
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and computational insights into the nature of weak intermolecular interactions in trifluoromethyl-substituted isomeric crystalline N-methyl-N-phenylbenzamides†
Piyush
Panini
and
Deepak
Chopra
*
Crystallography and Crystal Chemistry Laboratory, Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Madhya Pradesh, 462066 India. E-mail: dchopra@iiserb.ac.in; Fax: +91-0755-6692392
First published on 20th August 2015
Abstract
The knowledge about the prevalence of weak interactions in terms of the nature and energetics associated with their formation is of significance in organic solids. In the present study, we have systematically explored the existence of different types of intermolecular interactions in ten out of the fifteen newly synthesized trifluoromethyl derivatives of isomeric N-methyl-N-phenylbenzamides. Detailed analyses of all the crystalline solids were performed with quantitative inputs from interaction energy calculations using the PIXEL method. These studies revealed that in the absence of a strong hydrogen bond, the crystal packing is mainly stabilized by a cooperative interplay of weak C–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–H⋯π, and C(sp2)/(sp3)–H⋯F–C(sp3) hydrogen bonds along with other related interactions, namely, π⋯π and C(sp3)–F⋯F–C(sp3). It is of interest to observe the presence of short and directional weak C–H⋯OC hydrogen bonds in the packing, having a substantial electrostatic (coulombic + polarization) contribution towards the total stabilization energy. The C(sp3)–F group was recognized in the formation of different molecular motifs in the crystal packing as utilizing different intermolecular interactions. The contribution from electrostatics among the different weak hydrogen bonds was observed in the decreasing order: C–H⋯OC > C–H⋯F–C(sp3) > C–H⋯π. Furthermore, there was an increase in the electrostatic component with a concomitant decrease in the dispersion component for the shorter and directional hydrogen bonds.
C, C–H⋯π, and C(sp2)/(sp3)–H⋯F–C(sp3) hydrogen bonds along with other related interactions, namely, π⋯π and C(sp3)–F⋯F–C(sp3). It is of interest to observe the presence of short and directional weak C–H⋯OC hydrogen bonds in the packing, having a substantial electrostatic (coulombic + polarization) contribution towards the total stabilization energy. The C(sp3)–F group was recognized in the formation of different molecular motifs in the crystal packing as utilizing different intermolecular interactions. The contribution from electrostatics among the different weak hydrogen bonds was observed in the decreasing order: C–H⋯OC > C–H⋯F–C(sp3) > C–H⋯π. Furthermore, there was an increase in the electrostatic component with a concomitant decrease in the dispersion component for the shorter and directional hydrogen bonds.
Introduction
The study of intermolecular interactions, which link the molecules in the solid state, has been crucial and of prime focus in crystal engineering.1–6 In this regard, strong hydrogen bonds (e.g. N/O–H⋯N/O) along with weak hydrogen bonds (like C–H⋯O/N/C) are now well understood and recognized in chemistry and biology.7–11 Recent emphasis in crystal engineering is focused on gaining a greater understanding of weak intermolecular interactions, particularly those involving organic fluorine.12,13 The replacement of hydrogen with a fluorine atom is recognized to affect the physicochemical properties of a compound but without much change in the molecular size.14,15 It also shows a greater increase in stability, which results in the increased resistance of a compound towards metabolic degradation.16,17 20% of pharmaceuticals and 30% of agrochemicals are reported to possess organic fluorine, and this number is still increasing.18 The participation of organic fluorine in different intermolecular interactions was initially questioned by researchers.19 In the last decade, many studies on the participation of organic fluorine in different intermolecular interactions have been studied and their role in the formation of different supramolecular arrangements has been recognized.20–25 The significance of these interactions has been very well summarized in a recent review,26 in a perspective27 and in a highlight.28 The significance of such weak intermolecular interactions involving organic fluorine was studied both in the presence and absence of strong hydrogen bonds.29–35 However, the systematic study of these interactions in terms of the electronic nature of the participating fluorine atoms, i.e. fluorine atom connected to the C-atom of a different state of hybridization, was not reported. Most of the past investigations involved the presence of a fluorine atom present on the phenyl ring (C-atom in sp2 hybridized state). We recently investigated the capability of a fluorine atom connected to an sp3 hybridized C-atom in the trifluoromethyl group (–CF3 group) in the formation of different structural motifs and their influence on the crystal packing in a series of isomeric trifluoromethyl-substituted benzanilides.21 The presence of a –CF3 group on the molecule increases the acidity of the neighbouring H atoms and thus increases the possibility of its participation in the formation of a hydrogen bond. Furthermore, it was recently experimentally established from charge density analysis performed using high resolution X-ray data that there is an intrinsic polarization of the electron density on the fluorine atoms of the trifluoromethyl group.36 This further increases the possibility of its participation in different intermolecular interactions, such as C–H⋯F, C–F⋯F–C, and C–F⋯π, in the solid state. The role of the presence of a –CF3 group in different fields of chemistry and biology is very well recognized.37–40 The influence of the presence of a –CF3 group has also been observed in phase transitions41 and in crystal engineering.42 It is thus of interest to analyze the role and influence of the CF3 group [F–C(sp3)] in the participation of different intermolecular interactions in the absence of any strong hydrogen bonds. In the crystal structure analysis of –CF3-substituted benzanilides, these interactions were structurally analyzed in the presence of a N–H⋯OC hydrogen bond. The focus is now on the removal of the H-atom connected to the N-atom, which eliminates the presence of any strong donor in the molecule, and hence eliminates the possibility of the formation of the N–H⋯OC H-bond.
In this regard, a library of trifluoromethyl-substituted N-methyl-N-phenylbenzamides were synthesized (Scheme 1) by replacing the H-atom connected to a N-atom with a methyl group, and then their crystal structures were analyzed to investigate the nature and role of the weak interactions (of the type C–H⋯F–Csp3, Csp3–F⋯F–Csp3) in the crystal packing. Thus, these compounds now eliminate the possibility of formation of any strong H-bond. The substitution of the hydrogen atom with the methyl group completely alters the molecular conformation from a trans to cis geometry relative to that in benzanilides.43–45 The change in the fluorescence and luminescence behavior with the change in its molecular conformation has been very well studied by different research groups.46–48 To obtain a better understanding of the nature of the different intermolecular interactions, the evaluation of the stabilization energy of these interactions is of prime focus in the present study. The contribution of the possible different interactions towards the crystal packing was quantified by PIXEL.49 The PIXEL method provides important insights towards understanding the crystal packing by partitioning the interaction energy or cohesive energy into their coulombic, polarization, dispersion and repulsion contributions. To gain a better insight into the different weak intermolecular interactions present in the crystal packing, selected molecular pairs (extracted from the crystal packing connected with different intermolecular interactions) were analyzed. This is in contrast to the routine practice of providing details on the crystal packing related to the arrangement of molecules50,51 on the basis of pure geometry with no inputs from energetics. On the contrary, in reality, it is the latter that contributes immensely towards crystal formation.
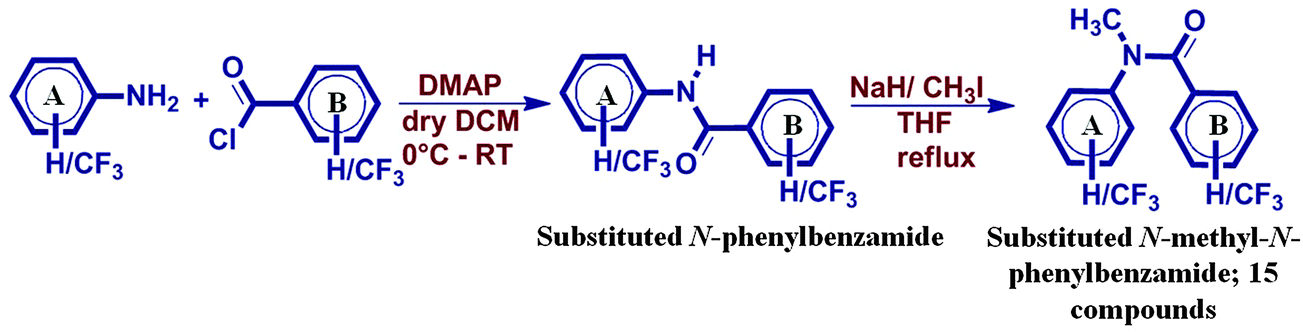 | ||
| Scheme 1 Synthetic route for all the compounds. ‘A’ and ‘B’ denote the two phenyl rings from the starting material, aniline and corresponding benzoyl chloride. Compound code is denoted as ‘NMAB’ in this study. ‘NM’ refers to N-methyl substitution on N-phenylbenzamide, A or B = 0 [no substitution on that ring] and A or B = 1, 2, 3 [o, m, p substitution of the –CF3 group on that ring, respectively]. | ||
Experimental section
The starting materials, trifluoromethyl (–CF3)-substituted anilines and benzoyl chloride, were obtained from Sigma Aldrich and were used directly as received. All the solvents and other reagents, namely, dimethylaminopyridine (DMAP), methyl iodide (CH3I) and sodium hydride [(NaH) 60% solution in oil], were of analytical grade. The solvents, dichloromethane (DCM) and tetrahydrofuran (THF), were dried before use for the chemical reactions. The intermediate compounds, namely, all the substituted N-phenylbenzamides, were synthesized in accordance with the procedure already reported in our earlier work.21Synthesis of substituted N-methyl-N-phenylbenzamides
In a two-neck round bottom flask containing 1.2 equivalents of NaH, 4 ml of dry THF was added with constant stirring using a magnetic stirrer. Then, 1 equiv. of the substituted N-phenyl benzamides was added slowly to the reaction mixture with constant stirring. Furthermore, 3–4 ml of dry THF was added and the entire reaction mixture was refluxed for 2 h. The reaction mixture was then allowed to cool to room temperature. Afterwards, cold methyl iodide, (excess amount, 0.6 ml added in all reactions) was added slowly to the reaction mixture with the whole flask kept over an ice bath with constant stirring. After the addition, the reaction mixture was continuously stirred at room temperature for 1–2 h. The progress of the reaction was monitored with thin layer chromatography (TLC). After the completion of the reaction, it was quenched with 20 ml of 5% HCl and extracted with ethyl acetate and then washed with brine solution three times. The organic extract was further washed with saturated solution of sodium sulphite to remove excess iodine. The organic extract was again washed with brine solution and then dried with anhydrous sodium sulphate. The crude product thus obtained was finally purified by column chromatography with ethyl acetate and hexane as the eluant. The polarity of the eluant was increased slowly from 0% to 10% while performing the column. The yield was recorded after evaporation of the solvent on completion of the column. Initially, after the column, all the compounds were obtained as a thick oil (Scheme 1). Compounds NM02, NM03, NM10, NM11, NM12, NM22, NM23, NM31 and NM33 became solid after one day when placed in the deep freeze section of the refrigerator or after recrystallization from hexane on scratching the walls of the container. Compound NM30 was observed as a low melting solid (melting point = 39 °C). The remaining five synthesized compounds (NM01, NM20, NM13, NM21, and NM32) remained as a thick oil even at −20 °C.All the synthesized compounds were characterized by FTIR [Fig. S1(a)–(o), ESI†], 1H NMR [Fig. S2(a)–(o), ESI†] spectroscopy. The melting points were recorded using DSC for all the solid compounds, and these are given in the ESI,† [Fig. S3(a)–(j)]. Powder X-ray diffraction (PXRD) data were recorded for all the solid compounds and then compared with the simulated PXRD patterns [Fig. S4(a)–(j), ESI†]. Details about the product yields, melting points and spectroscopy data of all the synthesized compounds are listed in Section S1 in the ESI.†
Details on all the crystallization experiments of all the solid compounds from the different solvents and solvent mixtures are presented in the ESI,† (Table S1). Single crystals of all the solids except NM30 were obtained from a slow evaporation method. Compound NM30 was observed to appear as a single crystal in the sample vial at a temperature below 25 °C.
Data collection, structure solution and refinement
Single crystal X-ray diffraction data were collected on a CrysAlis CCD Xcalibur, Eos (Nova), Oxford Diffraction using Mo Kα radiation (λ = 0.71073 Å) for all the compounds except NM03, NM30, NM22 and NM31. The single crystal X-ray diffraction data of NM03, NM22 and NM31 were collected on a Bruker D8 Venture diffractometer with a CMOS detector using graphite monochromated Mo Kα (λ = 0.7107 Å) radiation, whereas data for NM30 were collected on a Bruker APEX II three circle diffractometer with a CCD detector. All the data except for NM12 were collected at 120(2) K.All the crystal structures were solved by direct methods using SIR92.52 The non-hydrogen atoms were refined anisotropically and the hydrogen atoms bonded to C atoms were positioned geometrically and refined using a riding model with Uiso(H) = 1.2Ueq [C(sp2)] and Uiso(H) = 1.5Ueq [C(sp3)]. Compound NM30 was observed to be twinned in two orientations with the ratio for the BASF values being 0.59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.41. The twin law and the corresponding HKLF5 file were generated using the ‘TwinRotMat’ tool in WinGx53 and the refinement was performed with the HKLF5 file using SHELXL2013.
:0.41. The twin law and the corresponding HKLF5 file were generated using the ‘TwinRotMat’ tool in WinGx53 and the refinement was performed with the HKLF5 file using SHELXL2013.
The disorder associated with the CF3 group was modelled with the PART command in two independent orientations (the major component was labeled ‘A’) in SHELXL 2013.54 Molecular and packing diagrams were generated using Mercury 3.3.55 Geometrical calculations were carried out using PARST56 and PLATON.57Table 1 lists all the crystallographic and refinement data. Lists of selected dihedral angles are presented in Tables 2 and 3.
| DATA | NM02 | NM03 | NM10 | NM30 | NM11 |
|---|---|---|---|---|---|
| Formula | C15H12NOF3 | C15H12NOF3 | C15H12NOF3 | C15H12NOF3 | C16H11NOF6 |
| Formula weight | 279.26 | 279.26 | 279.26 | 279.26 | 347.26 |
| CCDC No. | 1025677 | 1025678 | 1025679 | 1027431 | 1025680 |
| Crystal system; space group | Monoclinic; C2/c | Monoclinic; P21/c | Orthorhombic; Pbca | Monoclinic; P21/c | Monoclinic; P21/c |
| a (Å) | 35.8393(17) | 15.1633(5) | 9.6555(7) | 23.731(5) | 8.9803(4) |
| b (Å) | 10.6270(4) | 9.9603(3) | 13.7420(11) | 8.3394(15) | 10.8526(4) |
| c (Å) | 15.1660(7) | 17.7768(6) | 19.3339(15) | 13.706(2) | 14.8181(5) |
| α (°)/β (°)/γ (°) | 90/114.399(5)/90 | 90/97.269(2)/90 | 90/90/90 | 90/106.707(12)/90 | 90/92.763(3)/90 |
| Volume (Å3)/density (g cm−3) | 5260.3(4)/1.410 | 2663.27(15)/1.393 | 2565.3(3)/1.446 | 2597.9(8)/1.428 | 1442.49(10)/1.599 |
| Z/Z′ | 16/2 | 8/2 | 8/1 | 8/2 | 4/1 |
| F(000)/μ (mm−1) | 2304/0.118 | 1152/0.116 | 1152/0.121 | 1152/0.119 | 704/0.153 |
| θ (min, max) | 2.34, 25.00 | 1.35, 27.58 | 2.79, 25.00 | 0.90, 25.01 | 2.75, 25.00 |
| h min,max, kmin,max, lmin,max | −42, 42; −12, 12; −18, 18 | −18, 19; −12, 11; −23, 18 | −11, 11; −16, 16; −22, 22 | −28, 28; −9, 9; −16, 16 | −10, 10; −12, 12; −17, 17 |
| No. of total ref./unique ref./obs. ref. | 25157/4632/3475 |
22406/6117/4629 |
12605/2262/1743 |
4556/4556/3759 | 13687/2534/2204 |
| No. of parameters | 391 | 395 | 211 | 364 | 236 |
| R_all, R_obs | 0.0635, 0.0417 | 0.0659, 0.0490 | 0.0580, 0.0398 | 0.0522, 0.0403 | 0.0434, 0.0368 |
| wR2_all, wR2_obs | 0.1008, 0.0888 | 0.1356, 0.1259 | 0.1028, 0.0901 | 0.0897, 0.0854 | 0.0982, 0.0933 |
| Δρmin,max(e Å−3) | −0.244, 0.215 | −0.357, 0.381 | −0.221, 0.207 | −0.182, 0.223 | −0.237, 0.308 |
| G. o. F | 1.050 | 1.061 | 1.055 | 1.063 | 1.043 |
| DATA | NM12 | NM22 | NM23 | NM31 | NM33 |
|---|---|---|---|---|---|
| Formula | C16H11NOF6 | C16H11NOF6 | C16H11NOF6 | C16H11NOF6 | C16H11NOF6 |
| Formula weight | 347.26 | 347.26 | 347.26 | 347.26 | 347.26 |
| CCDC No. | 1025681 | 1025682 | 1025683 | 1025684 | 1025685 |
| Crystal system; space group | Monoclinic; P21/c | Monoclinic; P21/c | Monoclinic; P21/c | Monoclinic; P21/c | Triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 9.0978(7) | 8.3764(5) | 11.2958(5) | 12.3185(10) | 8.9977(4) |
| b (Å) | 22.1735(15) | 23.2362(17) | 14.0246(4) | 7.9401(5) | 10.8088(5) |
| c (Å) | 7.9449(5) | 7.9755(6) | 10.4868(3) | 15.4912(12) | 16.6720(9) |
| α (°)/β (°)/γ (°) | 90/101.946(4)/90 | 90/103.567(2)/90 | 90/115.258(4)/90 | 90/90.168(4)/90 | 105.291(2)/98.901(2)/102.688(2) |
| Volume (Å3)/density (g cm−3) | 1568.02(19)/1.471 | 1509.00(18)/1.529 | 1502.48(9)/1.535 | 1515.19(19)/1.522 | 1486.36(12)/1.552 |
| Z/Z′ | 4/1 | 4/1 | 4/1 | 4/1 | 4/2 |
| F(000)/μ (mm−1) | 704/0.141 | 704/0.146 | 704/0.147 | 704/0.146 | 704/0.149 |
| θ (min, max) | 1.84, 25.00 | 2.65/25.00 | 2.47/25.00 | 2.63/25.00 | 1.30/30.67 |
| h min,max, kmin,max, lmin,max | −10, 9; −22, 26; −9, 9 | −9, 8; −27, 27; −9, 9 | −13, 13; −16, 16; −12, 12 | −14, 13; −9, 9; −18, 18 | −11, 12; −15, 15; −23, 14 |
| No. of total ref./unique ref./obs. ref. | 13654/2767/2117 |
21987/2652/2199 |
14260/2649/2238 |
11173/2683/2044 |
30219/9103/7006 |
| No. of parameters | 247 | 246 | 250 | 246 | 491 |
| R_all, R_obs | 0.0637, 0.0482 | 0.0534, 0.0410 | 0.0499, 0.0411 | 0.0590, 0.0384 | 0.0602/0.0430 |
| wR2_all, wR2_obs | 0.1388, 0.1285 | 0.1059, 0.0993 | 0.1065, 0.0993 | 0.0930, 0.0865 | 0.1143/0.1040 |
| Δρmin,max(e Å−3) | −0.222, 0.363 | −0.343, 0.497 | −0.339, 0.398 | −0.239, 0.215 | −0.380, 0.446 |
| G. o. F | 1.066 | 1.033 | 1.066 | 1.034 | 1.039 |
C hydrogen bond and their comparison with the values obtained from the DFT/B3LYP calculations (in italic)
| Angle 1 (°) plane1/2 | Angle 2 (°) plane1/3 | Angle 3 (°) plane2/3 | Geometry of C(sp3)–H⋯O (Å, °) | |
|---|---|---|---|---|
| (′) denotes the second molecule in the asymmetric unit. | ||||
| NM00 |
67.4(1)
67.3 |
47.1(1)
32.9 |
62.5(1)
60.1 |
2.47, 93
2.42, 90 |
| NM02 |
66.9(1)/64.9(1)′
70.3/69.9′ |
46.0(1)/53.5(1)′
34.3/36.6′ |
65.4(1)/62.6(1)′
70.9/62.1′ |
2.32, 102/2.39, 97′
2.50, 87/2.42, 91′ |
| NM03 |
58.6(1)/71.9(1)′
65.5/67.0 |
44.0(1)/43.5(1)′
39.2/38.8 |
59.8(1)/70.2(1)′
57.1/61.0 |
2.31, 100/2.27, 104′
2.23, 106/2.51, 86′ |
| NM10 |
69.8(1)
76.3 |
39.2(1)
38.9 |
67.9(1)
77.1 |
2.28, 102
2.33, 97 |
| NM30 |
57.4(1)/55.5(1)′
68.0/65.5 |
43.7(1)/43.4(1)′
36.4/32.7 |
48.8(1)/53.6(1)′
58.4/54.8 |
2.37, 94/2.65, 80′
2.37, 96/2.46, 87 |
| NM11 |
1.7(1)
4.6 |
84.9(1)
84.4 |
85.6(1)
85.5 |
— |
| NM12 |
73.7(1)
76.1 |
58.9(1)
39.4 |
84.4(1)
77.5 |
2.56, 87
2.32, 98 |
| NM22 |
62.3(1)
67.7 |
65.4(1)
33.9 |
64.8(1)
59.1 |
2.41, 97
2.42, 91 |
| NM23 |
69.1(1)
66.3 |
50.5(1)
34.6 |
66.3(1)
58.5 |
2.34, 102
2.43, 90 |
| NM31 |
61.4(1)
67.9 |
61.2(1)
59.8 |
61.5(1)
57.8 |
2.43, 92
2.24, 106 |
| NM33 |
69.5(1)/72.9(1)′
65.9/68.6 |
34.1(1)/62.4(1)′
38.3/46.5 |
56.7(1)/65.5(1)′
57.6/62.3 |
2.38, 94/2.40, 98′
2.37, 97/2.42, 98′ |
| Ref. code | Space group, Z | Cell parameters, a, b, c (Å)/α, β, γ (°) | Angle 1 (°) | Angle 2 (°) | Angle 3 (°) |
|---|---|---|---|---|---|
| 43Azumaya et al., 1994; 65Cockroft et al., 2007. | |||||
| JAZJOJ1043 (NM00) | Pbca, Z = 8 | 12.5881, 12.3092, 14.6542/90, 90, 90 | — | — | — |
| YEGKIE43 | P21nb, Z = 4 | 11.308(1), 15.878(2), 6.876(5)/90, 90, 90 | 70.8 | 36.7 | 65.0 |
| YEGLAX43 |
P, Z = 2 |
11.602, 12.766(4), 7.372(1)/92.19(3), 104.93(2), 137.31(1) | 72.6 | 62.1 | 83.3 |
| YEGKEA43 | P21/a, Z = 4 | 13.257(7), 13.234(11), 8.005(1)/90, 98.01(1), 90 | 65.9 | 76.6 | 62.8 |
| YEGKOK43 | P21/n, Z = 4 | 14.909(1), 6.795(2), 13.358(1)/90, 98.46(1), 90 | 67.5 | 43.7 | 75.2 |
| YEGJEY43 | Cc, Z = 4 | 15.250(3), 7.502(1), 13.733(3)/90, 106.77(2), 90 | 1.06 | 85.5 | 85.1 |
| DIBGIF65 | P21/n, Z = 4 | 7.123(3), 16.792(8), 13.785(7)/90, 102.881, 90 | 16.5 | 85.3 | 84.3 |
| DIBGAX65 | Pc, Z = 8, Z′ = 4 | 11.1542(10), 8.4970(7), 31.528(3)/90, 95.122(2), 90 | 11.0 | 87.7 | 76.9 |
Theoretical calculations
The molecular geometry of each compound was optimized by DFT/B3LYP calculation with 6-31G** basis set using TURBOMOLE.58 The experimentally obtained geometries were considered as the starting coordinates for the calculation. The molecular geometry thus obtained for the isolated molecule was compared with the experimentally observed solid state geometry (Table 2).The lattice energies (Table 4) of all the compounds were calculated by the PIXELC module in the CLP computer program package (version 10.2.2012), the total energy being partitioned into their coulombic (Ecoub), polarization (Epol), dispersion (Edisp) and repulsion (Erep) contributions. For the calculations, accurate electron densities of the molecules were obtained at MP2/6-31G** with GAUSSIAN0959 with H atoms at their neutron value. In the case of disordered molecules, the molecular conformation with the maximum population was considered for the calculation. The interaction energy of the selected molecular pairs, extracted from the crystal packing and related by the corresponding symmetry element, was also calculated by the PIXEL method (from the mlc file after the calculation). The total interaction energy was partitioned into their coulombic (Ecoub), polarization (Epol), dispersion (Edisp) and repulsion (Erep) contributions. These are listed in Table 5 along with the selected intermolecular interactions connecting the two molecules in the molecular pair. The % dispersion energy contribution (% Edisp) towards the total stabilization energy was calculated as follows:
| %Edisp = [Edisp/(Ecoub + Epol + Edisp)] × 100; |
| E Coul | E Pol | E Disp | E Rep | E Tot | |
|---|---|---|---|---|---|
| a Molecules exist in trans conformation. | |||||
| NM00 | −30.3 | −15.5 | −123.0 | 65.5 | −103.3 |
| NM02 | −38.0 | −14.8 | −120.7 | 66.3 | −107.2 |
| NM03 | −42.3 | −16.0 | −119.6 | 75.1 | −102.8 |
| NM10 | −45.5 | −16.4 | −122.4 | 69.4 | −114.9 |
| NM30 | −35.5 | −13.4 | −122.9 | 66.4 | −105.5 |
| NM11 | −34.6 | −13.9 | −122.9 | 64.3 | −107.1 |
| NM12 | −36.4 | −13.5 | −97.9 | 46.0 | −101.9 |
| NM22 | −46.7 | −19.7 | −119.6 | 77.2 | −108.7 |
| NM23 | −40.8 | −15.9 | −122.7 | 70.9 | −108.5 |
| NM31 | −38.6 | −12.8 | −111.2 | 57.2 | −105.4 |
| NM33 | −45.7 | −16.8 | −125.7 | 78.8 | −109.3 |
| YEGLAX | −35.2 | −13.5 | −129.2 | 67.0 | −117.8 |
| YEGKEA | −30.3 | −15.6 | −116.4 | 61.8 | −100.4 |
| YEGKOK | −35.9 | −14.8 | −127.4 | 71.9 | −106.2 |
| YEGJEYa | −32.8 | −13.6 | −142.6 | 78.5 | −110.5 |
| Pair/motifa | Symmetry code | Centroid-centroid distance (Å) | E Coul | E Pol | E Disp | E Rep | E Tot | DFT-D2/B97-D (BSSE corrected) | Possible involved interactionsc | Geometry (Å/°) D(D⋯A), d(H⋯A), ∠D–H⋯A |
|---|---|---|---|---|---|---|---|---|---|---|
| a Arranged in descending order of energy. b Values in parenthesis represent % dispersion energy contribution (%Edisp) towards the total stabilization, % electrostatic contribution (%Eelec) = 100 − %Edisp. c Cg1 and Cg2 refer to the centre of gravity for the phenyl rings C1–C6 and C7–C12, respectively. | ||||||||||
| NM00 (Pbca, Z′ = 1; ref. code: JAZJOJ10) | ||||||||||
| I | −x + 3/2, y + 1/2, z | 6.320 | −9.1 | −7.7 | −33.1 (66) | 25.4 | −24.5 | −28.4 |
C6–H6⋯O1
C14–H14C⋯C5 (π) |
3.271(1)/2.41/136
3.757(1)/2.98/129 |
| II | −x + 1/2, −y + 1/2, −z + 1 | 6.877 | −5.3 | −2.1 | −25.7 (78) | 13.6 | −19.6 | −24.3 |
C3–H3⋯C9 (π)
C2–H2⋯C11 (π) H14A⋯H9 |
3.911(1)/2.96/147
3.664(1)/2.99/121 2.38 |
| III | x−1/2, y, −z + 3/2 | 7.885 | −5.0 | −2.5 | −19.6 (72) | 9.8 | −17.2 | −19.7 |
C12–H12⋯C4 (π)
C14–H14A⋯C3 (π) |
3.880(1)/2.98/140
3.787(1)/2.97/133 |
| IV | x, −y + 1/2, z−1/2 | 7.397 | −4.5 | −2.5 | −11.5 (62) | 4.7 | −13.9 | −15.8 |
C9–H9⋯C2 (π)
C9–H9⋯O1 |
4.082(1)/3.14/147
3.361(1)/2.89/106 |
| V | −x + 2, y + 1/2, −z + 3/2 | 9.167 | −3.7 | −1.8 | −8.8 (62) | 4.2 | −10.1 | −11.3 | C4–H4⋯O1 | 3.907(1)/2.86/163 |
| VI | −x + 2, −y + 1, −z + 1 | 7.527 | 0.6 | −1.1 | −15.4 (97) | 5.8 | −10.1 | −11.2 | C10–H10⋯C4 (π) | 3.936(1)/3.18/128 |
| VII | −x + 3/2, −y + 1, z−1/2 | 9.065 | −1.8 | −0.6 | −7.9 (77) | 2.9 | −7.3 | −8.4 |
C10–H10⋯C6 (π)
C10–H10⋯C5 (π) |
4.037(1)/3.11/144
3.836(1)/3.18/120 |
| NM02 (C2/c, Z′ = 2) | ||||||||||
| I 1⋯1 | −x + 1/2, −y + 1/2, −z | 7.800 | −17.8 | −5.4 | −30.5 (57) | 18.6 | −35.1 | −43.2 |
C12–H12⋯O1
C14–H14B⋯O1 |
3.790(3)/2.77/158
3.610(3)/2.83/129 |
| II 2⋯2 | −x + 1, y, −z + 1/2 | 5.505 | −8.8 | −4.6 | −44.2 (77) | 26.1 | −31.5 | −31.0 |
C4′⋯C4′ (π⋯π)
C5′⋯C5′ (π⋯π) C5′–H5′⋯C11′ (π) |
3.431(2)
3.393(2) 3.772(2)/2.78/153 |
| III 1⋯2 | x, −y, z + 1/2 | 6.175 | −14.5 | −6.0 | −30.1 (59) | 19.5 | −31.1 | −35.4 |
C8–H8⋯O1′
C6′–H6′⋯F1 C8′––H8′⋯F1 |
3.393(2)/2.46/144
3.313(2)/2.43/138 3.400(3)/2.65/126 |
| IV 1⋯1 | −x + 1/2, y + 1/2, −z + 1/2 | 7.260 | −11.4 | −4.6 | −36.2 (69) | 22.2 | −30.0 | −35.3 |
C10–H10⋯O1
C5–H5⋯Cg2 (π) |
3.429(3)/2.67/127
3.670(2)/2.65/157 |
| V 1⋯2 | x, y, z | 5.450 | −6.3 | −4.8 | −23.7 (68) | 12.4 | −22.4 | −27.9 |
C2′–H2′⋯O1
C15–F1⋯F1A′–C15′ |
3.356(3)/2.45/141
2.823(2)/98(1)/158(1) |
| VI 1⋯2 | −x + 1/2, −y + 1/2, −z | 8.499 | −8.3 | −4.0 | −14.9 (55) | 9.5 | −17.6 | −18.2 |
C11–H11⋯O1′
C12–H12⋯O1′ C14–H14B⋯F1A′ |
3.312(3)/2.58/125
3.428(2)/2.84/115 3.623(7)/2.68/146 |
| VII 1⋯2 | x, y−1, z | 8.941 | −4.6 | −1.1 | −14.4 (72) | 5.4 | −14.7 | −18.4 |
C9′–H9′⋯F2
C4⋯C9′ (π⋯π) |
3.300(3)/2.71/114
3.802(2) |
| VIII 2⋯2 | x, −y, z + 1/2 | 7.593 | −5.6 | −2.2 | −17.7 (69) | 11.0 | −14.5 | −18.5 |
C10′–H10′⋯C4′ (π)
C11′–H11′⋯C1′ (π) |
3.609(2)/2.78/133
4.008(2)/2.99/157 |
| IX 2⋯2 | x, y−1, z | 10.627 | −5.4 | −1.7 | −10.2 (59) | 7.3 | −10.1 | −10.7 |
C14′–H14E⋯F1A′
C8′–H8′⋯F2A′ C8′–H8′⋯F3A′ |
3.541(8)/2.51/160
3.253(11)/2.58/120 3.797(9)/2.74/166 |
| X 1⋯2 | −x + 1, −y, −z + 1 | 10.441 | −2.3 | −0.6 | −6.2 (68) | 2.0 | −7.1 | −8.3 |
C10′–H10′⋯F3
C10′–H10′⋯F2 |
3.829(2)/2.86/149
3.800(2)/2.89/142 |
| XI 1⋯2 | −x + 1, y, −z + 1/2 | 9.365 | −2.6 | −1.0 | −7.6 (68) | 4.3 | −7.0 | −8.1 |
C4′–H4′⋯F2
C4′–H4′⋯F3 |
3.619(3)/2.66/147
3.306(3)/2.68/117 |
| NM03 (P21/c, Z′ = 2) | ||||||||||
| I 1⋯2 | x, y, z | 5.173 | −23.0 | −8.0 | −41.3 (57) | 33.0 | −39.3 | −52.0 |
C12–H12⋯O1′
C14–H14B⋯O1′ C6⋯C5′ (π⋯π) C6⋯C6′ (π⋯π) |
3.297(2)/2.26/161
3.448(2)/2.66/129 3.387(1) 3.481(1) |
| II 2⋯2 | −x, y−1/2, −z + 1/2 | 6.506 | −17.2 | −7.3 | −46.7 (65) | 36.1 | −35.1 | −42.1 |
C10′–H10′⋯O1′
C5′–H5′⋯Cg2′ (π) C8′–H8′⋯C11′ (π) |
3.340(2)/2.45/139
3.523(2)/2.49/159 3.799(2)/2.80/154 |
| III 1⋯1 | −x + 1, −y, −z | 6.565 | −9.7 | −4.4 | −41.5 (75) | 27.9 | −27.6 | −33.4 |
C10–H10⋯Cg1(π)
C9⋯C9 (π⋯π) C9⋯C10 (π⋯π) |
3.834(1)/2.89/146
3.245(1) 3.430(1) |
| IV 1⋯2 | x, −y + 1/2, z + 1/2 | 7.758 | −8.3 | −3.7 | −28.6 (70) | 17.0 | −23.6 | −28.6 | C3′–H3′⋯C10 (π) | 3.860(1)/2.79/170 |
| V 1⋯2 | −x, −y, −z | 8.970 | −9.9 | −4.7 | −20.4 (58) | 14.7 | −20.3 | −21.8 | C12′–H12′⋯O1 | 3.404(2)/2.33/173 |
| VI 1⋯2 | −x, y + 1/2, −z + 1/2 | 8.380 | −7.8 | −2.9 | −16.1 (60) | 10.9 | −15.9 | −18.6 |
C9′–H9′⋯O1
C10′–H10′⋯C3(π) |
3.614(2)/2.69/143
3.605(1)/2.71/139 |
| VII 1⋯2 | x, y−1, z | 8.775 | −8.0 | −2.3 | −8.8 (46) | 4.6 | −14.5 | −16.7 |
C3–H3⋯O1′
C2′–H2′⋯F2A |
3.393(2)/2.57/132
3.721(3)/2.69/160 |
| VIII 1⋯2 | x, −y−1/2, z−1/2 | 8.192 | −4.5 | −1.4 | −9.3 (61) | 6.0 | −9.2 | −10.7 |
C2–H2⋯F1A′
C3–H3⋯F1A′ |
3.111(3)/2.54/112
3.241(2)/2.81/104 |
| IX 1⋯1 | −x + 1, y + 1/2, −z + 1/2 | 8.594 | −2.2 | −0.9 | −8.2 (73) | 3.7 | −7.6 | −10.7 | C5–H5⋯F3A | 3.527(4)/2.46/172 |
| X 1⋯1 | −x, −y, −z | 10.564 | −2.6 | −1.6 | −4.1 (49) | 1.0 | −7.3 | −8.8 | C14–H14C⋯O1 | 3.987(2)/2.96/159 |
| XI 1⋯1 | x, −y−1/2, z−1/2 | 9.340 | −2.8 | −0.7 | −5.7 (62) | 3.2 | −6.0 | −7.2 |
C9–H9⋯F2A
C8–H8⋯F2A |
3.259(3)/2.60/118
3.386(3)/2.89/109 |
| XII 1⋯2 | −x + 1, y + 1/2, −z + 1/2 | 10.943 | −2.8 | −0.4 | −3.8 (54) | 1.4 | −5.6 | −6.8 | C11–H11⋯F2A′ | 3.555(3)/2.64/142 |
| NM10 (Pbca, Z′ = 1) | ||||||||||
| I | −x + 1, −y, −z + 1 | 5.968 | −16.2 | −6.2 | −34.2 (60) | 20.2 | −36.2 | −41.3 | C11–H11⋯C5 (π) | 3.775(1)/2.74/161 |
| II | x−1/2, y, −z + 3/2 | 7.490 | −11.3 | −5.8 | −31.4 (65) | 20.5 | −28.1 | −33.4 |
C14–H14B⋯O1
C8–H8⋯C6 (π) |
3.382(2)/2.59/130
3.951(1)/2.90/164 |
| III | −x + 1/2, y−1/2, z | 7.881 | −9.1 | −2.6 | −18.7 (62) | 9.9 | −20.6 | −22.9 |
C9–H9⋯F3A
C10–H10⋯F3A C14–H14C⋯C10 (π) |
3.378(15)/2.69/121
3.409(15)/2.76/119 3.974(2)/3.05/144 |
| IV | x−1/2, −y + 1/2, −z + 1 | 6.713 | −4.8 | −1.5 | −16.5 (72) | 5.4 | −17.4 | 23.0 |
C14–H14C⋯F2A
C2–H2⋯F1A C3–H3⋯F1A |
3.670(10)/2.72/146
3.335(11)/2.79/111 3.338(12)/2.79/112 |
| V | −x + 1, y−1/2, −z + 3/2 | 8.997 | −7.2 | −3.7 | −10.8 (50) | 10.1 | −11.7 | −11.4 | C5–H5⋯O1 | 3.310(2)/2.42/139 |
| VI | −x + 3/2, y−1/2, z | 8.989 | −4.4 | −2.1 | −8.0 (55) | 4.9 | −9.6 | −11.2 | C4–H4⋯O1 | 3.567(2)/2.50/169 |
| VII | x + 1, y, z | 9.655 | −0.5 | −1.7 | −11.8 (84) | 7.1 | −6.9 | −10.8 | C3–H3⋯C8 (π) | 3.839(2)/2.83/155 |
| NM30 (P21/c, Z′ = 2) | ||||||||||
| I 1⋯2 | x, y, z | 7.321 | −11.4 | −5.4 | −31.2 (65) | 22.4 | −25.6 | −32.3 |
C3′–H3′⋯O1
C2′–H2′⋯C1 C13⋯C3 (π⋯π) |
3.403(4)/2.65/127
3.727(3)/2.97/127 3.575(3) |
| II 2⋯2 | x, −y + 1/2, z + 1/2 | 8.623 | −8.0 | −4.1 | −17.7 (59) | 11.0 | −18.8 | −19.6 | C8′–H8′⋯O1′ | 3.359(3)/2.53/133 |
| III 1⋯1 | x, −y + 3/2, z + 1/2 | 8.686 | −8.4 | −3.9 | −17.0 (58) | 11.0 | −18.3 | −19.6 | C12–H12⋯O1 | 3.352(4)/2.49/136 |
| IV 2⋯2 | x, −y + 3/2, z−1/2 | 7.524 | −5.2 | −2.0 | −23.9 (77) | 12.8 | −18.3 | −21.8 |
C12′–H12′⋯F1′
C4′⋯C9′ (π⋯π) C5′⋯C9′ (π⋯π) |
3.385(3)/2.63/126
3.547(3) 3.408(3) |
| V 1⋯1 | x, −y + 1/2, z−1/2 | 7.482 | −5.8 | −2.3 | −25.0 (76) | 14.9 | −18.2 | −17.2 |
C8–H8⋯F2
C3⋯C11 (π⋯π) C4⋯C11 (π⋯π) |
3.379(4)/2.62/127
3.346(3) 3.528(3) |
| VI 1⋯2 | x, −y + 3/2, z + 1/2 | 8.400 | −4.6 | −3.4 | −22.2 (74) | 13.2 | −17.0 | −21.1 |
C4′–H4′⋯C6 (π)
C4′⋯C13 |
3.758(3)/2.83/144
3.685(3) |
| VII 2⋯2 | −x, −y + 1, −z + 1 | 7.998 | −6.1 | −1.6 | −18.3 (70) | 8.9 | −17.0 | −22.1 | C15′–F2′⋯C9′ (π) | 3.713(3)/3.155(2)/104(1) |
| VIII 1⋯1 | −x + 1, −y + 1, −z + 2 | 8.078 | −5.9 | −1.4 | −18.2 (71) | 9.2 | −16.4 | −21.3 | C15–F3⋯C11 (π) | 3.693(3)/3.161(2)/102 |
| IX 2⋯2 | −x, −y + 1/2, −z + ½ | 6.529 | −3.6 | −1.8 | −19.0 (78) | 8.4 | −16.0 | −19.8 |
C12′–H12′⋯F2′
C15′–F2′⋯C13′ |
3.338(3)/2.52/132
4.517(3)/3.203(2)/166(1) |
| X 1⋯1 | −x + 1, −y + 1/2, −z + 3/2 | 6.520 | −3.4 | −1.8 | −18.1 (78) | 7.8 | −15.5 | −19.3 |
C8–H8⋯F3
C15–F3⋯C13 |
3.330(3)/2.54/129
4.473(3)/3.154(2)/166(1) |
| XI 1⋯1 | x, y + 1, z | 8.339 | −6.0 | −2.1 | −11.7 (59) | 4.9 | −14.9 | −17.3 |
C4–H4⋯O1
C14–H14B⋯F1 |
3.547(4)/2.73/132
3.637(3)/2.68/147 |
| XII 2⋯2 | x, y−1, z | 8.339 | −6.2 | −2.5 | −12.3 (59) | 6.4 | −14.6 | −17.2 |
C4′–H4′⋯O1′
C14′–H14E′⋯F3′ |
3.515(4)/2.66/135
3.495(4)/2.59/141 |
| XIII 1⋯2 | x, −y + 1/2, z + 1/2 | 8.349 | −4.4 | −3.1 | −13.0 (63) | 7.9 | −12.6 | −13.8 |
C5–H5⋯C6′ (π)
C5′–H5′⋯O1′ |
3.722(3)/2.73/152
3.499(5)/2.91/114 |
| XIV 2⋯2 | −x, −y + 2, −z + 1 | 10.740 | 1.6 | −0.3 | −5.2 (95) | 2.1 | −1.8 | −2.5 |
C15′–F2′⋯F3′–C15′
C15′–F3′⋯F3′–C15′ |
3.104(2)/90(1)/128(1)
3.002(2)/94(1)/94(1) |
| XV 1⋯1 | −x + 1, −y, −z + 2 | 10.759 | 1.6 | −0.3 | −5.1 (94) | 1.9 | −1.8 | −2.6 | C15–F1⋯F1–C15 | 2.966(2)/94(1)/94(1) |
| NM11 (P21/c, Z′ = 1) | ||||||||||
| I | −x + 1, −y, −z + 2 | 7.697 | −27.0 | −7.9 | −29.4 (46) | 23.4 | −40.8 | −43.4 |
C9–H9⋯O1
C10–H10⋯F6 |
3.498(2)/2.46/160
3.296(2)/2.49/144 |
| II | x−1, y, z | 8.980 | −7.7 | −2.6 | −29.4 (74) | 17.6 | −22.0 | −31.3 |
C10⋯C3 (π⋯π)
C11⋯C4 (π⋯π) |
3.560(2)
3.579(2) |
| III | −x + 2, −y + 1, −z + 2 | 8.424 | 0.1 | −1.6 | −24.9 (94) | 8.6 | −17.8 | −23.3 |
C2⋯C3 (π⋯π)
C16⋯C4 (π⋯π) |
3.960(2)
3.955(2) |
| IV | x, −y + 1/2, z−1/2 | 7.409 | −4.6 | −1.5 | −17.4 (74) | 6.7 | −16.7 | −21.0 |
C5–H5⋯F6
C6–H6⋯F4 |
3.543(2)/2.79/127
3.773(2)/2.82/148 |
| V | −x + 2, y + 1/2, −z + 3/2 | 8.484 | −11.8 | −3.9 | −10.7 (41) | 9.9 | −16.6 | −18.2 |
C5–H5⋯O1
C4–H4⋯F3 |
3.287(2)/2.35/144
3.645(2)/2.65/154 |
| VI | −x + 1, y + 1/2, −z + 3/2 | 7.347 | 0.6 | −2.5 | −23.7 (93) | 9.8 | −15.8 | −21.0 |
C14–H14A⋯F1
C14–H14A⋯F3 C10–H10⋯C5 (π) |
3.305(2)/2.84/106
3.731(2)/2.87/137 3.871(2)/3.11/128 |
| VII | x−1, −y + 1/2, z−1/2 | 11.364 | −2.3 | −0.5 | −6.3 (69) | 3.8 | −5.3 | −7.5 |
C11–H11⋯F5
C3–H3⋯F1 |
3.600(2)/2.58/156
3.727(2)/2.78/147 |
| NM12 (P21/c, Z′ = 1) | ||||||||||
| I | −x + 2, −y + 2, −z | 5.674 | −6.7 | −3.9 | −41.4 (80) | 15.7 | −36.2 | −45.0 |
C10–H10⋯C3 (π)
C9–H9⋯C5 (π) C9⋯C9 (π⋯π) C15–F1⋯F4A–C16 |
3.821(2)/3.02/131
4.077(1)/3.04/160 3.624(2) 3.074(3)/140(1)/112(1) |
| II | x, −y + 3/2, z−1/2 | 7.061 | −12.9 | −7.1 | −21.8 (52) | 16.2 | −25.6 | −31.2 |
C12–H12⋯O1
C6–H6⋯O1 |
3.356(3)/2.32/160
3.562(3)/2.57/153 |
| III | x, y, z−1 | 7.945 | −8.9 | −2.5 | −16.5 (59) | 5.7 | −22.3 | −25.9 |
C11–H11⋯O1
C10–H10⋯F2 |
3.646(3)/2.66/152
3.446(3)/2.69/127 |
| IV | x−1, y, z | 9.098 | −4.0 | −1.6 | −17.5 (76) | 7.3 | −15.8 | −20.7 |
C14–H14B⋯C3 (π)
C3–H3⋯F1 |
4.000(2)/2.96/162
3.751(3)/2.78/149 |
| V | −x + 2, −y + 2, −z + 1 | 7.883 | −3.1 | −0.9 | −12.1 (75) | 4.1 | −12.1 | −16.0 | C2–H2⋯F1 | 3.427(3)/2.63/130 |
| VI | x−1, y, z−1 | 10.768 | 0.2 | −0.4 | −4.9 (96) | 1.3 | −3.8 | −6.1 |
C14–H14A⋯F5A
C15–F3⋯F4A–C16 |
3.814(9)/2.85/149
3.061(8)/166(2)/156(2) |
| NM22 (P21/c, Z′ = 1) | ||||||||||
| I | x, −y + 1/2, z + 1/2 | 7.291 | −24.7 | −10.9 | −32.9 (48) | 31.5 | −37.0 | −40.5 |
C2–H2⋯O1
C8–H8⋯O1 C6–H6⋯O1 |
3.500(2)/2.43/173
3.118(2)/2.21/140 3.679(3)/2.69/152 |
| II | −x + 1, −y + 1, −z | 5.937 | −2.9 | −4.7 | −42.8 (85) | 24.3 | −26.0 | −33.4 |
C10–H10⋯C5 (π)
C10⋯C10 (π⋯π) |
3.837(2)/2.79/163
3.500(1) |
| III | x−1, y, z | 8.376 | −10.2 | −4.6 | −30.0 (67) | 20.6 | −24.2 | −30.3 |
C4–H4⋯Cg2 (π)
C5–H5⋯F2A |
3.475(1)/2.51/148
3.583(14)/2.67/142 |
| IV | x, y, z + 1 | 7.976 | −6.2 | −2.6 | −17.9 (67) | 11.2 | −15.6 | −18.1 |
C12–H12⋯F1A
C6–H6⋯F6 |
3.145(15)/2.25/139
3.352(2)/2.58/128 |
| V | −x + 1, −y + 1, −z + 1 | 8.023 | −3.1 | −0.8 | −7.7 (66) | 1.4 | −10.2 | −14.6 |
C11–H11⋯F5
C11–H11⋯F6 |
3.815(2)/2.91/142
3.882(2)/2.99/140 |
| NM23 (P21/c, Z′ = 1) | ||||||||||
| I | −x + 1, −y + 1, −z | 5.065 | −15.0 | −9.3 | −58.3 (71) | 43.4 | −39.1 | −50.8 |
C10–H10⋯Cg1 (π)
C10⋯C11 (π⋯π) |
3.652(2)/2.61/161
3.331(2) |
| II | −x + 1, −y + 1, −z + 1 | 7.982 | −18.4 | −8.1 | −37.1 (58) | 25.0 | −38.7 | −42.4 |
C12–H12⋯O1
C2–H2⋯O1 |
3.652(2)/2.58/172
3.552(2)/2.80/126 |
| III | x, −y + 1/2, z−1/2 | 6.205 | −14.6 | −5.6 | −27.6 (58) | 16.3 | −31.4 | −38.7 |
C5–H5⋯O1
C2–H6⋯O1 |
3.314(2)/2.62/121
3.302(2)/2.60/122 |
| IV | −x + 1, y + 1/2, −z + 1/2 | 7.260 | −4.6 | −2.3 | −18.6 (73) | 11.3 | −14.2 | −19.0 |
C14–C14C⋯F5A
C5⋯C12 (π⋯π) |
3.636(2)/2.62/157
3.457(2) |
| V | x−1, −y + 1/2, z−1/2 | 10.749 | −2.0 | −0.7 | −8.4 (76) | 3.1 | −8.0 | −10.6 |
C8–C8⋯F4A
C15–F3A⋯F6A–C16 |
3.700(3)/2.64/166
2.948(2)/148(1)/96(1) |
| VI | x + 1, y, z + 1 | 11.682 | −2.1 | −0.6 | −6.7 (71) | 3.9 | −5.5 | −6.6 |
C3–H3⋯F1A
C15–F3A⋯F4A–C16 |
3.550(2)/2.48/173
3.004(2)/96(1)/145(1) |
| NM31 (P21/c, Z′ = 1) | ||||||||||
| I | −x + 1, y−1/2, −z + 1/2 | 7.187 | −11.9 | −4.9 | −32.8 (66) | 19.5 | −30.1 | −36.8 |
C4–H4⋯O1
C4⋯C1 (π⋯π) C3⋯C6 (π⋯π) |
3.410(2)/2.42/151
3.665(2) 3.656(2) |
| II | x, y−1, z | 7.940 | −12.5 | −3.6 | −17.2 (52) | 11.3 | −22.1 | −26.0 |
C9–H9⋯O1
C6–H6⋯F3A C12–H12⋯F1A C14–H14A⋯F1A |
3.508(2)/2.54/149
3.229(7)/2.38/135 3.699(6)/2.72/150 3.656(7)/2.74/143 |
| III | −x, −y, −z + 1 | 9.001 | −5.5 | −1.5 | −18.7 (73) | 6.2 | −19.6 | −24.9 |
C11⋯C11 (π⋯π)
C14–H14B⋯F2A |
3.595(2)
3.600(6)/2.90/123 |
| IV | x, −y + 1/2, z + −1/2 | 8.143 | −5.5 | −3.3 | −20.1 (70) | 10.8 | −18.2 | −19.5 |
C11–H11⋯O1
C5–H5⋯F6 C6–H6⋯F6 |
3.692(2)/2.76/145
3.244(2)/2.52/123 3.280(2)/2.61/120 |
| V | −x, y + 1/2, −z + 1/2 | 7.937 | −6.5 | −2.1 | −15.2 (64) | 9.6 | −14.2 | −19.4 | C14–H14C⋯Cg2 (π) | 3.939(2)/3.05/139 |
| VI | −x + 1, −y, −z + 1 | 8.313 | −0.7 | −0.8 | −9.6 (86) | 1.9 | −9.2 | −13.8 | C4–H4⋯F2A | 3.614(5)/2.80/133 |
| VII | x, −y−1/2, z− + 1/2 | 9.458 | −0.0 | −0.3 | −6.3 (95) | 1.7 | −4.9 | −7.6 | C15–F3A⋯F6–C16 | 2.918(2)/134(1)/111(1) |
|
NM33 (P, Z′ = 2) |
||||||||||
| I 1⋯1 | −x + 1, −y + 1, −z | 7.358 | −17.3 | −8.1 | −38.8 (60) | 26.4 | −37.8 | −40.8 | C8–H8⋯O1 | 3.562(1)/2.49/173 |
| II 1⋯2 | −x + 1, −y + 1, −z | 4.361 | −10.6 | −8.0 | −57.2 (75) | 40.6 | −35.3 | −49.8 |
C3′–H3′⋯Cg1 (π)
C9–H9⋯C8′ (π) C8⋯C3′ (π⋯π) C9⋯C3′ (π⋯π) |
3.664(2)/2.63/159
3.648(1)/2.95/122 3.486(2) 3.311(2) |
| III 2⋯2 | −x + 1, −y + 1, −z + 1 | 6.426 | −12.7 | −3.8 | −32.5 (66) | 16.0 | −33.0 | −37.1 |
C6′–H6′⋯F2A′
C6′–H6′⋯F1A′ C5′–H5′⋯F1A′ C11′⋯C11′ (π⋯π) |
3.772(10)/2.72/166
3.475(7)/2.72/127 3.544(8)/2.88/120 3.547(2) |
| IV 1⋯2 | x, y, z | 8.192 | −21.1 | −6.9 | −19.4 (41) | 19.2 | −28.2 | −29.3 |
C8′–H8′⋯O1
C2′–H2′⋯O1 |
3.370(2)/2.30/169
3.285(2)/2.42/136 |
| V 1⋯2 | −x, −y + 1, −z | 8.726 | −8.3 | −4.7 | −26.4 (67) | 17.6 | −21.9 | −28.2 |
C14–H14C⋯O1′
C14–H14B⋯C2′(π) C11⋯C5′ (π⋯π) |
3.636(2)/2.80/134
3.502(2)/2.63/138 3.674(2) |
| VI 1⋯1 | −x + 1, −y, −z | 7.963 | −5.6 | −2.4 | −28.8 (78) | 17.4 | −19.4 | −29.7 |
C4⋯C5 (π⋯π)
C16⋯C6 (π⋯π) |
3.541(1)
3.615(1) |
| VII 1⋯2 | x + 1, y, z | 10.529 | −14.9 | −5.0 | −10.5 (35) | 15.5 | −15.0 | −14.6 |
C3–H3⋯O1′
C14′–H14F⋯F5 |
3.167(1)/2.20/148
3.542(2)/2.81/125 |
| VIII 2⋯2 | x−1, y, z | 8.998 | −6.3 | −1.1 | −10.3 (58) | 3.8 | −14.0 | −17.7 |
C14′–H14D⋯F3A′
C15′–F3A′⋯C13′ |
3.275(11)/2.75/110
3.179(10)/177 |
| IX 1⋯1 | x−1, y, z | 8.998 | −3.1 | −1.5 | −11.6 (72) | 6.1 | −10.0 | −13.1 |
C12–H12⋯F6
C14–H14B⋯F6 |
3.397(2)/2.43/148
3.311(2)/2.77/111 |
| X 1⋯1 | −x, −y, −z−1 | 11.332 | −4.0 | −0.7 | −8.4 (64) | 3.2 | −10.0 | −11.5 |
C11–H11⋯F6
C15–F2⋯F3–C15 |
3.762(1)/2.78/151
3.009(1)/130(1)/99(1) |
| XI 1⋯2 | −x + 1, −y, −z | 8.377 | −4.4 | −1.5 | −11.9 (67) | 7.9 | −10.0 | −13.8 |
C14′–H14F⋯F6
C14′–H14F⋯F4 |
3.509(2)/2.48/160
3.654(2)/2.76/140 |
| XII 1⋯2 | x, y, z−1 | 10.301 | −3.8 | −1.1 | −7.9 (62) | 4.9 | −8.0 | −9.5 |
C11′–H11′⋯F1
C12′–H12′⋯F1 C12′–H12′⋯F2 |
3.210(2)/2.54/119
3.262(2)/2.66/115 3.899(1)/2.90/153 |
| XIII 2⋯2 | −x + 1, −y + 2, −z + 1 | 9.320 | −2.9 | −0.4 | −5.6 (63) | 1.8 | −7.2 | −10.7 | C5′–H5′⋯F5A′ | 3.682(7)/2.78/141 |
| XIV 1⋯1 | −x + 2, −y, −z | 13.180 | 1.1 | −0.2 | −4.7 (96) | 2.7 | −1.2 | −1.5 |
C16–F5⋯F6–C16
C16–F5⋯F5–C16 |
3.012(1)/139(1)/95(1)
2.889(1)/101(1)/101(1) |
Hence, % electrostatic contribution (coulombic + polarization), %Eelec = 100 − %Edisp
These values are reported in Table 5.
The PIXEL interaction energy was further compared with the interaction energies obtained from the theoretical calculations at the DFT + Disp/B97D60–62 level at a higher aug-cc-pVTZ basis set using TURBOMOLE.63 The hydrogen atoms were moved to neutron values (1.083 Å for C–H) before the calculations. The basis set superposition error (BSSE) for the interaction energies was corrected using the counterpoise method.64 The final interaction energies are listed along with the PIXEL interaction energies in Table 5.
Results and discussion
ORTEP of NM10 and NM11 are presented in Fig. 1(a) and (b) with the atom numbering scheme. The ORTEPs for the remaining compounds are deposited in the ESI,† [Fig. S5(a)–(j)]. The Cambridge Structural Database search (CSD version 5.35 updates Nov 2013) was performed for related structures to compare the molecular conformation and crystal packing of related molecules with the present series of compounds. The result gave only 8 hits [Fig. 1(d)]. The crystal structure of N-methyl-N-phenylbenzamide (labeled as ‘NM00’) is reported (ref. code: JAZJOJ10) in the CSD. The compounds in this series exist preferably in the cis-conformation [Fig. 1(c)]. Due to the presence or substitution of H by a functional group ortho to both the phenyl rings, the conformation may change to trans orientation on account of the role of sterics, as is observed in the case of NM11, YEGJEY, DIBGIF and DIBGAX. It is of interest to note that the methyl substitution ortho to both of the phenyl rings (YEGLAX) exhibits a cis-conformation, whereas substitution by a trifluoromethyl (–CF3) group at the same position in NM11 leads to a trans geometry [Fig. 1(b) and (d)]. The reason for this observation may be the possibility of the formation of two intramolecular weak C(sp3)–H⋯OC hydrogen bonds in the case of YEGLAX, which stabilizes the molecular conformation in the cis-geometry. The dihedral angles between the planes formed by the two phenyl rings and the central part (OC–N–CH3) of the molecules for all the structures are compared in Tables 2 and 3. The angle between the two phenyl rings for the molecule with the cis-geometry display similar values (angle 1, value more than 60°, Tables 2 and 3). The deviation of the phenyl ring (plane 2) on the nitrogen side (angle 3, the value being more than 56°) from OC–N–CH3 plane (plane 3) was observed to be more than for angle 2 (the value being less than 60° in most cases), which is the dihedral angle between plane 1 (phenyl ring on carbonyl side) and plane 3. No significant changes were observed between the solid state geometry and gaseous state geometry. The molecular conformation was observed to be stabilized by the presence of an intramolecular weak C(sp3)–H⋯OC hydrogen bond in both the solid state and the gas phase. In the case of the molecules with the trans conformation, both the phenyl rings were nearly orthogonal to the central plane 3 (OC–N–CH3 plane) in both the solid state and gas phase geometry to minimize the sterics in the molecule.
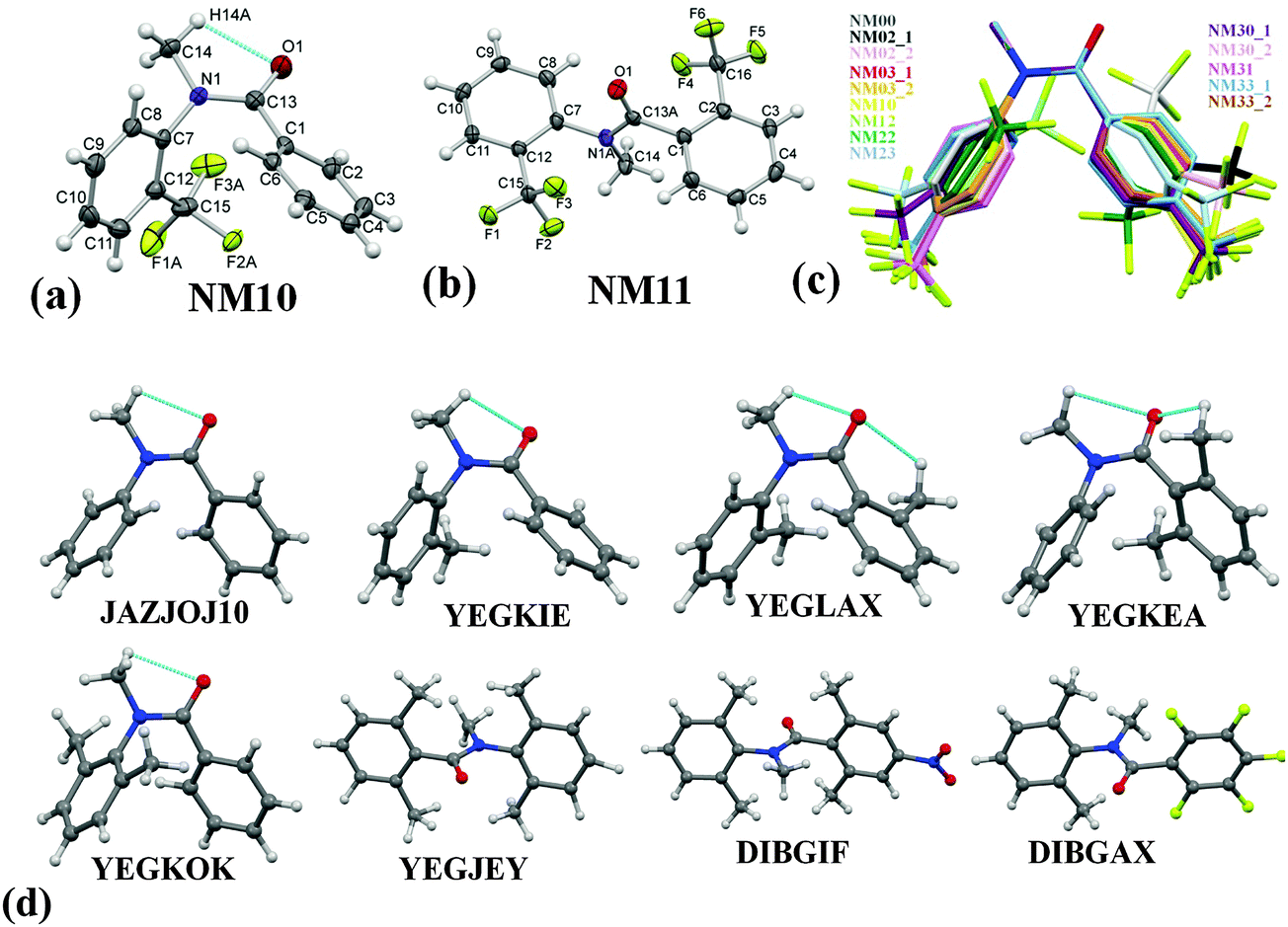 | ||
| Fig. 1 (a) and (b) ORTEP of NM10 and NM11 drawn with 50% ellipsoidal probability with an atom numbering scheme displaying two possible conformations in this class of compounds. A similar numbering scheme was followed in all the structures. Only the major component of the disordered part of the molecule is shown for clarity. The dotted lines indicates the presence of an intramolecular C(sp3)–H⋯OC hydrogen bond. ORTEPs of the other molecules are shown in Fig. S5 in the ESI.† (c) Overlay of all the structures in cis-geometry, drawn with Mercury 3.0. (d) Molecular diagram of related molecules reported in CSD with their reference code. Dotted lines indicate the presence of an intramolecular C–H⋯OC hydrogen bond. | ||
From the lattice energy calculations, using the PIXEL method for all the molecules (Table 4), it was observed that the values lie between 102 kJ mol−1 and 115 kJ mol−1, with the dispersion energy being the major component. The lattice energy of the four related molecules in CSD was also calculated using the PIXEL method (Table 4). The result demonstrated that the substitution of the methyl group on N-methyl-N-phenyl benzamide did not exhibit significant changes in the lattice energy.
Molecular pairs and crystal packing analysis
It was of interest in this study to analyze the crystal packing of N-methyl-N-phenyl benzamides (NM00) and to compare it with that of its trifluoromethyl-substituted analogues (Table 1). The molecular pairs extracted from the crystal packing of NM00 are shown in Fig. 2(a), along with the associated interaction energies. The analysis of the molecular pairs revealed that the crystal packing in NM00 is mainly stabilized by the presence of weak C–H⋯OC and C–H⋯π hydrogen bonds [Fig. 2(a) and Table 5]. The most stabilized motif I (I.E = −24.5 kJ mol−1) consists of a short C–H⋯OC and a C–H⋯π hydrogen bond, resulting in the formation of a molecular chain with the utilization of the b-glide plane perpendicular to the crystallographic a-axis [Fig. 2(b)]. Such chains are interconnected with the second most stabilized molecular motif II (I.E = −19.6 kJ mol−1), which consists of a pair of a C–H⋯π hydrogen bond and a short H⋯H contact. The important fact observed here is that the motif I, which consists of a short C–H⋯O hydrogen bond (dH⋯O = 2.41 Å), has a 34% electrostatic (coulombic + polarization) and 66% dispersion contribution out of the total stabilization, while the values corresponding to motif II are 22% and 78%, respectively, which primarily involves C–H⋯π hydrogen bonds. Similar trends were observed in the other motifs as well. Motifs involving C–H⋯π hydrogen bonds (III, VI, VII) display a dispersion contribution greater than 72%, the highest being in the case of motif VI (97%), wherein no interactions less than the sum of the van der Waals radii66 were observed. Furthermore, in the case of motifs IV and V, wherein weak C–H⋯O hydrogen bonds are present, the dispersion contribution decreases to 62%. Motif IV was observed to be utilized in the formation of a molecular chain along the c-axis (using the c-glide plane perpendicular to the a-axis) and such chains are interconnected with motifs V and VI [Fig. 2(c)].
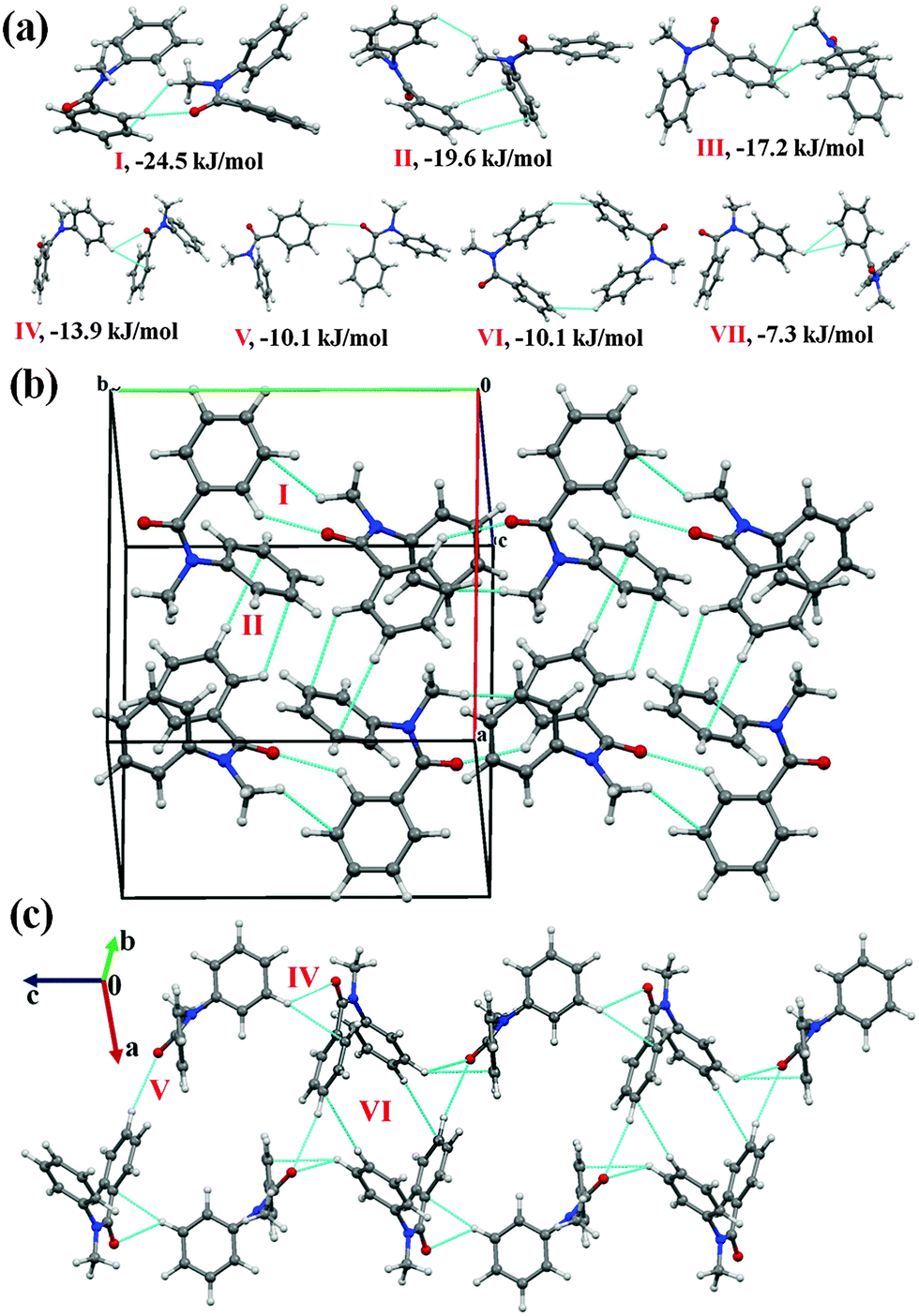 | ||
| Fig. 2 (a) Selected molecular pairs along with their PIXEL interaction energy in NM00. Roman numbers in red indicate the molecular pairs (in Table 5). (b) Packing of molecules via the utilization of weak C–H⋯OC and C–H⋯π hydrogen bonds in NM00. The molecular pairs in Table 5 are indicated with Roman numbers in red in all the figures in this study. (c) Weak C–H⋯OC and C–H⋯π hydrogen bonds in the packing of molecules in NM00 along the crystallographic c-axis. | ||
N-Methyl-N-phenyl-3-(trifluoromethyl)benzamide (NM02)
Compound NM02 crystallizes in the monoclinic centrosymmetric C2/c space group with two molecules in the asymmetric unit. The asymmetric unit (motif V, I.E = −22.4 kJ mol−1, Table 5) is held via a short C(sp2)–H⋯OC (2.45 Å/141°; involving acidic hydrogen, H2′) and a short type II C(sp3)–F⋯F–C(sp3) contact [2.823(2) Å, 98(1)°, 158(1)°]. The presence of a σ-hole on the fluorine atoms in the CF3 group has recently been revealed and is responsible for the formation of such interactions in the crystal packing.36 It was also well established that type II halogen–halogen contacts may be considered as a halogen bond.67,68 It can be noted here that the electrostatic contribution (coulombic + polarization) towards the total stabilization energy is 32% between the two interacting molecules in the asymmetric unit.
Furthermore, the analysis of the molecular pairs extracted [Fig. 3(a)] from the crystal packing of NM02 revealed that among the top six most stabilized motifs, five consists (motifs I, III–VI) of the presence of weak C–H⋯OC hydrogen bonds with stabilization energies ranging from 17.6 kJ mol−1 to 35.1 kJ mol−1 with substantial electrostatic contributions (in the range of 31 to 45%, Table 5). The highest stabilized (with a 43% electrostatic contribution) molecular motif was I, which included the presence of dimeric bifurcated weak C–H⋯OC hydrogen bonds with donor atoms from two different C–H bonds [C(sp2)–H and C(sp3)–H] in different electronic environments. Motifs II, III and IV were observed to provide similar stabilization (I.E: −31.5, −31.1 and −30.0 kJ mol−1, respectively) but differed in the nature of the participating interactions. In the case of motif II, the molecules interact via the existence of C–H⋯π hydrogen bonds and π⋯π interactions, with the % contribution from the dispersion being the highest (77%) among all the motifs. Motif III involved one short C(sp2)–H⋯OC (2.46 Å/144°) and two C(sp2)–H⋯F–C(sp3) hydrogen bonds (2.43 Å/138°; 2.65 Å/126°); the former being significantly short. The dispersion contribution was 59% with this being a significant contribution and comparable to related weak H-bonds. Furthermore, motif IV, which involved one C(sp2)–H⋯OC and a short C–H⋯π hydrogen bond (2.65 Å/157°, Table 5), showed a dispersion contribution (69%) in between that of motifs II and III. Two bifurcated C(sp2)–H⋯OC along with C(sp3)–H⋯F–C(sp3) were observed to hold the molecules in motif VI (I.E = −18.2 kJ mol−1) with the highest (45%) electrostatic contribution among all the motifs. Furthermore, in the case of motif VII (I.E = −14.7 kJ mol−1) and VIII (I.E = −14.5 kJ mol−1), wherein C–H⋯π hydrogen bonds and π⋯π interactions are present, the total stabilization was dominated from the contribution due to the dispersion interactions (72 and 69%, respectively). It is to be noted that the crystal packing in NM02 was also stabilized, albeit less, by the presence of weak C(sp2/sp3)–H⋯F–C(sp3) hydrogen bonds (motifs IX–XI). Motif IX (I.E = −10.1 kJ mol−1) showed the presence of one bifurcated C(sp2)–H⋯F–C(sp3) and a short and directional C(sp3)–H⋯F–C(sp3) (2.51 Å/160°) hydrogen bond with the electrostatic contribution being 41% of the total stabilization. Motifs X and XI [involving bifurcated C(sp2)–H⋯F–C(sp3) hydrogen bonds], which were observed to contribute similar stabilization (−7.1 and −7.0 kJ mol−1) towards the crystal packing, contain a 32% contribution from electrostatics. The stabilization energy for a C–H⋯F hydrogen bond was reported to be −0.40 kcal mol−1 (−1.6 kJ mol−1) by an ab initio theoretical calculation in the molecular crystal.69 It was observed in the same study that the stabilization energy for a C–H⋯F hydrogen bond was mainly dominated by electrostatic and dispersion components with the latter being more prominent. Fig. 3(b) and (c) display the packing of molecules in NM02 with the utilization of such weak interactions.
