, E. Muthusamy and S. Mann, Angew. Chem., Int. Ed., 2004, 43, 4928–4933 CrossRef CAS PubMed.
- N. T. Whilton, S. L. Burkett and S. Mann, J. Mater. Chem., 1998, 8, 1927–1932 RSC.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- A. R. Wright, M. Li, S. Ravula, M. Cadigan, B. El-Zahab, S. Das, G. A. Baker and I. M. Warner, J. Mater. Chem. C, 2014, 2, 8996–9003 RSC.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
- E. Dinda, M. H. Rashid, M. Biswas and T. K. Mandal, Langmuir, 2010, 26, 17568–17580 CrossRef CAS PubMed.
- C. Zhu, L. Han, P. Hu and S. Dong, Nanoscale, 2012, 4, 1641–1646 RSC.
- N. Li, M. Echeverría, S. Moya, J. Ruiz and D. Astruc, Inorg. Chem., 2014, 53, 6954–6961 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting figures and experimental details. See DOI: 10.1039/c4nr04544k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |