
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemistry of the pheromones of mealybug and scale insects
Yunfan
Zou
a and
Jocelyn G.
Millar
*ab
aDepartment of Entomology, University of California, Riverside, CA 92521, USA. E-mail: Jocelyn.millar@ucr.edu; Fax: +1 951 827 3086; Tel: +1 951 827 5821
bDepartment of Chemistry, University of California, Riverside, CA 92521, USA
First published on 20th March 2015
Abstract
Covering: 1979 to 2014
This article comprehensively reviews the syntheses of all known sex pheromones of scales and mealybugs, describes how they were identified, and how the synthetic pheromones are used in insect management.
 Yunfan Zou | Dr Yunfan Zou was born in Beijing, China, in 1975. After receiving his BSc in Chemistry and MSc in Organic Chemistry, both from Tsinghua University, he joined the group of Professor Michael Pirrung at Duke University, completing his doctoral studies in 2006. He has been working with Professor Jocelyn Millar at UC Riverside since 2007, first as a postdoctoral scholar, and currently as a research specialist. As a synthetic organic chemist, his research is focused on developing efficient and practical syntheses of newly discovered semiochemicals, both to confirm their identifications and to provide material for field trials. |
 Jocelyn G. Millar | Jocelyn Millar holds the rank of Distinguished Professor in the Department of Entomology and Cooperating Faculty Member in the Department of Chemistry at the University of California, Riverside. He obtained his PhD in organic chemistry with A.C. Oehlschlager at Simon Fraser University in Canada in 1983, then did postdoctoral studies with R. M. Silverstein in Syracuse, New York, and E. W. Underhill at the National Research Council of Canada before joining UC Riverside in 1988. He is the author or coauthor of >275 papers, 25 book chapters and reviews, and 4 edited books on insect chemical ecology. |
1 Introduction
Mealybugs and scales (Order Hemiptera, Superfamily Coccoidea) comprise a group of small herbivorous insects with sucking mouthparts that colonize a wide variety of plants. Heavy infestations may result in direct plant damage and yield losses. Equal or greater indirect damage may be caused by the proliferation of sooty molds and other fungi on the insects' honeydew excretions. More recently, their importance as major agricultural pests has substantially increased due to their role as vectors of plant pathogens, particularly leafroll viruses. Furthermore, quarantine restrictions may prohibit importation of potentially infested agricultural commodities, particularly fruits, unless they have undergone expensive disinfestation treatments. The systematics, biology, and economic importance of identified scales and mealybugs are comprehensively summarized on the Scale Net website (http://www.sel.barc.usda.gov/scalenet/scalenet.htm), and methods for their management, including the use of pheromone-based techniques, have been recently reviewed.1Most mealybugs and scales reproduce sexually, with the sedentary, wingless females producing powerful sex pheromones to attract the fragile and ephemeral males for mating. These pheromones have several interesting characteristics. First, they possess extraordinary biological activity, with lures baited with a few micrograms of pheromone remaining attractive for periods of at least several months under field conditions. Second, the structures of many of the pheromones are chemically interesting, usually consisting of irregular terpenoids, some of which possess unique terpenoid skeletons. These unique structures, coupled with the tiny amounts in which they are produced, have presented challenges for both their identification and their synthesis. Third, unlike most other insects, in which related species usually create unique pheromone signals by using different ratios or subsets of shared compounds, scales and mealybugs appear to create unique pheromone channels by producing species-specific structures. Several reviews of scale and mealybug pheromone chemistry and applications have been published, but all are outdated, and/or are focused on a subset of species.2–5 Thus, this review will summarize the literature on the identification and synthesis of all known scale and mealybug pheromones, and conclude with a short summary of practical applications and commercialization of the pheromones.
2 Methods for isolation and identification of scale and mealybug pheromones
Adult female scales and mealybugs can live for periods of several months, during which time they will continue to produce pheromone at the rate of a few ng per day until they are mated. Once mated, pheromone production is greatly reduced or ceases altogether, and so a key to identifying their pheromones is to produce large cohorts of virgin females which will release pheromone for weeks at a time. Thus, large colonies of the insects are established on factitious hosts such as potato sprouts or squash fruit, specifically chosen because they will last for a number of weeks without rotting. Cohorts of virgin females are then created in one of two ways. First, the developing males can be manually removed, often helped by the fact that they move off the host fruit to pupate.6 The second, and more efficient and convenient method, takes advantage of the fact that the males undergo a complete metamorphosis to the adult stage whereas the females do not. Thus, males are much more sensitive to insect growth regulating hormones (IGRs) than females, and treatment of mixed sex cohorts of immatures with a discriminating dose of an IGR by spraying or dipping infested host materials into an IGR solution, will selectively kill the males, leaving large cohorts of virgin females on the host.7 The female-infested material is then placed in glass chambers swept continuously with clean air, with the headspace volatiles collected on an adsorbent such as Porapak Q or activated charcoal, changed at daily to weekly intervals. The trapped volatiles are recovered by elution with solvents such as pentane or methylene chloride. Comparison of the profile of volatiles with that from collections from uninfested host material usually allows the insect-produced compounds to be singled out for identification. Alternatively, analysis of the extracts by gas chromatography coupled with electroantennogram detection, using antennae of males as living detectors, allows the unequivocal location of pheromone compounds in the extracts.8As with other insect pheromones that are usually present in only microgram amounts in even composite extracts, most identifications of scale and mealybug pheromones have used spectroscopic methods, particularly mass spectrometry, in combination with microscale chemical tests to determine the presence or absence of specific functional groups. Some of these tests have included catalytic hydrogenation to determine the numbers of rings and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, ozonolysis to determine number and positions of double bonds, base hydrolysis (sometimes followed by reesterification) to confirm presence of esters, and carbon skeleton determination by high-temperature reduction of all functional groups (for full descriptions of these and other microchemical tests applied to semiochemical identification, see ref. 9). Further fragments of information about the structures have been garnered from high resolution mass spectrometry to obtain molecular formulae, and from use of retention indices to estimate molecular size and the possible presence of polar functional groups. For some of the more complex or unusual structures, identification by microscale NMR was unavoidable, sometimes requiring several years of insect rearing to collect enough material to obtain spectra.8 In fact, reviewing some of the clever methods that have been used to isolate and identify microgram amounts of these volatile pheromones makes an interesting read.
C double bonds, ozonolysis to determine number and positions of double bonds, base hydrolysis (sometimes followed by reesterification) to confirm presence of esters, and carbon skeleton determination by high-temperature reduction of all functional groups (for full descriptions of these and other microchemical tests applied to semiochemical identification, see ref. 9). Further fragments of information about the structures have been garnered from high resolution mass spectrometry to obtain molecular formulae, and from use of retention indices to estimate molecular size and the possible presence of polar functional groups. For some of the more complex or unusual structures, identification by microscale NMR was unavoidable, sometimes requiring several years of insect rearing to collect enough material to obtain spectra.8 In fact, reviewing some of the clever methods that have been used to isolate and identify microgram amounts of these volatile pheromones makes an interesting read.
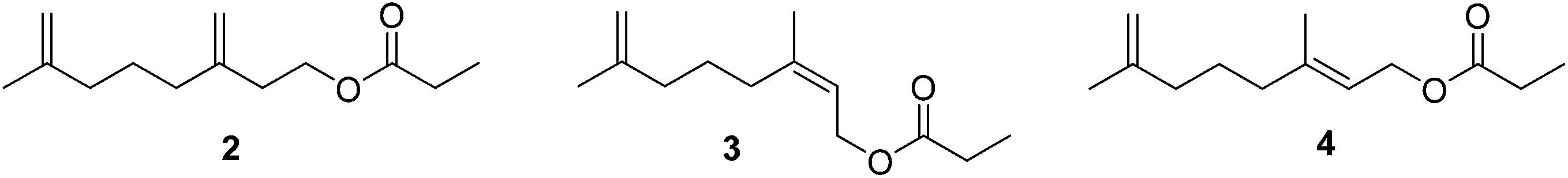
3 Overview of scale and mealybug pheromones
To date, sex pheromones of 24 scale and mealybug species have been identified, from the families Diaspididae (7 species), Margorodidae (3 species), and Pseudococcidae (1 sp. in the subfamily Phenacoccinae, 13 spp. in the subfamily Pseudococcinae). For diaspidid and pseudococcid pheromones, all known examples are terpenoid derivatives, with many of them constituting esters of irregular, non-head-to-tail terpene alcohols. They can be grouped into 4 structural categories: acyclic mono- or sesquiterpene alcohols, alcohols with a cyclopropane ring (chrysanthemol), mono- or sesquiterpenols with a cyclobutane ring, and monoterpene alcohols with a cyclopentane ring. The acid portion of these esters is variable, consisting of simple straight-chain acids such as acetic, propanoic, and butanoic acids, through to chiral acids such as 2-methylbutanoic and 2-acetoxy-3-methylbutanoic acids, and unsaturated acids such as senecioic and 3-methyl-3-butenoic acids. Exceptions include the hemiterpenol ester pheromone of the Matsumoto mealybug, and the degraded terpenoid ketone pheromone of Aulacaspis murrayae, which nevertheless contains the non-head-to-tail, 1′–2 linkage of two isoprene units that characterizes many of the acyclic pheromones. In contrast, the pheromones of margorodid scales are clearly of polyketide origin. In the descriptions of the identifications and syntheses, the pheromones are grouped by structural classes rather than by taxonomic relatedness, to highlight the structural similarities.3.1 Acyclic pheromones

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46.7:4.8 (Fig. 1).11–13
:46.7:4.8 (Fig. 1).11–13
 | ||
| Fig. 1 The three components of San Jose scale pheromone. | ||
Male scales were attracted to any one of the three compounds, and the compounds did not seem to act synergistically. The first synthesis was designed so that all three compounds could be made from a shared intermediate 9 (Scheme 1A).12 Thus, orthoester Claisen rearrangement of the enolate 6 from 2-methyl-2-propen-1-ol 5 gave ester 7, which was reduced and converted to the bromide 9. Copper-catalyzed regioselective addition of the corresponding Grignard reagent 10 to 3-butyn-1-yl propanoate 11 gave the pheromone 2. Alternatively, the Grignard reagent was added in the same fashion to TMS-protected 3-butyn-1-ol, followed by deprotection and esterification (not shown). Anderson subsequently developed a one-step procedure to the core alcohol structure 14 by alkylation of the dianion of 3-methyl-3-buten-1-ol 12 with 1-bromo-3-methyl-3-butene 13 (Scheme 1B).13 Chong's group, apparently unaware of this synthesis, subsequently published a very similar route.14
 | ||
| Scheme 1 Two syntheses of 2, the most abundant component of the San Jose scale pheromone. Reagents and conditions: (a) EtCOOH, (EtO)3CH, reflux, 68%; (b) LiAlH4, THF, 90%; (c) Ph3PBr2, pyridine, CH3CN, 70%; (d) Mg, ether; (e) CuBr, Me2S, −45–20 °C, 42%. (f) 2 BuLi, TMEDA, hexane, 0–20 °C, ∼40%. | ||
Weiler and coworkers (Scheme 2) alkylated the dianion of methyl acetoacetate 15 with 1-bromo-3-methyl-3-butene 13, then converted the resulting β-ketoester 16 to the enolate 17 to protect the ketone from reduction, and reduced the ester to an alcohol, with concomitant hydrolysis of the enolate during the workup to give 18. Esterification followed by conversion of the ketone to a methylene completed the synthesis of 2.15
 | ||
| Scheme 2 Weiler and coworkers' synthesis of 2. Reagents and conditions: (a) NaH, BuLi, THF, 70%; (b) TMSiCl, Et3N, ether, 97%; (c) LiAlH4, ether, 89%; (d) (EtCO)2O, pyridine, 90%; (e) CH2Br2, Zn, TiCl4, THF, 83%. | ||
Lombardo and Weedon attempted a photochemical deconjugation of ethyl 3,7-dimethyl-2,7-octadienoate, but the desired product was obtained only in low yield in a mixture of byproducts; the synthesis was not useful and is mentioned only for completeness.16 Veselovskii and coworkers also developed a low-yielding synthesis but there was insufficient detail given to determine whether the resulting product was pure or a mixture of isomers.17
The 2nd and 3rd pheromone components are (E)- and (Z)-isomers. Anderson and coworkers prepared the (Z)-isomer by regio- and stereoselective addition of Grignard reagent 10 to 1-propyne 19, and reaction of the resulting vinyl cuprate intermediate 20 with CO2, esterification with diazomethane, reduction of the ester 22, and acylation (Scheme 3A).13 The (E)-isomer was prepared by analogous copper-catalyzed reaction of 4-methyl-4-pentenyllithium 24 with methyl 2-butynoate 25, reduction of the ester 26, and acylation (Scheme 3B).13
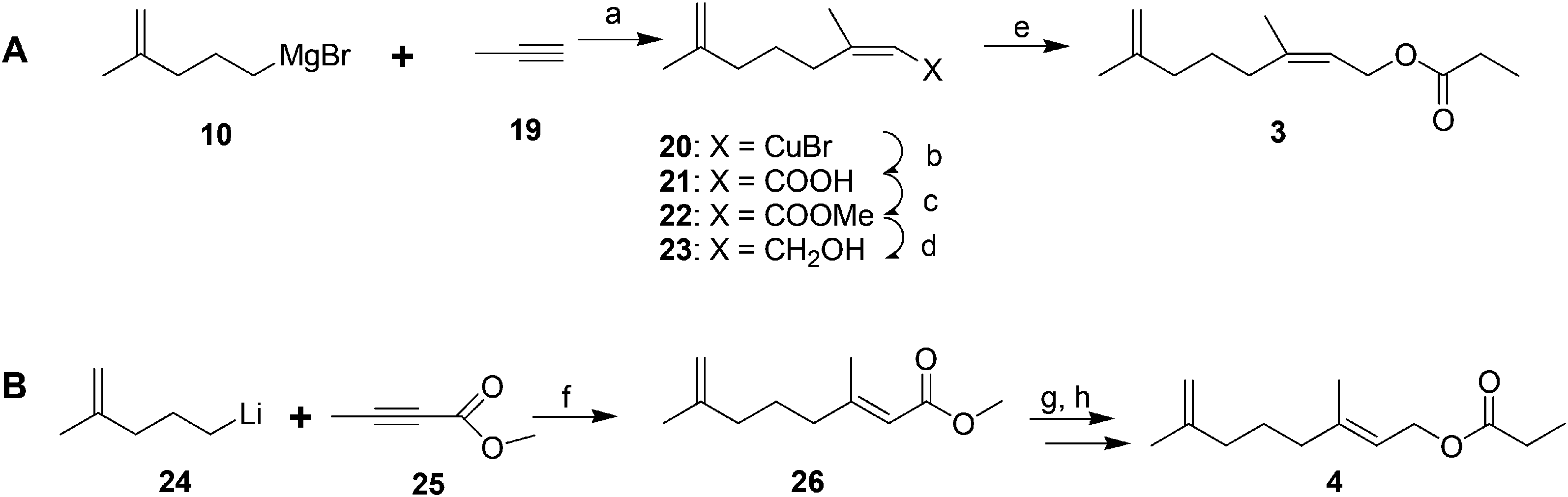 | ||
| Scheme 3 Anderson et al. synthesis of the 2nd and 3rd components of the San Jose scale pheromone. Reagents and conditions: (a) CuBr, Me2S, ether; (b) HMPA, CO2, (EtO)3P, −15 °C; (c) CH2N2, ether, 47% from 10; (d) DIBAL, benzene, quant.; (e) (EtCO)2O, pyridine, 20 °C, 62%. (f) CuI, TMEDA, ether, −60 °C, 85%; (g) DIBAL, benzene, 20 °C, 90%; (h) (EtCO)2O, pyridine, 85 °C, 79%. | ||
In another synthesis of the two isomers using synthons with preset stereochemistry, Moiseenkov and coworkers alkylated 1-iodo-3-methyl-3-butene with an allylic (E)-hydroxysulfone or a related (Z)-hydroxysulfonamide, but because the preparation of these two synthons was not described, the overall utility of the syntheses is not clear.18 A related scheme formed the carbon skeleton by sequential alkylation of bis(arylsulphonyl)methane with (E–Z)-4-chloro-3-methyl-2-buten-1-ol and ethyl 2-methylprop-2-en-1-yl carbonate but suffered from low overall yield and incomplete control of the alkene stereochemistry.19 A 12-step synthesis of a mixture of the two isomers starting from methyl 4-oxopentanoate is mentioned only for completeness.20
In contrast, Weiler's group prepared the (E)- and (Z)-isomers by stereoselective formation of the (E)- and (Z)-enolates of β-ketoester 16 (the intermediate from the synthesis of the first pheromone component), trapping the enolates with diethylchlorophosphate, followed by alkylation with dimethyllithium cuprate with no loss of stereochemical integrity (Scheme 4).15 Thus, treatment of the ketoester 16 with NaH in THF gave, after trapping with diethyl chlorophosphate, the (Z)-enol phosphate 27, and reaction with MeMgCl with copper catalysis gave the trisubstituted alkene 22 as a 98E/2Z mixture. Choice of the reaction conditions and Grignard reagent counterion were critical to the maintenance of high stereochemical integrity. In contrast, formation of the enolate with Et3N in HMPA, trapping as the (E)-enol phosphate 29, and alkylation gave the Z-isomer 26 (98Z/2E). The syntheses were completed by reduction of the terminal esters 22 and 26 and acylation of the resulting alcohols 23 and 28.
 | ||
| Scheme 4 Weiler's synthesis of the (E)- and (Z)-isomers constituting the minor components of the San Jose scale pheromone by selective formation of the (Z)- and (E)-enolates respectively. Reagents and conditions: (a) NaH, (EtO)2POCl, THF, 90–95%; (b) MeMgCl, MeCu, THF, 68–70%; (c) DIBAL, ether, 98% for (E)-isomer, 82% for (Z)-isomer; (d) (EtCO)2O, pyridine, DMAP, ether, 96% for (E)-isomer, 92% for (Z)-isomer; (e) Et3N, DMAP, HMPA, (EtO)2POCl, 97%. | ||
Rather than trying to make the trisubstituted alkenes stereoselectively, Novak et al. used readily available geraniol 30 or its acetate 31 or propanoate esters 32 as starting materials (Scheme 5).21 Thus, regioselective chlorination with t-butyl hypochlorite with migration of the double bond produced the corresponding allylic chlorides 33–35 with a terminal methylene, with no loss of the stereochemical integrity of the other double bond. Alcohol 33 and acetate 34 were reduced with LiAlH4, followed by esterification with propanoyl chloride, whereas the chloride 35 from geranyl propanoate was reduced directly to the final products with NaBH4 and NaI in DMF. The same series of reactions using nerol as starting material produced the (Z)-alkene analogs. Minor variations involving different methods of removing the allylic leaving group were later published.22
 | ||
| Scheme 5 Synthesis of one of the minor components of San Jose scale pheromone from geraniol or geranyl esters. Reagents and conditions: (a) t-BuOCl, silica gel, hexane, 0–20 °C, 61–78%; (b) for alcohol and acetate, LiAlH4, THF, then EtCOCl, Et3N, CH2Cl2, ∼52–56%; (c) for propanoate ester, NaBH4, NaI, DMF, 58%. | ||
 | ||
| Fig. 2 Esters of lavandulol and related compounds. | ||
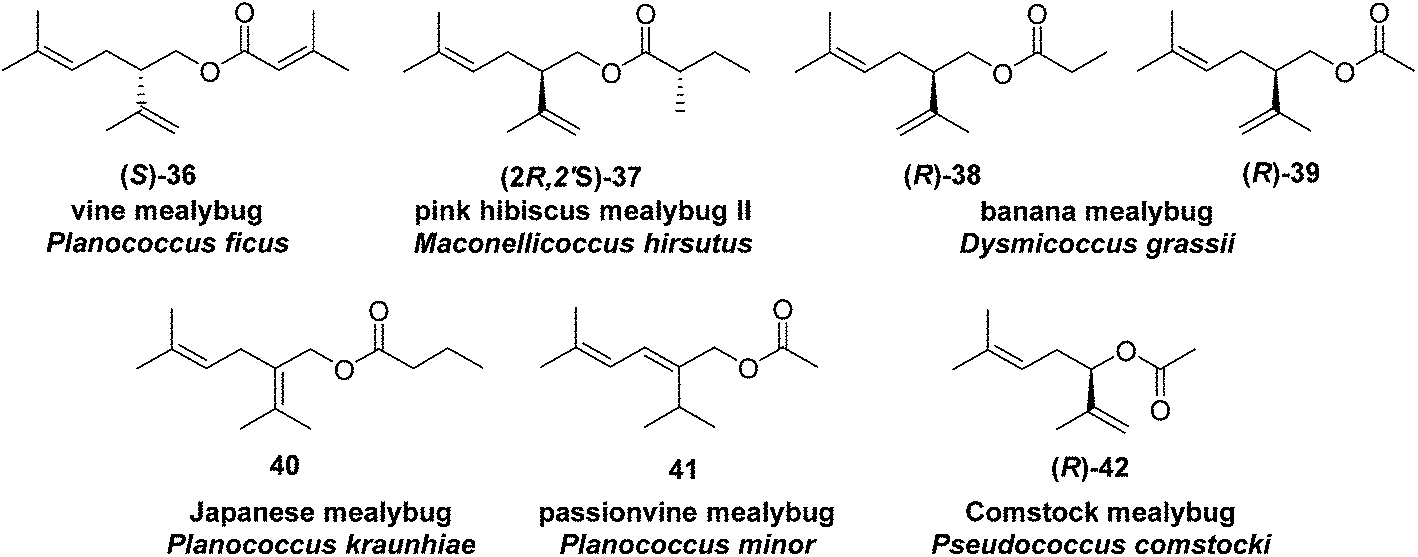
Vine mealybug pheromone. The sex pheromone of the vine mealybug, Planococcus ficus, was identified from mass spectrometry and microchemical tests as (S)-(+)-lavandulyl senecioate 36.27 Racemic lavandulyl senecioate proved to be as attractive to male mealybugs as the insect produced (S)-enantiomer, indicating no inhibition by the unnatural enantiomer. Lavandulol also was identified as a minor component in extracts from virgin females, but it was not attractive alone, and inhibited attraction at higher doses.24 In Israeli populations, Zada and coworkers found another compound in headspace volatiles, (S)-(+)-lavandulyl isovalerate.28 As in California, males were only attracted to the same major component 36. During esterification of lavandulol with senecioyl chloride, choice of base was important; use of Et3N resulted in extensive deconjugation of the senecioate, whereas pyridine yielded the desired ester cleanly.27
Pink hibiscus mealybug pheromone minor component. The sex pheromone of the pink hibiscus mealybug, Maconellicoccus hirsutus (Green), was identified by Zhang and coworkers as a 1
:5 mixture of (R)-lavandulyl (S)-2-methylbutanoate 37 and (R)-2,2-dimethyl-3-(1-methylethylidene)cyclobutylmethyl (S)-2-methylbutanoate [(R)-maconelliyl (S)-2-methylbutanoate (see Fig. 6, compound 257).29 The alcohol portion of the minor component 37 is the enantiomer of the alcohol moiety of vine mealybug pheromone 36; it was prepared by a known synthesis.23 The chirality–bioactivity relationship of 36 is discussed below with the description of the major component 257.
Banana mealybug pheromone. The sex pheromone of the banana mealybug, Dysmicoccus grassii Leonardi, was identified by de Alfonso and coworkers as (R)-(−)-lavandulyl propionate 38 and acetate 39 in a 6
:1 ratio.30 The major component 38 was more active than the minor component 39, and the two components acted additively rather than synergistically. The unnatural (S)-enantiomers were neither attractive nor inhibitory. The compounds were synthesized by kinetic resolution of racemic lavandulol,25 followed by esterification with the appropriate acid chloride or anhydride.
Japanese mealybug pheromone. The sex pheromone of the Japanese mealybug, Planococcus kraunhiae (Kuwana), was identified by Sugie and coworkers as 2-isopropyliden-5-methyl-4-hexen-1-yl butyrate (fujikonyl butyrate) 40.31 The alcohol portion, fujikonol, is a regioisomer of lavandulol, and the pheromone was generated in 4 steps from racemic lavandulol 43 by oxidation to aldehyde 44, acid catalyzed isomerization to the conjugated aldehyde 45, reduction, and esterification (Scheme 6).32
 | ||
| Scheme 6 Synthesis of the Japanese mealybug pheromone 40. Reagents and conditions: (a) Dess-Martin periodinane, CH2Cl2, room temp, overnight, 77%; (b) 1 M aq. HCl, THF, 50 °C, 24 h, 78%; (c) NaBH4, EtOH, room temp, 4 h, 87%; (d) butyric anhydride, pyridine, 40 °C, overnight, 83%. | ||
Passionvine mealybug pheromone. The sex pheromone of the passionvine mealybug, Planococcus minor (Maskell), was identified by Ho and coworkers as (E)-2-isopropyl-5-methyl-2,4-hexadienyl acetate 41.33 To confirm the gross structure and determine the stereochemistry, a nonstereoselective synthesis based on Wittig reaction of a semi-stabilized allylic ylide was used to produce both isomers, which could only be separated on milligram scale by HPLC. The (E)-isomer attracted male mealybugs in laboratory bioassays, whereas the (Z)-isomer was antagonistic. Because this route was not suitable for production of the pheromone for practical use, Millar developed a short, stereoselective synthesis of 41 (Scheme 7)34 by recognizing that the substitution pattern of the trisubstituted alkene 51 was ideally set up for copper-catalyzed, regio- and stereoselective anti-addition of an isopropyl Grignard reagent to propargylic alcohol 49. The stereoselectivity was proposed to result from formation of cyclic intermediate 50.35 In the event, addition of isopropylmagnesium bromide to enynol 49 gave dienol 51 with >99% (E)-selectivity. Acetylation completed the 3-step synthesis.
 | ||
| Scheme 7 Synthesis of the passionvine mealybug pheromone 41. Reagents and conditions: (a) CuI, (Ph3P)2PdCl2, pyrrolidine, 40%; (b) i-PrMgBr, CuI, THF, 64%; (c) AcCl, pyridine, Et2O, 97%. | ||
Comstock mealybug pheromone. The Comstock mealybug pheromone was the first such compound identified from a Pseudococcidae species, and so in what follows, we have described the identification in some detail to give the reader a sense of the methods that were available at that time. The pheromone was identified independently and virtually simultaneously by two groups. The first team, led by Barbara Bierl-Leonhardt, used the insect growth regulator method to generate cohorts of virgin females, collecting ∼30 mg of pheromone over 2–3 years (∼5 million female-day-equivalents).36 The pheromone was identified primarily by mass spectral interpretation and microchemical tests, including hydrolysis and reacetylation to prove it was an acetate ester, catalytic reduction to determine that there were 2 C
C double bonds, ozonolysis to determine their positions, carbon skeleton determination by high temperature reduction to remove all functional groups, determination that the alcohol was a secondary alcohol by use of retention indices, and silylation of the alcohol portion followed by mass spectrometry to determine the position of the secondary alcohol.
The racemic pheromone was readily synthesized (Scheme 8),37 with the key step being the rearrangement of epoxide 55 to the secondary allylic alcohol 56. Derivatization with Mosher's acid chloride gave two diastereomeric esters which were separable by GC, and corresponding derivatization of the alcohol from the natural pheromone gave one peak, verifying that the insect produced only one enantiomer, but it was not yet possible to determine which enantiomer it was, although the natural pheromone was shown to have a positive optical rotation. Fortunately, the racemic material was as attractive to males as the purified natural compound or as virgin females, which also suggested that the pheromone was a single component.
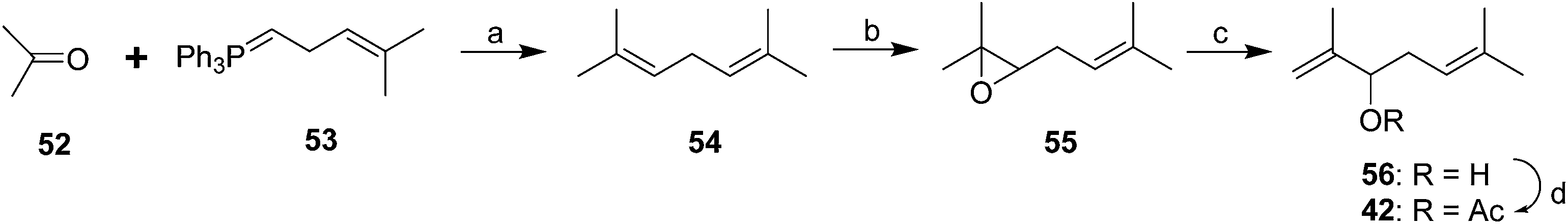 | ||
| Scheme 8 First racemic synthesis of the Comstock mealybug pheromone 42. Reagents and conditions: (a) Solvent not stated; (b) m-CPBA, solvent not stated; (c) Al(i-PrO)3, toluene, reflux; (d) Ac2O, pyridine. | ||
The parallel identification effort of Negishi et al. generated cohorts of virgin females by physical removal of males.6 The pheromone structure was narrowed down to 6 possibilities by mass spectral interpretation and microchemical tests, followed by NMR of 6–7 μg of pheromone isolated from 600000 females. The correct structure was determined by a two-step synthesis, albeit in low yield (Scheme 9A).38 Thus, slow addition of a mixture of methacrolein 57 and 3-methyl-3-butenyl bromide 58 to Li metal in THF at room temp gave an 11.4% yield of the alcohol precursor 56 of the pheromone. In a subsequent improvement, reaction of 2-methylpropenyllithium 60 with 2-(methylethenyl)oxirane 59 proceeded in much better (60%) yield (Scheme 9B).39
 | ||
| Scheme 9 Racemic syntheses of the Comstock mealybug pheromone 42. Reagents and conditions: (a) Li, THF, room temp; 11.4%; (b) Ac2O, pyridine, 77%. (c) THF, 60%; (d) AcCl, pyridine, ether, 77%. | ||
The first chiral syntheses of the pheromone enantiomers were accomplished with Sharpless asymmetric epoxidation of an appropriate trisubstituted olefin alcohol 64, followed by rearrangement and opening of the two enantiomeric epoxides 65 to produce the desired secondary allylic alcohol enantiomers in ∼90% ee (Scheme 10).40 The alcohol precursor 64 was produced in three steps from β-methallyl 2-pyridyl sulfide 61. The (R)-enantiomer of 42 had a positive optical rotation, and was much more active in bioassays, indicating that the natural pheromone was (R). Another synthesis of the (R)-enantiomer was accomplished by kinetic resolution of the alcohol precursor 56 to the pheromone (generated in 5 steps) by selective asymmetric epoxidation of the (S)-alcohol, leaving the (R)-enantiomer unchanged (not shown).41
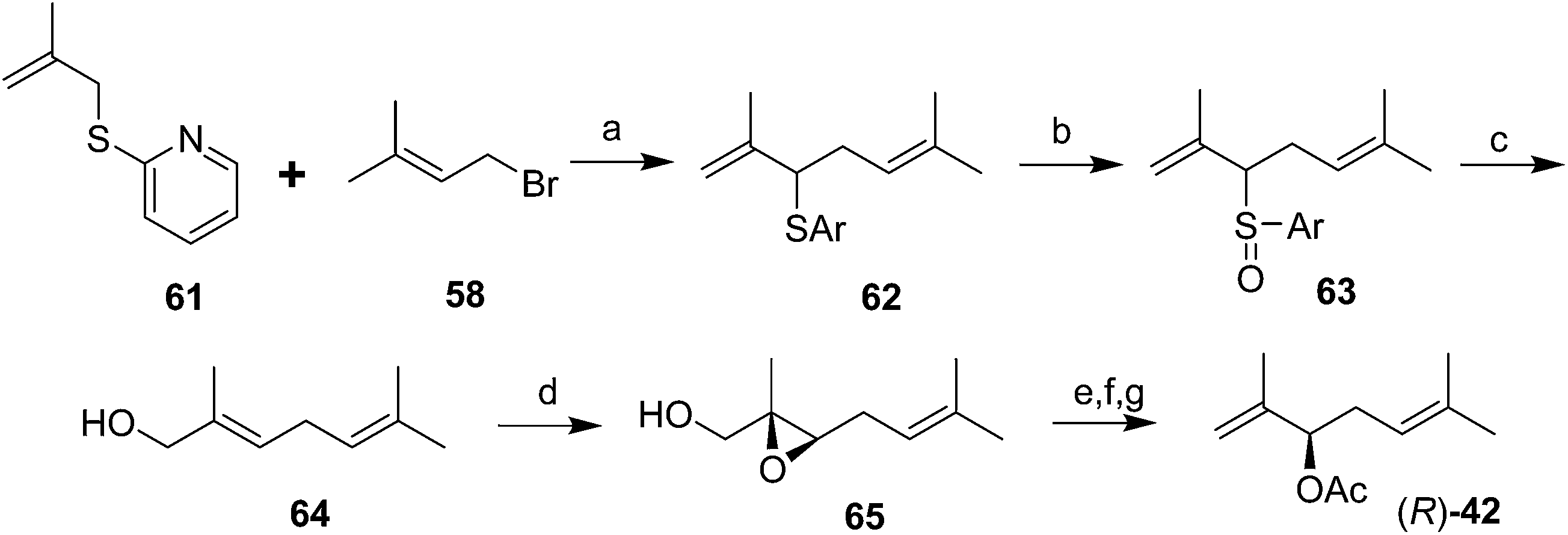 | ||
| Scheme 10 Mori and Ueda's chiral synthesis of the Comstock mealybug pheromone. Reagents and conditions: (a) BuLi, THF, −70 °C-room temp, 85%; (b) m-CPBA, CH2Cl2, −30–10 °C, quant.; (c) Et2NH, MeOH, 46%; (d) t-BuOOH, Ti(i-PrO)4, diethyl (D)-(−)-tartrate, CH2Cl2, −23 °C; aq. tartartic acid, 59%; (e) TosCl, pyridine; NaI, acetone, reflux, 82%; (f) Zn, AcOH, 0 °C-room temp, 79%; (g) Ac2O, pyridine, 80%. | ||
Larchevêque and Petit devised a shorter and more efficient synthesis of either enantiomer, using chiral ethyl glycidates 66 (readily prepared from the enantiomers of serine in three steps) as chiral synthons (Scheme 11).42 Thus, regioselective opening of the epoxide in (R)-66 with diisobutenyllithium cuprate 67 gave the chiral hydroxyester 68. MEM protection of the alcohol followed by low temperature reaction of 69 with MeLi gave ketone 70. Straightforward Wittig olefination, removal of the protecting group, and acetylation completed the synthesis. Use of the TBDMS protecting group instead of MEM resulted in lower yields.42
 | ||
| Scheme 11 Larchevêque and Petit's synthesis of one enantiomer of the Comstock mealybug pheromone from ethyl (R)-glycidate. Reagents and conditions: (a) Ether, −60 °C, 90%; (b) MEMCl, diisopropylethylamine, CH3CN, 85%; (c) MeLi, TMEDA, ether/pentane, −100 °C, 80%; (d) Ph3PCH3Br, BuLi, ether, 50%; (e) pyridinium p-toluenesulfonate, EtOH, reflux, 88%; (f) Ac2O, pyridine, 90%. | ||
Several relatively short and efficient syntheses of the racemate were subsequently developed. For example, base-catalyzed tandem Michael addition/cyclization of methyl chloroacetate 72 and methyl acrylate 73 followed by equilibration of the crude product in MeOH/MeONa gave a 66% yield of the trans-diester cyclopropane 74 (Scheme 12A).43 Treatment of the diester with MeLi gave the crystalline diol 75 almost quantitatively, reaction of which with AcOH/Ac2O gave the racemic pheromone 42 (74%, ∼50% overall). Alternatively, condensation of 3-methyl-3-buten-2-one 76 and methacrolein 57, acetylation, and removal of the ketone with concomitant migration of a double bond provided another short route to racemic 42 in ∼50% yield (Scheme 12B).44
 | ||
| Scheme 12 Several short syntheses of racemic Comstock mealybug pheromone. Reagents and conditions: (a) NaOMe, then NaOMe, MeOH, 66%; (b) MeLi, ether, quant; (c) AcOH, Ac2O, 80 °C, 74%. (d) LDA, ether, −60 °C; (e) Ac2O, pyridine; (f) p-TsNHNH2, NaBH3CN, AcOH, 70 °C. (g) Li 1-(dimethylamino)naphthalenide, THF, −78 °C; (h) CeCl3; (i) Ac2O, pyridine. | ||
A third synthesis produced alcohol 56 from reaction of 1-methylethenylmagnesium bromide with 4-methyl-3-pentenal (generated from acrolein in 3 steps), for an overall 30% yield (not shown).45 A fourth carried out a 4-step sequence in one pot, with 45% isolated yield (Scheme 12C).46 The critical feature was the transmetallation of the initial lithium anion with CeCl3 to produce the cerium complex 80 in order to direct attack of the least substituted carbon towards the carbonyl of methacrolein 57, giving alcohol 56 in 54% yield.
Other, less efficient approaches that have been explored include the photooxidation of 2,6-dimethyl-2,5-heptadiene47 and a rather long 9-step route centered on a sila-Cope rearrangement.48
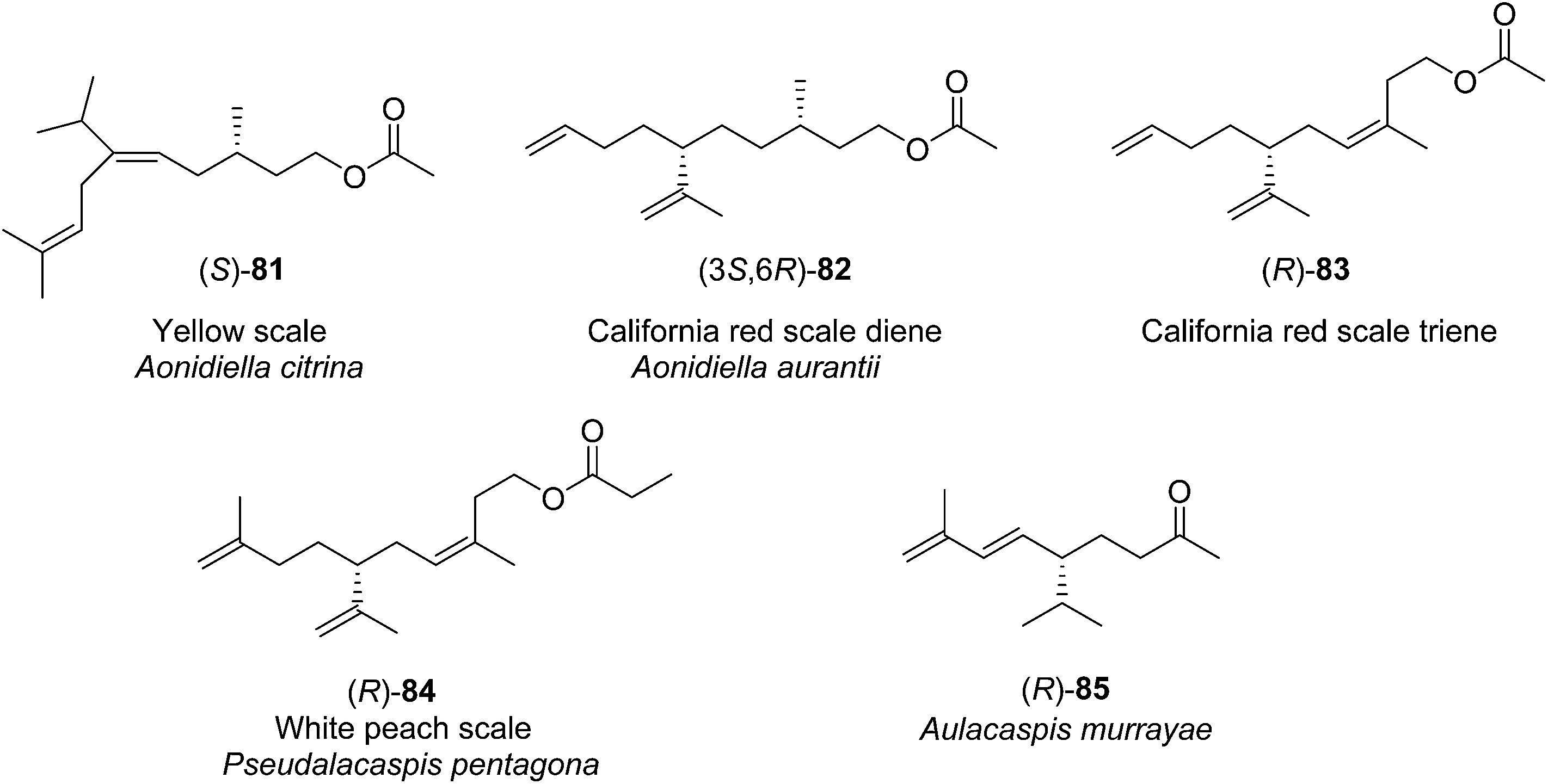 | ||
| Fig. 3 Structures of the pheromones of California red scale, yellow scale, white peach scale, and the scale Aulacaspis murrayae. | ||
Yellow scale pheromone. The basic structure of the yellow scale pheromone 81 was identified via a combination of mass and 1H NMR spectral data, and the results of microchemical tests (catalytic hydrogenation, LiAlH4 reduction and reacetylation, ozonolysis), but there was insufficient data to determine the absolute configuration or the stereochemistry of the double bond.49 Thus, the first nonstereoselective synthesis by Anderson and Henrick was designed to provide both the (Z)- and (E)-isomers (Scheme 13).50 Monoprotection of 3-methyl-1,5-pentanediol 86, pyridinium chlorochromate oxidation, and reaction of the resulting aldehyde 88 with the anion from diethyl 1-ethoxycarbonyl-2-methylpropylphosphonate 89 gave trisubstituted alkene 90 as a 4
:1 mixture of (Z)- and (E)-isomers. Reduction of the ester, acetylation of the resulting allylic alcohol 91, and alkylation of 92 with diisobutenyllithium cuprate completed the carbon skeleton. After deprotection and acetylation, the resulting isomers were readily separable by preparative GC, with the (E)-isomer 81 matching the insect-produced compound.
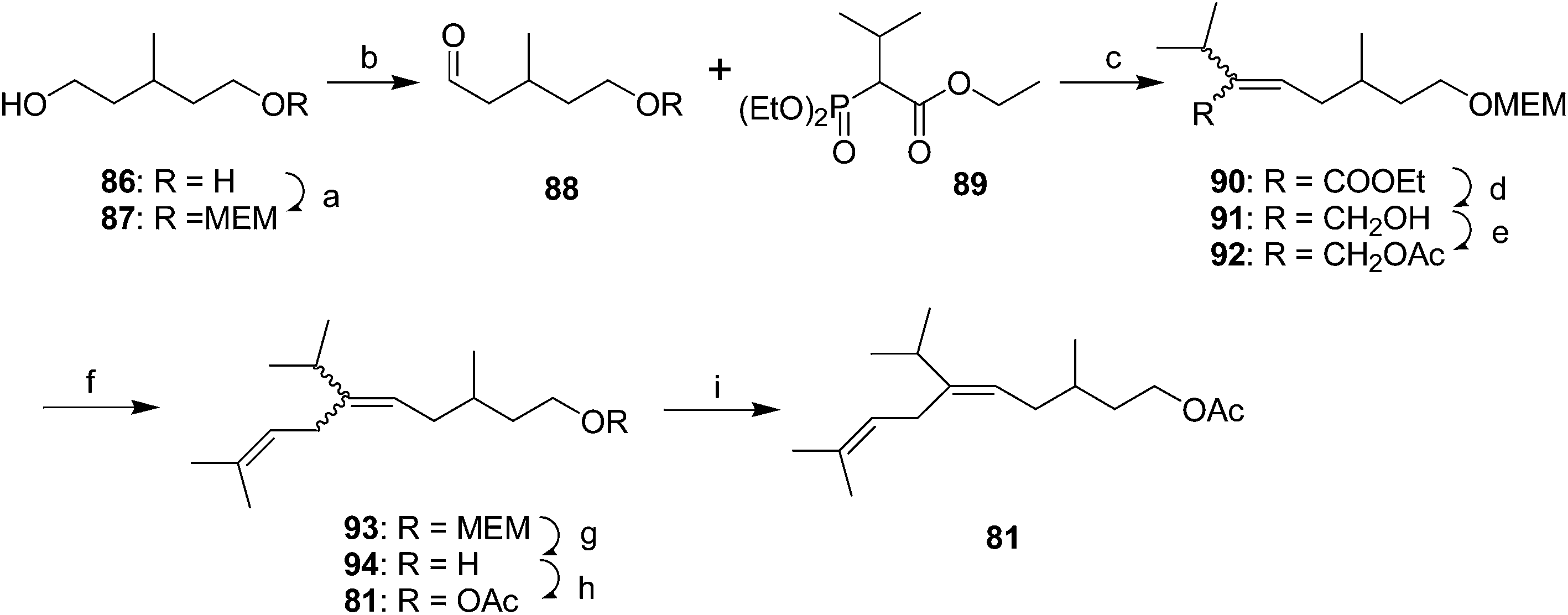 | ||
| Scheme 13 First nonstereoselective synthesis of yellow scale pheromone. Reagents and conditions: (a) BuLi, THF, MEMCl, 47%; (b) PCC, NaOAc, CH2Cl2, 82%; (c) NaH, THF, 58%, 4:1 (Z–E); (d) DIBAL, benzene, 90%; (e) Ac2O, pyridine, quantitative; (f) diisobutenyllithium cuprate, ether, −25 °C, 60%; (g) Cl3CCOOH, EtOH/H2O, 70 °C; (h) Ac2O, pyridine, 65% over 2 steps; (i) preparative GC. | ||
Mori and coworkers developed three syntheses of the pheromone in rapid succession, with the first aimed at determining the double bond geometry,51 the second at developing a chiral synthesis producing one enantiomer,52 and the third at providing both enantiomers.53 These syntheses were long and low-yielding, have been previously reviewed in detail, and so they are only summarized here. In the first synthesis (Scheme 14A),51 reaction of the Grignard reagent prepared from 1-bromo-2-methyl-3-butene 95 with 2-(1-methylethyl)acrolein 96 followed by orthoester Claisen rearrangement gave ester 98 which was elaborated to the (Z)-isomer of 81 in 4 steps. The correct (E)-isomer 81 was generated from ester 98 in 11 steps, using a reaction sequence that included inversion of the double bond by the sequence of epoxidation, opening the epoxide with lithium diphenylphosphide, methylation of the alkyldiphenylphosphine intermediate to form the phosphonium salt, and elimination of methyldiphenylphosphine oxide.
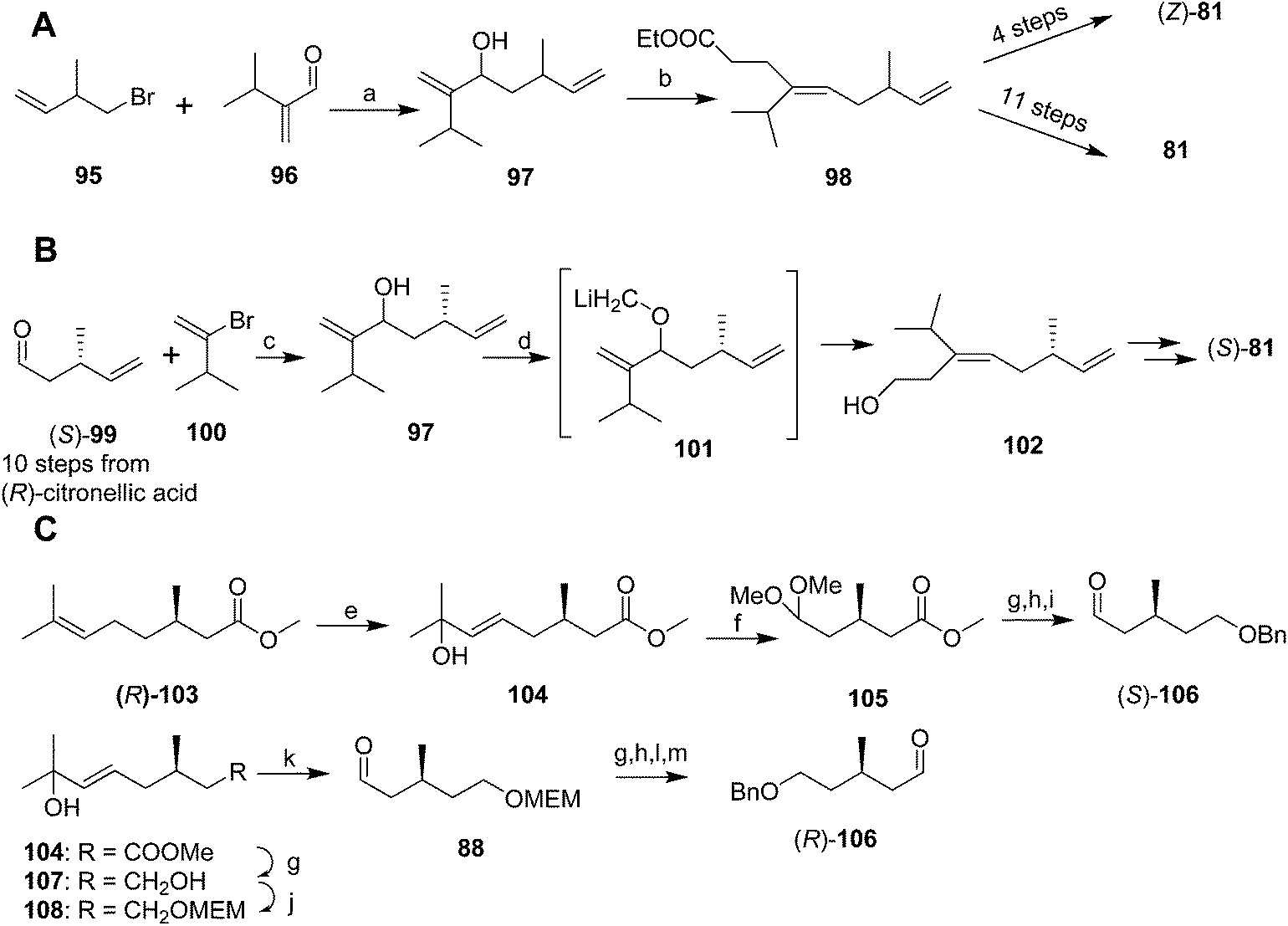 | ||
| Scheme 14 Three syntheses of yellow scale pheromone by Mori's group. Reagents and conditions: (a) Mg, ether, 34%; (b) ethyl orthoacetate, H+, 135 °C, 63%. (c) Mg, THF, 30%; (d) KH, 18-crown-6, THF, then ICH2SnBu3, then −78 °C, BuLi, 44%. (e) (PhSe)2, H2O2, CH2Cl2, then t-BuOOH, 83%; (f) O3, MeOH, −78 °C, then Me2S, then MeOH, PTSA, 83%; (g) LiAlH4, ether, 87%; (h) NaH, DME, PhCH2Br; (i) HClO4, THF/H2O, ∼quantitative. (j) NaH, MEMCl; (k) O3, MeOH, −78 °C, then Me2S; (l) MeOH, PTSA, 40 °C, 36% over 6 steps; (m) CrO3·2Py. | ||
Mori's second synthesis used enantiomerically pure (R)-citronellic acid, in turn generated from (R)-pulegone, as the source of chirality (Scheme 14B).52 Thus, aldehyde 99, generated in 10 steps from (R)-citronellic acid, was reacted with the Grignard reagent from 3-methyl-2-bromo-1-butene 100 (generated in 2 steps and 9% yield from 3-methyl-1-butene, and requiring spinning band distillation purification) to give chiral alcohol 97. O-Alkylation with iodomethyltributyltin and transmetallation with lithium to give 101 then induced a 2,3-Wittig rearrangement to yield (E)-alcohol 102 stereoselectively, which was elaborated to the (S)-enantiomer (S)-81 in the same sequence of steps used in their previous synthesis.
The third synthesis produced both pheromone enantiomers from methyl (R)-citronellate 103 (Scheme 14C).53 Thus, regiospecific Markovnikov addition of phenylselenenic acid (generated in situ from diphenyldiselenide and H2O2) to the alkene, followed by oxidation of the alkylphenylselenium intermediate and elimination gave allylic alcohol 104. Ozonolysis and reductive quenching with dimethylsulfide, and protection of the aldehyde as the dimethylacetal, followed by reduction of the ester and protection of the resulting alcohol as the benzyl ether yielded a chiral synthon 105 with differentiated terminals. Deprotection of the dimethyl acetal and reaction of the aldehyde (S)-106 with the Grignard reagent from vinyl bromide 100 provided the key intermediate required for the 2,3-Wittig rearrangement used in the previous synthesis to establish the (E)-trisubstituted double bond, and further elaboration to the final product. Alternatively, the primary alcohol of diol 107 was MEM protected, followed by ozonolysis, reduction of the resulting aldehyde (S)-88 with LiAlH4 to the alcohol, and protection as the benzyl ether, giving a second chiral synthon with differentiated ends. Removal of the MEM protecting group and oxidation gave the (R)-enantiomer of aldehyde 106, which was then carried through to (R)-81 as described above.
A synthesis from Julia's group controlled the stereochemistry of the trisubstituted double bond by stereoselective elimination of AcOH from a vicinal acetoxysulfone 114 (Scheme 15).54 Thus, (R)-3-methylvalerolactone 109, generated by desymmetrization of 3-methylpentanedioc acid dimethyl ester with pig liver esterase to give the (R)-half-ester, followed by selective reduction of the free acid with borane and acid-catalyzed closure of the lactone, was alkylated with the anion of sulfone 110 with attack at the carbonyl of the lactone, giving hydroxyketone 111. Protection of the alcohol, reduction of the ketone and acetylation gave vicinal acetoxysulfone 114, which stereoselectively eliminated AcOH when treated with powdered NaOH in ether to give the (E)-trisubstituted alkene 115 in 98% stereochemical purity. Reaction of the sulfone acetate 114 with isopropylmagnesium bromide with FeCl3 catalysis unfortunately gave an ∼1:1 mixture of the (E)-alkene 94 (with no loss of stereochemical purity) and the product from hydrogenolysis instead of alkylation. Deprotection and acetylation completed the synthesis of (S)-81, with separation of the desired trisubstituted alkene from the disubstituted byproduct by flash chromatography on silica gel impregnated with silver nitrate.
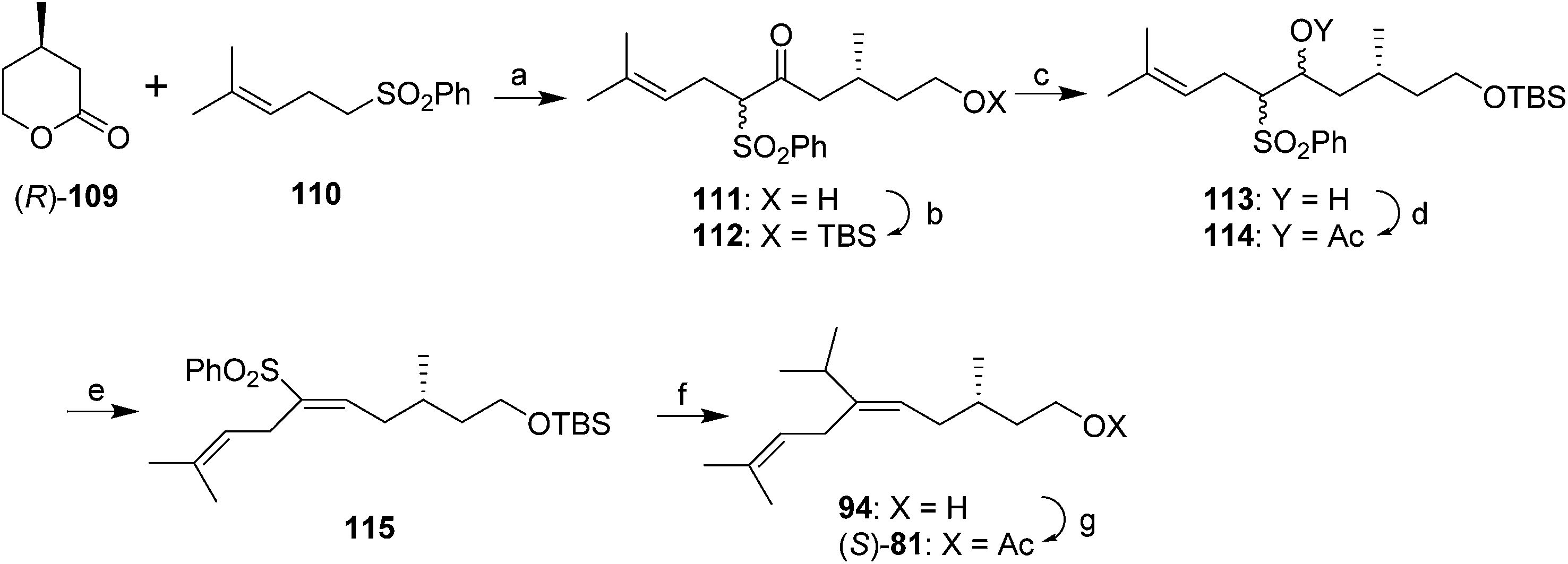 | ||
| Scheme 15 Julia's synthesis of yellow scale pheromone. Reagents and conditions: (a) BuLi, THF/TMEDA, 62%; (b) TBDMSiCl, Et3N, DMAP, CH2Cl2, 97%; (c) NaBH4, MeOH/H2O, 92%; (d) Ac2O, Et3N, DMAP, CH2Cl2, 95%; (e) powdered NaOH, ether, 94%; (f) i-PrMgCl, FeCl3, ether, −78–20 °C, then TBAF·3H2O, THF, 36%; (g) Ac2O, Et3N, DMAP, CH2Cl2, 96%. | ||
Millar developed a short, convergent, and highly stereoselective synthesis via silylcupration of an acetylene (Scheme 16).55 Thus, regiospecific addition of bis(phenyldimethylsilyl) lithium cuprate to 3-methyl-1-butyne 116 and trapping of the intermediate vinyl cuprate 117 with prenyl bromide gave 118 in 77% yield. Replacement of the R3Si group with iodine, and reaction of the resulting unstable vinyl iodide 119 with 4-chloro-2-methylbutylzinc iodide with (Ph3P)4Pd catalysis, and conversion of the crude coupling product 120 to the acetate 81 completed the synthesis, with complete control of the alkene stereochemistry. Although the racemate was prepared, the synthesis could be readily modified to produce either enantiomer by using chiral 4-chloro-2-methylbutyl iodide, preparable in a number of ways, to form the organozinc coupling partner.
 | ||
| Scheme 16 Millar's synthesis of yellow scale pheromone. Reagents and conditions: (a) (PhMe2Si)2CuLi, THF, −78 °C, then prenyl bromide, 77%; (b) I2, MeCN, 51%; (c) 4-chloro-2-methylbutylzinc iodide, (Ph3P)4Pd, THF; (d) NaOAc, HMPA, 75 °C, 42% from the vinyl iodide 119. | ||
In another approach to forming the trisubstituted double bond with excellent (E)-stereoselectivity, reaction of aldehyde 121 (made from (S)-citronellol in a route very similar to that used by Mori, see Scheme 14) with the t-butyl triethylsilylimine 122, prepared in two steps from 5-methylhex-4-enal, followed by elimination of triethylsilanol from the resulting β-hydroxysilane, gave alkenal 123 as an 85:15 E/Z mixture (Scheme 17).56 However, the mixture was readily isomerized to the pure (E)-isomer by treatment with pyridinium hydrochloride in CH2Cl2. 1,2-Addition of MeMgI to the aldehyde 123, oxidation of the resulting secondary alcohol to the ketone, and Wittig olefination gave the conjugated terminal diene 124. Regioselective hydrogenation of the terminal double bond in 124 with Wilkinson's catalyst and hydrogen, deprotection, and acetylation gave the pheromone (S)-81 in high stereochemical purity.
 | ||
| Scheme 17 Baudouy and Sancho's synthesis of yellow scale pheromone. Reagents and conditions: (a) s-BuLi, THF, −78–0 °C, then CF3COOH and water, 66%; (b) pyridinium hydrochloride, CH2Cl2, 100%; (c) MeMgI, ether, −70 to −35 °C; (d) (COCl)2, DMSO, Et3N, CH2Cl2; (e) MePh3PBr, BuLi, THF, 20 °C-reflux; 90% from the aldehyde; (f) DIBAL, solvent not stated, quantitative; (g) (Ph3P)3RhCl, H2, benzene, quantitative; (h) Ac2O, Et3N, solvent not stated, 94%. | ||
Harusawa et al. published essentially the same route three times as a demonstration of their method of synthesizing alkenes of defined stereochemistry by [3,3]-sigmatropic rearrangements of cyclic 8-membered thiocarbonate precursors (Scheme 18).57 Thus, methyl 4-methyl-2-butynoate 126 was stereoselectively converted to vinyl stannane 127. After reduction of the ester and protection of the resulting alcohol 128, the vinylstannane was converted to the corresponding iodide 130. After lithium–iodine exchange, the vinyllithium was reacted with aldehyde 131 (prepared in 3 steps and 54% yield from 3-methylglutaric anhydride) to give allylic alcohol 132. Treatment with LiHMDS resulted in formation of the 8-membered thiocarbonate 133 which immediately underwent stereoselective [3,3]-sigmatropic rearrangement to give the 10-membered thiocarbonate ring structure 134 with exclusively (Z)-stereochemistry. The high stereoselectivity was postulated to arise from the rearrangement proceeding via a less sterically congested chairlike transition state. The nascent prenyl sidechain was then constructed by deprotection of the allylic alcohol, oxidation to the aldehyde 136, and Wittig reaction with isopropenyltriphenylphosphorane. Reductive desulfurization by lithium in ammonia with a proton source, or lithium p,p′-di-t-butylbiphenylide and HMPA gave the desired 1,4-diene 96 along with ∼20–33% of the conjugated diene 138. Acetylation completed the synthesis, and the pheromone 81 was separated from the conjugated impurity 139 by medium pressure chromatography. In their third publication (not shown),57c the authors streamlined the route somewhat by substituting the (R)-enantiomer of aldehyde 131 (generated in 4 steps by desymmetrization/ring opening of 3-methylglutaric anhydride with lipase PS and n-BuOH to give the (R)-half-ester in 86% ee, and functional group manipulations), altering the order of steps, and by allowing the alkoxide from reaction of a vinyl anion with aldehyde 131 to warm to 0 °C so that the thiocarbonate ring formed and spontaneously underwent the [3,3]-sigmatropic rearrangement.
 | ||
| Scheme 18 Synthesis of racemic yellow scale pheromone via [3,3]-sigmatropic rearrangement of a cyclic thiocarbonate. Reagents and conditions: (a) Me3SnCu·SMe2, THF, −78 °C, 87%; (b) DIBAL, pentane/toluene, ∼-45 °C, 82%; (c) TBDMSiOTf, pyridine, quantitative; (d) I2, ether, quantitative; (e) t-BuLi, ether, −78 °C; (f) LiHMDS, THF, 72% from stannane 128; (g) TBAF, THF, 97%; (h) (COCl)2, DMSO, CH2Cl2, −78 °C, then Et3N; (i) i-PrPh3PBr, BuLi, ether, 46–60% from alcohol 135; (j) Li, NH3 or LDBB, THF/HMPA; (k) Ac2O, DMAP, CH2Cl2, 60% over 2 steps. | ||
Finally, Negishi and coworkers developed an efficient, convergent synthesis that exploited two synthetic methods developed by their group (Scheme 19).58 First, they used their ZACA reaction (zirconium-catalyzed asymmetric carboalumination of alkenes) to enantioselectively add a methyl group to silyl-protected 3-buten-1-ol 140. The product 141, obtained in 72% ee, was readily upgraded to 98% ee by a lipase-catalyzed kinetic resolution. The resulting alcohol (R)-141 was then converted to an iodide, ready for coupling. To construct the key trisubstituted alkene, addition of BBr3 across the triple bond of 3-methyl-1-butyne 116, Pd catalyzed replacement of the bromide on carbon 2 with prenylzinc bromide, and conversion of the resulting alkenyl boron dibromide to the vinyl iodide 119 proceeded with complete regio- and stereochemical control, in 77% yield. The first synthon was then lithiated and transmetallated with ZnBr2 to give 142. Pd-catalyzed reaction between organozinc 142 and vinyl iodide 119 then provided the completed carbon skeleton in very high stereochemical purity. Removal of the silyl group and acetylation completed the synthesis, providing (S)-81 in 34% yield over 6 steps. As an interesting side note, during the course of the development and optimization of the coupling between allylzinc bromides and bromoborane intermediates from addition of BBr3 to terminal alkynes (e.g., 143 + 144 → 119), the authors found that allylzinc bromide itself did not react, but any other allylzinc bromide with substituents on either the 2 or 3 positions, reacted readily.
 | ||
| Scheme 19 Negishi's synthesis of yellow scale pheromone. Reagents and conditions: (a) (−)-dichlorobis-(neomenthylindenyl)zirconium [(NMI)2ZrCl2], Me3Al, CH2Cl2, −78–23 °C, then O2, 82%, 72% ee; (b) Amano lipase PS, vinyl acetate, CH2Cl2, 63%, 98% ee; (c) I2, imidazole, Ph3P, CH2Cl2, 97%; (d) t-BuLi, ether, −100 °C, then ZnBr2, THF, −78–23 °C; (e) BBr3, CH2Cl2, −78–23 °C; (f) (Ph3P)2PdCl2; (g) I2 in THF, then aq. NaOAc, THF, 77%; (h) (Ph3P)4Pd, THF, 23 °C; (i) TBAF, THF, 74% from iodide; (j) Ac2O, pyridine, CH2Cl2, 92%. | ||
Pheromone components of California red scale. The pheromone of California red scale was the first pheromone identified from the insect order Hemiptera, using material from an astonishing 400 million female-days of collection.59 The identification made extensive use of microchemical tests, along with 1H NMR. The pheromone consists of diene 82 and triene 83. The insect produces the (3S,6R)-enantiomer of 82, but the (3S,6S)-diastereomer is also active, and in fact, males respond well to the full blend of 4 stereoisomers, indicating that the three unnatural isomers are not inhibitory. The more unsaturated compound 83 also has the (6R)-configuration.
The challenges in synthesizing the two compounds are different because the diene component has two chiral centers but no stereochemistry associated with the double bonds, whereas the triene component has a single chiral center, and a trisubstituted double bond in which the stereochemistry must be controlled. Consequently, despite their superficial similarity, quite different synthetic strategies and intermediates were used in their syntheses.
Diene component of the California red scale pheromone. The first reported synthesis of the diene component 82 as a diastereomeric mixture used an AlCl3-catalyzed ene reaction of citronellyl acetate 146 and methyl propiolate 147 to give diester 148 (Scheme 20).60a Selective reduction of the conjugated double bond, followed by a 4-step sequence to add the additional carbon and convert it to a terminal double bond (reaction of the methyl ester with phenylsulfonylmethyllithium to add one carbon, reduction of the resulting ketone to the secondary alcohol, acetylation, and reduction of the geminal sulfone acetate with sodium amalgam in EtOH) and reacetylation to replace the acetate which had come off during this sequence, completed the synthesis of 82 in about 17% overall yield. Another route using an ene reaction between citronellyl acetate and formaldehyde gave the desired alcohol product in good yield, but attempts to convert this alcohol to the pheromone produced only complex mixtures of products (not shown).60b
 | ||
| Scheme 20 First synthesis of the diene component of the red scale pheromone. Reagents and conditions: (a) AlCl3, benzene, 55%; (b) Fe(CO)5, NaOH, MeOH/H2O, 80%; (c) PhSO2CH2Li, 80%; (d) NaBH4, EtOH, 86%; (e) acetylation, conditions not stated; (f) Na amalgam, EtOH; (g) acetylation, conditions not stated, 56% over 3 steps. | ||
A cleaner and more practical synthesis of 82 by the authors who identified the pheromone made clever use of available chiral synthons by first developing a short nonstereoselective synthesis from racemic citronellol to prove the basic structure (Scheme 21A).61 Thus, the epoxide of (±)-citronellyl acetate 150 (or citronellyl pivalate) was reacted with di(3-butenyl)lithium cuprate to regioselectively open the epoxide, and the resulting tertiary alcohol 151 was then mesylated followed by elimination in the same pot, giving a 4:1 mixture of the desired isomer 82 with the tetrasubstituted double bond isomer. The latter was removed by selective epoxidation of the more electron rich tetrasubstituted double bond and chromatographic separation of the epoxide 152 from the desired product 82. When the acetate ester was used in the alkylation step, the acetate was partially removed, requiring reacetylation of the crude product, whereas this was not the case with the pivalate ester, but with the pivalate route, the pivalate had to be removed later and replaced with acetate.
 | ||
| Scheme 21 Anderson's syntheses of racemic and chiral diene 82. Reagents and conditions: (a) di(3-butenyl)lithium cuprate, ether, −20–4 °C, then Ac2O, pyridine, 46%; (b) MesCl, Et3N, 0 °C-room temp; (c) m-CPBA, CHCl3, room temp, prep. TLC, 73%. (d) HClO4, THF/H2O, 67%; (e) Mosher's acid chloride, DMAP, MeCN, then HPLC; (f) LiAlH4, THF, 73%; (g) MesCl, Et3N, CH2Cl2; (h) KOH, MeOH, 62% over 2 steps; (i) di(3-butenyl)lithium cuprate, ether, –20–4 °C, 70%; (j) MesCl, Et3N; 97%; (k) BuLi, hexane, then H3O+, quant.; (l) Ac2O, Et3N, ether, then m-CPBA, CH2Cl2, room temp, prep. TLC, 37%. | ||
The synthesis was then repeated using (R)- and (S)-citronellols, giving diastereomeric mixtures of (3R,6R/S)- and (3S,6R/S)-isomers respectively. Only the latter was attractive to male scales, and so the epoxide from MEM-protected (S)-citronellol 153 was opened to give diastereomeric diols 154, the Mosher's ester derivatives of which (155) were separable by HPLC (Scheme 21B). Reduction of the purified (3S,6R)-hydroxy ester to the diol 154, mesylation, and ring closure to reform the epoxide (3R,6R)-153, followed by the alkylation and elimination sequence as before, gave intermediate 157. Although removal of the MEM protecting group by standard methods failed, the authors developed a two-step sequence of treatment with strong base followed by aqueous acid that delivered the alcohol (3S,6R)-158 in quantitative yield. Routine acetylation completed the synthesis, and the resulting (3S,6R)-82 exactly matched the naturally produced compound. The (3S,6S)-diastereomer was made in analogous fashion from the other hydroxyester diastereomer.
Baudouy and Maliverney exploited (−)-dihydrocarvone 159 as a source of both stereocenters in the pheromone (Scheme 22).62 Thus, straightforward manipulations transformed (−)-dihydrocarvone 159 into bromoester 161. DIBAL reduction of the ester yielded the corresponding alcohol with no epimerization at the adjacent chiral center. The authors initially attempted to alkylate the bromoalcohol with allylmagnesium chloride prepared from the typical 98% pure Mg sold for making Grignard reagents, with CuI catalysis, resulting in 65% of the desired coupling product plus 20% of a byproduct from transmetallation of the bromide. This byproduct was completely eliminated by using high purity Mg (99.8%) to prepare the Grignard reagent, and by carrying out the coupling in refluxing DME without the CuI catalyst. The synthesis was then completed by a straightforward sequence (conversion of the alcohol to iodide 162, one-carbon chain extension with NaCN, hydrolysis to the acid, reduction, and acetylation). Alternatively, iodide 162 was chain extended by reaction with the anion of phenylmethylsulfide, followed by reaction with MeI, NaI, and DMF to give the chain-extended iodide, which was converted to acetate (3S,6R)-82 by treatment with tetramethylammonium acetate in DMF.
 | ||
| Scheme 22 Synthesis of (3S,6R)-82 from (−)-dihydrocarvone. Reagents and conditions: (a) LDA, Me3SiCl, DME; (b) O3, CH2Cl2/MeOH, then NaBH4; (c) CH2N2, ether, 90% over 3 steps; (d) MesCl, Et3N, ether; (e) Bu4NBr, benzene; (f) DIBAL, heptane, 0 °C; (g) allyl MgCl, DME, reflux, 72%; (h) I2, imidazole, Ph3P, MeCN, 98%; (i) NaCN, DMF, 95%; (j) NaOH, EtOH/H2O; (k) LiAlH4, ether, 90% over 2 steps; (l) Ac2O, pyridine, 94%. | ||
Another stereoselective synthesis used (R)-limonene as the source of the chiral isopropenyl group at C6, with the second chiral center at C3 being generated by an asymmetric Michael reaction, giving a product with a 9:1 d.r. (not shown).63
Dragan and coworkers used some interesting and unusual chemistry to devise a synthesis of the racemic pheromone from citronellyl acetate 146 (Scheme 23).64 Thus, treatment of acetate 146 with DMSO and triflic anhydride gave allylic sulfide 163, presumably via nucleophilic attack of the alkene on the activated acylsulfoxonium cation from DMSO. Treatment with strong base then resulted in a 1,2-Stevens rearrangement, possibly as shown in Scheme 23. Oxidation of the resulting sulfide 165 to the sulfone 166, reduction of the acetate to the alcohol 167, deprotonation with 5 equivalents of BuLi and reaction with 5 equivalents of allyl bromide gave the trialkylated product 168. The large excesses were necessary in order to produce a reasonable monoalkylation yield at the desired methylene position. Reductive removal of the sulfone and acetylation completed the synthesis.
 | ||
| Scheme 23 Synthesis of racemic red scale diene component from citronellyl acetate. Reagents and conditions: (a) DMSO, (CF3CO)2O, CH2Cl2, −10 °C, 90%; (b) MeONa, MeOH, reflux, 59%; (c) reacetylate, conditions and yield not given; (d) m-CPBA, ether, −40 °C-room temp, 77%; (e) LiAlH4, THF, −20 °C, 95%; (f) Excess BuLi, excess allyl bromide, THF/HMPA, −60 °C, 61%; (g) Na, NH3/THF, 52%; (h) conditions and yield not stated. | ||
A few additional modifications have appeared since 1989. For example, Zhang et al. stereoselectively dihydroxylated (S)-citronellyl N-phenylcarbamate with the fungus Aspergillus niger.65 The resulting diol could be converted in several steps to the (3S,6S)-epoxide of citronellyl acetate, an intermediate used in previous syntheses (150, Scheme 21). Auer et al. developed a 14-step synthesis from (R)-(+)-limonene in which a key step was electrolysis of an iodide intermediate derived from limonene with a very large excess (160 equiv) of methyl crotonate.66 The resulting 2:1 mixture of the desired product with a cyclic byproduct were only separable by HPLC, limiting the utility of the synthesis. Kefalas and Ragoussis reported a 3-step synthesis from (S)-citronellyl acetate 146 (Scheme 24),67 using regioselective allylic oxidation, conversion of the resulting alcohol 169 to chloride 170, and SN2′ alkylation of the chloride using 3-butenylmagnesium bromide with copper catalysis, giving the SN2′ (82) and SN2 products in a 20:1 ratio. The stereochemistry at C6 was not controlled, but reports had indicated that male scale were attracted to both the (3S,6R)- and (3S,6S)-diastereomers, so this did not appear to be an issue for practical use.
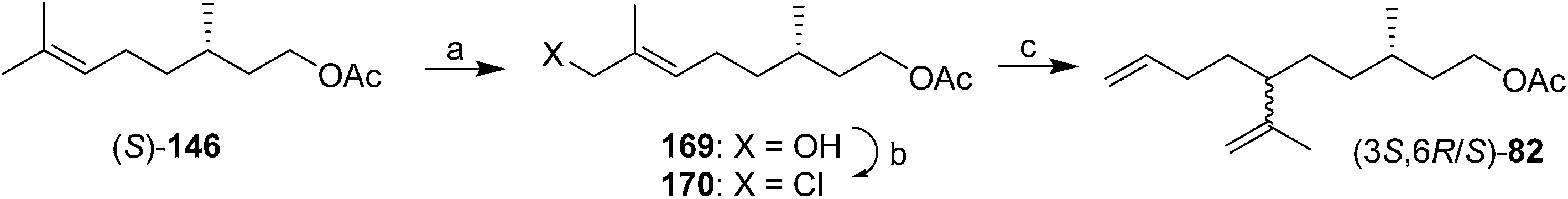 | ||
| Scheme 24 3-step synthesis of technical grade diene 82. Reagents and conditions: (a) SeO2, t-BuOOH, then NaBH4, 52%; (b) Ph3P, CCl4, 75%; (c) 3-butenylmagnesium bromide, CuCN·2LiCl, THF, −78 °C, 78%. | ||
Triene component of the California red scale pheromone. The first synthesis of the triene component of the red scale pheromone 83 was designed to be flexible enough to produce all 4 stereoisomers (Scheme 25).59 (R)- and (S)-carvone were chosen as suitable sources of the chiral center. Thus, epoxidation of (S)-carvone 171 followed by hydrolysis of the epoxide 172 and Pb(OAc)4 oxidation gave ester acetal 174. Treatment with triethyl orthoformate and acid in EtOH opened the ring and gave a synthon 175 with differentiated ends. Reduction of the ester, conversion of the resulting alcohol to bromide 176, and two-carbon chain extension with ethenyllithium and deprotection gave dienal 177, which was subjected to Wittig reaction to complete the carbon skeleton, followed by acetylation. The (Z)- and (E)-isomers of 83 were separated by preparative GC, and the opposite enantiomers were generated from (R)-carvone. The (R,Z)-isomer matched the insect-produced compound.
 | ||
| Scheme 25 First nonstereoselective synthesis of the triene component of red scale pheromone. Reagents and conditions: (a) H2O2, NaOH, MeOH, 77%; (b) HClO4, THF/H2O, 65 °C, 27%; (c) Pb(OAc)4, EtOH/benzene; 98%; (d) HC(OEt)3, PTSA, EtOH, 94%; (e) LiAlH4, THF, 98%; (f) TosCl, pyridine, 90%; (g) NaBr, HMPA; 93% (h) CH2CHLi, THF, 78%; (i) PTSA, acetone/water, 96%; (j) Ph3PC(CH3)CH2CH2OLi, THF, 49%, Z/E ∼ 1:1; (k) Ac2O, pyridine, 92%. | ||
Since that first synthesis, a number of other strategies have been explored, using synthons derived from the chiral pool (e.g., enantiomers of carvone or limonene), or induction of asymmetry in achiral precursors. Control of the stereochemistry of the trisubstituted alkene also has been addressed by a number of different approaches.
In the first general strategy exploiting the chiral pool, dienal 177 or the corresponding alcohol have been used in several routes. For example, two routes used chiral aldehyde 177 generated from (R)-limonene as a key intermediate. In the first, Wittig reaction between aldehyde 177 and the ylide from 3-hydroxy-1-methylpropyltriphenylphosphonium iodide, analogous to the route used by Roelofs et al.59 gave a 67:33 mixture of the (Z)- and (E)-isomers of the penultimate alcohol in 75% yield (not shown).63 The second route achieved excellent control over the alkene geometry, but at the cost of a considerably longer route (Scheme 26).68 Thus, reaction between aldehyde 177 and silylimine 179 (generated in 4 steps from 1,4-butanediol in 63% yield) followed by hydrolysis gave α,β-unsaturated aldehyde 180 in an 87:13 ratio favoring the desired (E)-isomer, and the undesired (Z)-isomer was cleanly isomerized to the (E)-isomer by treatment of the mixture with pyridinium hydrochloride. After reduction of the aldehyde to the corresponding alcohol 181, the synthesis was completed by one of two sequences in similar yields, i.e., reductive removal of the benzyl protecting group, selective conversion of the allylic alcohol of 182 to the chloride, reduction of the chloride to the methyl group, and acetylation, or conversion of the allylic alcohol to a sulfate leaving group, reduction to the methyl, and simultaneous removal of the benzyl protecting group and acetylation with acetic anhydride with perchloric acid catalysis (not shown). Chiral dienal 177 also has been produced from camphor, but in a very long route (13 steps).69
 | ||
| Scheme 26 Baudouy and Prince's route to (R)-83. Reagents and conditions: (a) s-BuLi, THF, −78–0 °C, then aq. oxalic acid; pyridinium hydrochloride, CH2Cl2, 66%; (b) LiAlH4, ether, 100%; (c) Li/NH3/ether, 88%; (d) NCS, Me2S/CH2Cl2; (e) LiAlH4, ether; 85% over 2 steps; (f) Ac2O, pyridine, 94%. | ||
In an interesting comparative study, Whittaker and coworkers used three different methods of asymmetric induction to generate key dienal 177 enantioselectively.70 The first (Scheme 27A) began with two-carbon chain extension of 4-pentenal 183 with malonic acid and pyridine/pyrrolidine. Although the acid 184 was obtained as an E/Z mixture, the conditions used for conversion to the acid chloride 185 resulted in isomerization of the (Z)-isomer, producing pure (E)-2,6-heptadienoyl chloride 185. Reaction with (L)-ephedrine in the presence of 1,8-bis(dimethylamino)naphthalene (proton sponge®) gave α,β-unsaturated amide 186. 1,4-Addition of 2-propenylmagnesium bromide and cleavage of the amide with DIBAL gave the aldehyde (R)-177, with an unfortunate 21% yield in the cleavage step. The second route (Scheme 27B) proceeded via addition of lithiated 2,4,4,6-tetramethyl-5,6-dihydro-1,3-oxazine 188 to 4-pentenal 183. Reduction of the oxazine 189 to the cyclic aminal 190 and acid hydrolysis gave 2,6-heptadienal 191 as the pure (E)-isomer. This route was selected from among several tried because the others had problems with low yield, awkward byproducts, or incomplete control of the double bond stereochemistry. Reaction of enal 191 with t-butyl (S)-2-amino-3,3-dimethylbutyrate 192 gave conjugated imine 193. Enantioselective addition of isopropenylmagnesium bromide created the chiral center, followed by hydrolysis of the imine to aldehyde (R)-177. A third route (not shown) used an oxazoline chiral auxiliary to successfully introduce the chiral center, but it proved impossible to remove the auxiliary group without degradation of the product.
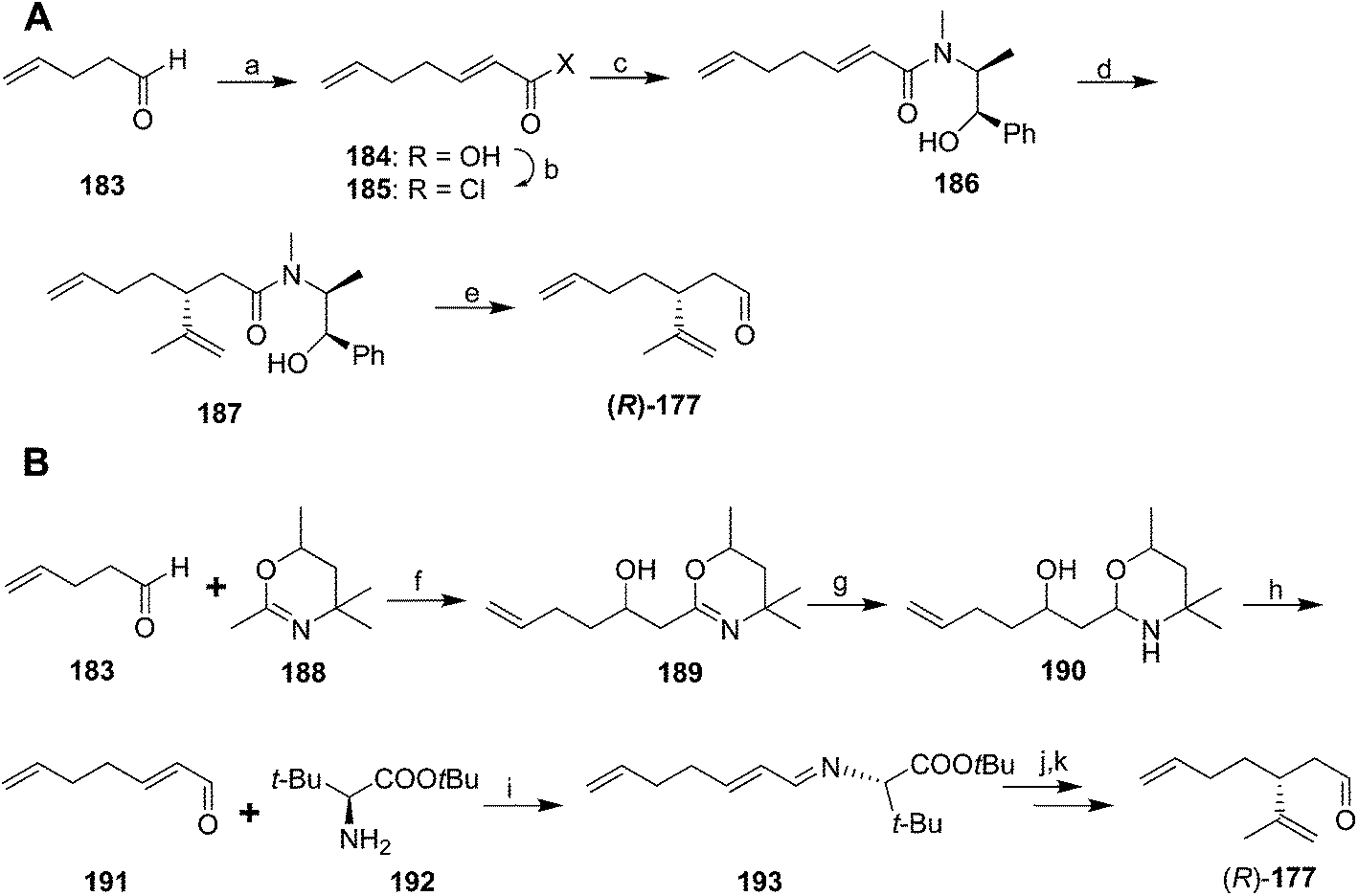 | ||
| Scheme 27 Two routes to chiral aldehyde (R)-177 based on asymmetric induction. Reagents and conditions: (a) malonic acid, pyridine, pyrrolidine, reflux, 79%; (b) SOCl2, 0 °C-reflux, 94%; (c) (L)-ephedrine, proton sponge®, THF, 88%; (d) isopropenylmagnesium bromide, THF/ether, −60 to −20 °C, 51–69%; (e) DIBALH, hexane, then MeOH, H3O+, 21%. (f) BuLi, THF, −78 °C to room temp, 83%; (g) NaBH4, pH 6–8, 98%; (h) H3O+, reflux, 45%; (i) t-butyl (S)-2-amino-3,3-dimethylbutyrate, benzene, 0 °C; (j) isopropenylmagnesium bromide, THF/ether, −70 to −15 °C; (k) H3O+, 43% over 3 steps. | ||
In yet another variation, Normant's group formed a chiral acetal with a twofold axis of symmetry by transacetalization of (E)-2,6-heptadienal diethyl acetal with (2R,3R)-2,3-butanediol. 1,4-Addition of isopropenyl copper in the presence of LiBr, BF3, and Bu3P followed by acidic hydrolysis gave key aldehyde 177, but in only 85% ee (not shown).71
Racemic dienal 177 has been generated by reaction of 4-pentenal 183 with the stabilized ylid from triethylphosphono-2-propionate, reduction to the alcohol 195, Claisen orthoester rearrangement with triethylorthoacetate via196, reduction to the alcohol 198, and PCC oxidation to dienal 177 (Scheme 28).72
 | ||
| Scheme 28 Synthesis of racemic aldehyde 177 by Claisen rearrangement. Reagents and conditions: (a) triethyl phosphono-2-propionate, NaH, ether, 0 °C, 74%; (b) LiAlH4, ether, 0 °C, 81%; (c) (EtO)3CH, H+, 140 °C, 82%; (d) LiAlH4, ether, 0 °C, 81%; (e) PCC, CH2Cl2, 55%. | ||
A second group of strategies focused on controlling the stereochemistry of the trisubstituted double bond. In the first synthesis of this type, Still and Mitra developed a short and efficient synthesis in which the stereochemistry of the trisubstituted double bond was controlled by the geometry of the transition state of a 2,3-Wittig rearrangement (Scheme 29A).73 Thus, ethyl 3,3-dimethacrylate 199 was deconjugatively alkylated at the α-position with 1-bromo-3-butene, and the ester 200 was reduced to the alcohol 201 which was converted to the corresponding bromide 202. Reaction of the Grignard reagent from bromide 202 with methacrolein 57 gave allylic alcohol 203, the anion of which was alkylated with iodomethyltributyltin to give 204. Transmetallation with BuLi then induced a 2,3-sigmatropic rearrangement, giving the desired (Z)-isomer of the alcohol precursor 178 to the pheromone with 95–96% isomeric purity. The high stereoselectivity was suggested to be a consequence of the large R-group in the 5-membered transition state adopting a pseudoaxial orientation for steric reasons. The synthesis was completed by acetylation.
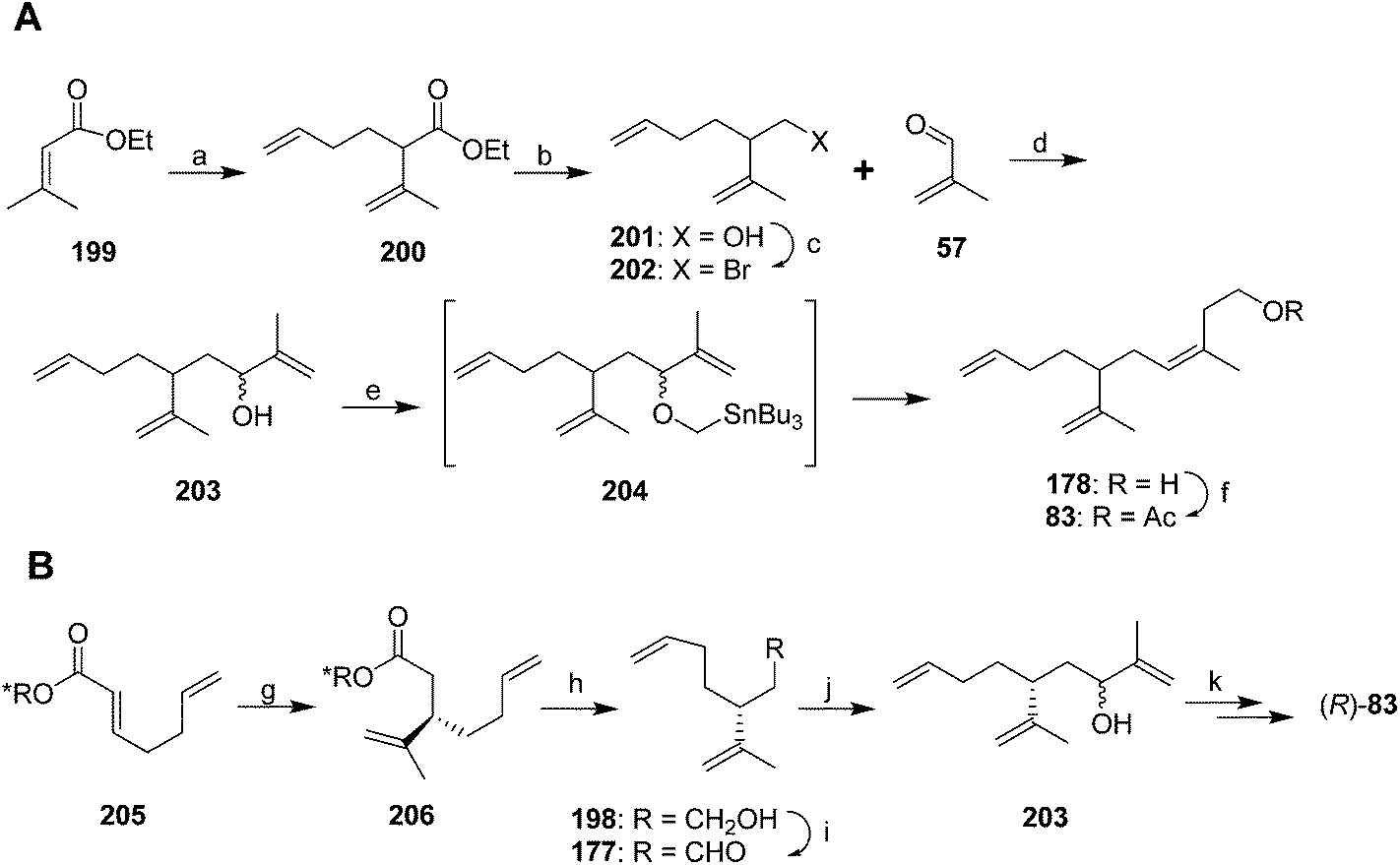 | ||
| Scheme 29 Syntheses of racemic and chiral triene 83 based on 2,3-Wittig rearrangements. Reagents and conditions: (a) LDA, THF/HMPA, 1-bromo-3-butene, −78 °C, 80%; (b) LiAlH4, solvent not given; (c) Ph3P, NBS, solvent not given, 93% over 2 steps; (d) Mg, ether, −20 °C, 52%; (e) KH, Bu3SnCH2I, THF, then BuLi, −78 °C; (f) Ac2O, pyridine, 83% over 2 steps. (g) CH2CH(Me)Cu·PBu3, BF3, −78–40 °C, >90%; (h) LiAlH4, 96%; (i) (COCl)2, DMSO, −78 °C, then Et3N, 85%; (j) CH2CCH3Li, ether, -78 °C, 85%; (k) steps, ∼71%. | ||
Oppolzer and Stevenson generated intermediate 203 by enantioselective alkylation of (E)-acid 205 esterified with a Re-face directing chiral auxiliary (Scheme 29B).74 Reaction with CH2CH(Me)Cu·PBu3 and BF3, followed by reduction to the alcohol 198, oxidation to the aldehyde 177, and reaction with 2-propenyllithium gave the key intermediate 203 from the Still and Mitra synthesis but with the chiral center fixed. Alcohol 203 was then carried through to the final product (R)-83 (98.2% ee) via the (Z)-selective 2,3-sigmatropic rearrangement developed by Still and Mitra (see above). The key allylic alcohol 203 has also been generated in 14 steps from camphor.69
Cooke and Burman used a trisubstituted alkene synthon with the desired stereochemistry in place rather than trying to control the stereochemistry during reactions (Scheme 30).75 Thus, spinning band distillation of dimethyl glutaconate gave the (Z)-isomer 207 in high purity, which was reduced to diol 208. Regioselective halogenation and protection of the homoallylic alcohol gave key intermediate 209. The second required intermediate, ylide 212, was formed by acylation of stabilized triphenylphosphorane 211, followed by elimination of HCl to give the terminal enone 212 as a methyl vinyl ketone equivalent. Sequential conjugate addition of allyllithium and alkylation of the resulting enolate 213 with bromide 209 inserted two of the three alkenes. Decarbalkoxylation and simultaneous replacement of the THP with acetate by treatment with hot AcOH, followed by cleavage of the acetate with K2CO3 in MeOH and the ylide with aqueous ethanolic NaOH gave ketone 215. Acetylation and Wittig reaction to install the terminal methylene completed the synthesis.
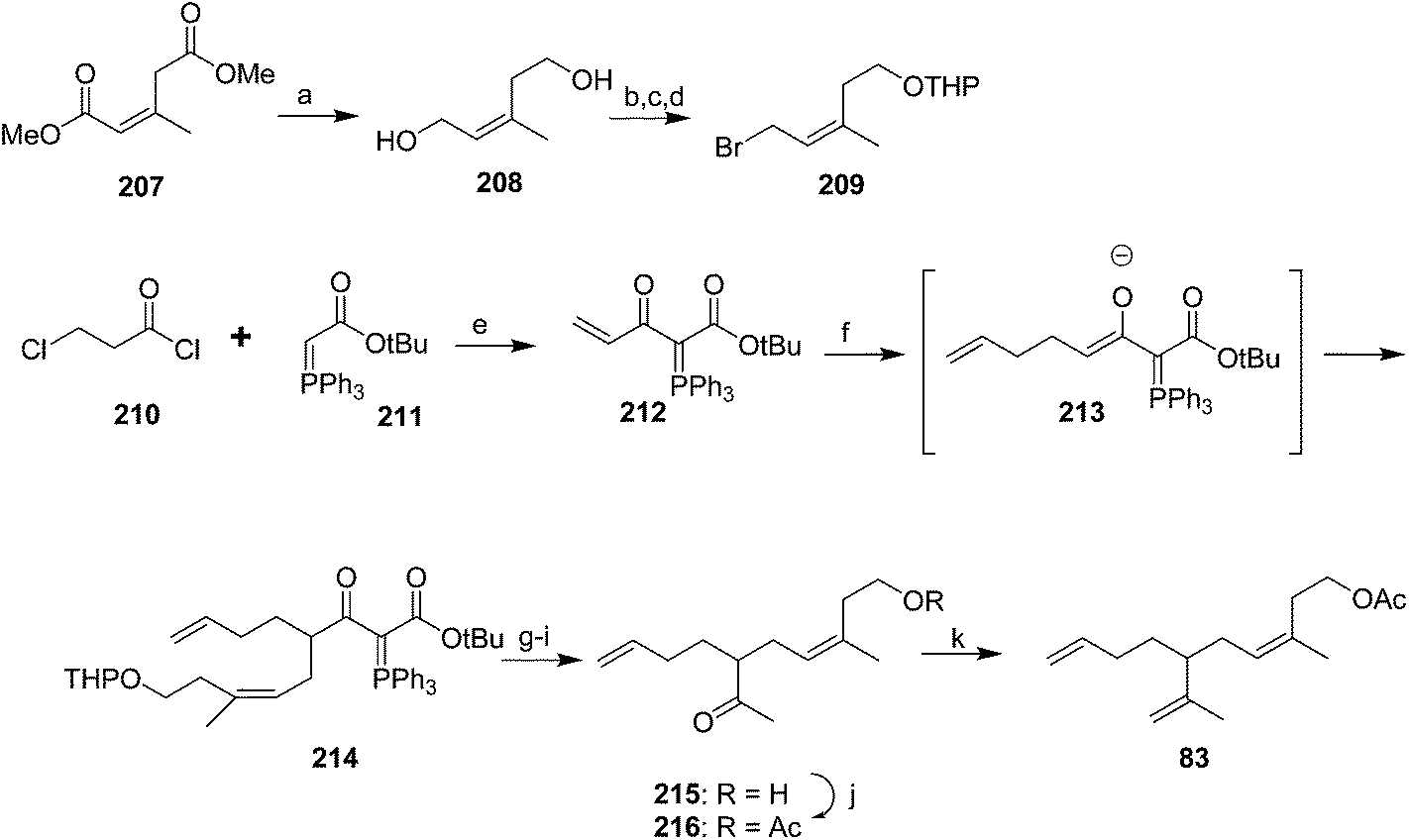 | ||
| Scheme 30 Synthesis of triene 83 from a synthon with the double bond geometry already in place. Reagents and conditions: (a) AlH3, solvent not given; (b) NCS, Me2S; (c) dihydropyran, H+, 74%; (d) NaBr, N-methylpyrrolidinone, 72%; (e) benzene, 5–25 °C, 91%, then NaOMe, MeOH, 74%; (f) (allyl)4Sn, BuLi, THF, −78–0 °C, then 212, 0–27 °C, 87%; (g) AcOH, 90 °C, 86%; (h) K2CO3, MeOH, 97%; (i) aq. EtOH, pH 9–10, reflux, 84%; (j) Ac2O, pyridine, 92%; (k) Ph3PCH3Br, BuLi, THF, 57%. | ||
In their stereoselective synthesis of the trisubstituted alkene (R)-83 (Scheme 31),76 Caine and Crews regioselectively opened the epoxide ring of epoxidized (S)-carvone 172 with thiophenol, and under the conditions of a Wittig olefination, the resulting keto alcohol underwent an in situ retroaldol condensation, with the resulting aldehyde being captured by the ylide from 3-hydroxy-1-methylpropyltriphenylphosphonium bromide. The high (Z)-selectivity was attributed to unfavorable electrostatic interactions between three negatively charged groups in the betaine intermediate that would lead to the (E)-isomer. Although the stereochemistry of the trisubstituted alkene was controlled, 5 further steps were required to remove the keto and sulfide groups and place the terminal alkene, the final product was contaminated with 11% of the isomer with the terminal double bond moved in one position, and yields were not given for all steps, so the efficiency of the synthesis is unclear.
 | ||
| Scheme 31 Synthesis of triene (R)-83 from a synthon with the double bond geometry already in place. Reagents and conditions: (a) PhSH, Et3N, MeCN; (b) 3-hydroxy-1-methylpropyltriphenyl-phosphonium bromide, BuLi, THF/HMPA, 0 °C, 83%; (c) Ac2O, pyridine, room temp; (d) m-CPBA, CH2Cl2, −5 °C, 85% over 2 steps; (e) 135 °C, 0.15 mm Hg; (f) NaBH4, CeCl3·7H2O; (g) Ac2O, pyridine, 52% over 3 steps; (h) HCO2NH4, PdCl2·(PPh3)2, dioxane, reflux. | ||
Celebuski and Rosenblum exploited the ability of (η1-allyl) ferrocenyliron complexes to undergo electrophilic addition to form a dihapto iron-olefin cation, followed by regio- and stereoselective addition of a nucleophile, to simultaneously install both the trisubstituted double bond and one of the two terminal double bonds (Scheme 32).77 Thus, reaction of allylic chloride 221, prepared in one step from commercial diene 220, with C5H5Fe(CO)2Na (FpNa, prepared from Fp2 by reduction with Na/Hg amalgam) gave allylic organometallic 222via SN2′ and/or spontaneous rearrangement of the product from SN1 reaction. The second synthon was prepared in two steps from 3,6-dihydro-4-methyl-2H-pyran 223, by ring-opening with acetyl chloride catalyzed by Zeise's salt, and conversion of the resulting acetoxychloride 224 to the iodide 225 with NaI in acetone. Fortuitously, stirring the 1:1 mixture of (E/Z)-isomers of 225 with the Fp-diene complex 222 in nitromethane for several days resulted in exclusive production of the desired (Z)-isomer of the pheromone because only the (Z)-isomer of iodide 225 reacted with the complex, and the iodide apparently isomerized under the reaction conditions so that as the (Z)-isomer was consumed, it was replaced by continuing isomerization of the (E)-isomer. The specificity of the alkylation reaction was proposed to be due to anchimeric assistance to ionization by the acetoxy group, which would be possible with the (Z)- but not the (E)-isomer of iodide 225.
 | ||
| Scheme 32 Synthesis of racemic triene 83 from a synthon with the double bond geometry already in place. Reagents and conditions: (a) Ca(OCl)2, CH2Cl2/H2O, 73%; (b) Fp2, Na/Hg, DME, 57%; (c) AcCl, toluene, K[PtCl3(C2H4)], 75%; (d) NaI, acetone, no yield reported; (e) 225 + 222, nitromethane, room temp, 42%. | ||
McCullough et al. cleverly manipulated the regiochemistry of alkylation of allyl anion intermediates in their synthesis of racemic 83 (Scheme 33).46 Thus, deprotonation of methallyl phenyl sulfide and regioselective α-alkylation of the anion 226 with 1-bromo-3-butene gave 227. Reductive lithiation with lithium 4,4′-di(tert-butyl)biphenyl (LDBB), transmetallation with titanium, and reaction with formaldehyde resulted in regioselective alkylation at the most substituted terminus of the metallated intermediate. Conversion of the resulting alcohol 201 to bromide 202, followed by another regioselective alkylation with lithiated methallyl phenyl sulfide 226 at the α-position gave allylic sulfide 228. Reductive lithiation with LDBB, transmetallation with CeCl3, and reaction with formaldehyde resulted in both regioselective alkylation at the least substituted terminus this time, and high selectivity for the (Z)-alkene, due to the configuration of the anion remaining fixed by complexation with cerium. Acetylation completed the synthesis.
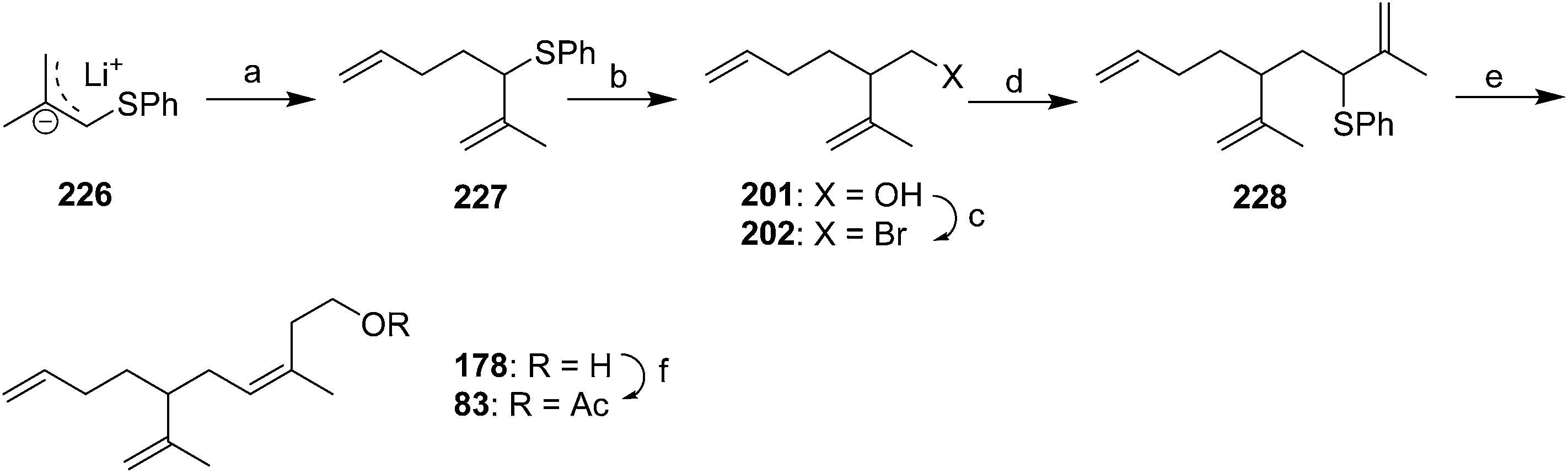 | ||
| Scheme 33 Synthesis of racemic triene 83 by regiocontrol of alkylation. Reagents and conditions: (a) 1-bromo-3-butene, s-BuLi, THF, −78 °C, 85%; (b) lithium 4,4′-(di-t-butyl)biphenyl (LDBB), THF, −78 °C, then Ti(i-PrO)4, then CH2O, 79%; (c) CBr4, PPh3, MeCN, 81%; (d) methallyl phenyl sulfide, s-BuLi, THF, −78 °C, 78%; (e) LDBB, THF −78 °C, then CeCl3, then CH2O; (f) Ac2O, pyridine, 54%. | ||
Vasil'ev et al. addressed the alkene stereochemistry problem with a (Z)-selective, 1,4-dihydrogenation of a conjugated diene precursor (Scheme 34).78 Thus, aldehyde 229, generated in ∼34% yield in 4 steps from ethyl acetoacetate, subjected to Wadsworth–Horner–Emmons olefination gave triene 230 in ∼79% yield, as a 65:35 mixture of (2E,4E)- and (2Z,4E) isomers. 1,4-Dihydrogenation of this mixture with a methyl benzoate-chromium tricarbonyl catalyst gave the desired (3Z)-alkene 231 as the sole product, which was carried through to the pheromone 83 in several straightforward steps.
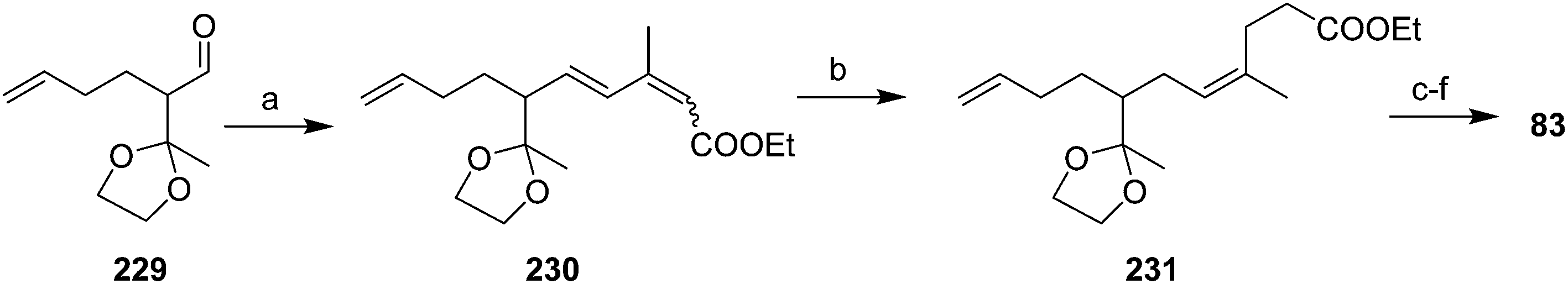 | ||
| Scheme 34 Synthesis of racemic triene 83 by a (Z)-selective, 1,4-dihydrogenation. Reagents and conditions: (a) triethyl 3-methyl-4-phosphonocrotonate, KOH, 18-crown-6, benzene, 69%; (b) H2, 80 atm, (PhCOOMe)Cr(CO)3, acetone, 120 °C, 52%; (c) LiAlH4, ether; (d) oxalic acid, H2O/acetone, reflux, 52% over 2 steps; (e) Ph3PCH2, THF/DMSO; (f) Ac2O, Na2CO3, 100 °C, 45% over 2 steps. | ||
Most recently, Hesse et al. constructed the trisubstituted alkene with the desired (Z)-stereochemistry using stereoselective protodeboronation and conversion of an (E)-allylic boronic ester into a (Z)-trisubstituted alkene (Scheme 35).79 Thus, vinylboronate ester 234 was prepared in two steps from 2-butyn-1-ol 232 by TBDMS-protection of the alcohol followed by copper-catalyzed hydroboration with pinacolatoborane. Deprotonation of the 2,4,6-triisopropylbenzoate ester 235 (prepared in 4 steps and 63% yield from (R)-limonene) and reaction with the boronic ester 234 gave the expected ate complex, which upon heating rearranged to the allylic intermediate 236. Treatment with TBAF·3H2O and heating resulted in sequential removal of the protecting group and protodeboronation to give alcohol 178, with >20:1 (Z/E) selectivity. The stereoselectivity was attributed to steric interactions disfavoring the (E)-isomer in the proposed six-membered ring transition state during protodeboronation. Acetylation completed the synthesis.
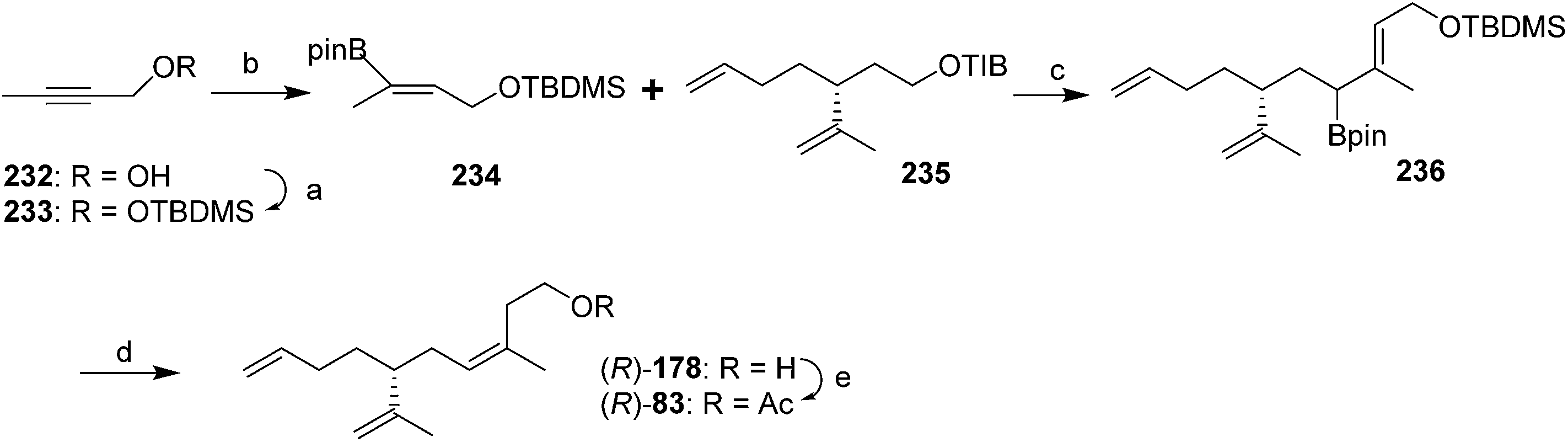 | ||
| Scheme 35 Synthesis of triene (R)-83 from a synthon with the double bond geometry already in place. Reagents and conditions: (a) TBDMSiCl, imidazole, DMAP, CH2Cl2, 99%; (b) CuCl (5%), PPh3 (6%), t-BuOK (20%), B2pin2, MeOH/THF, room temp; (c) s-BuLi, TMEDA, Et2O, −78 °C-reflux; (d) TBAF·3H2O, room temp-45 °C, 73% over 2 steps; (e) Ac2O, pyridine, 99%. | ||
Pheromone of white peach scale. The pheromone of the white peach scale was originally identified as propionate ester (R)-84, using a combination of liquid and preparative gas chromatography to isolate the active compound, and spectroscopic data with microchemical tests to identify it.80 However, there may be different pheromone races or possibly even cryptic species of white peach scale. Specifically, populations from France were found to produce the corresponding alcohol 241 in larger quantities than the propanoate ester, and males from this population required both compounds for optimal response.81 In contrast, Florida males responded best to the propanoate alone, and responses were decreased by higher doses of the alcohol in combination with the ester.82 Unfortunately, the original paper describing the identification of the propanoate from the Florida population did not mention whether the alcohol was present in the aeration extracts.
The core structure of the white peach scale pheromone alcohol 241 and ester 84 differs from that of the triene ester component of the red scale pheromone 83 by a single methyl group on carbon 9, and so the syntheses of white peach scale pheromone usually involved minor modifications to those routes. In fact, the initial syntheses were somewhat easier than those for red scale pheromone because the product from ozonolysis of limonene was perfectly set up for straightforward elaboration to the desired structure. Thus, limonene 237 was ozonized in MeOH with a trace of acid and quenching with Me2S, producing protected ketoaldehyde 238 in one step (Scheme 36A).80 Wittig reaction to convert the ketone to a terminal methylene, deprotection of the aldehyde, and a second Wittig reaction with the lithium salt of 3-triphenylphosphoranylidene-1-butanol completed the carbon skeleton, and the (E)- and (Z)-isomers were separated by HPLC before final esterification with propanoic anhydride. Thus, by using both enantiomers of limonene as starting materials, all possible isomers were generated, and the insect-produced compound was determined to be the (Z)-isomer by GC, and the (R)-enantiomer via bioassays.
 | ||
| Scheme 36 Syntheses of white peach scale pheromone enantiomers from either enantiomer of limonene. Reagents and conditions: (a) O3, MeOH, Me2S, H+, 80%; (b) Ph3PCH2, THF, 75%; (c) PTSA, acetone/H2O, 96%; (d) Ph3PC(CH3)CH2CH2OLi, THF, 48%; (e) (EtCO)2O, pyridine, 90%. (f) BuLi, THF, Ph3PEtBr, −78 °C (g) BuLi, −78 °C; (h) ethylene oxide, −5 °C, 15%. | ||
In a significant improvement to this route, generation of Ph3PC(CH3)CH2CH2OLi by reaction of ethylidenetriphenylphosphorane with ethylene oxide followed by deprotonation, as opposed to generation of the phosphoranylidene by reaction of the lithium salt of 3-triphenylphosphoranylidene-1-propanol with MeI gave a much cleaner and higher yielding Wittig reaction.83 In an interesting final modification, the authors discovered that deprotonation of the betaine 242 initially formed by reaction between ethylidenetriphenylphosphorane and aldehyde 240, followed by reaction with ethylene oxide and elimination of triphenylphosphine oxide, gave 98% (Z)-selectivity, albeit in low (15%) yield (Scheme 36B).83 Because male scale appeared to be indifferent to the (E)-isomer, a recently filed patent used essentially the same route to prepare the pheromone as blends of the (E)- and (Z)-isomers for field use.84
The synthesis of Vasil'ev et al. was identical to their synthesis of the red scale pheromone (See Scheme 34) with the exception that 1-bromo-3-methyl-3-butene was used to make the protected ketoaldehyde starting material in place of 1-bromo-3-butene.78
Pheromone of the scale Aulacapis murrayae. Ho and coworkers very recently identified the pheromone of A. murrayae as the (R)-enantiomer of the degraded irregular terpenoid solanone.85 This compound was originally identified from tobacco, and several syntheses of the racemate have been published.86 However, only chiral syntheses will be described because male scales are inhibited by the (S)-enantiomer.85
In a communication on a method for regio- and stereoselective preparation of conjugated dienes from allylic alcohols, Mori and coworkers prepared racemic solanone, and multigram quantities of a precursor 245 that could be transformed into (S)-solanone in two easy steps (Scheme 37).87 However, the paper did not describe how the precursor 244 was prepared, nor, surprisingly, did it report carrying the precursor through to (S)-solanone (S)-85. Thus, the utility of this synthesis is hard to assess, nor is it clear whether it could be readily adapted to the synthesis of the (R)-enantiomer.
 | ||
| Scheme 37 Mori and coworkers' partially described synthesis of (S)-solanone. Reagents and conditions: (a) MesCl, Et3N, CH2Cl2, then diisopropylethylamine, HMPA, 140–180 °C, 64%. | ||
The only published synthesis of (R)-solanone 85 began with asymmetric induction using Evans' chiral oxazolidinone chemistry to place the chiral center (Scheme 38).85 Dihydroxylation of the intermediate 246 resulted in spontaneous lactonization to the γ-lactone, followed by acetylation to give 247. Reduction of the ester and lactone simultaneously, and selective formation of the acetonide from the vicinal diol gave 248. Swern oxidation of the primary alcohol followed by Horner–Wadsworth–Emmons olefination with diethyl(2-methylallyl)phosphonate gave the 1,3-diene 249 as one stereoisomer. One-pot cleavage of the acetonide and oxidative cleavage of the resulting diol gave an aldehyde which was reduced to alcohol 250 and converted to the tosylate. Displacement of the tosylate with cyanide, followed by reaction of the resulting nitrile 251 with MeMgBr and aqueous workup gave a few milligrams of (R)-solanone (R)-85, sufficient for bioassays to verify its bioactivity.
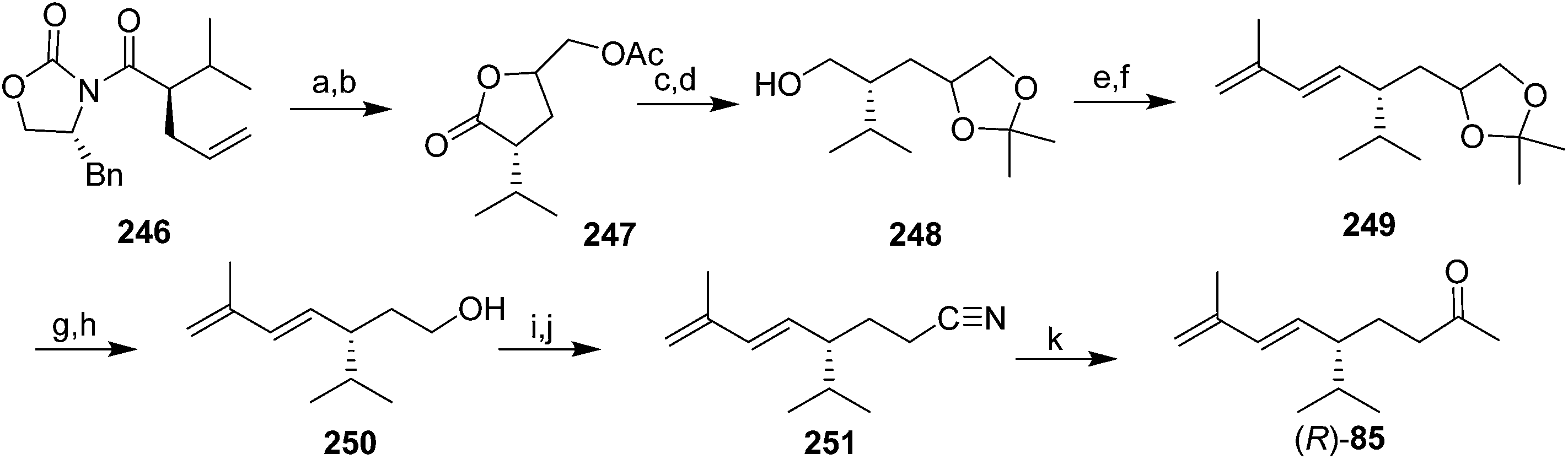 | ||
| Scheme 38 Synthesis of (R)-solanone 85. Reagents and conditions: (a) OsO4, NMO, MeCN, room temp; (b) Ac2O, pyridine, DMAP, 0 °C, 98% over 2 steps; (c) LiBH4, THF, room temp; (d) 2,2-dimethoxypropane, H+, DMF, room temp, 84% over 2 steps; (e) (COCl)2, DMSO, CH2Cl2, −78 °C, then Et3N; (f) diethyl (2-methylallyl)phosphonate, BuLi, THF, HMPA, −78 °C-room temp, 84% over 2 steps; (g) NaIO4, AcOH, H2O, room temp; (h) NaBH4, MeOH, 0 °C, 86% over 2 steps; (i) TosCl, Et3N, DMAP, CH2Cl2, room temp; (j) NaCN, DMSO, room temp, 73% over 2 steps; (k) CH3MgBr, toluene, reflux, then H2O, 53%. | ||
3.2 Cyclic pheromones
 | ||
| Fig. 4 Cyclopropane-containing pheromones (esters of chrysanthemol). | ||
Madeira mealybug pheromone. The sex pheromone of the Madeira mealybug, Phenacoccus madeirensis Green, was identified by Ho and coworkers as (1R,3R)-[2,2-dimethyl-3-(2-methylprop-1-enyl)cyclopropyl]methyl (R)-2-methylbutanoate [(1R,3R)-chrysanthemyl (R)-2-methylbutanoate] 252.88 The absolute configuration was determined by comparison of retention times of the acid and alcohol portions with those of enantiopure standards on a chiral column. (1R,3R)- and (1S,3S)-Chrysanthemol were obtained by resolution of racemic trans-chrysanthemol by lipase-catalyzed acylation with succinic anhydride.26 2-Methylbutanoic acid was resolved by formation of diastereomeric amides with (S)-α-methylbenzylamine, separation by semipreparative HPLC, and hydrolysis of the separated diastereomers in refluxing 70% H2SO4, without epimerization. (R)-Lavandulyl (R)-2-methylbutanoate was identified as a minor component in extracts from virgin females, but it was not attractive to males, nor did it act synergistically or additively with the main component 252.
Ho and coworkers also studied the relationship between chirality and bioactivity of the stereoisomers of 252 (Fig. 5).89 The insect-produced (1R,3R,2′R)-isomer was most attractive, whereas the (1R,3R,2′S)-diastereomer (opposite stereochemistry in the acid portion) was inhibitory. In contrast, the (1S,3S,2′R)-isomer (opposite stereochemistry in the alcohol portion) was neither attractive nor inhibitory. The diastereomers prepared from the cis-alcohol (1R,3S,2′R and 1S,3R,2′R) were also inhibitory.
 | ||
| Fig. 5 Stereoisomers of Madeira mealybug pheromone. | ||
Citrophilous mealybug pheromone. The sex pheromone of the citrophilous mealybug, Pseudococcus calceolariae, was identified jointly by New Zealand and Chilean teams as [2,2-dimethyl-3-(2-methylprop-1-enyl)cyclopropyl]methyl 2-acetoxy-3-methylbutanoate (chrysanthemyl 2-acetoxy-3-methylbutanoate) 253, and readily synthesized by esterification of trans-chrysanthemol with 2-acetoxy-3-methylbutyryl chloride.90 Unelius and coworkers subsequently determined the absolute configuration to be (1R,3R)-chrysanthemyl (R)-2-acetoxy-3-methylbutanoate by comparison of the retention times of the acid and alcohol portions with synthetic standards on a chiral column.91 The correct stereoisomer of the pheromone was readily prepared from (1R,3R)-chrysanthemol (from LiAlH4 reduction of commercial (1R,3R)-chrysanthemic acid) and (R)-2-acetoxy-3-methylbutyryl chloride (from acetylation of the hydroxyacid with Ac2O and pyridine, then conversion to the acid chloride with oxalyl chloride). The (R)-configuration in the acid moiety was necessary for activity. The non-natural stereoisomers were at most slightly inhibitory, so that relatively inexpensive racemic chrysanthemol could be used for commercial synthesis of the pheromone.
 | ||
| Fig. 6 Scale and mealybug pheromones with a cyclobutane-containing monoterpenol core. | ||
Citrus mealybug pheromone. The sex pheromone of the citrus mealybug, Planococcus citri (Risso), was identified by Bierl-Leonhardt and coworkers as (1R,3R)-3-isopropenyl-2,2-dimethylcyclobutylmethyl acetate [(1R,3R)-planococcyl acetate] 254.92 The natural enantiomer and the racemate were equally active, indicating no inhibition by the unnatural (1S,3S)-enantiomer.93 The trans isomers were much less active than 254.92 Several syntheses have been reported, most of which used (+)-α-pinene 258 as an obvious starting material because it contains the required cyclobutane skeleton, and precursors to all the functional groups with the correct substitution pattern and stereochemistry. Thus, (+)-α-pinene 258, or (R)-(+)-verbenone 259 prepared by allylic oxidation of (+)-α-pinene, were subjected to oxidative ring opening. Syntheses reported before 1989 have been thoroughly reviewed2 and will not be discussed further here.
Scheme 39 summarizes Passaro's synthesis of (R)-254,94 which incorporated two major improvements to a previous route.95 Specifically, the crucial step of ruthenium-catalyzed oxidative ring opening with concomitant decarboxylation of (R)-(+)-verbenone 259 was carried out in aqueous tert-butanol instead of CCl4:CH3CN:H2O (2:1:4). This modified procedure afforded (+)-cis-pinononic acid 260 in excellent yield (94%), was easier to perform, and simplified product isolation. Secondly, methylenation of 260 using Zn/CH2Br2/TiCl4 (ref. 96) was superior to Wittig reactions in larger scale preparations. LiAlH4 reduction of acid 261 and acetylation of the resulting alcohol 262 gave the desired pheromone 254 in >40% overall yield from verbenone.
 | ||
| Scheme 39 Passaro's synthesis of the citrus mealybug pheromone (1R,3R)-254 from (+)-α-pinene 258. Reagents and conditions: (a) RuCl3, NaIO4, t-BuOH, H2O, 94%; (b) Zn, CH2Br2, TiCl4; (c) LiAlH4, Et2O; (d) Ac2O, pyridine, 44% for 3 steps. | ||
Kukovinets et al. used essentially the same route and intermediates, but slightly different reagents, for example, employing ozonolysis for the oxidative cleavage of the ring.97
Another route began with direct oxidative cleavage of the ring in (+)-α-pinene 258 by ozonolysis to give ketoaldehyde 263 (Scheme 40).93 Selective conversion of the aldehyde to enol acetate 264 followed by a second ozonolysis removed one carbon, and chemoselective reduction98 of the aldehyde of 265 with Zn(BH4)2 in mixed ether solvents gave pinononic alcohol 266, which was subjected to Wittig reaction and acetylation to afford the pheromone 254. All intermediates were used as crude materials, and only the final product was purified by chromatography to provide technical grade (85% pure) pheromone, which proved to be as active as the pure pheromone in field trials.
 | ||
| Scheme 40 Zada's synthesis of the citrus mealybug pheromone (1R,3R)-254 from (+)-α-pinene 258. Reagents and conditions: (a) O3, NaHCO3 (cat.), then Me2S, 78%; (b) Ac2O, Et3N, DMAP, 100% (E–Z = 1:1); (c) O3, NaHCO3 (cat.), then Me2S, 86%; (d) Zn(BH4)2, DME, Et2O, THF; (e) Ph3PCH2, THF; (f) Ac2O, pyridine, 35% for 3 steps. | ||
Citriculus mealybug pheromone. The sex pheromone of the citriculus mealybug, Pseudococcus cryptus Hempel, was identified by Arai and coworkers as (1R,3R)-3-isopropenyl-2,2-dimethylcyclobutylmethyl 3-methyl-3-butenoate 255 [(1R,3R)-planococcyl 3-methyl-3-butenoate].99 The alcohol portion was identical to that of the citrus mealybug pheromone.
Scheme 41 summarizes Nakahata's 5-step synthesis that utilized all ten carbon atoms of the starting material (+)-α-pinene 258.100 Thus, treatment of (+)-α-pinene with Pb(OAc)4 gave 267 with a small amount of allylic rearrangement product 268 (95:5 ratio). The order of addition and careful drying of the Pb(OAc)4 were crucial for minimizing the formation of 268. Ozonolysis with reductive workup, followed by methanolysis of the acetate gave triol 269. 269 was converted into a mixture of acetate 254 and formate 272 (3:4 ratio) by conversion of the vicinal diols into olefins via 2-methoxy-1,3-dioxolane derivatives 270 and 271 and subsequent treatment with Ac2O.101 The mixture of 254 and 272 was hydrolyzed to alcohol 262 in a one-pot operation. The alcohol 262 was esterified with 3-methyl-3-butenoic acid in the presence of DCC and DMAP. The use of only a catalytic amount of DMAP (about 0.1 equivalent) was essential for preventing migration of the double bond of the acid portion into conjugation.
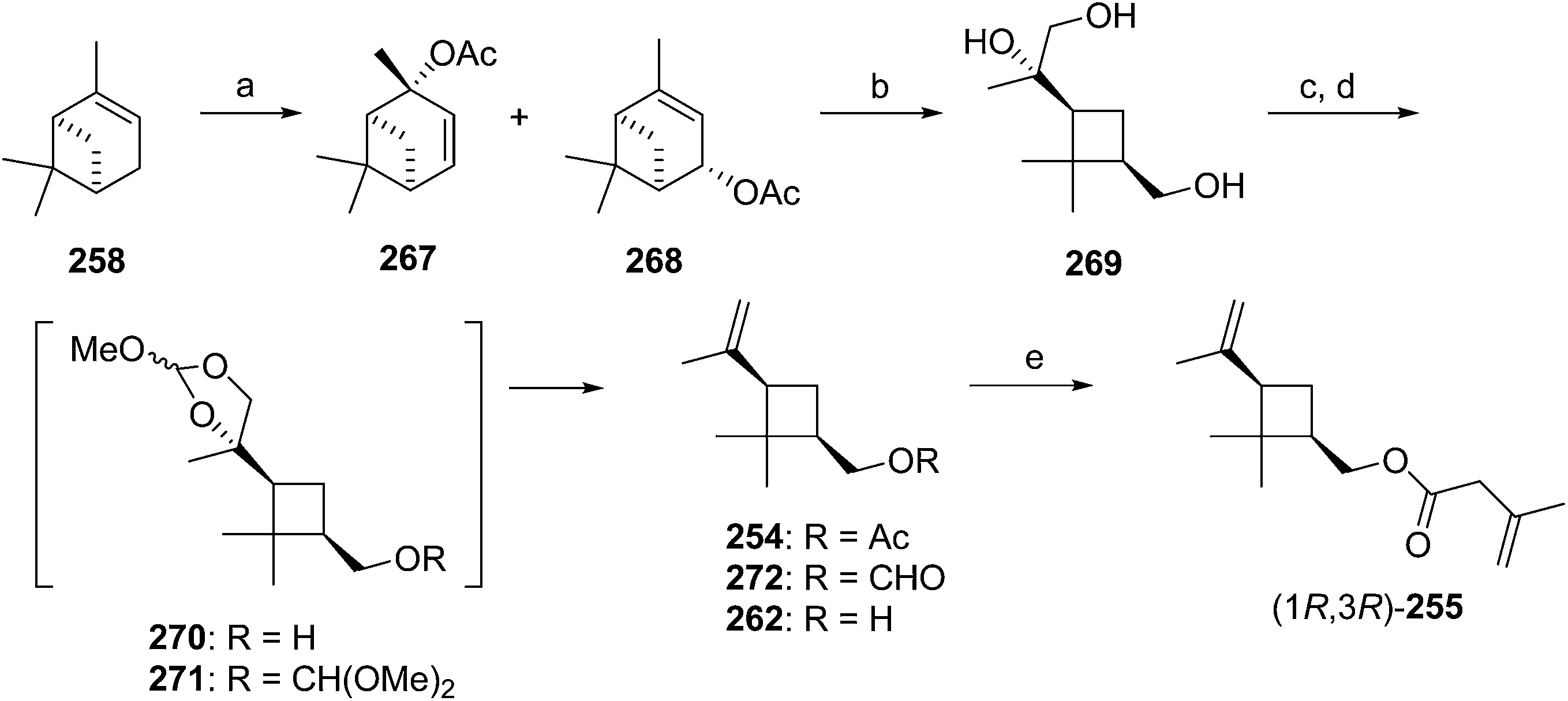 | ||
| Scheme 41 Synthesis of the citriculus mealybug pheromone (1R,3R)-255 from (+)-α-pinene 258. Reagents and conditions: (a) Pb(OAc)4, C6H6, 75%, 267:268 = 95:5; (b) O3, MeOH, then NaBH4, and finally K2CO3, 72%; (c) HC(OMe)3, PPTS, THF; (d) Ac2O, then aq. NaOH, 88% for 2 steps; (e) 3-methyl-3-butenoic acid, DCC, DMAP, 91%. | ||
Albopicta scale pheromone. The pheromone of the invasive scale Acutaspis albopicta was recently identified as (1S,3S)-3-isopropenyl-2,2-dimethylcyclobutylmethyl (R)-2-methylbutanoate 256, in which the alcohol portion is the enantiomer of that found in the pheromones of the citrus and citriculus mealybugs.102 The alcohol portion was synthesized from (S)-(−)-verbenone by the method of Passaro et al. (see Scheme 39),94 the acid portion was made by kinetic resolution of 2-methylbutanol with Amano lipase PF, followed by Jones oxidation of the resolved alcohols, and the ester linkage was made by treatment of a CH2Cl2 solution of the acid and alcohol portions with the coupling reagent 1-ethyl-3-[(3-dimethylamino)propyl]carbodiimide hydrochloride (EDC) and DMAP.102 The other stereoisomers were made by synthesis of the enantiomeric alcohol portion from (R)-(+)-verbenone, and esterifications with commercially available (S)-2-methylbutanoic acid or (R)-2-methylbutanoic prepared as described above. Bioassays of all 4 stereoisomers determined that the absolute configuration of the monoterpenol portion was critical, with the (1R,3R,2′R) and (1R,3R,2′S) diastereomers being inactive, whereas the (1S,3S)-alcohol esterified with either enantiomer of 2-methylbutanoic acid were equally active, indicating that the male scale did not discriminate the absolute configuration of the acid portion of the pheromone.
Pink hibiscus mealybug pheromone (major component). As mentioned earlier, the sex pheromone of the pink hibiscus mealybug, Maconellicoccus hirsutus (Green), was identified by Zhang and coworkers as a 1
:5 mixture of (R)-lavandulyl (S)-2-methylbutanoate 37 and (R)-maconelliyl (S)-2-methylbutanoate 257.29 Maconelliol 276, the alcohol portion of 257, is structurally similar to the planococcyl alcohol portion of the citrus and citriculus mealybug pheromones, with a methylethylidene group on the ring instead of an isopropenyl group. Zhang et al. developed an enantioselective synthesis of 257 (Scheme 42)103 starting from (+)-cis-pinononic acid 260, a key intermediate in the citrus mealybug pheromone synthesis (see Scheme 39). Reaction of 260 (94% ee) with 3 equivalents of MeMgCl resulted in partial loss of enantiomeric purity (80% ee) due to double epimerization during nucleophilic addition. However, reaction with 1 equivalent of MeLi followed by 1.6 equivalents of MeMgCl gave tertiary alcohol 273 with no loss of chirality. Thinking to prevent decarboxylation during dehydration, 273 was converted to its methyl ester, but when the ester was subjected to conventional dehydration methods, the desired methylethylidene product was contaminated with the isopropenyl isomer, which was difficult to separate. However, treatment of the free acid 273 under the same conditions resulted in lactone 274 as the only product, which was cleanly converted to the desired methylethylidene product 275 by treatment with acid. LiAlH4 reduction and condensation of the resulting alcohol 276 with (S)-2-methylbutanoic acid gave the desired ester (1R,2′S)-257 in good yield.
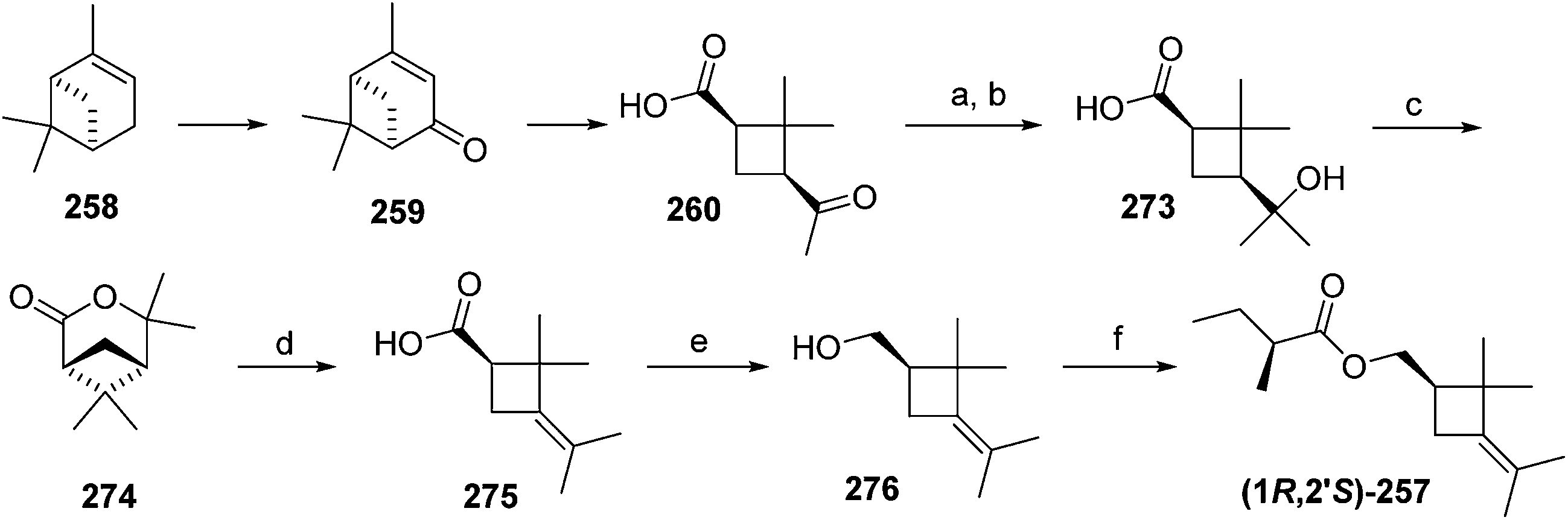 | ||
| Scheme 42 Synthesis of (1R,2′S)-257, the major component of the pink hibiscus mealybug pheromone from (+)-α-pinene 258. Reagents and conditions: (a) MeLi/Et2O, 1.0 equiv, THF, −20 °C, 15 min; (b) MeMgCl/THF, 1.6 equiv, −10 °C, 30 min, room temp, 1 h, 89%; (c) POCl3, pyridine, room temp, 24 h, 75%; (d) PTSA, C6H6, reflux, 24 h, 78%; (e) LiAlH4, Et2O, room temp, overnight, 88%; (f) (S)-2-methylbutanoic acid, (COCl)2, DMF, C6H6, room temp, 1.5 h, 79%. | ||
Zhang and coworkers carried out a systematic study of the relationship between chirality and biological activity of the stereoisomers of 257.104 Their results suggested that the stereochemistry of the acid portion is more critical than that of the alcohol portion. Thus, the (R)-alcohol esterified with the (S)-acid was attractive, whereas the (R,R)-diastereomer was inhibitory. The chiral center in the alcohol portion was less important to attraction or inhibition. Importantly, this was the first report of enantiomeric antagonism in any mealybug or scale insects. Furthermore, optimal attraction was critically dependent on dose, with doses above 10 μg resulting in decreased attraction.
Ester of a cyclobutane-containing sesquiterpenol: oleander scale pheromone. The oleander scale pheromone is the only example so far of a scale or mealybug pheromone incorporating a sesquiterpenoid cyclobutane structure. Its identification required the integration of fragments of information from a variety of analysis methods.105 High resolution tandem MS provided the molecular formula (C17H28O2) and the formulae of a number of fragments obtained from the parent molecule and some derivatives. Microchemical reactions showed the presence of two C
C double bonds, one ring, and an acetate ester, but this still did not allow determination of the complete structure. Subsequently, 1H and 13C spectra acquired on 900 μg of pheromone accumulated over several years yielded the basic structure but not the relative and absolute stereochemistries, nor were the authors successful in preparing crystalline derivatives. Thus, the final structure was proven by synthesis of isomers of known configuration, and bioassays of the two possible enantiomers showed that the insect-produced compound likely had the (1R,2S)-configuration (Fig. 7).
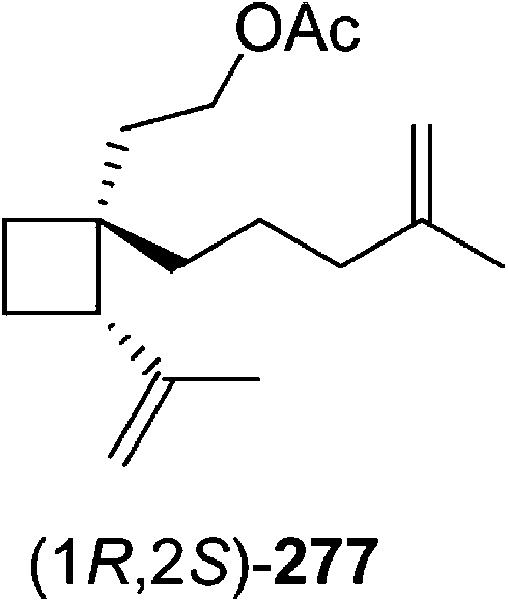 | ||
| Fig. 7 Oleander scale pheromone. | ||
The goal of the first synthetic effort was to develop stereoselective syntheses of all four possible stereoisomers with known absolute configurations, in large enough amounts to provide material for bioassays, and without having to resolve scalemic intermediates (Scheme 43).106 (R)- and (S)-carvone were chosen as readily accessible chiral synthons, and the initial steps of the synthesis were modeled after those used in the synthesis of grandisol, a monoterpenoid analog containing the same 1,1,2-trisubstituted cyclobutane motif. Thus, (R)-(−)-carvone 171 was converted to acetal ester 175 in 3 steps, followed by alkylation of the corresponding enolate to install the longest side chain. Hydrolysis of the acetal, reduction of the aldehyde, and tosylation of the resulting alcohol then yielded the key intermediate 278 for formation of the cyclobutane ring. After careful optimization of the conditions, the cyclization proceeded in 46% yield, giving an unexpectedly high 19:1 ratio of the two possible diastereomers of 279. The order of the steps was critical because it proved impossible to install the long side chain if the cyclization was performed first. With the 1,1,2-trisubstituted cyclobutane core assembled, straightforward functional group manipulations then gave (1S,2S)-277, in 14 steps from (R)-carvone.
 | ||
| Scheme 43 First syntheses of stereoisomers of the oleander scale pheromone. Reagents and conditions: (a) 3 steps from (R)-carvone, 34%; (b) KHMDS, THF/HMPA, 5-iodo-2-methyl-1-pentene, −20–0 °C, 60% based on recovered starting material; (c) PTSA, acetone/water 1:1; (d) NaBH4, EtOH, 0 °C; (e) TosCl, pyridine, 43% over 3 steps; (f) LiHMDS, THF/HMPA, −10 °C, 46%; (g) LiAlH4, THF, 86%; (h) TosCl, DMAP, CH2Cl2, 0 °C, 86%; (i) NaCN, H2O/HMPA, 87%; (j) DIBAL, CH2Cl2, −20 °C, aq. workup; (k) NaBH4, EtOH, 0 °C, 44% over 2 steps; (l) Ac2O, pyridine; 70%. (m) OsO4, NMO, acetone/H2O; (n) NaIO4, MeOH/H2O, 64%; (o) DBU, benzene, reflux, 2:3 mixture of epimers; (p) Ph3PCH3Br, BuLi, THF, −70 °C-room temp, 75% over 2 steps; (q) DIBAL, CH2Cl2,−20 °C, then H3O+; (r) NaBH4, EtOH, 32% from nitrile; (s) Ac2O, pyridine, 70%. | ||
The authors then tried to take advantage of the high stereocontrol obtained in the cyclization step to develop a synthesis of the other diastereomer, by installing the acetoxyethyl sidechain or a suitable precursor before cyclizing, and then converting the carboethoxy group to the 4-methyl-4-pentenyl side-chain. However, several variations on this theme were not successful. Instead, (1S,2S)-280 from the first synthesis was oxidized to the diketone 281, and the ketone directly attached to the ring was epimerized with base, giving an inseparable 2:3 mixture of diastereomers of 281. Wittig reactions then installed the two terminal methylenes, followed by conversion of the nitrile to the alcohol diastereomers 282, which were separable by flash chromatography. Acetylation of each alcohol then completed the syntheses. The other two stereoisomers were obtained with the same sequences of reactions from (S)-carvone. With all stereoisomers available, the insect-produced compound was shown to have the (1R*,2S*) relative configuration, the two enantiomers of which did not resolve by chiral stationary phase chromatography. However, bioassays showed that the natural compound was almost certainly the (1R,2S)-enantiomer.
The key step in a second synthesis of the racemic pheromone was a regio- and stereocontrolled intramolecular 4-exo-tet-cyclization of a cis-epoxynitrile precursor 285 (Scheme 44).107a The cis-stereochemistry of the epoxide was important to ensure high regioselectivity, allowing preferential formation of the cyclobutane ring. Thus, nitrile 283 was alkylated with MOM-protected 2-bromoethanol, then epoxidized with m-CPBA. Use of MOM rather than other more bulky protecting groups proved crucial to later steps in the synthesis. Optimization of the epoxide opening and cyclization conditions resulted in a 60% yield of 286, with a 79:21 ratio favoring the desired trans product. Protection of the resulting alcohol, conversion of the nitrile to an aldehyde, and Wittig reaction then installed the precursor to the third side chain, giving 289. Removal of the TBDMS protecting group, PDC oxidation, and removal of the ketal protecting group gave the diketone 290. Double Wittig reaction with Ph3PCH2 then gave a precursor with the two terminal methylene groups installed. However, the final MOM protecting group could not be removed cleanly under a variety of conditions. Instead, the MOM group was removed from diketone 290 by treatment with an acid catalyst in MeOH, followed by hydrolysis of the resulting acetal to the hemiacetal 291. Double Wittig reaction of this precursor with a large excess of Ph3PCH2 then gave the penultimate precursor (1R,2S)-282, and acetylation completed the synthesis. In an alternative sequence, all three protecting groups were removed from 289 to give the ketonediol, followed by selective acetylation of the primary alcohol, oxidation of the secondary alcohol, and double Wittig olefination. The latter step resulted in partial removal of the acetate ester, which was easily reinstalled with acetic anhydride, Et3N, and DMAP. This alternative sequence (not shown) produced a similar yield from 289 as the first series of steps.
 | ||
| Scheme 44 Petschen's synthesis of racemic and chiral forms of oleander scale pheromone. Reagents and conditions: (a) LDA, Br(CH2)2OMOM, THF/HMPA, 61%; (b) m-CPBA, CH2Cl2, 92%; (c) LiHMDS, THF, 0 °C-reflux, 60%; (d) TBDMSiCl, imidazole, DMF, 98%; (e) DIBAL, hexane, then H3O+, 90%; (f) Ph3PCHCH2C(OCH2CH2O)CH3, THF, 82%; (g) H2, Pd/C, EtOH, 90%; (h) TBAF, THF, 98%; (i) PDC, DMF, 97%; (j) Amberlyst A15 resin, acetone/H2O, 95%; (k) Dowex 50W-X4, MeOH, then SiO2, hexane, acetone/H2O; (l) excess Ph3PCH2, THF, 45% over 2 steps; (m) Ac2O, Et3N, DMAP, CH2Cl2, 98%. | ||
With a synthesis of the racemic pheromone in hand, the authors then carried out kinetic resolutions of intermediate 286 with lipase PS or PFL and vinyl acetate to provide the enantiomers of 286, each in >98% ee, which were then carried through to the final products by the route described above. Both enzymes selectively esterified the (1R,2S,5R)-enantiomer of alcohol 286.107b
In an entirely different approach, Schmidt et al. used a TiCl4-promoted [2 + 2] cycloaddition to construct trisubstituted cyclobutane intermediate 294 in 96% yield and an anti/syn ratio of 2:1 (Scheme 45).108a Reduction of the esters and protection of the resulting alcohols, followed by a modified Tamao–Fleming oxidation gave diprotected triol 297. Several straightforward steps then gave advanced intermediate 299. At any stage the diastereomers were apparently separable, so the diastereomers could be carried forward as a mixture or as separate isomers. However, the synthesis does not yet seem to have been carried forward to completion.108b
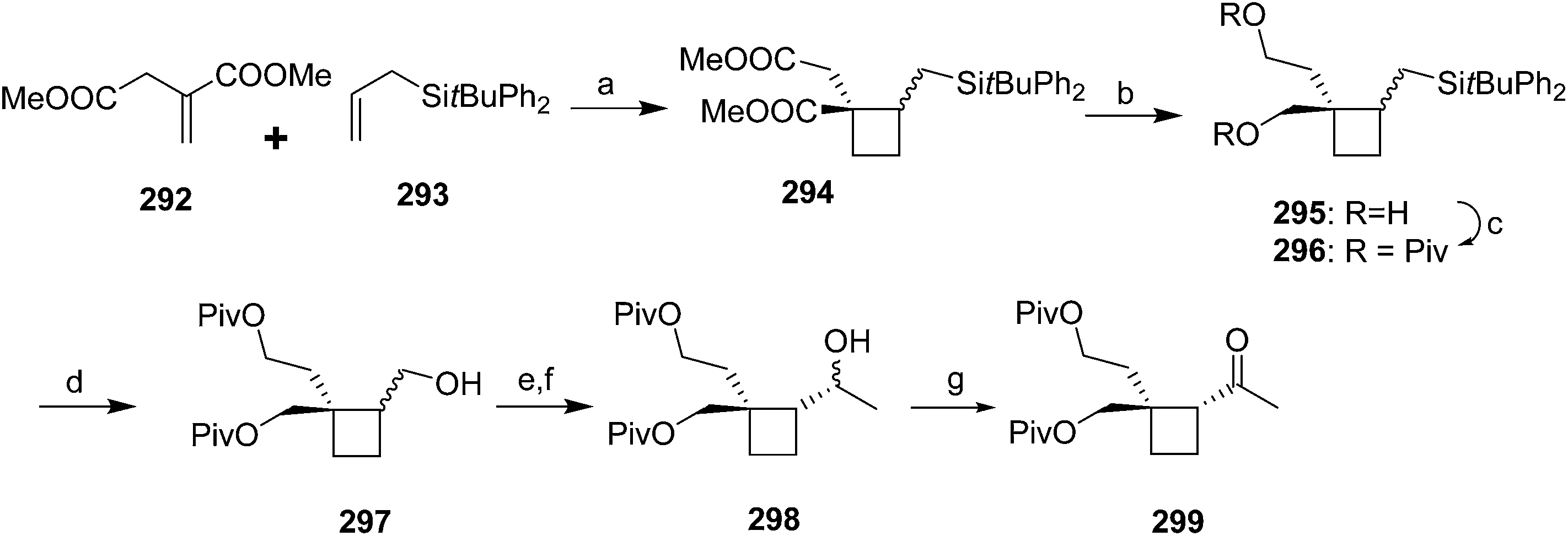 | ||
| Scheme 45 Synthesis of an advanced intermediate for the oleander scale pheromone. Reagents and conditions: (a) TiCl4, CH2Cl2, room temp – reflux, 96%; (b) LiAlH4, THF, 93%; (c) PivCl, Et3N, DMAP, CH2Cl2, 90%; (d) BF3·2AcOH, ClCH2CH2Cl, reflux, then KF, NaHCO3, H2O2, THF/MeOH, 1:1, 36–48% over 2 steps; (e) PDC, molecular sieve; (f) MeMgBr, THF, −10–25 °C, ∼72%, anti and syn isomers separated by chromatography; (g) TPAP, NMO, 4A molecular sieve, CH2Cl2, −10 °C-room temp, 95%. | ||
 | ||
| Fig. 8 Mealybug pheromones with cyclopentanyl and cyclopentenyl substructures. | ||
Longtailed mealybug pheromone. The pheromone of the longtailed mealybug, Pseudococcus longispinus, was identified by Millar and coworkers as 2-(1,5,5-trimethylcyclopent-2-en-1-yl)ethyl acetate 300.110 The first synthesis of the racemate used a 2,3-Wittig rearrangement to set the two contiguous quarternary centers while simultaneously placing the double bond correctly (Scheme 46).110 Thus, cyclization of isobutyl 2-butenoate 303 in hot polyphosphoric acid yielded multigram quantities of cyclopentenone 304. This interesting reaction represents an example of a general rearrangement of α,β-unsaturated esters to cyclopentenones, and presumably proceeds via a fragmentation-recombination-Nazarov cyclization pathway.111 Reduction and O-alkylation with Bu3SnCH2I, followed by transmetallation with BuLi at low temperature, gave the rearranged cyclopentenol 307, albeit in low yield. A straightforward series of reactions extended the chain by one carbon via aldehyde 310, followed by reduction of the aldehyde and acetylation.
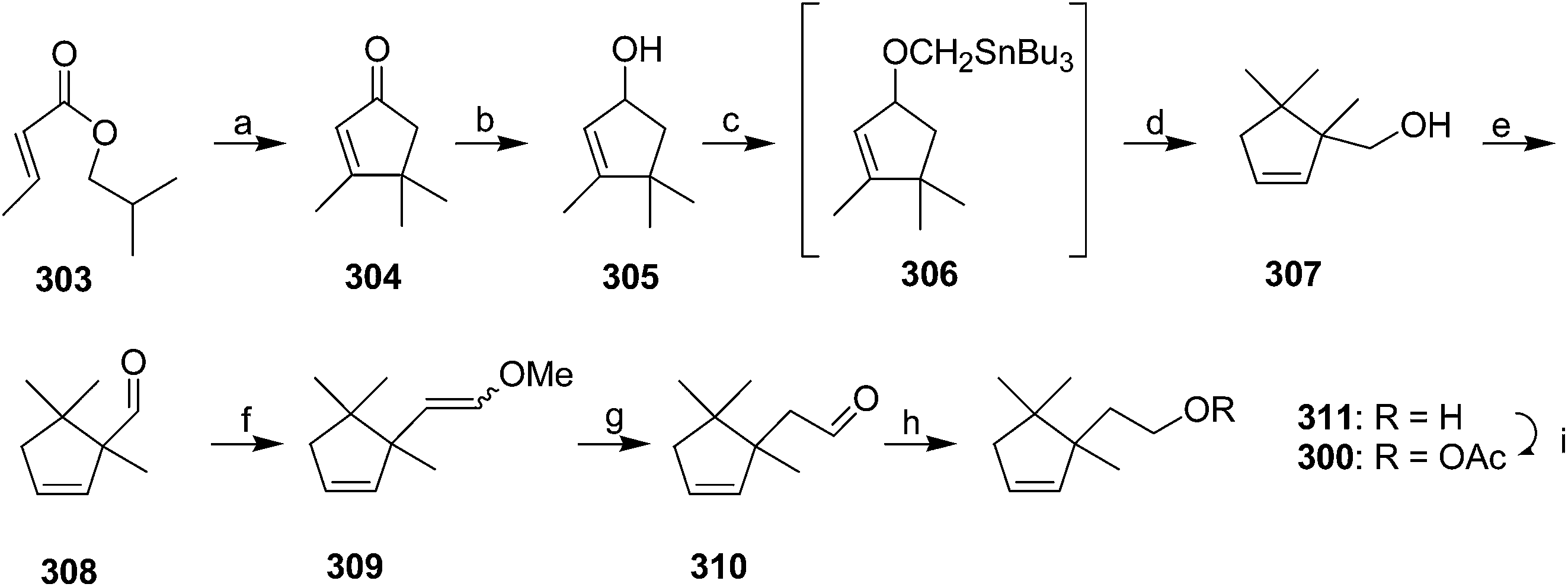 | ||
| Scheme 46 Millar group's first synthesis of longtailed mealybug pheromone via 2,3-Wittig rearrangement. Reagents and conditions: (a) polyphosphoric acid, 100 °C; (b) LiAlH4, ether, 89%; (c) KH, THF, Bu3SnCH2I, 91%; (d) BuLi, THF, −100 °C, 25%; (e) PCC, CH2Cl2; (f) MeOCH2PPh3Cl, BuLi, THF, 53% over 2 steps; (g) H3O+, 86%; (h) NaBH4, EtOH, 95%; (i) AcCl, pyridine, ether, 77%. | ||
The second synthesis used an entirely different strategy to construct the cyclopentane ring from an acylic precursor, via insertion of a carbene into a methine C–H bond to close the ring (Scheme 47).112 Thus, β-ketoester 313, from γ-alkylation of methylacetoacetate 15 with isobutyl iodide 312, was converted to diazo ester 314. Rh-catalyzed cyclization proceeded in excellent yield, followed by alkylation of the resulting ketoester 315 with methyl iodide to complete the assembly of the vicinal quarternary carbons. Conversion of the ketone of 316 to the enol triflate 317 and Pd-catalyzed reduction placed the double bond, giving 318. The remainder of the synthesis used the same series of steps described above, i.e., one-carbon extension of the side chain, reduction of the resulting aldehyde, and acetylation, producing >5 g of the racemic pheromone.
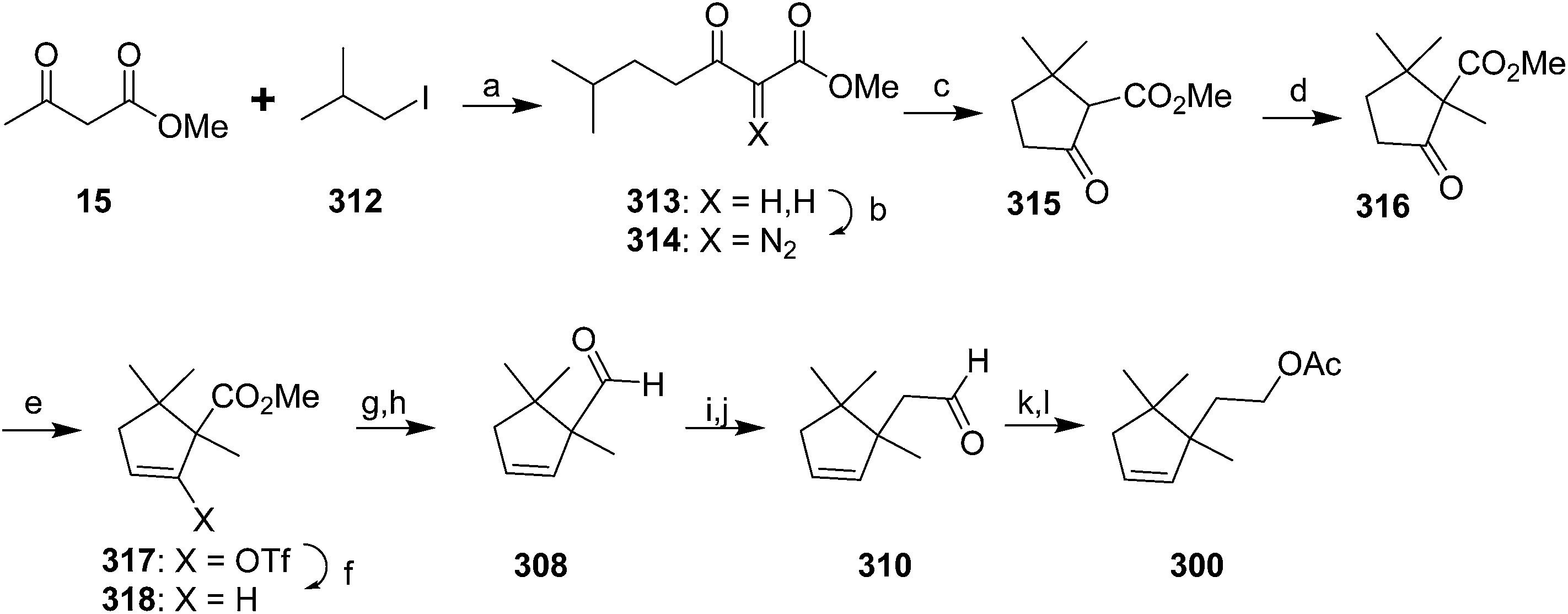 | ||
| Scheme 47 Millar group's 2nd synthesis of longtailed mealybug pheromone, based on an intramolecular cyclization. Reagents and conditions: (a) NaH, BuLi, THF, 43%; (b) p-acetamidobenzenesulfonyl azide, Et3N, MeCN, 96%; (c) Rh2(OAc)4, CH2Cl2, 91%; (d) K2CO3, MeI, acetone, 84%; (e) LDA, N-phenyl-bis(trifluoromethanesulfonimide), THF, 90%; (f) Pd(OAc)2, Ph3P, Bu3N, HCOOH, DMF, 82%; (g) LiAlH4, Et2O; (h) (COCl)2, DMSO, CH2Cl2, Et3N; (i) MeOCH2PPh3Cl, NaHMDS, THF, 72% over 3 steps; (j) H3O+, THF; (k) LiAlH4, Et2O, 85% over 2 steps; (l) AcCl, pyridine, Et2O, 95%. | ||
To encourage commercialization of the pheromone, a shorter 6-step sequence was then developed (Scheme 48).113 The first two steps were the same as those used in the first synthesis, but then instead of using a 2,3-Wittig rearrangement, an Ireland-Claisen rearrangement was used. This produced the complete carbon skeleton of the pheromone, in contrast to the 2,3-Wittig rearrangement route which required several extra steps to add an additional carbon. The Ireland-Claisen rearrangement of 319 proceeded smoothly through the silyl ketene acetal 320 to give γ,δ-unsaturated carboxylic acid 321 in respectable yield. LiAlH4 reduction of acid 321 and acetylation completed the synthesis.
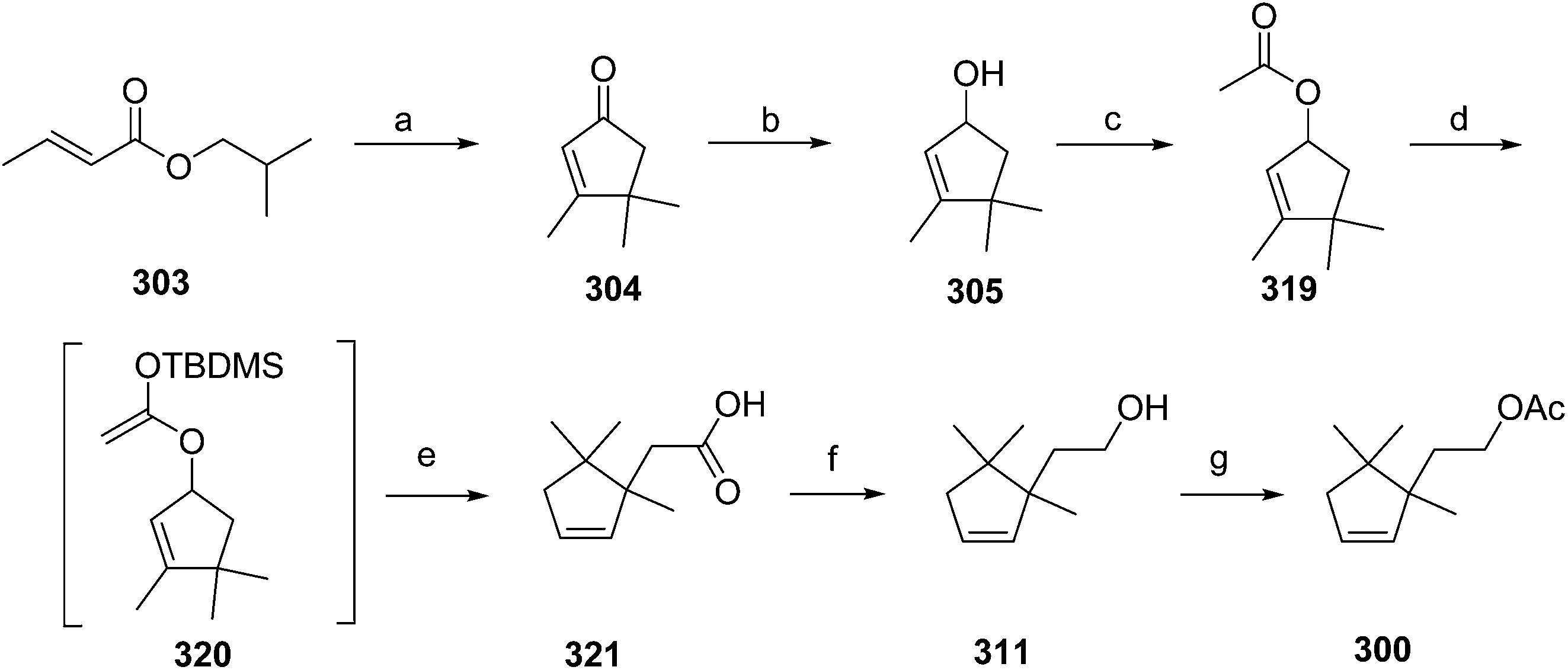 | ||
| Scheme 48 Synthesis of the longtailed mealybug pheromone 300 based on Ireland-Claisen rearrangement. Reagents and conditions: (a) polyphosphoric acid; (b) LiAlH4, Et2O; (c) Ac2O, DMAP, pyridine, CH2Cl2, 78% for 2 steps; (d) LDA, TBDMSiCl, THF; (e) reflux, then NaOH, acidic workup, 50% from 319; (f) LiAlH4, Et2O, 95%; (g) AcCl, pyridine, Et2O, 95%. | ||
Bailey and Bakonyi recently developed a concise synthesis of 300 based on intramolecular carbolithiation (Scheme 49).114 Alkylation of the lithium enolate derived from 322 gave ketone 323. When standard Wittig reaction conditions were employed to convert 323 to 324, a large quantity of alkyne was generated as a side product by dehydrohalogenation of the vinyl iodide. In contrast, Utimoto's olefination procedure,115 using a CH2Br2–TiCl4–Zn system with catalytic PbCl2 cleanly afforded (Z)-1-iodo-4,4,5-trimethyl-1,5-hexadiene 324. The key 5-exo-trig ring closure via lithiation, cyclization, and trapping the resulting anion with paraformaldehyde proceeded efficiently (65%) in one pot to give 311, and acetylation completed the synthesis.
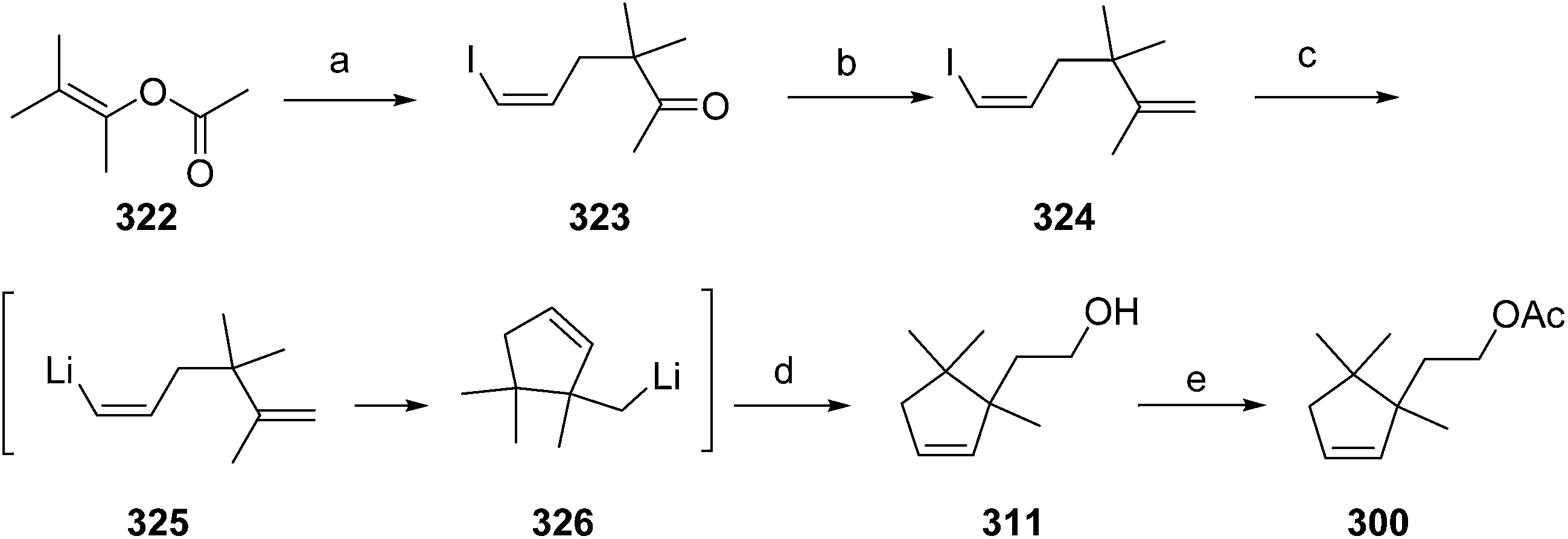 | ||
| Scheme 49 Synthesis of the longtailed mealybug pheromone 300 based on intramolecular carbolithiation. Reagents and conditions: (a) MeLi, DME, (Z)-3-bromo-1-iodopropene, 64%; (b) Zn, cat. PbCl2, CH2Br2, THF, then TiCl4, 53%; (c) 2.2 eq. t-BuLi, −78 °C, 10 min, then TMEDA, room temp, 1 h; (d) (CH2O)n, −78 °C, then room temp, 1 h, finally H3O+, 65% over 2 steps; (e) AcCl, pyridine, Et2O, 86%. | ||
The Reddy group developed a short and efficient synthesis of 300 based on ring-closing metathesis (Scheme 50).116 The overall strategy is similar to the total synthesis of racemic herbertenediol.117 Thus, olefination of sterically hindered ketone 327 led to α,β-unsaturated ester 328. Because the Wittig reaction and its Horner–Wadsworth–Emmons variant were not successful, a two-step Meyer–Schuster rearrangement118 was utilized for the desired transformation. DIBAL reduction, followed by Hg-promoted vinylation gave allyl vinyl ether 330. Treatment of 330 with DIBAL at −40 °C led to clean Claisen rearrangement and in situ reduction of the resulting aldehyde to give the key intermediate 331. Acetylation and ring-closing metathesis then afforded the target 300. Alternatively, alcohol 331 was subjected to ring-closing metathesis followed by acetylation to give 300 in slightly lower yield over the two steps (53 vs. 63%).
 | ||
| Scheme 50 Synthesis of the longtailed mealybug pheromone 300 based on ring-closing metathesis. Reagents and conditions: (a) ethoxyacetylene, n-BuLi, THF, −78 °C; (b) Sc(OTf)3, CH2Cl2–EtOH, room temp, 71% for 2 steps; (c) DIBAL, CH2Cl2, −20 °C, 92%; (d) ethyl vinyl ether, Hg(OAc)2, 47% (77% based on recovered starting material); (e) DIBAL, CH2Cl2, −40 °C, 69%; (f) Ac2O, Et3N, CH2Cl2; (g) Grubbs II catalyst (10 mol%), CH2Cl2, room temp, 63% for 2 steps. | ||
The Reddy group then modified the synthesis to prepare both enantiomers of the pheromone (Scheme 51).119 Thus, racemic alcohol 331 was esterified with Harada's camphorsultam phthalic (CSP) acid to produce a mixture of diastereomers 333 and 334, which were separated by preparative HPLC. The relative and absolute configurations of 334 were determined through X-ray crystal structure analysis, revealing that the chiral center in the intermediate had the (R)-configuration. Each enantiomer was then hydrolyzed and converted to the respective pheromone enantiomers. In field trials, the (S)-(+)-enantiomer was highly attractive to male mealybugs, indicating that this was almost certainly the insect-produced enantiomer. The (R)-enantiomer was neither attractive nor inhibitory, so the racemic pheromone is entirely sufficient for practical purposes.
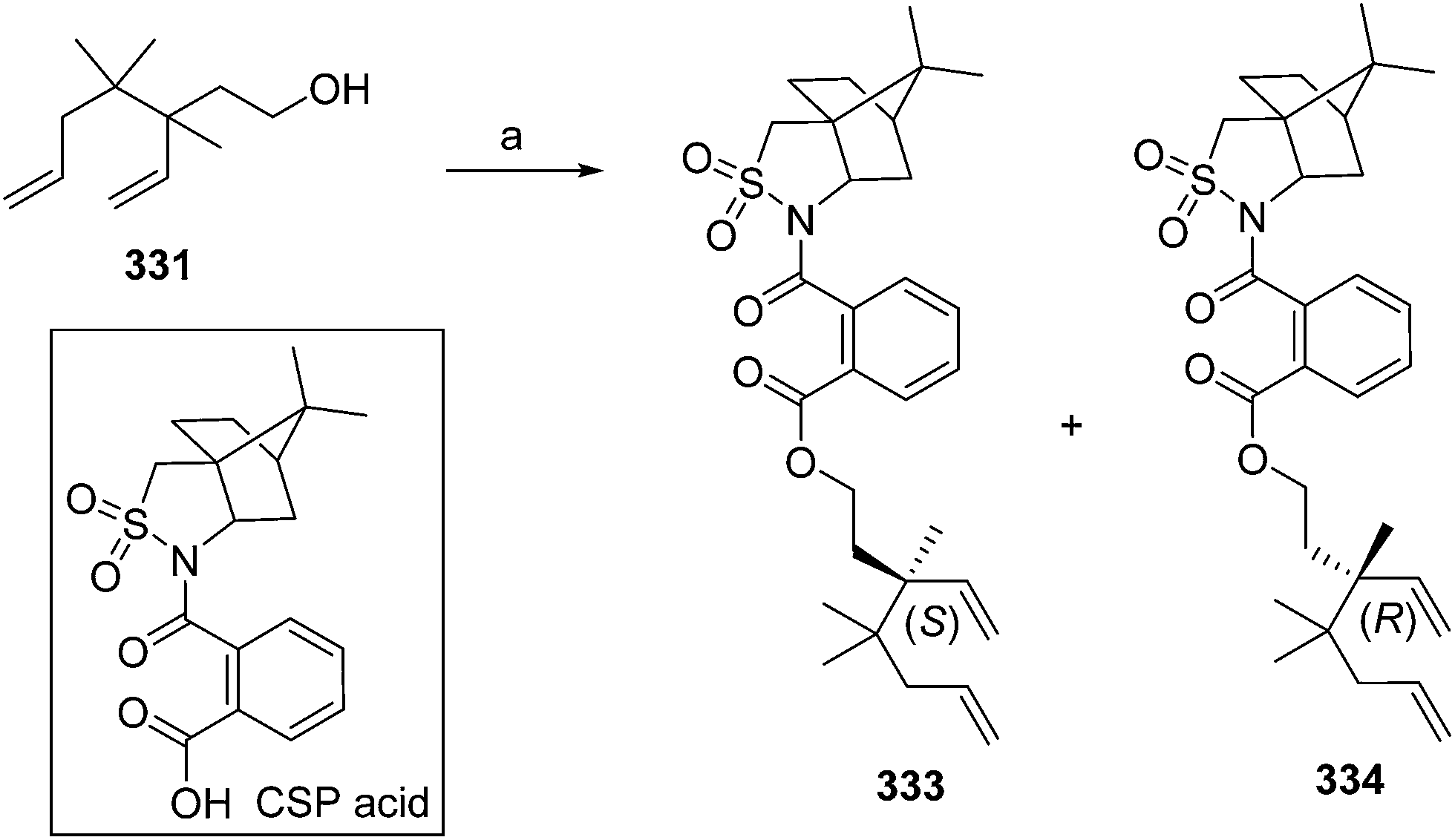 | ||
| Scheme 51 Formation of separable diastereomeric derivatives of 331 en route to synthesis of the (R)- and (S)-enantiomers of the longtailed mealybug pheromone 300. Reagents and conditions: (a) dicyclohexylcarbodiimide, DMAP, CSP acid, CH2Cl2. | ||
Grape mealybug pheromone. The sex pheromone of the grape mealybug, Pseudococcus maritimus, was identified by the Millar group as trans-α-necrodyl isobutyrate 301.8 Because the insect is difficult to maintain in culture, it took several years to obtain enough purified pheromone to determine the structure by microprobe NMR. Furthermore, the pheromone was produced in very small quantities so gas chromatography coupled to electroantennogram detection had to be used to locate the pheromone as a small peak in a complex background.8 The core monoterpene alcohol structure trans-α-necrodol 342 had been previously identified from a carrion beetle, and synthesized several times. Scheme 52 summarizes the synthesis reported by the Millar group,120 which combined and optimized elements from previous syntheses of necrodol isomers, including a diastereoselective synthesis of β-necrodol 341,121 and an efficient isomerization of β-necrodol to α-necrodol 342,122 the core structure of the pheromone 301. Thus, alkylation of ethyl acetoacetate 336 with bromide 335 followed by intramolecular Knoevenagel condensation gave 332, followed by regioselective decarboxylation to ketoester 338. Conjugate addition of dimethylzinc to 338 with nickel catalysis, and trapping the resulting enolate with methyl iodide established the trans-relative stereochemistry at C1 and C3 in 339. Methylenation under neutral conditions using Zn/CH2Br2/TiCl4 (ref. 96) followed by LiAlH4 reduction gave trans-β-necrodol 341. Upon brief exposure to lithium ethylenediamide (LEDA), the exo double bond of 341 isomerized to the trisubstituted endo double bond to give trans-α-necrodol 342 as the kinetic product. Timing of this transformation was crucial because prolonged treatment resulted in further isomerization of 342 to the thermodynamically most stable tetrasubstituted alkene. The selectivity in the isomerization presumably arose from the orientation of the hydroxymethyl group, which shielded one of the two diastereotopic faces of 341 and induced the allylic deprotonation to proceed across the more accessible face. The synthesis was completed by esterification with isobutyryl chloride.
 | ||
| Scheme 52 Synthesis of the racemic grape mealybug pheromone 301. Reagents and conditions: (a) Br2, CHCl3; (b) NaH, THF, 335; (c) NaI, AcOH, diglyme, 28% over 3 steps; (d) Me2Zn, Ni(acac)2, THF, then HMPA, MeI, 64%; (e) TiCl4, CH2Br2, Zn, THF, CH2Cl2, 67%; (f) LiAlH4, Et2O, 97%; (g) Li, ethylenediamine, 65%; (h) isobutyryl chloride, Et3N, DMAP (cat.), CH2Cl2, 93%. | ||
The synthesis was readily adapted to produce both enantiomers of the pheromone via lipase-catalyzed kinetic resolution of the alcohol 343 generated by reduction of ketone 339 (Scheme 53), as previously described.121 With both enantiomers in hand, the insect-produced pheromone was determined to be a 85:15 mixture of the (R,R)- and (S,S)-enantiomers. Nevertheless, in bioassays, the more easily made racemate was highly attractive to male mealybugs and satisfactory for detection and monitoring of this insect.
 | ||
| Scheme 53 Synthesis of the (1R,4R)- and (1S,4S)-enantiomers of the grape mealybug pheromone 301. Reagents and conditions: (a) NaBH4, EtOH; (b) vinyl acetate, Amano AK lipase; (c) K2CO3, MeOH; (d) DMSO, (COCl)2, Et3N, CH2Cl2. | ||
Obscure mealybug pheromone. The sex pheromone of the obscure mealybug, Pseudococcus viburni, was identified by the Millar group as (1R*,2R*,3S*)-1-acetoxymethyl-2,3,4,4-tetramethylcyclopentane 302.123 The small amount of purified material was insufficient to determine the relative stereochemistry and so the first synthesis was designed to be nonstereoselective, in order to produce all four possible diastereomers as standards so that the relative stereochemistry could be determined.123 After the relative stereochemistry was conclusively assigned as (1R*,2R*,3S*), modification of the second step to make it diastereoselective gave a preponderance of the desired diastereomer (Scheme 54).124 Thus, cyclization of cheap isobutyl methacrylate 345 in hot polyphosphoric acid gave trisubstituted cyclopentenone 346 in multigram quantities. Conjugate addition of Me2CuLi, quenching the resulting enolate at −78 °C with a chelating proton donor (ethyl salicylate),125 produced the thermodynamically disfavored cis-isomer 347 as the major product (cis–trans = 72
:28). Methylenation with no epimerization at C-2 using Zn/CH2Br2/TiCl4,96 followed by hydroboration/oxidation gave alcohol 349 with the wrong configuration at C-1. After oxidation to aldehyde 350, base-catalyzed epimerization readily inverted the configuration, followed by reduction back to alcohol 349 and acetylation to give (1R*,2R*,3S*)-302.
 | ||
| Scheme 54 Millar's diastereoselective synthesis of racemic obscure mealybug pheromone 302. Reagents and conditions: (a) polyphosphoric acid, 100 °C, 34%; (b) Me2CuLi, ethyl salicylate quench, −78 °C, 89%, (cis–trans = 72:28); (c) Zn, CH2Br2, TiCl4, THF, CH2Cl2; (d) BH3·Me2S, then NaOH, H2O2, 75% for 2 steps; (e) PCC, CH2Cl2, 81%; (f) NaOMe, MeOH; (g) NaBH4, EtOH, 93% for 2 steps; (h) AcCl, pyridine, 81%. | ||
Because this synthesis gave only modest cis–trans selectivity during establishment of the relative stereochemistry at C-2 and C-3 (346 → 347), and because the desired stereochemistry was cis, an improved synthesis based on stereoselective reduction of a tetrasubstituted alkene was developed (Scheme 55).126 Thus, reaction of (E)-crotonyl chloride 351 with 2-methybutane 352 in the presence of AlCl3 gave cyclopentenone 353, presumably via acylation of the alkene generated in situ by hydride transfer from the isoalkane followed by Nazarov cyclization of the divinylketone intermediate.127 The cis relative stereochemistry at C-2 and C-3 was set by rhodium-catalyzed catalytic hydrogenation of tetrasubstituted alkene 353 (cis/trans = 95:5), following general conditions developed by Paquette and coworkers.128 The synthesis was then completed as described in Scheme 54 above.
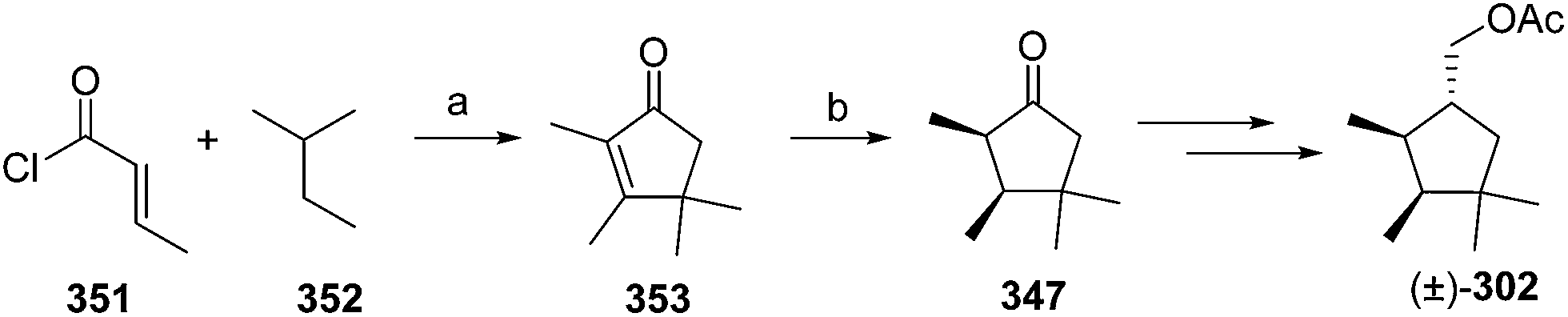 | ||
| Scheme 55 Improved diastereoselective synthesis of the obscure mealybug pheromone (±)-302. Reagents and conditions: (a) AlCl3, CH2Cl2, 55%; (b) H2 (1 atm), Rh/C, Na2CO3, pentane, 81%, (cis/trans = 95:5). | ||
The insect-produced compound was determined to be the (1S,2S,3R)-enantiomer by lipase-catalyzed kinetic resolution of the racemate, followed by vibrational circular dichroism analysis to assign the absolute stereochemistry of each enantiomer.129 The assignment was subsequently confirmed by enantioselective synthesis of the natural (1S,2S,3R)-enantiomer by Hashimoto and coworkers starting from known carboxylic acid 355, prepared from prenyl alcohol 354 in two steps (Scheme 56).130 The stereocenter at C-3 was established by asymmetric methylation of 356, directed by Evans' chiral auxiliary. Reductive cleavage of the chiral auxiliary, and Swern oxidation followed by Wittig olefination in one pot produced γ-alkyl-α,β-unsaturated ester 359. The stereocenter at C-2 was established by diastereoselective conjugate addition of an organocopper reagent to 359. After several failed attempts, Yamamoto's method131 using Me2CuLi/TMSiCl in CH2Cl2 gave the desired product 360 with complete diastereoselectivity, rationalized by a Felkin-Anh type transition state model. Compound 360 was ozonized with a reductive workup to afford alcohol 361, followed by triflation. The stereocenter at C-1 was established in the final cyclization step. A single stereoisomer was obtained, probably due to the greater thermodynamic stability of 362 as compared to the corresponding epimer. DIBAL reduction of ester 362 and acetylation of the resulting alcohol gave (1S,2S,3R)-302.
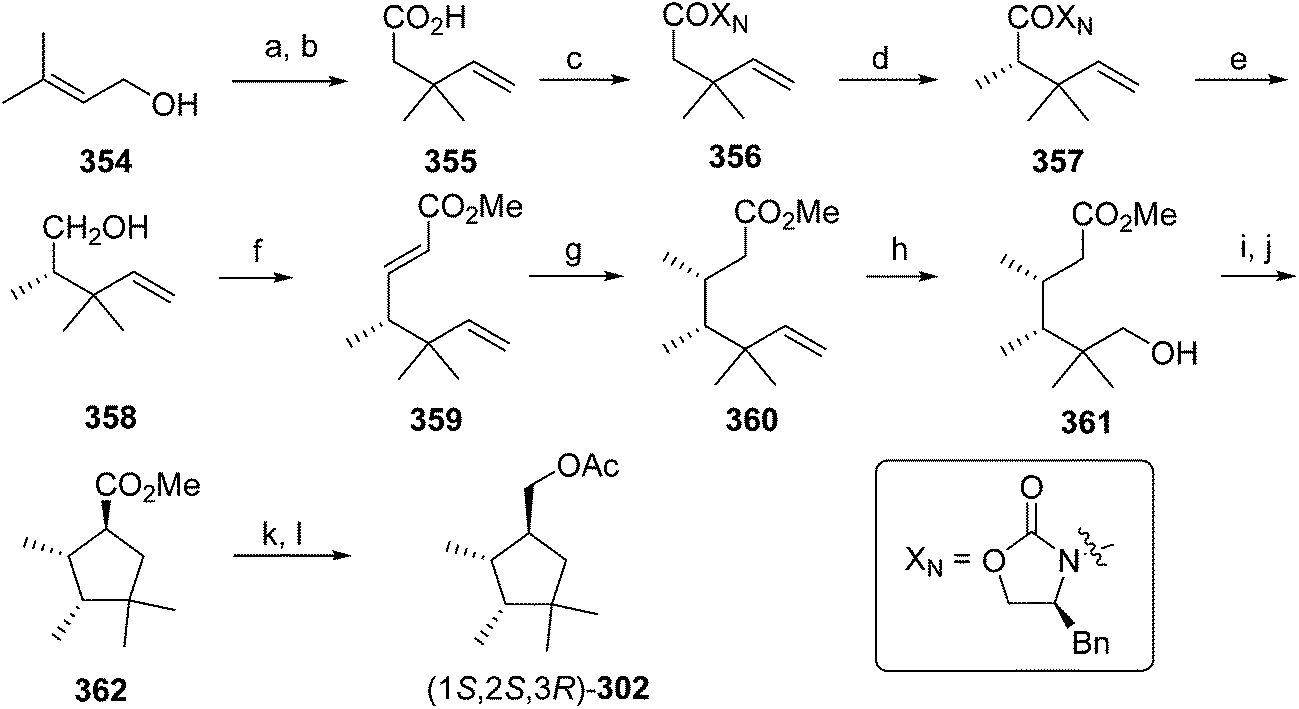 | ||
| Scheme 56 Enantioselective synthesis of the obscure mealybug pheromone (1S,2S,3R)-302. Reagents and conditions: (a) orthoester Claisen rearrangement; (b) alkaline hydrolysis, 71% over 2 steps; (c) PivCl, Et3N, THF, then XN-Li, 83%; (d) NaHMDS, MeI, THF, 74%; (e) NaBH4, THF/H2O, 71%; (f) Swern oxidation, CH2Cl2, then Ph3PCHCO2Me (one pot), 83%; (g) Me2CuLi, TMSiCl, CH2Cl2, 81%; (h) O3, MeOH/CH2Cl2, then NaBH4, 80%; (i) Tf2O, 2,6-lutidine, CH2Cl2; (j) NaHMDS, 72% for 2 steps; (k) DIBAL, CH2Cl2, 94%; (l) Ac2O, pyridine, 85%. | ||
The Reddy group subsequently reported an efficient synthesis of the (1R,2R,3S)-enantiomer from commercial D-(−)-pantolactone 363 (Scheme 57).132 Thus, benzyl-protected lactol 364 was converted into α,β-unsaturated ester 365 by Wittig olefination and tosylation of the resulting alcohol. The two chiral centers at C-1 and C-2 were installed in one pot by a tandem conjugate addition/cyclization reaction, giving 366. The alcohol from LiAlH4 reduction of ester 366 was protected with the TBDPS group, followed by removal of the benzyl group, oxidation of the resulting secondary alcohol to ketone 368, and Tebbe methylenation, furnishing alkene 369. Hydrogenation of the exocyclic double bond using Wilkinson's catalyst established the chiral center at C-3 diastereoselectively giving 370. Deprotection and acetylation of the resulting alcohol gave (1R,2R,3S)-302.
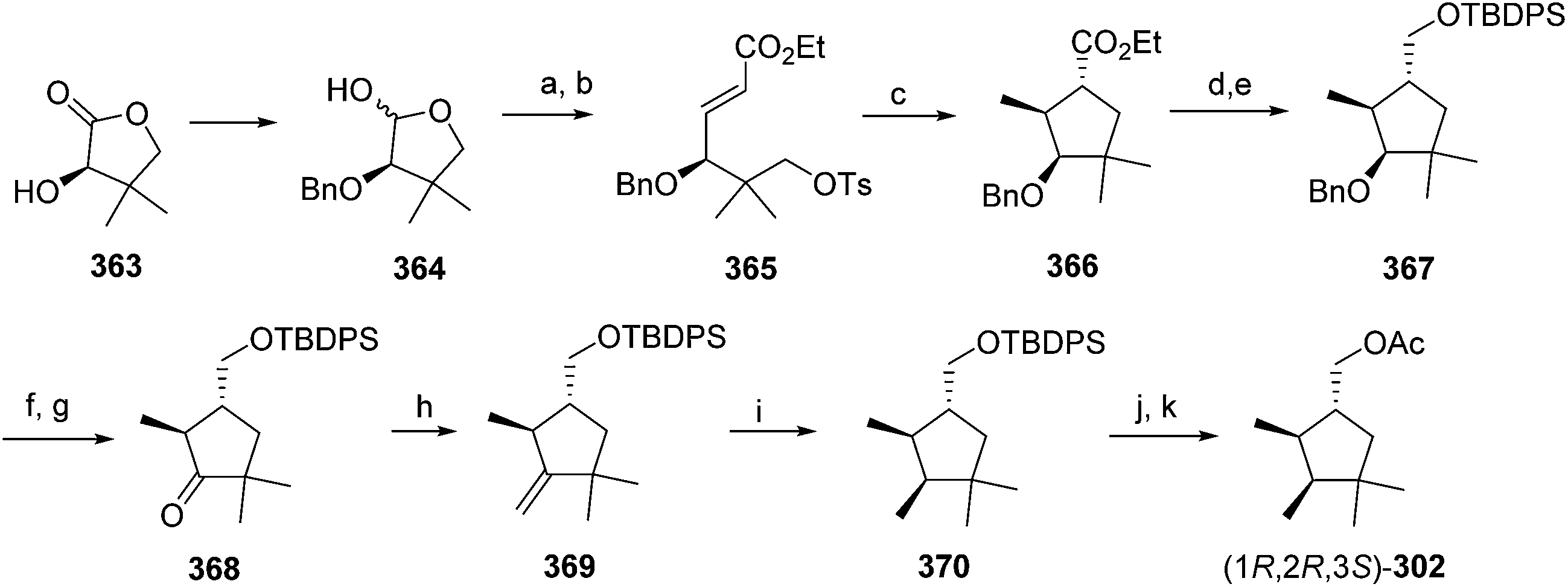 | ||
| Scheme 57 Enantioselective synthesis of the unnatural, (1R,2R,3S)-enantiomer of the obscure mealybug pheromone. Reagents and conditions: (a) Ph3PCHCO2Et, PhMe, reflux, 8 h, 75%; (b) TosCl, pyridine, DMAP, rt, 24 h, 88%; (c) Me2CuLi, TMSiCl, THF, −78 °C to room temp, 24 h, 94%; (d) LiAlH4, THF, 0 °C, 0.5 h, 95%; (e) TBDPSiCl, imidazole, pyridine, room temp, 93%; (f) 10% Pd/C, H2, MeOH, 91%; (g) Dess-Martin periodinane, CH2Cl2, 0 °C, 1 h, 89%; (h) Tebbe's reagent, THF, 0 °C, 45 min, 89%; (i) Rh(PPh3)3Cl, H2, THF, t-BuOH (1:1), room temp, 85%; (j) TBAF, THF, 60 °C, 1 h, 86%; (k) Ac2O, pyridine, DMAP, CH2Cl2, room temp, 1 h, 87%. | ||
3.3 Polyketide pheromones of pine bast scales
As their names suggest, pine bast scales in the genus Matsucoccus develop exclusively on pine, and their pheromones are markedly different than those of other scale and mealybug species. Specifically, the pattern of methylation for all of the five pheromone compounds identified from pine bast scales clearly indicates that they are products of the polyketide rather than the terpenoid biosynthetic pathway. As might be expected for pheromones of congeneric species, the structures share a common structural motif on the left hand side of the molecules as drawn in Fig. 9, with variable chains appended on the right hand side, and two of the structural pairs vary only in the stereochemistry of one double bond. Because most of the synthetic strategies used to make these compounds have taken advantage of the shared structural motif, the syntheses will be described collectively rather than being broken out by species as was done in earlier sections. | ||
| Fig. 9 Structures of pheromone components of pine bast scales. | ||
The first pine bast scale pheromone was identified by a joint effort among US, Chinese, and Korean scientists as (2E,4E)-4,6,10,12-tetramethyltrideca-2,4-dien-7-one (matsuone) 371, for the red pine bast scale Matsucoccus resinosae in the US, and two related and possibly synonymous species indigenous to Asia known as black pine bast scale, M. matsumurae in China and M. thunbergianae in Korea.133 Subsequently, the sex pheromone of the maritime pine bast scale M. feytaudi, native to Africa and Europe, was identified as a mixture of (8E,10E)-3,7,9-trimethyldodeca-8,10-dien-6-one 372 and its (8Z,10E)-isomer 373 (373 is ∼3% of 372).134 The pheromone of the Israeli pine bast scale M. josephi was identified as a 75:25 mixture of (2E,6E,8E)-5,7-dimethyldeca-2,6,8-trien-4-one 374 and its (2E,6Z,8E)-isomer 375.135 Enantioselective syntheses of stereoisomers of 371, 372, and 374, followed by bioassays or GC comparison with natural pheromones determined the absolute stereochemistry of 371 as (6R,10R), 372 as (3S,7R), and 374 as 5R (Fig. 9). Thus, all these Matsucoccus pheromones share the same (R)-configured ketone motifs, with the remaining parts of the molecules differing and conferring species specificity. The diastereomeric (6R,10S)-371 was also active, whereas the other two diastereomers were not.136,137b Similarly, the unnatural diastereomer (3R,7R)-372 was attractive to male M. feytaudi, whereas the other two diastereomers of 372 were inactive.138 The unnatural (5S)-374 also was not attractive to M. josephi.139
Cywin and coworkers reported the first enantioselective synthesis of two diastereomers of 371, (6R,10R)-371 and (6R,10S)-371 respectively (Scheme 58).140 The convergent synthesis started from commercially available (S)-(−)-glycidol 376. Routine transformations gave methyl ketone 378. Chelation-controlled addition of propynyl magnesium bromide, followed by LiAlH4 reduction afforded tertiary allylic alcohol 379 with the desired stereochemistry. O-Alkylation with 2-(chloromethyl)-4,5-dihydro-4,4-dimethyloxazole and treatment of the resulting ether 380 with n-BuLi resulted in [2,3]-Wittig rearrangement to give a mixture of 381 (major product) and its (Z)-isomer (minor product). The mixture was converted to alcohol 382 and its (Z)-isomer, which were readily separable by flash chromatography. After conversion of 382 to epoxide 383, reductive removal of the (benzyloxy)methyl (BOM) protecting group and dehydration using the Burgess protocol installed the second (E)-double bond, giving 384. The copper-catalyzed coupling of (E,E)-diene epoxide (2S,3R)-384 with (S)- and (R)-2,4-dimethylpentylmagnesium bromide, followed by oxidation, afforded (6R,10R)-371 and (6R,10S)-371. Comparison of the 500 MHz 1H NMR spectra of the two synthetic diastereomers with that of the natural pheromone established the relative stereochemistry of 371 as 6R*,10R* (6,10-syn).
 | ||
Scheme 58 Cywin et al. enantioselective synthesis of (6R,10R)-371 and (6R,10S)-371. Reagents and conditions: (a) TBDMSiCl, imidazole, CH2Cl2, 0 °C; (b) MeMgBr, CuI, THF; (c) PhCH2OCH2Cl, i-PrNEt2, LiAlH4; (d) TBAF, THF, 58% over 4 steps; (e) Swern oxidation; (f) MeMgBr, THF, then e, 85% over 3 steps; (g) MeC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMgBr, THF; (h) LiAlH4, THF; 81% over 2 steps; (i) 2-(chloromethyl)-4,5-dihydro-4,4-dimethyloxazole, KH, DME; (j) n-BuLi, THF, −78 °C; (k) p-MeOPhCH2Cl, KH, DME; (l) TFA, H2O, 24 h, then LiAlH4, THF, 44% over 4 steps; (m) TosCl, pyridine; (n) 1M aq. Ce(NH4)2(NO3)6, MeCN; (o) NaOMe, MeOH; 74% over 3 steps; (p) Na, NH3, −78 °C; (q) MeOC(O)NSO2NEt3, benzene, 50 °C; 92% over 2 steps; (r) (S)-2,4-dimethylpentylMgBr, CuI, THF, 47%; (r′) (R)-2,4-dimethylpentylMgBr, CuI, THF, 47%; (s) (n-Pr)4NRuO4, NMO, 4A sieves, CH2Cl2, 98%. CMgBr, THF; (h) LiAlH4, THF; 81% over 2 steps; (i) 2-(chloromethyl)-4,5-dihydro-4,4-dimethyloxazole, KH, DME; (j) n-BuLi, THF, −78 °C; (k) p-MeOPhCH2Cl, KH, DME; (l) TFA, H2O, 24 h, then LiAlH4, THF, 44% over 4 steps; (m) TosCl, pyridine; (n) 1M aq. Ce(NH4)2(NO3)6, MeCN; (o) NaOMe, MeOH; 74% over 3 steps; (p) Na, NH3, −78 °C; (q) MeOC(O)NSO2NEt3, benzene, 50 °C; 92% over 2 steps; (r) (S)-2,4-dimethylpentylMgBr, CuI, THF, 47%; (r′) (R)-2,4-dimethylpentylMgBr, CuI, THF, 47%; (s) (n-Pr)4NRuO4, NMO, 4A sieves, CH2Cl2, 98%. | ||
Following the same route, Cywin and coworkers reported the first enantioselective synthesis of two diastereomers of the major component of the sex pheromone of M. feytaudi, (3S,7R)-372 and (3R,7R)-372 (Scheme 59).141 The convergent synthesis employed the advanced intermediate described in Scheme 58, epoxide (2S,3R)-384 and (S)- and (R)-2-methylbutylmagnesium bromide. Comparison of the 500 MHz 1H NMR spectra of the two synthetic diastereomers with that of the natural pheromone established the relative stereochemistry of 372 as 3S*,7R* (3,7-syn), the same as that of matsuone 371.
 | ||
| Scheme 59 Cywin's enantioselective synthesis of (3S,7R)-372 and (3R,7R)-372. Reagents and conditions: (a) (S)-2-methylbutylMgBr, CuI, THF, 60%; (a′) (R)-2-methylbutylMgBr, CuI, THF, 58%; (b) (n-Pr)4NRuO4, NMO, 4A sieves, CH2Cl2. | ||
Shi and coworkers used an analogous diastereoselective [2,3]-Wittig rearrangement of tertiary bisallylic ether 390 to provide a direct entry to conjugated diene systems containing an (E)-disubstituted double bond, a stereochemically defined trisubstituted double bond, and a homoallylic hydroxyl group, key structural elements of 371, 372, and 374 (Scheme 60).137a Rearrangement of easily prepared 390 favored the formation of syn-391 under all reaction conditions, but the ratio of products was clearly influenced by reaction conditions. Use of a lithium base at low temperature with THF/HMPA solvent produced optimum yields of syn-391 (syn-391: 93.6%, anti-391: 6.4%).