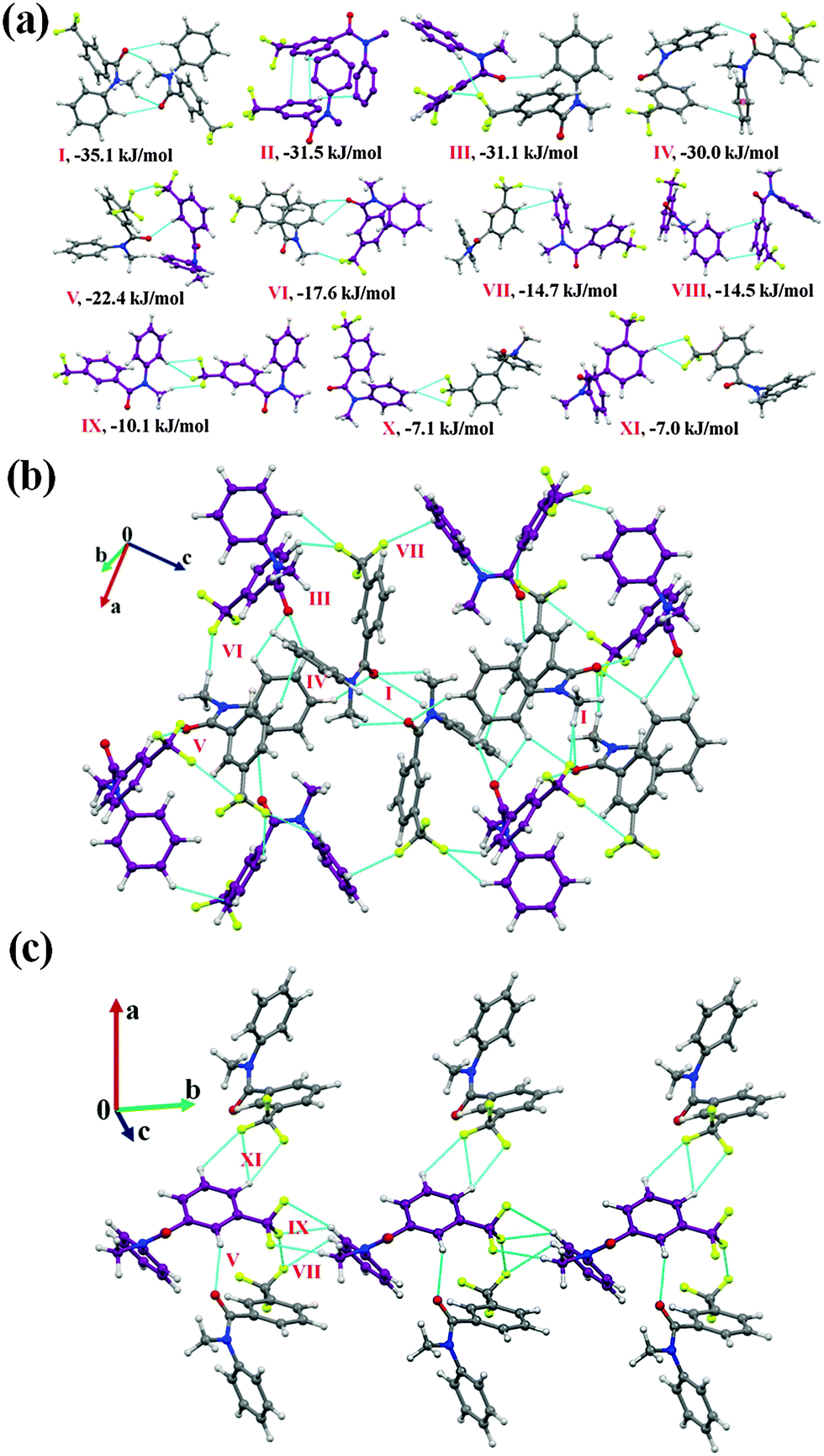 | ||
| Fig. 3 (a) Selected molecular pairs, along with their PIXEL interaction energy in NM02. C atoms are in purple and represent the second molecule in the asymmetric unit. (b) Packing of molecules in NM02 with the presence of weak C–H⋯OC, C–H⋯F and C–H⋯π hydrogen bonds. (c) Part of the crystal packing down the ab plane in NM02, displaying the presence of weak C–H⋯OC and C–H⋯F–C(sp3) hydrogen bonds along with C(sp3)–F⋯F–C(sp3) interactions. | ||
N-Methyl-N-phenyl-4-(trifluoromethyl)benzamide (NM03)
Compound NM03 crystallizes in the monoclinic centrosymmetric P21/c space group with Z′ = 2. A bifurcated weak C(sp3)/(sp2)–H⋯OC hydrogen bond [this includes a short and highly directional C(sp2)–H⋯OC; 2.26 Å, 161°, Table 5] along with π⋯π interactions were observed to link the molecules in the asymmetric unit. This molecular motif has the highest stability [motif I, I.E = −39.3 kJ mol−1, Fig. 4(a)] in the crystal packing of NM03 [Fig. 4(b) and (c)] with the electrostatic contribution being 43%. Although motif I primarily consists of π⋯π interactions, a relatively high electrostatic contribution towards the total stabilization (in comparison to related molecular motifs, wherein C–H⋯π or π⋯π are present, the electrostatic contribution was observed to be less than 30%) is due to the presence of short C(sp3)/(sp2)–H⋯OC hydrogen bonds. Similarly, in the case of the second most stabilized molecular pair (motif II, I.E = −35.1 kJ mol−1), wherein the molecules are linked with a short C(sp2)–H⋯OC (2.45 Å, 139°) and two (including one at a short distance) directional C(sp2)–H⋯π (2.49 Å, 159°; 2.80 Å, 154°) hydrogen bonds, the electrostatic contribution is 35% [Table 5]. It can be noted here that motif I [consisting of highly short and directional C(sp2)–H⋯O] has approximately 6 kJ mol−1 more coulombic contribution than that in motif II, whereas the opposite situation was observed in the case of the dispersion contribution with a similar magnitude of approximately 6 kJ mol−1. Motif III (I.E = −27.6 kJ mol−1) and IV (I.E = −23.6 kJ mol−1) are characterized by the presence of weak C(sp2)–H⋯π and π⋯π interactions, and the dispersion energy contribution in them exceeds 75% and 70% respectively. Furthermore, a short and highly directional C(sp2)–H⋯O hydrogen bond (2.33 Å, 173°, motif V) was observed to provide 20.3 kJ mol−1 stabilization towards the crystal packing in NM03 with the contribution from electrostatics being 42%. Similar trends were observed in the case of motifs VI and VII [Fig. 4(a)], wherein the molecules are held via the presence of C(sp2)–H⋯O hydrogen bonds, along with the other interactions (Table 5). Moreover, the packing of molecules in NM03 were also observed to be stabilized by the presence of weak C(sp2)–H⋯F–C(sp3) hydrogen bonds (motifs VIII–XII, except X), which consists of long C(sp2)–H⋯O (2.96 Å/159°) with stabilization energies ranging from 9.2 kJ mol−1 to 5.6 kJ mol−1 with %Eelec in the range from 27% to 46% (Table 5). Fig. 4(c) shows that the highly stabilized motif I and III are interlinked via the presence of weak C(sp2)–H⋯F–C(sp3) hydrogen bonds down the (110) plane in the molecular packing of NM03.
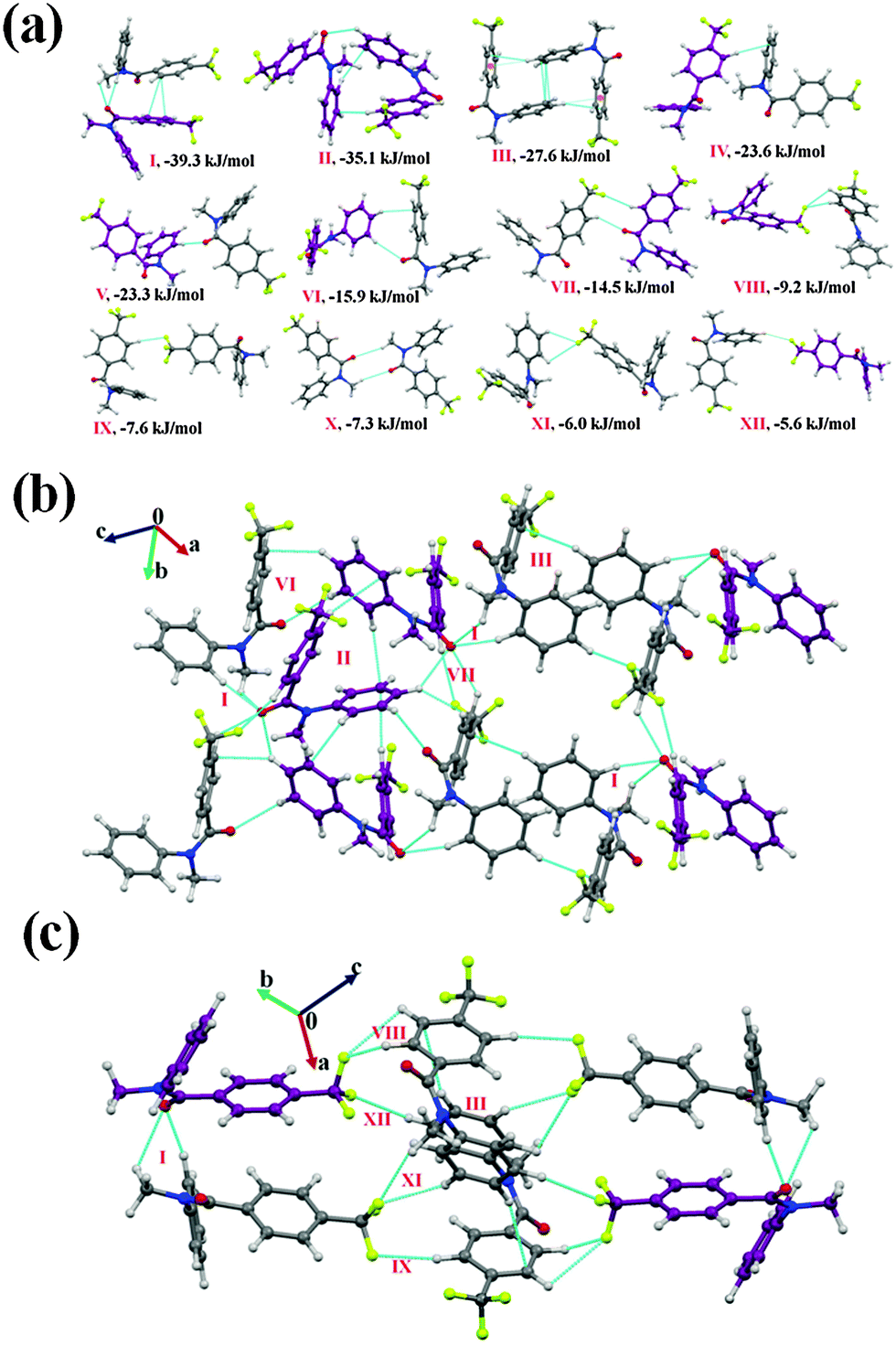 | ||
| Fig. 4 (a) Selected molecular pairs, along with their PIXEL interaction energy in NM03. C atoms are in purple and represent the second molecule in the asymmetric unit. (b) Packing of molecules down the (101) plane in NM03, displaying the presence of weak C–H⋯OC, C–H⋯π and C–H⋯F–C(sp3) hydrogen bonds. (c) Part of the crystal packing displaying motifs I and III (Table 5) connected via weak C(sp2)–H⋯F–C(sp3) hydrogen bonds down the (110) plane in NM03. | ||
N-Methyl-N-(2-(trifluoromethyl)phenyl)benzamide (NM10)
Compound NM10 crystallizes in the orthorhombic centrosymmetric Pbca space group with Z = 8. Molecular pairs extracted from the crystal packing in NM10 have been highlighted [Fig. 5(a)] along with their interaction energies. The highest stabilized molecular motif I (I.E = −36.2 kJ mol−1) is similar to motif II in NM02 and motif III in NM03 [Fig. 5(a)]. As in the previous case, the molecules are linked via the presence of a short C–H⋯π with %Edisp = 60, which is 15–17% less than in the previous case (Table 5). This may be due to the absence of C⋯C (π⋯π) interactions, in the present case, at a distance less than 4 Å. It can be observed, on viewing down the crystallographic bc plane [Fig. 5(b)], that the molecular chains formed with the utilization of motif III (I.E = −20.6 kJ mol−1) and motif VI (I.E = −9.6 kJ mol−1) along the b-axis are interconnected with motifs II (I.E = −28.1 kJ mol−1) and V (I.E = −11.7 kJ mol−1). Motif II, consists of a short C(sp3)–H⋯OC (2.59 Å, 130°) and C(sp2)–H⋯π at longer distances [%Edisp being 65%]. Furthermore, motifs III and IV, which involve weak C–H⋯F–C(sp3) hydrogen bonds at a distance greater than the sum of the van der Waals radii of H and F (2.67 Å), were observed to provide more stabilization in comparison to motifs V and VI, which consist of a short C(sp2)–H⋯OC hydrogen bond (Table 5). Differences among them appear in the nature of the individual components of the total stabilization energy. In the case of III and IV, this is of a dispersive origin (more than 62%), while motif V (%Eelec = 50) and motif VI (%Eelec = 45) show a very significant contribution from electrostatics. In the crystal packing of NM10, a less stabilized molecular motif (motif VII, −6.9 kJ mol−1), involving weak C(sp2)–H⋯π hydrogen bond, was also observed, with %Edisp = 84.
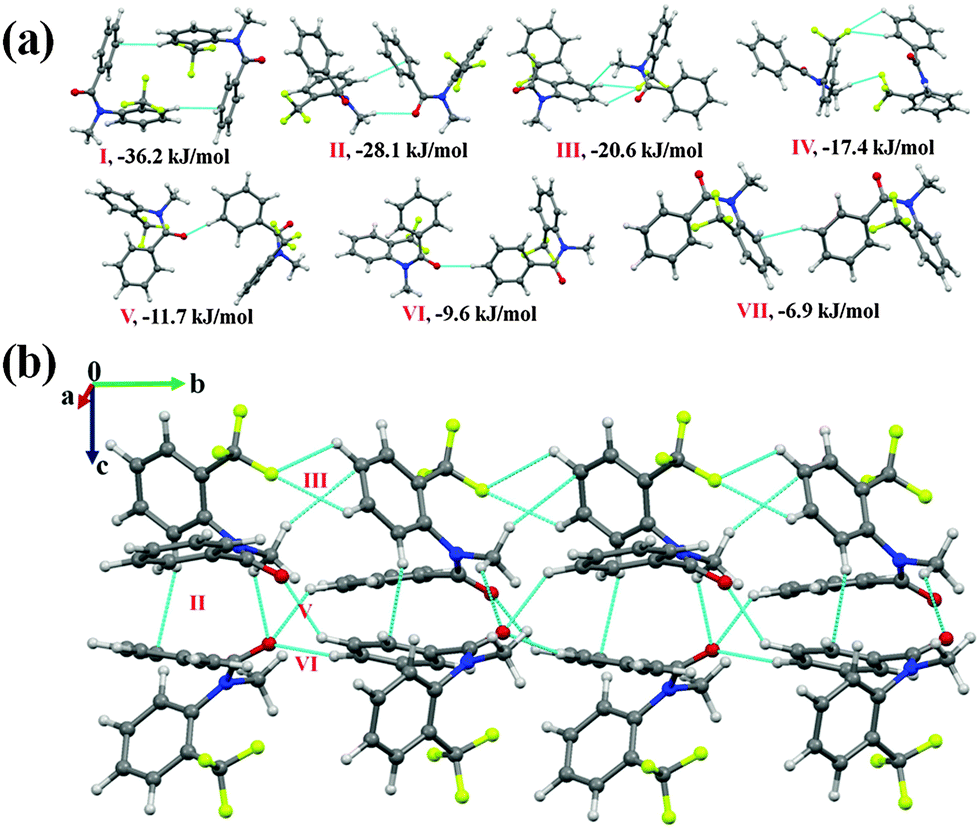 | ||
| Fig. 5 (a) Molecular pairs, along with their interaction energies. extracted from the crystal packing in NM10. (b) Packing of molecules down the bc plane via weak C–H⋯OC, C–H⋯π and C–H⋯F–C(sp3) hydrogen bonds in NM10. | ||
N-Methyl-N-(4-(trifluoromethyl)phenyl)benzamide (NM30)
Compound NM30 crystallizes in the centrosymmetric monoclinic space group P21/c with two molecules in the asymmetric unit. The asymmetric unit was observed to be a highly stabilized molecular pair (I.E = −25.6 kJ mol−1 with %Edisp = 65) in the crystal packing involving weak C(sp2)–H⋯OC and C(sp2)–H⋯π hydrogen bonds, along with the presence of a π⋯π interaction. The molecular motifs II to V were observed to provide similar stabilization (Table 5, I.E of approximately 18.2 to 18.8 kJ mol−1) towards the crystal packing. Among these, motifs II and III were found to be involved in the formation of a short C(sp2)–H⋯OC hydrogen bond (2.53 Å, 133°; 2.49 Å, 136°) with an electrostatic contribution of 41% and 42%, respectively. Motifs IV and V were involved in the formation of a weak C(sp2)–H⋯F bond along with the π⋯π interaction, and hence show a high dispersion contribution (77 and 76%, respectively). The weak C(sp2)–H⋯π hydrogen bond, along with the π⋯π interactions, were observed to connect to two symmetry independent molecules in the crystal packing in motif VI (I.E = −17.0 kJ mol−1 with %Edisp of 74). Moreover, the dimeric C(sp3)–F⋯π interaction was found to link two molecules in the crystal packing with motifs VII (I.E = −17.0 kJ mol−1) and VIII of similar stabilization (Table 5), together with a substantial dispersion contribution (more than 70%). The interaction energy of the C(sp3)–F⋯π interaction (for one interaction, the approximate value will be −8.5 kJ mol−1; herein, a phenyl group, which is involved in the interaction, is attached with an electron withdrawing –CF3 group) is similar to the value for the C–F⋯πF interaction (−2.43 kcal mol−1 for the interaction of fluoromethane with hexafluorobenzene) by MP2/aug-cc-pVDZ calculation.70 In motifs IX and X (I.E being −16.0 and 15.5 kJ mol−1, respectively), a weak C(sp2)–H⋯F hydrogen bond along with a C(sp3)–F⋯CO interaction were observed to connect the molecules. Furthermore, weak C(sp2)–H⋯OC and C(sp3)–H⋯F–C(sp3) hydrogen bonds were observed to be involved in two similarly stabilized molecular pairs (motifs XI and XII) in the crystal packing. A short and directional C(sp2)–H⋯π (2.73 Å, 152°) along with weak C(sp2)–H⋯OC hydrogen bonds were recognized to be involved in connecting the two symmetry independent molecules in the crystal packing in motif XIII (I.E = −12.6 kJ mol−1, with %Edisp = 63%). Moreover, type I C(sp3)–F⋯F–C(sp3) interactions were observed to connect the molecules in the weakly stabilized molecular motifs XIV and XV with a positive coulombic contribution. The stabilization in these motifs is mainly of a dispersion origin (more than 94%, Table 5) with the overall stabilization energy being 1.8 kJ mol−1. This stabilization energy is comparable with the value reported in a recent analysis (by an ab initio method and by symmetry-adapted perturbation theory (SAPT)) on the nature of C–F⋯F–C for the all unique dimers extracted from the crystal structure of CF4, C2F4 and C6F6.71 From the SAPT analysis, it was observed that the total stabilization energy was mainly dominated by the dispersion energy component and the electrostatic component can be stabilizing or destabilizing depending on the orientation of the interacting dimers. Fig. 6(b) represents the packing of molecules in NM30 down the crystallographic ac plane.