 | ||
| Scheme 60 Preparation and [2,3]-Wittig rearrangement of tertiary bis-allyllic ether 390. Reagents and conditions: (a) LiAlH4, 98%; (b) LiAlH4, 89%; (c) 2-(chloromethyl)-4,5-dihydro-4,4-dimethyloxazole, KH, 90%; (d) KH, 100%. | ||
This transformation could be applied in two ways. First, carrying out the [2,3]-Wittig rearrangement under the conditions described above gave (±)-syn-391 in excellent yield, which could be isolated in over 99% purity by recrystallization from ether. Then, derivatization of the racemate with (S)-camphanyl chloride gave a mixture of two diastereomers, from which (2S,3R)-393 could be selectively crystallized. Derivatization of racemic (±)-syn-391 with (R)-camphanyl chloride similarly gave (2R,3S)-393 in > 99% de. The two enantiomers were then transformed to epoxides (2S,3R)-384 and (2R,3S)-384, respectively. The copper-catalyzed coupling of the epoxides with (S)- and (R)-2,4-dimethylpentylmagnesium bromide, respectively, followed by oxidation, afforded the four stereoisomers of 371 (Scheme 61).137b
 | ||
| Scheme 61 Shi's enantioselective synthesis of all four stereoisomers of 371. Reagents and conditions: (a) n-BuLi, THF/HMPA = 7:3, −100 °C, 0.5 h, 95%; (b) (S)-camphanyl chloride and (R)-camphanyl chloride, recrystallization; (c) TFA, H2O, THF, 25 °C, 24 h; (d) LiAlH4, THF, 0 °C, 4 h, 87% for 2 steps; (e) TosCl, pyridine, CH2Cl2, −30 °C, 48 h; (f) NaOMe, MeOH, 0 °C, 2 h, 63% for 2 steps; (g) (S)-2,4-dimethylpentylMgBr, CuI, THF; (g′) (R)-2,4-dimethylpentylMgBr, CuI, THF; (h) (n-Pr)4NRuO4, NMO, 4A molecular sieves, 25 °C, 0.5 h, 82% for 2 steps for (6R,10R)-371. | ||
This transformation was then extended to generate the (E,E)- and (Z,E)-conjugated dienes in one pot, providing an easy access to the geometric isomers of all three of the Matsucoccus pheromones (Scheme 62).137c In the presence of two equivalents of KH, the tertiary allylic alcohol 389 was alkylated to afford ether 390, which without isolation underwent the desired [2,3]-Wittig rearrangement to give a mixture of four racemic, conjugated dienes (syn-391:anti-391:syn-392:anti-392 = 7:1:1:1). The mixture was converted directly into the corresponding mixture of four dienediols 394 and 395. Periodate cleavage then afforded a mixture of aldehydes 396 and 397 (4:1 ratio), which were separated by flash chromatography. Addition of the appropriate Grignard or lithium reagents, followed by oxidation, yielded geometrically pure final products. This geometrically selective synthesis provided the naturally occurring sex pheromone components 371–375, along with 399, the geometric isomer of 371, each as a racemic and diastereomeric mixture. Whereas 373 and 375 were found as trace components in the insects, 399 has not yet been reported as a natural product.
 | ||
| Scheme 62 Shi's geometrically selective synthesis of all the Matsucoccus pheromones 371-375 and 399 as racemic and diastereomeric mixtures. Reagents and conditions: (a) TFA, H2O, THF, 25 °C, 24 h; (b) LiAlH4, THF, 0 °C, 4 h, 87% for 2 steps; (c) NaIO4, dioxane:H2O (1:1), room temp, 2 h, 55%; (d) 3,5-dimethylhexylMgBr, THF, room temp, 1 h; (e) 3-methylpentylMgBr, THF, room temp, 1 h; (f) (E)-CH3CHCHBr, t-BuLi, THF, −115 °C, 1 h; (g) Jones' reagent, 0 °C, 5 min; (h) (n-Pr)4NRuO4, NMO, CH2Cl2, room temp, 0.5 h. | ||
Mori's synthesis of all four stereoisomers of 372, the major component of the sex pheromone of M. feytaudi, built up the chain from the opposite end (Scheme 63).142 Thus, synthesis of (3R,7S)-372 started from (S)-(−)-citronellol 400, which provided the required chiral center at C-3 in 372. Routine transformations led to allylic alcohol 403. Sharpless asymmetric epoxidation of 403 with diethyl L-(+)-tartrate, formation and recrystallization of the dinitrobenzoate derivatives 405 to improve the enantiomeric purity, oxidation of the resulting epoxyalcohol 404 and Horner–Wittig olefination gave epoxy-ester 406. Palladium-catalyzed reductive cleavage of the epoxide gave 407, establishing the chiral center at C-7 in 372. Standard chain elongation steps and removal of the terminal, allylic functional group completed the carbon skeleton, and deprotection and oxidation of the resulting alcohol completed the synthesis, giving (3R,7S)-372. Following the same route, (3R,7R)-372 was synthesized from (S)-(−)-citronellol 400 employing diethyl D-(−)-tartrate in the Sharpless asymmetric epoxidation step. Similarly, (3S,7R)-372 and (3S,7S)-372 were synthesized from (R)-(+)-citronellol (R)-400, employing diethyl D-(−)-tartrate and diethyl L-(+)-tartrate in the asymmetric epoxidation steps, respectively.
 | ||
| Scheme 63 Mori's enantioselective synthesis of all four stereoisomers of 372. Reagents and conditions: (a) TosCl, pyridine; (b) LiAlH4, THF, 79% over 2 steps; (c) O3, NaHCO3, MeOH/CH2Cl2; (d) Ph3PC(Me)CO2Et, benzene, 81% over 2 steps; (e) DIBALH, Et2O, 95%; (f) (iPrO)4Ti, diethyl L-(+)-tartrate, t-BuOOH, 63%; (g) 3,5-DNBCl, pyridine, Et2O, recrystallization, 45%; (h) NaOH, MeOH/THF/H2O, 98%; (i) Swern oxidation, then d, 87%; (j) Pd2(dba)3·CHCl3, Ph3P, HCO2H, Et3N, dioxane, 78%; (k) TBDMSiCl, imidazole, DMF, then e, then i, 81%; (l) (MeO)2P(O)CH2CO2Me, n-BuLi, THF, 96%; (m) DIBALH, Et2O, 91%; (n) SO3·pyridine, THF, then LiAlH4, THF; 59%; (o) TBAF, THF, prep. HPLC, 71%; (p) Swern oxidation, 94%. | ||
Following a similar route, Mori's group synthesized (6S,10S)-matsuone 371 from (S)-citronellol (S)-400 and (6R,10R)-371 from (R)-citronellol (R)-400 (Scheme 64).143
 | ||
| Scheme 64 Mori's enantioselective synthesis of (6S,10S)-371. Reagents and conditions: (a) TosCl, pyridine; (b) NaCN, DMF; 95% over 2 steps; (c) LDA, MeI, THF; 80%; (d) DIBALH, Et2O; (e) H2NNH2·H2O, KOH, H2O/ethylene glycol, 89% over 2 steps; (f) O3, NaHCO3, MeOH/CH2Cl2, then Me2S; (g) Ph3PC(Me)CO2Et, benzene, 85% over 2 steps; (h) DIBALH, Et2O, 99%; (i) (iPrO)4Ti, diethyl L-(+)-tartrate, t-BuOOH, CH2Cl2, 81%. | ||
Mori's group developed an improved synthesis of (3S,7R)-372 from 420, a versatile building block in pheromone synthesis (Scheme 65).144 The epoxy alcohol 420 was prepared from optically active epoxide 419, derived from pig pancreatic lipase (PPL)-catalyzed asymmetric hydrolysis of meso-diacetate 418.145 Stereoselective cleavage of the epoxy ring with trimethylaluminum gave 421 as the major regioisomer, which was converted to epoxide 422 by selective tosylation of the primary alcohol and base induced closure of the epoxide ring.
 | ||
| Scheme 65 Preparation of intermediate epoxide 422. Reagents and conditions: (a) Pig pancreatic lipase, phosphate buffer/isopropyl ether, 0 °C, 71%; (b) TBDPSiCl, Et3N, DMAP, CH2Cl2, quant; (c) K2CO3, MeOH, 0 °C, 98%; (d) Me3Al, pentane, CH2Cl2, 62%; (e) TosCl, pyridine; (f) K2CO3, MeOH, 85% for 2 steps. | ||
The copper-catalyzed coupling of epoxide 422 with (S)-2-methylbutylmagnesium bromide gave 423, which after several standard operations afforded 424, which was carried through to (3S,7R)-372 as described in Mori's previous synthesis (Scheme 66).
 | ||
| Scheme 66 Mori's improved synthesis of (3S,7R)-372. Reagents and conditions: (a) (S)-2-methylbutylmagnesium bromide, CuBr, THF, 93%; (b) dihydropyran, PPTS, CH2Cl2, 95%; (c) TBAF, THF, 95%; (d) Swern oxidation; (e) Ph3PC(Me)CO2Et, benzene; (f) PTSA, EtOH, 79% over 3 steps. | ||
Mori's group used a similar route to synthesize (6R,10R)-371 (Scheme 67).146 Thus, reaction of epoxide 422 with (S)-2,4-dimethylpentylmagnesium bromide gave alcohol 426. An improvement over the previous synthesis was a more efficient removal of the primary hydroxyl group of 427, which was converted to the corresponding mesylate, and then reduced with lithium triethylborohydride (Super-hydride) to 428.
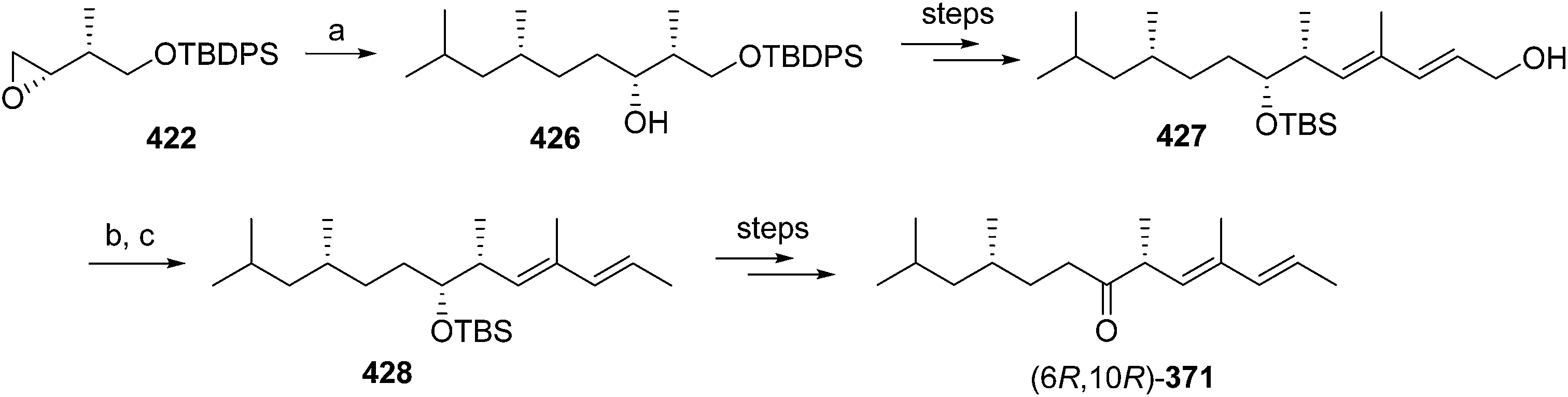 | ||
| Scheme 67 Mori's improved synthesis of (6R,10R)-371. Reagents and conditions: (a) (S)-2,4-dimethylpentylMgBr, CuBr, THF, 96%; (b) n-BuLi, Mes2O, THF; (c) LiBEt3H, THF, 73% for 2 steps. | ||
Lin and Xu developed enantioselective syntheses of all four stereoisomers of matsuone 371 from (R)- or (S)-citronellal, which provided the chiral center at C-10 in 371 (Scheme 68).147 Several straightforward steps converted (R)-citronellal 429 to aldehyde (R)-414, which was then subjected to asymmetric Aldol reactions to insert the second stereocenter. The (R)- or (S)- configuration at C-6 of 371 was determined in the transition state by employing the (Z)- or (E)-boronates 432 and 433, respectively. After protection of the hydroxyl group, alcohol 434 was subjected to ozonolysis and Wittig reaction to afford α,β-unsaturated ester 436. Reaction of 436 with the anion of ethyldiphenylphosphine oxide gave the keto-phophonate 437 which was reduced with DIBAL to give alcohol 438 with good selectivity (threo–erythro = 6:1). The threo isomer underwent base-induced elimination and concomitant deprotection to give, after oxidation, the desired pheromone (6R,10R)-371. The other three stereoisomers were prepared in the same manner from alcohol (3S,4R,7R)-434 and the corresponding alcohols from (S)-citronellal (S)-429.
 | ||
| Scheme 68 Lin and Xu's enantioselective synthesis of all four stereoisomers of 371. Reagents and conditions: (a) MeMgI, Et2O, 98%; (b) PCC, CH2Cl2, room temp, 96%; (c) i. MeMgI, Et2O, ii. n-BuLi, THF, −20 to 0 °C, iii. ClPO(OEt)2, TMEDA, −20 °C to room temp, 95%; (d) Li, EtNH2, −30 °C, quant.; (e) O3, CH2Cl2, −78 °C, then Ph3P; (f) TESiCl, imidazole, DMF, quant.; (g) O3, CH2Cl2, −78 °C, then Ph3P, room temp; (h) Ph3PC(Me)CO2Et, PhMe, 80 °C, 36 h, 76% over 2 steps; (i) Ph2P(O)Et, n-BuLi, THF, −78 °C, 95%; (j) DIBAL, Et2O, −78 °C, 74%; (k) NaH, DMF, 40 °C, 65%; (l) Swern oxidation, 90%. | ||
Lin and Xu reported a second approach to the synthesis of (6R,10R)-371, in which both chiral centers of 371 were controlled via manipulation of a bifunctional building block 440, obtained by the lipase-catalyzed transesterification of meso-2,4-dimethyl-1,5-propanediol 439 (Scheme 69).148 Thus, bromide 443 was prepared from 440 in five routine steps. Synthesis of the second required synthon 450 also started with 440. Thus, methyl ester 445, obtained from 440 in three steps, was subjected to phenylselenenylation and oxidative elimination to afford alkene 447, along with a small amount of isomeric 446, which was converted quantitatively to 447 by refluxing in anhydrous ethanol with catalytic RhCl3. Treatment of α,β-unsaturated ester 447 with the lithium salt of ethyl diphenylphosphine oxide, followed by regioselective reduction with NaBH4/CeCl3 furnished 449 with good selectivity (threo–erythro = 4:1). After removal of the erythro isomer by flash chromatography, the threo isomer underwent base-induced elimination and concomitant deprotection to give the diene homoallylic alcohol 450. Oxidation to the aldehyde, copper-catalyzed reaction with the Grignard reagent derived from 443, and Swern oxidation gave (6R,10R)-371.
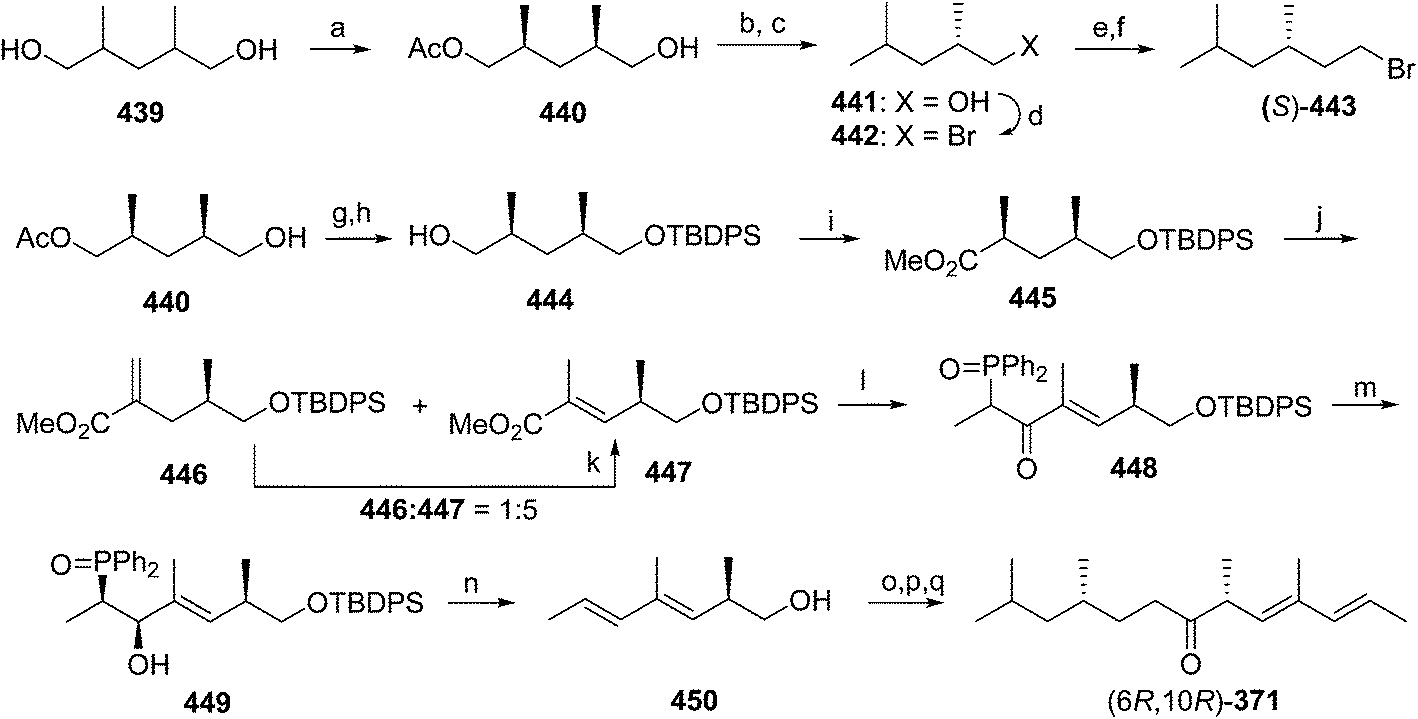 | ||
| Scheme 69 Lin and Xu's enzyme-based synthesis of (6R,10R)-371. Reagents and conditions: (a) vinyl acetate, pig pancreatic lyase, wet THF, 28 °C; (b) TosCl, pyridine, 92%; (c) LiAlH4, Et2O, reflux, 90%; (d) Ph3P, CBr4, CH2Cl2, 79%; (e) Mg, Et2O, reflux, then (HCHO)n, reflux, 74%; (f) Ph3P, CBr4, CH2Cl2, 82%; (g) TBDPSiCl, imidazole, DMF, 98%; (h) K2CO3, MeOH, 40 °C, 95%; (i) i. Jones oxidation, 0 °C, ii. CH2N2, Et2O, 0 °C, 83%; (j) i. LDA, THF, 0 °C; ii. PhSeBr, −78 °C, iii. H2O2, pyridine, CH2Cl2, 85%; (k) RhCl3·3H2O, EtOH, reflux, quant.; (l) Ph2P(O)Et, n-BuLi, −78 °C, 98%; (m) NaBH4, CeCl3, EtOH, −78 °C, 92%; (n) NaH, DMF, 40 °C, 90%; (o) Swern oxidation; (p) (S)-1-bromo-3,5-dimethylhexane, Mg, THF, CuI, reflux, 55% over 2 steps; (q) Swern oxidation, 92%. | ||
Zegelman and coworkers reported a relatively short synthesis of (±)-374 and (±)-375, the racemates of both components of the sex pheromone of M. josephi (Scheme 70).149 Reformatsky reaction between crotonaldehyde 451 and ethyl 2-bromopropanoate 452 gave hydroxyester 453, which was protected and reduced to aldehyde 454. Wittig reaction of 454 with the anion from allylic phosphonium salt 455 gave a mixture of (E)- and (Z)-isomers. Removal of the TBDMS protecting group followed by oxidation with pyridinium chlorochromate produced a mixture of (±)-374 and (±)-375 in a ratio of 56:44.
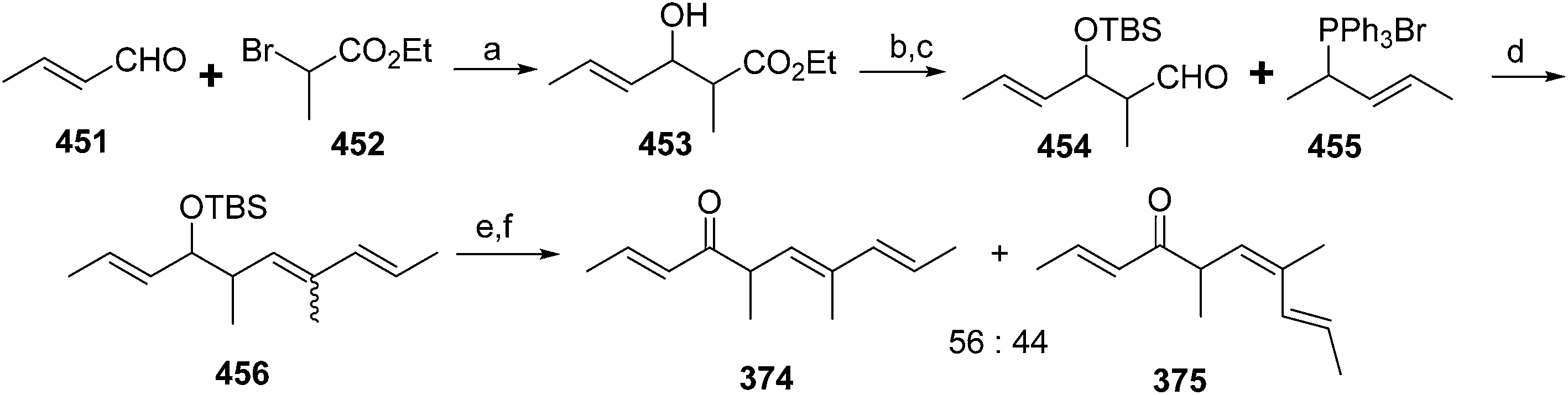 | ||
| Scheme 70 Zegelman's synthesis of (±)-374 and (±)-375. Reagents and conditions: (a) Zn–Cu, Et2O, room temp, 84%; (b) TBDMSiCl, imidazole, DMF, room temp; (c) DIBAL, PhMe, −78 °C, 63% over 2 steps; (d) n-BuLi, THF, 0–10 °C, 72%; (e) TBAF, THF, room temp; (f) PCC, NaOAc, CH2Cl2, room temp, 54% over 2 steps. | ||
A modified route led straight from the Wittig product to the pheromone (Scheme 71).149
 | ||
| Scheme 71 Zegelman's modified synthesis of (±)-374 and (±)-375. Reagents and conditions: (a) PCC, NaOAc, CH2Cl2, room temp, 62%; (b) HO(CH2)2OH, PTSA, CHCl3, reflux; (c) DIBAL, PhMe, −78 °C, 48% for 2 steps; (d) n-BuLi, THF, 0–10 °C; (e) aq. HCl, THF, room temp, 51% over 2 steps. | ||
Bioassays showed that pheromonal activity was due mainly to the E-isomer (±)-374, whereas the minor Z-isomer (±)-375 was of low activity, but not inhibitory.
Mori and Amaike reported an enantioselective synthesis of both enantiomers of 374 (Scheme 72),150 which used many of the same steps and synthons as in his syntheses of matsuone 371. Thus, the reactive ends of 420 were interchanged by sequential protection of the free hydroxyl and deprotection of the other hydroxyl, followed by Swern oxidation and Horner–Wittig reaction. In a key step, cleavage of the epoxy ring of 461 with trimethylaluminum in the presence of a small amount of water regio- and stereoselectively placed the chiral methyl group, giving 462 in excellent yield. Chain elongation, removal of the two terminal hydroxyls, deprotection and oxidation of the resulting secondary alcohol afforded (R)-374. (S)-374 was prepared in analogous fashion from the antipode of 460, in turn prepared from 420 in 4 simple steps.
 | ||
| Scheme 72 Mori's enantioselective synthesis of (R)-374. Reagents and conditions: (a) PivCl, pyridine, CH2Cl2, 93%; (b) TBAF, HF, H2O/THF, 95%; (c) Swern oxidation; (d) Ph3PC(Me)CO2Et, THF, 63% over 2 steps; (e) Me3Al (10 equiv.), H2O (6 equiv.), CH2Cl2, 89%; (f) TBDMSiCl, imidazole, DMF, 91%; (g) DIBAL, Et2O, 88%; (h) Swern oxidation, (i) (EtO)2P(O)CH2CO2Et, n-BuLi, THF, 61% over 2 steps; (j) DIBAL, Et2O, 98%; (k) n-BuLi, Mes2O, THF; (l) LiBEt3H, THF; (m) TBAF, THF, SiO2–AgNO3 chromatography; (n) 3,5-dinitrobenzoyl chloride, pyridine, recrystallization; (o) K2CO3, MeOH/THF; (p) Swern oxidation, 92%. | ||
Because the unnatural isomers were not inhibitory, Watanabe developed short syntheses of Matsucoccus sex pheromones 371, 372, and 374 as mixtures of isomers for a study of practical pest control (Scheme 73).151 The key reaction was Lewis acid-mediated SN2′ cyanation of symmetric tertiary alcohol 389 to afford common intermediate 467 as the major product, which could be separated from the undesired minor isomer 468 by flash chromatography. Nucleophilic addition of (±)-3,5-dimethylhexyllithium 469 and (±)-3-methylpentyllithium 470 followed by aqueous hydrolysis of the resulting imines afforded 371 and 372 respectively, as racemic and diastereomeric mixtures. However, reaction of 467 with (E)-1-propenyllithium resulted in the recovery of 467, presumably due to the lower nucleophilicity of the vinyl anion. Therefore, 467 was reduced to aldehyde 396, which then underwent nucleophilic addition of (E)-1-propenyllithium, followed by PDC oxidation to give racemic 374.
 | ||
| Scheme 73 Watanabe's concise syntheses of 371, 372, and 374 as racemic and diastereomeric mixtures. Reagents and conditions: (a) 1-propenylmagnesium bromide, 95%; (b) TMSCN, TMSOTf, 56% (467:468 = 5.3:1); (c) Et2O, then H3O+, 70%; (c′) Et2O, then H3O+, 73%; (d) DIBAL, then H3O+; (e) (E)-1-propenyllithium, THF, −78 °C, 67% over 2 steps; (f) PDC, DMF, 49%. | ||
Mori's group developed yet another synthetic route to (6R,10R)-371, (3S,7R)-372, and (5R)-374 with a synthon from the chiral pool, methyl (S)-2-methyl-3-hydroxypropanoate 471 (Scheme 74).152 The route incorporated a number of steps from previous routes. Thus, several routine steps gave α,β-unsaturated ester 474, which was converted to diene homoallylic alcohol 450 as described by Lin and Xu (Scheme 69). Two steps converted 450 to the key intermediate, Weinreb amide 479, followed by coupling with appropriate organometallic reagents to give (6R,10R)-371, (3S,7R)-372, and (5R)-374.
 | ||
| Scheme 74 The Mori group's syntheses of (6R,10R)-371, (3S,7R)-372, and (5R)-374 from one key intermediate. Reagents and conditions: (a) TBDMSiCl, imidazole, 98%; (b) DIBAL, hexanes/CH2Cl2; (c) Ph3PC(Me)CO2Et, benzene, 83% over 2 steps (E–Z = 95:5); (d) Ph2P(O)Et, n-BuLi, THF, 98%; (e) NaBH4, CeCl3·7H2O, EtOH, then silica gel chromatography, 66%; (f) NaH, DMF; (g) TBAF, THF, 90% over 2 steps; (h) PDC, DMF; (i) HNMe(OMe)·HCl, EDC·HCl, DMAP, diisopropylethylamine, CH2Cl2, 38% over 2 steps; (j) (S)-3,5-dimethylhexylMgBr, THF, 89%; (k) (S)-3-methylpentylMgBr, THF, 90%; (l) (E)-propenylMgBr, THF, 80%. | ||
Somewhat surprisingly, given the interesting challenges in the syntheses of the pine bast scale pheromones, there have been no further syntheses reported since 2003.
4 Practical applications of scale and mealybug pheromones
Pheromones can be used for detection and sampling of scales and mealybugs, and potentially for direct control. In practice, the pheromones are ideal for detection and sampling because they are powerful and species-specific attractants, very small amounts of pheromone (usually a few micrograms) are required per lure, and the pheromone structures are quite stable so that pheromone lures can have field lifetimes of several months or more. Because such small doses of pheromone are required per lure, relatively expensive, multistep syntheses of the pheromones are still feasible. For example, at a dose rate of 20 micrograms per lure, 1 g of pheromone is sufficient for 50000 lures. Furthermore, in most cases, male scales and mealybugs are insensitive to the presence of the unnatural enantiomers or other stereoisomers, and so less expensive racemic pheromone can be used for most applications. Consequently, pheromone lures for many species are available from commercial suppliers. Furthermore, because the pheromones are species-specific, no special expertise is required to identify the trapped insects; if male scales or mealybugs are detected in a pheromone-baited trap, it is almost certain that they are males of the species that produces that particular pheromone. The one documented exception has been the cross-attraction of the scale Quadraspidiotus zonatus to the pheromone of the San Jose scale, Q. perniciosus.153 Conversely, field trials with composite lures containing the pheromones of citrus, obscure, and longtailed mealybugs showed that there was minimal interference among these pheromones, suggesting that it may be possible to use “generic” mealybug lures that attract several species simultaneously.154 This may be useful in crops that can be infested with several species simultaneously because the control measures are usually the same no matter what species is present.
In terms of detection, pheromone-baited sticky traps can detect very low level and dispersed populations that would be virtually impossible to find by any other method. For example, the pheromone of the passionvine mealybug was used to show that this species had invaded Florida from the Caribbean islands, but populations were extremely low, possibly as a result of control by natural enemies.155 Pheromone-baited traps were also used as a sensitive method of tracking the expanding ranges of the invasive vine mealybug as it invaded California vineyards,24 and the pink hibiscus mealybug in several US states.156 As a proactive measure against another invasive species, the pheromone of the albopicta scale was identified so that it could be used as a tool for early detection of this insect if it invaded California from Mexico.102 As a further example of the powerful attraction of these pheromones, when developing protocols for using yellow scale pheromone to detect infestations, lure doses had to be lowered to 1–5 micrograms per lure, both to prevent traps from becoming completely covered with male scales and making them very difficult to count, and because higher doses apparently attracted male scales from substantial distances, providing the false impression that the orchard in which the trap was hung was infested, when the actual infestation might be some distance away.157
Pheromone-baited traps have also been used extensively in monitoring established populations of scales and mealybugs, particularly for monitoring the flight phenology of adults as an aid to making control decisions. It has proven more difficult to correlate trap catches with infestation levels to develop economic thresholds that, when exceeded, trigger control measures, for two interlinked reasons. First, as mentioned above, the pheromones are so powerful that they can attract males from some distance away, and second, scales and mealybugs tend to form highly clumped distributions, rather than being randomly distributed throughout a crop. Thus, high trap catches may not necessarily indicate high populations throughout the crop. Nevertheless, by judicious use of lower doses of pheromone to decrease the range of attraction of pheromone lures, in some cases it has been possible to develop good correlations between pheromone trap catches and infestation levels as determined by visual counts or other means.24,154,158 The interested reader is referred to a more extensive discussion of the development of pheromone-baited traps for monitoring purposes.3
Pheromones can be used to control insect populations in at least three ways: (1) disruption of mating by permeating the atmosphere of a crop with pheromone so that males cannot find females, (2) mass trapping, in which sufficient numbers of pheromone-baited traps are deployed to remove a large percentage of males, and (3) attract-and-kill, in which pheromone is mixed with a toxicant in a matrix applied in multiple droplets throughout a crop, so that males that are attracted and contact the droplets are killed. The feasibility of using these three methods for controlling scales and mealybugs hinges on a number of economic and biological factors. First and foremost, if the pheromones cannot be made in multikilo scale at a price that is competitive with other control measures, mating disruption at least will not be economically feasible. For mass trapping and attract-and-kill, which use much smaller quantities of pheromone, the costs of deploying and servicing large arrays of traps or attract-and-kill droplets may still present significant barriers to implementation. For attract-and-kill, there is also the additional requirement that formulations containing a toxicant may be subject to additional registration requirements. A number of other points also must be considered, including:
1. The number of generations per year, and the overall adult activity periods. Clearly the costs and technical issues relating to control of a species with a single generation per year and a relatively short adult activity period will be different than those involved in controlling a multivoltine species that is present for many months.
2. Female scales and mealybugs can live for several months, during which time they will continue to produce pheromone until they are mated. Thus, even if a substantial percentage of the available males is removed, the remaining males may still mate many of the available females because males can mate multiple times.159 Thus, almost all of the males must be removed or otherwise prevented from mating in order to achieve good control.
3. Conversely, adult males live for at most a few days under field conditions.
4. The highly clumped, non-random distribution of scales and mealybugs will also hinder pheromone-based control measures, because males and females will be close together in clumps, particularly in heavy infestations. Thus, males may serendipitously find females.
5. Pheromone formulations must have effective field lifetimes of many weeks. However, these issues are readily dealt with because scale and mealybug pheromones are relatively stable, and longevity of lures or other formulations can be readily adjusted by appropriate choice of release device.
6. The size of the market for any one pheromone is relatively small. That is, the species specificity of pheromones is a major advantage from the viewpoint of nontarget effects, and a major disadvantage because, unlike a broad-spectrum insecticide which is effective against multiple insects on numerous crops, a scale or mealybug pheromone will only affect a single target species.
In practice, mass trapping has only been attempted with the citrus mealybug. Whereas numbers of males were reduced in treated blocks relative to controls, damage levels were unaffected. Furthermore, it appeared that the traps were attracting more males into the treated areas, partially counteracting male removal by the traps.160
Mating disruption was first explored for California red scale in the 1980s,161 and has been followed up in a series of studies in the last decade162 which demonstrated decreased infestation levels and significantly less crop damage in treated areas versus untreated controls. Thus, this system has considerable promise biologically, if the economic hurdles to producing large quantities of the pheromone can be surmounted. Similarly, a mating disruption trial with San Jose scale was moderately successful but was not pursued further because of the prohibitive cost of the pheromone.163
The vine mealybug represents the most successful and to date only commercially viable use of mating disruption for control of scales or mealybugs, primarily because the pheromone can be made economically in large scale. Initial results from trials using a microencapsulated formulation were reported in 2006,164,165 and since then, other dispenser types have been developed, including reservoir dispensers and “puffers” that release puffs of pheromone at timed intervals.166–168 Vine mealybug mating disruption is most effective when used in combination with early-season insecticide treatments to reduce initial populations to low levels, which are then maintained with the disruptant treatment.167 In California, mating disruption of vine mealybug is now used on thousands of acres of wine grapes annually, and the active formulation is being registered for use in 8 additional countries by the manufacturer (Suterra LLC, Bend OR, USA).