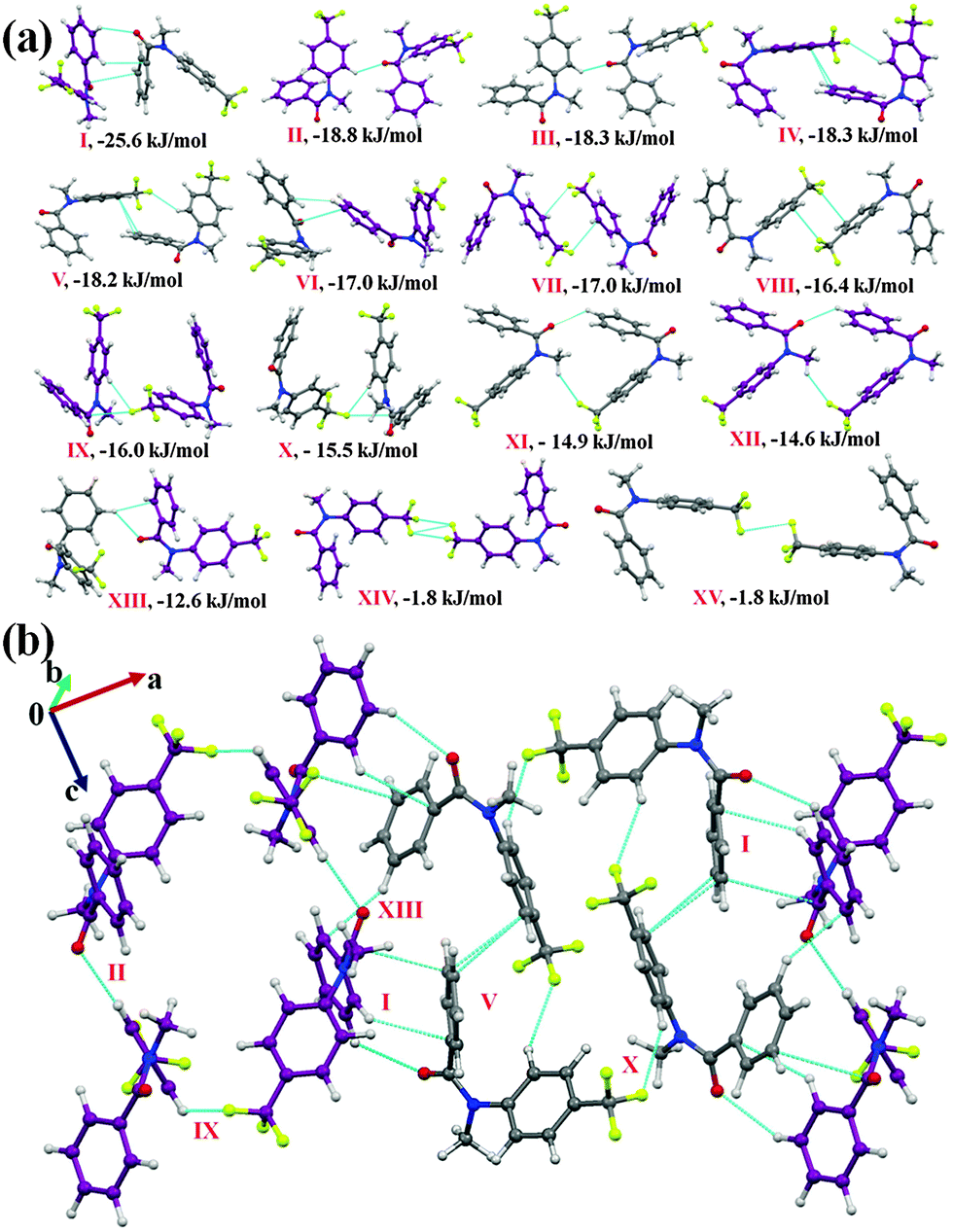 | ||
| Fig. 6 (a) Selected molecular pairs in NM30 along with their interaction energies. (b) Packing of molecules down the ac plane via weak C(sp2)–H⋯OC, C(sp2)–H⋯F–C(sp3), C(sp2)–H⋯π hydrogen bonds and π⋯π interactions in NM30. | ||
N-Methyl-2-(trifluoromethyl)-N-(2-(trifluoromethyl)phenyl)benzamide (NM11)
Compound NM11 crystallizes in a centrosymmetric monoclinic space group P21/c with Z = 4. Unlike other molecules in this series, the molecular structure is observed to be in the trans conformation with CO and N–C bonds oriented opposite to each other. This may be due to the minimization of the steric effect when two CF3 groups are present at the ortho position of the two phenyl rings in the molecule. (CH3)N–CO was observed to be disordered at two positions with the occupancy ratio of 0.939(3): 0.061(3) [modeled with PART command in the SHELXL 2013 at two orientations: ‘A’ (for higher occupancy) and ‘B’]. Selected molecular pairs extracted from the crystal packing are given in Fig. 7(a). A dimeric molecular motif, consisting of a pair of short and directional C(sp2)–H⋯OC (2.46 Å, 160°) and C(sp2)–H⋯F–C(sp3) (2.49 Å, 144°) hydrogen bonds along with offset π⋯π stacking interactions (motif I, I.E = −40.8 kJ mol−1), was observed to provide the highest stabilization towards the crystal packing. It is to be noted here that the %Eelec contribution was 54% with a coulombic contribution of 42%. The next two stabilized motifs (II and III) were involved in the formation of π⋯π stacking interactions between pair of molecules, the I.E being −22.0 and −17.8 kJ mol−1, respectively, with the stabilization being mainly dispersive in origin. It was observed that with an increase in the interacting distance of the Ph-ring (from motif II to motif III), the dispersion contribution towards the total stabilization increased from 74% to 94% with no stabilization from coulombic (positive coulombic contribution, Table 5) in the case of the latter. Furthermore, motifs IV and V were observed to contribute similar stabilization towards the crystal packing (−16.7 kJ mol−1 and −16.6 kJ mol−1) but were different in the nature of the involved interactions. Motif IV appeared to be connected via a long C(sp2)–H⋯F–C(sp3) hydrogen bond with %Eelec being 26%, whereas in motif V, the molecules were connected with short C(sp2)–H⋯OC (2.35 Å, 144°) and C(sp2)–H⋯F–C(sp3) (2.65 Å, 154°) hydrogen bonds. As expected, this results in the increase of the %Eelec contribution to 59% with 45% coulombic contribution (Table 5). The packing of molecules in NM11 was recognized to involve the formation of molecular networks wherein motif I was connected with motif V [Fig. 7(b)]. Moreover, motif VI [consisting of the pair of weak C(sp3)–H⋯F–C(sp3) and a C–H⋯π hydrogen bonds at distances longer than the sum of the van der Waals radii of the involved atoms, I.E = −15.8 kJ mol−1, %Edisp = 93%] generate a molecular chain with the utilization of a 21-screw along the b-axis [Fig. 7(c)]. Such a chain was observed to be linked via the weakly stabilized molecular motif VII (I.E = 5.3 kJ mol−1, %Edisp = 69%) down the bc plane, which involved two weak C(sp2)–H⋯F–C(sp3) (2.58 Å, 156°; 2.78 Å, 147°) hydrogen bonds (Table 5).
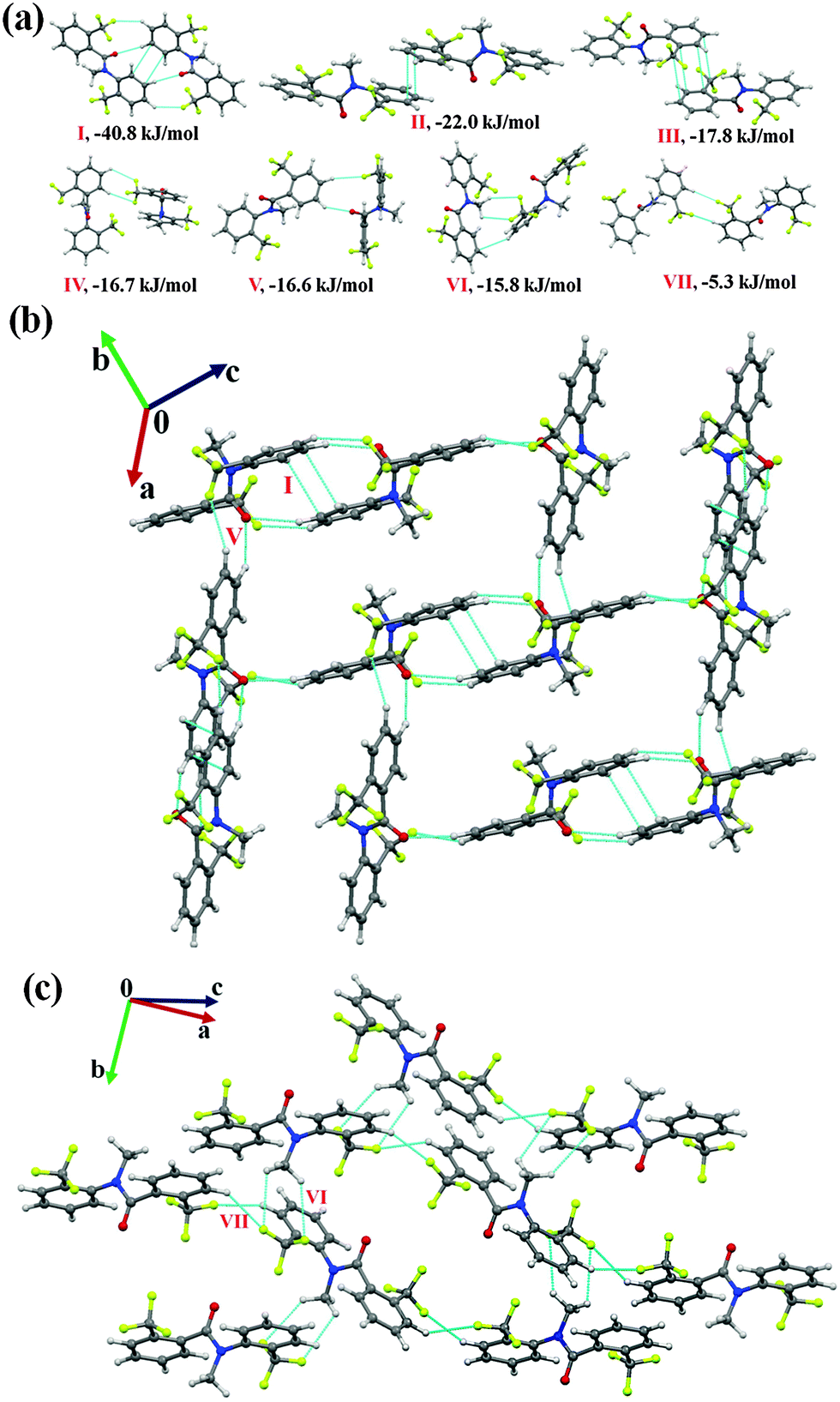 | ||
| Fig. 7 (a) Selected molecular pairs extracted from the crystal packing in NM11 along with their interaction energies. (b) Molecular network formed with the utilization of weak C(sp2)–H⋯OC, C(sp2)–H⋯F–C(sp3) hydrogen bonds and π⋯π interactions in NM11. (c) Packing of molecules via C(sp2)/(sp3)–H⋯F–C(sp3) hydrogen bonds in NM11. | ||
N-Methyl-3-(trifluoromethyl)-N-(2-(trifluoromethyl)phenyl)benzamide (NM12)
Compound NM12 crystallizes in the monoclinic P21/c space group with Z = 4. The analysis of the molecular pairs extracted from the crystal packing [Fig. 8(a)] showed that the highest stabilized molecular motif I [−36.2 kJ mol−1 with %Edisp = 80%; involves C(sp2)–H⋯π hydrogen bonds and π⋯π interactions] appears to be a robust motif in this series of compounds as also previously recognized in NM02, NM03 and NM10. However, it can also be noted here that this was not observed in the molecular packing of NM00. The packing of molecules down the bc plane in NM12 displays the formation of a molecular chain along the crystallographic c-axis via motif III (I.E = 22.3 kJ mol−1), which was observed to be interlinked with motif II (I.E = −25.6 kJ mol−1) and motif V (I.E = −12.1 kJ mol−1) [Fig. 8(b)]. Motif II consists of two short and directional C(sp2)–H⋯OC hydrogen bonds (2.32 Å, 160°; 2.57 Å, 153°) with a 48% contribution from electrostatics (Table 5). In the case of motif III (I.E = −25.6 kJ mol−1; %Eelec = 41), a weak C(sp2)–H⋯OC along with a C(sp2)–H⋯F–C(sp3) hydrogen bond was observed to connect the molecules, displaying a slightly less stabilization and electrostatic contribution than motif II (Table 5). Furthermore, dimeric C(sp2)–H⋯F–C(sp3) hydrogen bonds (2.63 Å, 130°) were recognized to link the molecules in motif V (with %Edisp = 75%). Moreover, a weak C(sp2)–H⋯π along with a weak C(sp2)–H⋯F–C(sp3) hydrogen bond (2.78 Å, 149°) were also observed to stabilize the crystal packing in NM12 (motif IV, −15.8 kJ mol−1; %Edisp = 76%). A weakly stabilized molecular pair (motif VI, I.E = −3.8), involving weak C(sp2)–H⋯F–C(sp3) hydrogen bonds along with a type I C(sp3)–F⋯F–C(sp3) interaction (Table 5), were also recognized in the crystal packing.