5 Conclusions
We hope that this review has given the reader an appreciation of the fascinating chemistry of scale and mealybug pheromones. Although the biosyntheses of these compounds have not yet been studied, it is clear that they fall into two major groups, based on irregular terpenoid biosynthetic pathways for the diaspidid and pseudococcid species, and polyketides for the margorodid species. Synthesis of these pheromones in pure form can be challenging, requiring control of one to several chiral centers and the geometries of di- and trisubstituted alkenes. However, because most species are not inhibited by the presence of unnatural isomers, much shorter, nonstereoselective syntheses can be used to prepare “technical” grade pheromones which are usually adequate for practical purposes. Although numerous stereoselective and nonstereoselective syntheses of the various compounds have been developed, faster, more efficient, and more economical syntheses are still highly desirable in order to accelerate the development and adoption of these useful natural products for pest management.Following the progression of syntheses from the late 1970s to the present is also an interesting study in the development of synthetic chemistry methodology. For example, syntheses of chiral pheromones in the 1980s made extensive use of chiral synthons available from natural sources, or derivatives thereof. Alternatively, diastereomeric derivatives were formed from reaction of a chiral derivatizing agent with the racemate of the desired synthon, followed by separation of the diastereomers by fractional crystallization or chromatography (Schemes 21, 51 and 61). The desired chiral synthon was then released from the purified diastereomer by hydrolysis or some other appropriate reaction. Both of these methods were limited by the number and types of chiral synthons or reagents that were available from natural sources.
This pool was then expanded somewhat by using enzymes to produce chiral synthons, either by kinetic resolution of racemates (i.e., enzyme-catalyzed reactions selective for one enantiomer) (Schemes 19, 53, and Madeira mealybug pheromone synthesis), or the related enzyme-catalyzed desymmetrization of achiral compounds containing two or more chiral centers such as meso compounds (Schemes 15, 65, 69 and 72).
However, the great leap forward that largely freed synthetic chemists from the limitations of the chiral synthons available from natural sources came with the introduction and widespread adoption of methods for asymmetric induction in achiral precursors, by a variety of methods. For example, temporarily attached chiral auxiliaries could be used to direct the stereoselective formation of new bonds (Schemes 27A, 27B, 29B, 38 and 56). Alternatively, chiral catalysts were developed that favored the production of one particular stereoisomer due to the three-dimensional assembly of the various reactants in the transition state (Scheme 19). In this day and age when the use of chiral catalysts is commonplace, it may be hard for younger chemists to appreciate the impact of a reaction such as the Sharpless asymmetric epoxidation on the field of organic synthesis. It was truly a turning point, in terms of demonstrating that generation of chirality by asymmetric induction was not only possible but practically feasible. In the intervening years, literally thousands of stereoselective reactions and applications have been developed, that have revolutionized the science of organic synthesis, both for small-scale applications in research laboratories, and in large-scale industrial applications.
Another area in which the advances in synthetic methodology are apparent from this review is in the formation of carbon–carbon bonds. Thus, in the syntheses from the 1980s, we see some of the first applications of copper-catalyzed reactions, or reactions using stoichiometric amounts of organocuprates. Subsequent years illustrate not only the expansion of the breadth and scope of these reactions, but introduction of additional coupling methods using other organometallics such as organostannanes (Schemes 14, 18, 29A and 46) and organozinc reagents (Schemes 7, 16 and 19), and reactions catalyzed by palladium complexes (Scheme 19). These include not only nucleophilic displacements, but also the regio- and stereoselective addition of organometallics to alkenes and alkynes (Schemes 1, 3, 7, 19 and 49). Olefin metathesis has also been used in pheromone synthesis (Scheme 50).
However, it is also useful to keep in mind that old reactions are not necessarily outdated or inefficient, particularly given that chiral catalysts containing rare elements such as palladium, gold, and ruthenium can be breathtakingly expensive. Rather, the different routes to a target compound, and the reagents required, should be carefully considered in light of the particular requirements of a project, including the required amount and stereochemical purity of a target compound.
To date, many of the pheromones described above have proven enormously useful for detection of infestations, particularly for invasive species. They have also found widespread use in monitoring scale and mealybug populations and their seasonal dynamics in numerous crops, providing valuable data on population cycles that inform pest management decisions. However, the use of pheromone-based methods for direct crop protection, particularly on a large scale, will not be competitive with other crop protection methods for a number of species because of the prohibitive cost of producing the required pheromones in sufficient quantities. This barrier to implementation may be insurmountable for some species because of the structural complexity of their pheromones.
References
- J. C. Franco, A. Zada and Z. Mendel, in Biorational control of arthropod pests, ed. I. Ishaaya and A. R. Horowitz, Springer Science + Business Media B.V., 2009, pp. 233–278 Search PubMed.
- K. Mori, in The Total Synthesis of Natural Products, ed. J. ApSimon, John Wiley & Sons, New York, 1992, vol. 9, pp. 280–332 Search PubMed.
- E. Dunkelblum, in Pheromones of Non-Lepidopteran Insects Associated with Agricultural Plants, ed. J. Hardie and A. K. Minks, CABI publishing, 1999, pp. 251–276 Search PubMed.
- J. G. Millar, K. M. Daane, J. S. McElfresh, J. A. Moreira and W. J. Bentley, in Semiochemicals in Pest and Weed Control, ed. R. J. Petroski, M. R. Tellez and R. W. Behle, ACS Symposium Series 906, American Chemical Society, Washington, DC, 2005, pp 11−27 Search PubMed.
- Y. Zou, S. P. Chinta and J. G. Millar, in Pest Management with Natural Products, ed. J. J. Beck, J. R. Coats, S. O. Duke and M. E. Koivunen, ACS Symposium Series 1141, American Chemical Society, Washington, DC, 2013, pp. 125–143 Search PubMed.
- T. Negishi, M. Uchida, Y. Tamaki, K. Mori, T. Ishiwatari, S. Asano and K. Nakagawa, Appl. Entomol. Zool., 1980, 15, 328 CAS.
- D. S. Moreno, J. Fargerlund and J. G. Shaw, J. Econ. Entomol., 1976, 69, 292 CrossRef CAS PubMed.
- B. A. Figadère, J. S. McElfresh, D. Borchardt, K. M. Daane, W. Bentley and J. G. Millar, Tetrahedron Lett., 2007, 48, 8434 CrossRef PubMed.
- A. B. Attygalle, in Methods in Chemical Ecology. Volume 1. Chemical Methods, ed. J. G. Millar and K. F. Haynes, Chapman and Hall, Norwell, MA, 1998, pp. 207–294 Search PubMed.
- J. Tabata, Y. Narai, N. Sawamura, S. Hiradate and H. Sugie, Naturwissenschaften, 2012, 99, 567 CrossRef CAS PubMed.
- M. J. Gieselmann, R. E. Rice, R. A. Jones and W. L. Roelofs, J. Chem. Ecol., 1979, 5, 891 CrossRef CAS.
- R. J. Anderson, H. R. Chinn, K. Gill and C. A. Henrick, J. Chem. Ecol., 1979, 5, 919 CrossRef CAS.
- R. J. Anderson, M. J. Gieselmann, H. R. Chinn, K. G. Adams, C. A. Henrick, R. E. Rice and W. L. Roelofs, J. Chem. Ecol., 1981, 7, 695 CrossRef CAS PubMed.
- K. H. Yong, J. A. Lotoski and J. M. Chong, J. Org. Chem., 2001, 66, 8248 CrossRef CAS.
- (a) M. Alderdice, C. Spino and L. Weiler, Tetrahedron Lett., 1984, 25, 1643 CrossRef CAS; (b) M. Alderdice, C. Spino and L. Weiler, Can. J. Chem., 1993, 71, 1955 CrossRef CAS.
- D. A. Lombardo and A. C. Weedon, Tetrahedron Lett., 1986, 27, 5555 CrossRef CAS.
- V. V. Veselovskii, S. P. Skorobogatov, M. A. Novikova and A. M. Moiseenkov, Izv. Akad. Nauk SSSR, Ser. Khim., 1990, 591 CAS , English translation: Russ. Chem. Bull., 1990, 39, 513.
- A. M. Moiseenkov, R. I. Ishchenko, V. V. Veselovskii, V. N. Odinokov, E. V. Polunin, B. G. Kovalev, B. A. Cheskis and G. A. Tolstikov, Khim. Prir. Soedin., 1989, 422 CAS , English translation: Chem. Nat. Compd., 1989, 25, 366.
- D. Ferroud, J. M. Gaudin and J. P. Genet, Tetrahedron Lett., 1986, 27, 845 CrossRef CAS.
- U. P. Dhokte and A. S. Rao, Synth. Commun., 1988, 18, 811 CrossRef CAS.
- L. Novak, L. Poppe, C. Szantay and E. Szabo, Synthesis, 1985, 939 CrossRef CAS.
- V. N. Odinokov, O. S. Kukovinets, R. A. Zainullin, E. Yu. Tsyglintseva, V. R. Sultanmuratova, V. V. Veselovskii, B. A. Dragan, T. Ya. Rubinskaya, B. A. Cheskis, A. M. Moiseenkov and G. A. Tolstikov, Khim. Prir. Soedin., 1989, 419 CAS , English translation: Chem. Nat. Compd., 1989, 25, 364.
- G. Cardillo, A. D'Amico, M. Orena and S. Sandri, J. Org. Chem., 1988, 53, 2354 CrossRef CAS.
- J. G. Millar, K. M. Daane, J. S. McElfresh, J. A. Moreira, R. Malakar-Kuenen, M. Guillen and W. J. Bentley, J. Econ. Entomol., 2002, 95, 706 CrossRef CAS PubMed.
- A. Zada and M. Harel, Tetrahedron: Asymmetry, 2004, 15, 2339 CrossRef CAS PubMed.
- A. Zada and E. A. Dunkelblum, Tetrahedron: Asymmetry, 2006, 17, 230 CrossRef CAS PubMed.
- D. M. Hinkens, J. S. McElfresh and J. G. Millar, Tetrahedron Lett., 2001, 42, 1619 CrossRef CAS.
- A. Zada, E. Dunkelblum, F. Assael, M. Harel, M. Cojocaru and Z. Mendel, J. Chem. Ecol., 2003, 29, 977 CrossRef CAS.
- A. Zhang, D. Amalin, S. Shirali, M. S. Serrano, R. A. Franqui, J. E. Oliver, J. A. Klun, J. R. Aldrich, D. E. Meyerdirk and S. L. Lapointe, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9601 CrossRef CAS PubMed.
- I. de Alfonso, E. Hernandez, Y. Velazquez, I. Navarro and J. Primo, J. Agric. Food Chem., 2012, 60, 11959 CrossRef CAS PubMed.
- H. Sugie, M. Teshiba, Y. Narai, T. Tsutsumi, N. Sawamura, J. Tabata and S. Hiradate, Appl. Entomol. Zool., 2008, 43, 369 CrossRef CAS.
- J. Tabata, Appl. Entomol. Zool., 2013, 48, 229 CrossRef CAS PubMed.
- H.-Y. Ho, C.-C. Hung, T.-H. Chuang and W.-L. Wang, J. Chem. Ecol., 2007, 33, 1986 CrossRef CAS PubMed.
- J. G. Millar, Tetrahedron Lett., 2008, 49, 315 CrossRef CAS PubMed.
- J. G. Duboudin and B. Jousseaume, J. Organomet. Chem., 1979, 168, 1 CrossRef CAS.
- B. A. Bierl-Leonhardt, D. S. Moreno, M. Schwarz, H. S. Forster, J. R. Plimmer and E. D. DeVilbiss, Life Sci., 1980, 27, 399 CrossRef CAS.
- B. A. Bierl-Leonhardt, D. S. Moreno, M. Schwarz, H. S. Forster, J. R. Plimmer and E. D. DeVilbiss, J. Chem. Ecol., 1982, 8, 689 CrossRef CAS PubMed.
- M. Uchida, K. Nakagawa, T. Negishi, S. Asano and K. Mori, Agric. Biol. Chem., 1981, 45, 369 CrossRef CAS.
- R. I. Ishchenko, V. V. Veselovskii, A. M. Moiseenkov, B. A. Cheskis and B. G. Kovalev, Khim. Prir. Soedin., 1989, 132 CAS , English translation: Chem. Nat. Compd., 1989, 25, 118.
- K. Mori and H. Ueda, Tetrahedron, 1981, 37, 2581 CrossRef CAS.
- N. Nakagawa and K. Mori, Agric. Biol. Chem., 1984, 48, 2799 CrossRef CAS.
- M. Larcheveque and Y. Petit, Bull. Soc. Chim. Fr., 1989, 130 CAS.
- L. Skattebol and Y. Stenstrom, Acta Chem. Scand., 1989, 43, 93 CrossRef PubMed.
- P. Baeckstroem and L. Li, Synth. Commun., 1990, 20, 1481 CrossRef CAS.
- S. K. Kang and C. S. Park, Org. Prep. Proced. Int., 1990, 22, 627 CrossRef CAS.
- (a) T. Cohen and M. Bhupathy, Acc. Chem. Res., 1989, 22, 152 CrossRef CAS; (b) D. W. McCullough, M. Bhupathy, E. Piccolino and T. Cohen, Tetrahedron, 1991, 47, 9727 CrossRef CAS.
- P. Baeckstrom, F. Bjokling, H.-E. Hogberg and T. Norin, Acta Chem. Scand., 1984, B38, 779 CrossRef PubMed.
- Y. Fall, N. V. Bac and Y. Langlois, Tetrahedron Lett., 1986, 27, 3611 CrossRef CAS.
- M. J. Gieselmann, D. S. Moreno, J. Fargerlund, H. Tashiro and W. L. Roelofs, J. Chem. Ecol., 1979, 5, 27 CrossRef CAS.
- R. J. Anderson and C. A. Henrick, J. Chem. Ecol., 1979, 5, 773 CrossRef CAS.
- T. Suguro, W. L. Roelofs and K. Mori, Agric. Biol. Chem., 1981, 45, 2509 CrossRef CAS.
- S. Masuda, S. Kuwahara, T. Suguro and K. Mori, Agric. Biol. Chem., 1981, 45, 2515 CrossRef CAS.
- K. Mori and S. Kuwahara, Tetrahedron, 1982, 38, 521 CrossRef CAS.
- E. Alvarez, T. Cuvigny, C. Harve du Penhoat and M. Julia, Tetrahedron, 1988, 44, 119 CrossRef CAS.
- J. G. Millar, Tetrahedron Lett., 1989, 30, 4913 CrossRef CAS.
- R. Baudouy and M.-R. Sancho, Tetrahedron, 1991, 47, 10015 CrossRef CAS.
- (a) S. Harusawa, H. Osaki, S. Takemura, R. Yoneda and T. Kurihara, Tetrahedron Lett., 1992, 33, 2543 CrossRef CAS; (b) S. Harusawa, S. Takemura, H. Osaki, R. Yoneda and T. Kurihara, Tetrahedron, 1993, 49, 7657 CrossRef CAS; (c) S. Harusawa, S. Takemura, R. Yoneda and T. Kurihara, Tetrahedron, 1993, 49, 10577 CrossRef CAS.
- Z. Xu and E. Negishi, Org. Lett., 2008, 10, 4311 CrossRef CAS PubMed.
- W. Roelofs, M. Gieselmann, A. Carde, H. Tashiro, D. S. Moreno, C. A. Henrick and R. J. Anderson, J. Chem. Ecol., 1978, 4, 211 CrossRef CAS.
- (a) B. B. Snider and D. Rodini, Tetrahedron Lett., 1978, 19, 1399 CrossRef; (b) B. B. Snider and G. B. Phillips, J. Org. Chem., 1983, 48, 464 CrossRef CAS.
- R. J. Anderson, K. G. Adams, H. R. Chinn and C. A. Henrick, J. Org. Chem., 1980, 45, 2229 CrossRef CAS.
- R. Baudouy and C. Maliverney, Tetrahedron, 1988, 44, 471 CrossRef CAS.
- D. Becker and Y. Sahali, Tetrahedron, 1988, 44, 4541 CrossRef CAS.
- V. A. Dragan, V. V. Veselovskii and A. M. Moiseenkov, Izv. Akad. Nauk SSSR, Ser. Khim., 1989, 1143 CAS , English translation: Russ. Chem. Bull., 1989, 38, 1038.
- X.-M. Zhang, A. Archelas and R. Furstoss, Tetrahedron: Asymmetry, 1992, 3, 1373 CrossRef CAS.
- L. Auer, C. Weymuth and R. Scheffold, Helv. Chim. Acta, 1993, 76, 810 CrossRef CAS.
- P. Kefalas and N. Ragoussis, Synthesis, 1995, 644 CrossRef PubMed.
- R. Baudouy and P. Prince, Tetrahedron, 1989, 45, 2067 CrossRef CAS.
- J. H. Hutchinson and T. Money, Can. J. Chem., 1985, 63, 3182 CrossRef CAS.
- M. Whittaker, C. R. McArthur and C. C. Leznoff, Can. J. Chem., 1985, 63, 2844 CrossRef CAS.
- P. Mangeney, A. Alexakis and J. F. Normant, Tetrahedron Lett., 1987, 28, 2363 CrossRef CAS.
- S. M. Kher and G. H. Kulkarni, Synth. Commun., 1990, 20, 495 CrossRef CAS.
- W. C. Still and A. Mitra, J. Am. Chem. Soc., 1978, 100, 1927 CrossRef CAS.
- W. Oppolzer and T. Stevenson, Tetrahedron Lett., 1986, 27, 1139 CrossRef CAS.
- M. P. Cooke Jr and D. L. Burman, J. Org. Chem., 1982, 47, 4955 CrossRef.
- D. Caine and E. Crews, Tetrahedron Lett., 1984, 25, 5359 CrossRef CAS.
- J. Celebuski and M. Rosenblum, Tetrahedron, 1985, 41, 5741 CrossRef CAS.
- A. A. Vasil'ev, A. L. Vlasyuk, G. V. Kryshtal and E. P. Serebryakov, Izv. Akad. Nauk SSSR, Ser. Khim., 1995, 2026 Search PubMed , English translation: Russ. Chem. Bull., 1995, 44, 1946.
- M. J. Hesse, C. P. Butts, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2012, 51, 12444 CrossRef CAS PubMed.
- R. R. Heath, J. R. McLaughlin, J. H. Tumlinson, T. R. Ashley and R. E. Doolittle, J. Chem. Ecol., 1979, 5, 941 CrossRef CAS.
- J. Einhorn, H. Bianchi and C. Benassy, C. R. Acad. Sci., Ser. III, 1983, 296, 861 Search PubMed.
- J. R. McLaughlin, J. Chem. Ecol., 1990, 16, 749 CrossRef CAS PubMed.
- R. R. Heath, R. E. Doolittle, P. E. Sonnet and J. H. Tumlinson, J. Org. Chem., 1980, 45, 2910 CrossRef CAS.
- N. Ishibashi, M. Nakamura, F. Mochizuki and T. Fukumoto, U.S. Pat. Appl. Publ.,20110172304 A1 20110714, 2011.
- H.-Y. Ho, B. S. Kuarm, C.-H. Ke, Y.-K. Ma, H.-J. Lee, C.-C. Cheng, K. K. Liu and J. G. Millar, J. Chem. Ecol., 2014, 40, 379 CrossRef CAS PubMed.
- (a) R. R. Johnson and J. A. Nicholson, J. Org. Chem., 1965, 30, 2918 CrossRef CAS; (b) A. Kohda, K. Nagayoshi, K. Maemoto and T. Sato, J. Org. Chem., 1983, 48, 425 CrossRef CAS; (c) O. S. Park, H. J. Kim, W. K. Chae and W. Y. Lee, Bull. Korean Chem. Soc., 1993, 14, 639 CAS.
- T. Kitahara, T. Matsuoka, H. Kiyota, Y. Warita, H. Kurata, A. Horiguchi and K. Mori, Synthesis, 1994, 692 CrossRef CAS PubMed.
- H.-Y. Ho, Y.-T. Su, C.-H. Ko and M.-Y. Tsai, J. Chem. Ecol., 2009, 35, 724 CrossRef CAS PubMed.
- H.-Y. Ho, C.-H. Ko, C.-C. Cheng and Y.-T. Su, J. Econ. Entomol., 2011, 104, 823 CrossRef CAS.
- A. M. El-Sayed, C. R. Unelius, A. Twidle, V. Mitchell, L.-A. Manning, L. Cole, D. M. Suckling, M. F. Flores, T. Zaviezo and J. Bergmann, Tetrahedron Lett., 2010, 51, 1075 CrossRef CAS PubMed.
- C. R. Unelius, A. M. El-Sayed, A. Twidle, B. Bunn, T. Zaviezo, M. F. Flores, V. Bell and J. Bergmann, J. Chem. Ecol., 2011, 37, 166 CrossRef CAS PubMed.
- B. A. Bierl-Leonhardt, D. S. Moreno, M. Schwarz, J. Fargerlund and J. R. Plimmer, Tetrahedron Lett., 1981, 22, 389 CrossRef CAS.
- A. Zada, E. Dunkelblum, M. Harel, F. Assael, S. Gross and Z. Mendel, J. Econ. Entomol., 2004, 97, 361 CrossRef CAS PubMed.
- L. C. Passaro and F. X. Webster, J. Agric. Food Chem., 2004, 52, 2896 CrossRef CAS PubMed.
- P. H. J. Carlsen and W. Odden, Acta Chem. Scand., Ser. B, 1984, 38, 501 CrossRef PubMed.
- L. Lombardo, Org. Synth., 1987, 65, 81 CrossRef CAS.
- O. S. Kukovinets, T. I. Zvereva, V. G. Kasradze, F. Z. Galin, L. L. Frolova, A. V. Kuchin, L. V. Spirikhin and M. I. Abdullin, Chem. Nat. Compd., 2006, 42, 216 CrossRef CAS PubMed.
- B. C. Ranu and R. Chakraborty, Tetrahedron Lett., 1990, 31, 7663 CrossRef CAS.
- T. Arai, H. Sugie, S. Hiradate, S. Kuwahara, N. Itagaki and T. Nakahata, J. Chem. Ecol., 2003, 29, 2213 CrossRef CAS.
- T. Nakahata, N. Itagaki, T. Arai, H. Sugie and S. Kuwahara, Biosci., Biotechnol., Biochem., 2003, 67, 2627 CrossRef CAS PubMed.
- M. Ando, H. Ohhara and K. Takase, Chem. Lett., 1986, 15, 879 CrossRef.
- J. G. Millar, S. P. Chinta, J. S. McElfresh, L. J. Robinson and J. G. Morse, J. Econ. Entomol., 2012, 105, 497 CrossRef CAS.
- (a) A. Zhang, J. Nie and A. Khrimian, Tetrahedron Lett., 2004, 45, 9401 CrossRef CAS PubMed; (b) A. Zhang and J. Nie, J. Agric. Food Chem., 2005, 53, 2451 CrossRef CAS PubMed.
- (a) A. Zhang and D. Amalin, Environ. Entomol., 2005, 34, 264 CrossRef CAS PubMed; (b) A. Zhang, S. Wang, J. Vitullo, A. Roda, C. Mannion and J. C. Bergh, Chem. Senses, 2006, 31, 621 CrossRef CAS PubMed.
- J. Einhorn, A. Guerrero, P.-H. Ducrot, F.-D. Boyer, M. Gieselmann and W. Roelofs, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9867 CrossRef CAS.
- F.-D. Boyer and P.-H. Ducrot, Eur. J. Org. Chem., 1999, 1201–1211 CrossRef CAS.
- (a) I. Petschen, A. Parrilla, M. P. Bosch, C. Amela, A. A. Botar, F. Camps and A. Guerrero, Chem.–Eur. J., 1999, 5, 3299 CrossRef CAS; (b) I. Petschen, M. P. Bosch and A. Guerrero, Tetrahedron: Asymmetry, 2000, 11, 1691 CrossRef CAS.
- (a) A. W. Schmidt, J. R. Suresh, G. Theumer and H.-J. Knolker, Chem. Lett., 2007, 36, 1478 CrossRef CAS; (b) A. W. Schmidt and H.-J. Knolker, Synlett, 2010, 2207 CAS.
- T. Eisner, M. Deyrup, R. Jacobs and J. Meinwald, J. Chem. Ecol., 1986, 12, 1407 CrossRef CAS PubMed.
- J. G. Millar, J. A. Moreira, J. S. McElfresh, K. M. Daane and A. S. Freund, Org. Lett., 2009, 11, 2683 CrossRef CAS PubMed.
- J. M. Conia and M. L. Leriverend, Bull. Soc. Chim. Fr., 1970, 2981 CAS.
- Y. Zou and J. G. Millar, J. Org. Chem., 2009, 74, 7207 CrossRef CAS PubMed.
- Y. Zou and J. G. Millar, Synlett, 2010, 2319 CrossRef CAS PubMed.
- W. F. Bailey and J. M. Bakonyi, J. Org. Chem., 2013, 78, 3493 CrossRef CAS PubMed.
- K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, J. Org. Chem., 1994, 59, 2668 CrossRef CAS.
- S. E. Kurhade, V. Siddaiah, D. Bhuniya and D. S. Reddy, Synthesis, 2013, 45, 1689 CrossRef CAS PubMed.
- A. Srikrishna and G. Satyanarayana, Tetrahedron, 2006, 62, 2892 CrossRef CAS PubMed.
- D. A. Engel and G. B. Dudley, Org. Biomol. Chem., 2009, 7, 4149 CAS.
- R. Ramesh, P. S. Swaroop, R. G. Gonnade, C. Thirupathi, R. A. Waterworth, J. G. Millar and D. S. Reddy, J. Org. Chem., 2013, 78, 6281 CrossRef CAS PubMed.
- Y. Zou, K. M. Daane, W. J. Bentley and J. G. Millar, J. Agric. Food Chem., 2010, 58, 4977 CrossRef CAS PubMed.
- J.-M. Galano, G. Audran, L. Mikolajezyk and H. Monti, J. Org. Chem., 2001, 66, 323 CrossRef CAS.
- H. Pamingle, R. L. Snowden and K. H. Schulte-Elte, Helv. Chim. Acta, 1991, 74, 543 CrossRef CAS.
- J. G. Millar, S. L. Midland, J. S. McElfresh and K. M. Daane, J. Chem. Ecol., 2005, 31, 2999 CrossRef CAS PubMed.
- J. G. Millar and S. L. Midland, Tetrahedron Lett., 2007, 48, 6377 CrossRef CAS PubMed.
- (a) N. Krause, Angew. Chem., Int. Ed., 1994, 33, 1764 CrossRef; (b) N. Krause, S. Ebert and A. Haubrich, Liebigs Ann./Recl., 1997, 2409 CrossRef CAS.
- Y. Zou and J. G. Millar, Tetrahedron Lett., 2011, 52, 4224 CrossRef CAS PubMed.
- C. Morel-Fourrier, J.-P. Dulcère and M. Santelli, J. Am. Chem. Soc., 1991, 113, 8062 CrossRef CAS.
- (a) W. Barth and L. A. Paquette, J. Org. Chem., 1985, 50, 2438 CrossRef CAS; (b) F. Kazmierczak and P. Helquist, J. Org. Chem., 1989, 54, 3988 CrossRef CAS.
- B. Figadere, F. J. Devlin, J. G. Millar and P. J. Stephens, Chem. Commun., 2008, 1106 RSC.
- K. Hashimoto, A. Morita and S. Kuwahara, J. Org. Chem., 2008, 73, 6913 CrossRef CAS PubMed.
- N. Asao, S. Lee and Y. Yamamoto, Tetrahedron Lett., 2003, 44, 4265 CrossRef CAS.
- A. K. Hajare, L. S. Datrange, S. Vyas, D. Bhuniya and D. S. Reddy, Tetrahedron Lett., 2010, 51, 5291 CrossRef CAS PubMed.
- G. N. Lanier, Y.-T. Qi, J. R. West, S. C. Park, F. X. Webster and R. M. Silverstein, J. Chem. Ecol., 1989, 15, 1645 CrossRef CAS PubMed.
- J. Einhorn, P. Menassieu, C. Malosse and P. H. Ducrot, Tetrahedron Lett., 1990, 31, 6633 CrossRef CAS.
- E. Dunkelblum, Z. Mendel, F. Assael, M. Harel, L. Kerhoas and J. Einhorn, Tetrahedron Lett., 1993, 34, 2805 CrossRef CAS.
- S. C. Park, A. J. Wi and K. Mori, Kor. J. Appl. Entomol., 1994, 33, 250 Search PubMed.
- (a) X. Shi, F. X. Webster, J. Kallmerten and J. Meinwald, Tetrahedron Lett., 1995, 36, 7197 CrossRef CAS; (b) X. Shi, F. X. Webster and J. Meinwald, Tetrahedron Lett., 1995, 36, 7201 CrossRef CAS; (c) X. Shi, F. X. Webster and J. Meinwald, Tetrahedron, 1995, 51, 10433 CrossRef CAS.
- H. Jactel, P. Menassieu, M. Lettere, K. Mori and J. Einhorn, J. Chem. Ecol., 1994, 20, 2159 CrossRef CAS PubMed.
- E. Dunkelblum, R. Gries, G. Gries, K. Mori and Z. Mendel, J. Chem. Ecol., 1995, 21, 849 CrossRef CAS PubMed.
- C. L. Cywin, F. X. Webster and J. Kallmerten, J. Org. Chem., 1991, 56, 2953 CrossRef CAS.
- C. L. Cywin and J. Kallmerten, J. Nat. Prod., 1991, 54, 1664 CrossRef CAS.
- K. Mori and S. Harashima, Liebigs Ann. Chem., 1993, 391 CrossRef CAS.
- K. Mori and S. Harashima, Liebigs Ann. Chem., 1993, 993 CrossRef CAS.
- K. Mori, T. Furuuchi and H. Kiyota, Liebigs Ann. Chem., 1994, 971 CrossRef CAS.
- J.-L. Brevet and K. Mori, Synthesis, 1992, 1007 CrossRef CAS.
- K. Mori, T. Furuuchi and H. Kiyota, Liebigs Ann. Chem., 1995, 2093 CrossRef CAS.
- G.-Q. Lin and W.-C. Xu, Tetrahedron Lett., 1993, 34, 5931 CrossRef CAS.
- G.-Q. Lin and W. C. Xu, Bioorg. Med. Chem., 1996, 4, 375 CrossRef CAS.
- L. Zegelman, A. Hassner, Z. Mendel and E. Dunkelblum, Tetrahedron Lett., 1993, 34, 5641 CrossRef CAS.
- K. Mori and M. Amaike, J. Chem. Soc., Perkin Trans. 1, 1994, 2727 RSC.
- H. Watanabe, T. Watanabe, T. Kitahara and K. Mori, Biosci., Biotechnol., Biochem., 1997, 61, 127 CrossRef CAS.
- S. Kurosawa, M. Takenaka, E. Dunkelblum, Z. Mendel and K. Mori, ChemBioChem, 2000, 1, 56 CrossRef CAS.
- J. E. Frey and B. Frey, Mol. Ecol., 1995, 4, 777 CrossRef CAS PubMed.
- R. A. Waterworth, R. A. Redak and J. G. Millar, J. Econ. Entomol., 2011, 104, 555 CrossRef.
- A. Roda, J. G. Millar, J. Rascoe, S. Weihman and I. Stocks, J. Econ. Entomol., 2012, 105, 2052 CrossRef.
- A. Francis, K. A. Bloem, A. L. Roda, S. L. Lapointe, A. Zhang and O. Onokpise, Fla. Entomol., 2007, 90, 440 CrossRef.
- E. E. Grafton-Cardwell, J. G. Millar, N. V. O'Connell and L. M. Hanks, J. Agric. Urban Entomol., 2000, 17, 75–88 CAS.
- V. M. Walton, K. M. Daane and K. L. Pringle, Crop Prot., 2004, 23, 1089 CrossRef PubMed.
- R. A. Waterworth, I. M. Wright and J. G. Millar, Ann. Entomol. Soc. Am., 2011, 104, 249 CrossRef.
- J. C. Franco, S. Gross, E. B. da Silva, P. Suma, A. Russo and Z. Mendel, An. Inst. Super. Agron., Univ. Tec. Lisboa, 2003, 49, 353 Search PubMed.
- A. Hefetz, S. Kronenberg, B. A. Peleg and I. Bar-Zakay, in Citriculture: Proc. 6th Int. Citrus Cong., ed. R. Goren, N. Goren and K. Mendel, Middle-East, Tel Aviv, Israel, March 6-11, 1988 Search PubMed.
- (a) S. Vacas, C. Alfaro, V. Navarro-Llopis and J. Primo, Bull. Entomol. Res., 2009, 99, 415–423 CrossRef CAS PubMed; (b) S. Vacas, C. Alfaro, V. Navarro-Llopis and J. Primo, Pest Manage. Sci., 2010, 66, 745 CrossRef CAS PubMed; (c) S. Vacas, C. Alfaro, J. Primo and V. Navarro-Llopis, J. Pest Sci., 2014 DOI:10.1007/s10340-014-0623-1; (d) S. Vacas, P. Vanaclocha, C. Alfaro, J. Primo, M. J. Verdú, A. Urbaneja and V. Navarro-Llopis, Pest Manage. Sci., 2012, 68, 142 CrossRef CAS PubMed.
- R. E. Rice, C. A. Atterholt, M. J. Delwiche and R. A. Jones, IOBC-WPRS Bulletin, 1997, 20, 151 Search PubMed.
- K. Daane, W. Bentley, V. Walton, R. Malakar-Kuenen, J. Millar, C. Ingels, E. Weber and C. Gispert, Calif. Agric., 2006, 60, 31 CrossRef.
- V. M. Walton, K. M. Daane, W. J. Bentley, J. G. Millar, T. E. Larsen and R. Malakar-Kuenen, J. Econ. Entomol., 2006, 99, 1280 CrossRef PubMed.
- A. Cocco, M. Coinu, A. Lentini, G. Serra and G. Delrio, IOBC-WPRS Bulletin, 2011, 67, 215 Search PubMed.
- K. M. Daane, W. J. Bentley, R. J. Smith, D. R. Haviland, E. A. Wever, M. Battany, C. Gispert and J. G. Millar, Planococcus mealybugs, in Grape Pest Management, ed. L. J. Bettiga, Publication 3343, University of California Agriculture and Natural Resources Publication 3343, Oakland CA, 3rd edn, 2013, pp. 246–260 Search PubMed.
- D. J. Langone, Efficacy of pheromone mating disruption for vine mealybug control, MSc. Thesis, Fresno State University, Fresno CA, 2014, p. 75, http://hdl.handle.net/10211.3/105368.
| This journal is © The Royal Society of Chemistry 2015 |