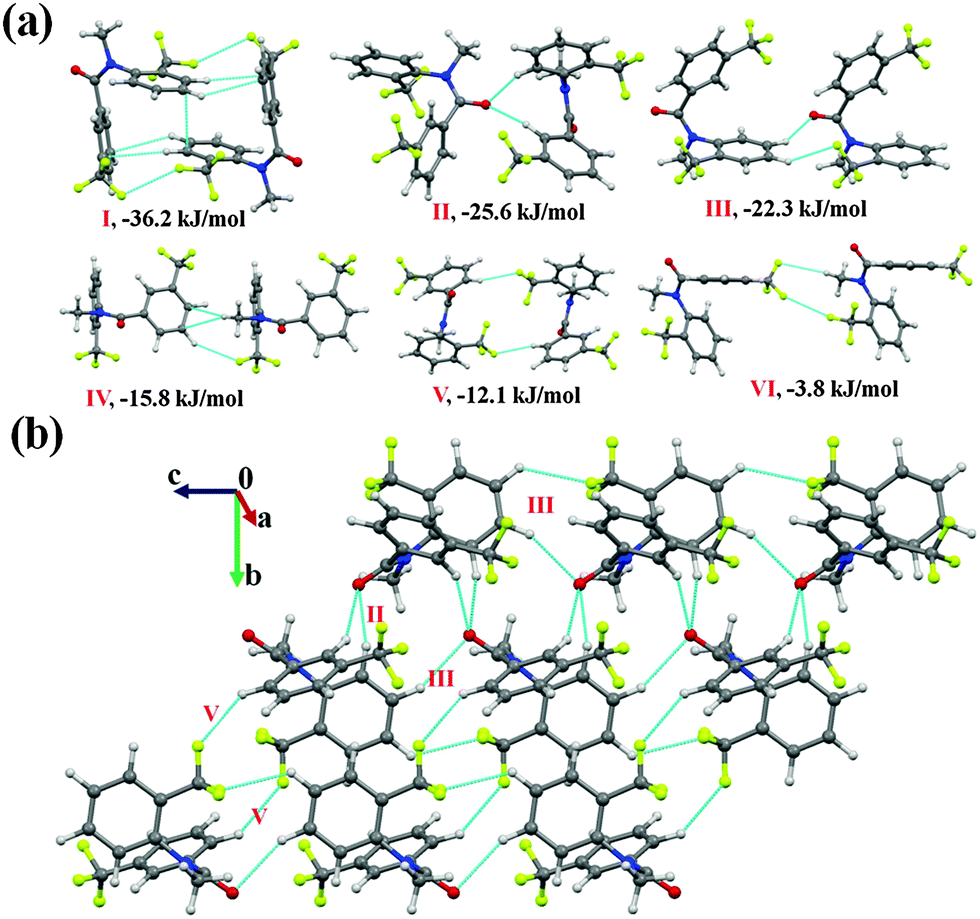 | ||
| Fig. 8 (a) Displaying molecular pairs extracted from molecular packing in NM12. (b) Packing of molecules down the bc plane with the utilization of weak C(sp2)–H⋯OC and C(sp2)–H⋯F–C(sp3) hydrogen bonds in NM12. | ||
N-Methyl-3-(trifluoromethyl)-N-(3-(trifluoromethyl)phenyl)benzamide (NM22)
Compound NM22 crystallizes in the centrosymmetric monoclinic space group (P21/c) with Z = 4. The molecular pairs, extracted from the crystal packing, are presented in Fig. 9(a). Three possible short and/or directional C(sp2)–H⋯OC (2.21 Å, 140°; 2.43 Å, 173°; 2.69 Å, 152°) hydrogen bonds, with motif I, involving the acidic hydrogen atoms, form the most stabilized (I.E = −37.0 kJ mol−1) pair in the crystal packing with the total stabilization being a 52% electrostatic (coulombic + polarization) contribution (Table 5). Motif II (I.E = 26 kJ mol−1), being the most common in this series of structures and consisting of a weak C(sp2)–H⋯π hydrogen bond and π⋯π interaction, was observed to provide stabilization to the crystal packing, which was primarily of a dispersive (85%) origin. The packing of molecules in NM22 was observed to form a zig-zag chain via motif I with the utilization of a c-glide perpendicular to the b-axis. Such a chain is connected via the utilization of motifs III and IV [Fig. 9(b)] down the bc plane. Motif III (I.E = −24.2 kJ mol−1) was found to involve a short C(sp2)–H⋯π (2.51 Å, 148°) along with a weak C(sp2)–H⋯F–C(sp3) hydrogen bond, whereas motif IV (I.E = −15.6 kJ mol−1) consisted of two C(sp2)–H⋯F–C(sp3) interactions. Both the motifs showed a similar contribution (67%) from dispersion towards the total stabilization. Moreover, a pair of bifurcated weak C(sp2)–H⋯F–C(sp3) hydrogen bonds were also recognized to stabilize (motif V, I.E being −10.2 kJ mol−1) the crystal packing in NM22.
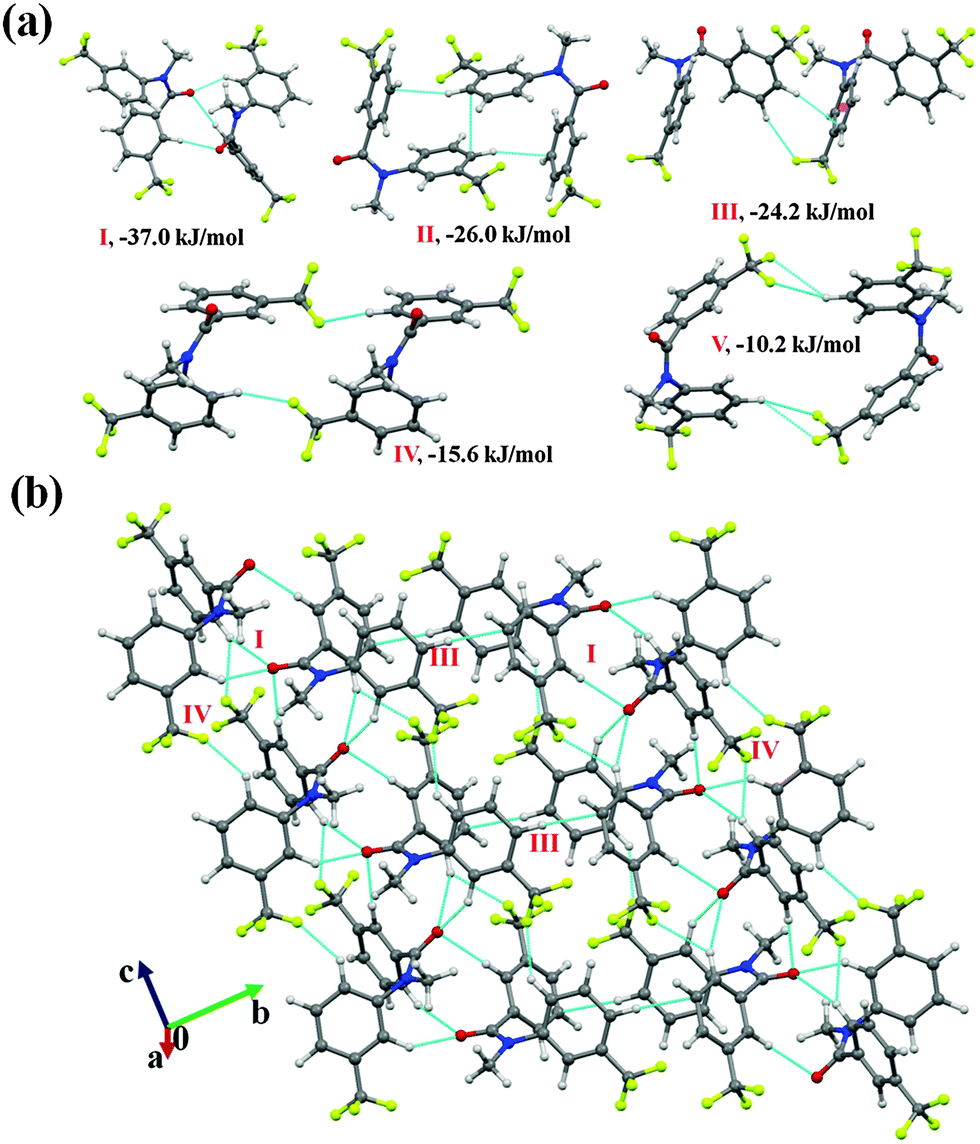 | ||
| Fig. 9 (a) Selected molecular pairs extracted from the crystal packing in NM22. (b) Network of weak C(sp2)–H⋯OC and C(sp2)–H⋯F–C(sp3) hydrogen bonds in the crystal packing down the bc plane in NM22. | ||
N-Methyl-4-(trifluoromethyl)-N-(3-(trifluoromethyl)phenyl)benzamide (NM23)
Compound NM23 crystallizes in the centrosymmetric monoclinic space group (P21/c) with Z = 4. Molecular pairs, extracted from the crystal packing of NM23, along with their stabilization energies are presented in Fig. 10(a). The analysis of the results depicts the presence of two similar dimeric stabilizing pairs [motif I (observed to be robust in this series) and motif II] in the crystal packing. Motif I (I.E = −39.1 kJ mol−1, %Edisp of 71%) was recognized to involve a short C(sp2)–H⋯π (2.61 Å, 161°) bond along with the presence of a weak offset π⋯π stacking interactions, whereas a dimeric bifurcated weak C(sp2)–H⋯OC interaction was observed to stabilize (I.E = −38.7 kJ mol−1 with %Edisp reduced to 58%) motif II in NM23 (Table 5). Both dimeric motifs I and II were found to be connected via motif III and IV in the formation of a molecular layer down the bc plane [Fig. 10(b)]. Bifurcated weak C(sp2)–H⋯OC hydrogen bonds, involving acidic hydrogens, were recognized to link the molecules in motif III (I.E = −31.4 kJ mol−1, %Edisp of 58%), whereas in the case of motif IV (I.E = −14.2 kJ mol−1 with %Edisp increased to 73%), a weak C(sp3)–H⋯F–C(sp3) hydrogen bond along with π⋯π stacking interaction were observed. Furthermore, a weak C(sp2)–H⋯F–C(sp3) hydrogen bond along with the type II C(sp3)–F⋯F–C(sp3) interaction [in motif V (−8.0 kJ mol−1) and VI (−5.5 kJ mol−1)] were also found to stabilize the crystal packing in NM23 [Fig. 10(a) and Table 5].
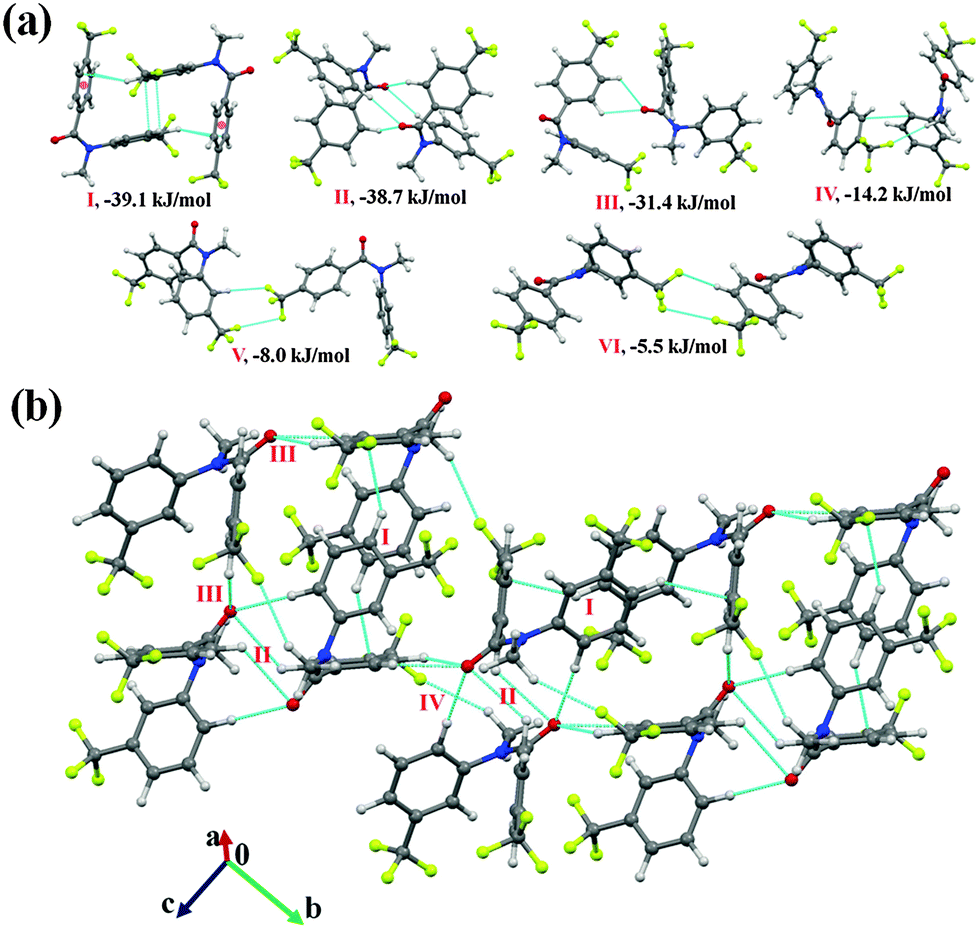 | ||
| Fig. 10 (a) Selected molecular pairs, along with their interaction energies, in NM23. (b) Packing view down the bc plane in NM23, depicting network of weak C(sp2)–H⋯OC, C(sp2)–H⋯π and C(sp3)–H⋯F–C(sp3) hydrogen bonds. | ||
N-Methyl-2-(trifluoromethyl)-N-(4-(trifluoromethyl)phenyl)benzamide (NM31)
Compound NM31 also crystallizes in the P21/c space group with Z = 4. Fig. 11(a) depicts the extracted molecular pairs from the crystal packing in NM31, along with their stabilizing energy. All the molecular motifs were observed to be stabilized by the presence of weak intermolecular interactions. The highest stabilized motif I (I.E = −30.1 kJ mol−1) was found to involve a short C(sp2)–H⋯OC (2.42 Å, 151°) hydrogen bond along with π⋯π stacking with the dispersion contribution being 66%. Motif I connects the molecule along the b-axis, utilizing a 21-screw in the formation of molecular chains in the crystal packing [Fig. 11(b)]. The chain is further stabilized via motif II (I.E = −22.1 kJ mol−1 with %Edisp being 52%), which involves a short C(sp2)–H⋯OC (2.54 Å, 149°) bond along with a short C(sp2)–H⋯F–C(sp3) (2.38 Å, 135°) bond and a bifurcated weak C(sp2)/(sp3)–H⋯F–C(sp3) hydrogen bond. Furthermore, a weak C(sp2)–H⋯OC bond with support from a bifurcated C(sp2)–H⋯F–C(sp3) hydrogen bond (motif IV, −18.2 kJ mol−1) was involved in the formation of a molecular chain with the utilization of the c-glide perpendicular to the b-axis. The chain was observed to be connected with motif I and motif III down the ac plane [Fig. 11(c)]. Motif III (I.E = −19.6 kJ mol−1, with %Edisp of 73%) consists of a dimeric weak C(sp3)–H⋯F–C(sp3) hydrogen bond along with π⋯π stacking. The packing of molecules in NM31 was also observed to involve the formation of a molecular motif V with weak C(sp3)–H⋯π interactions [the stabilization energy is −14.2 kJ mol−1]. Furthermore, weakly stabilized molecular motif VI [−9.2 kJ mol−1; involving a dimeric C(sp2)–H⋯F–C(sp3) hydrogen bonds] and motif VI [−4.9 kJ mol−1; involving C(sp3)–F⋯F–C(sp3) interactions] were also recognized in the crystal packing of NM31.
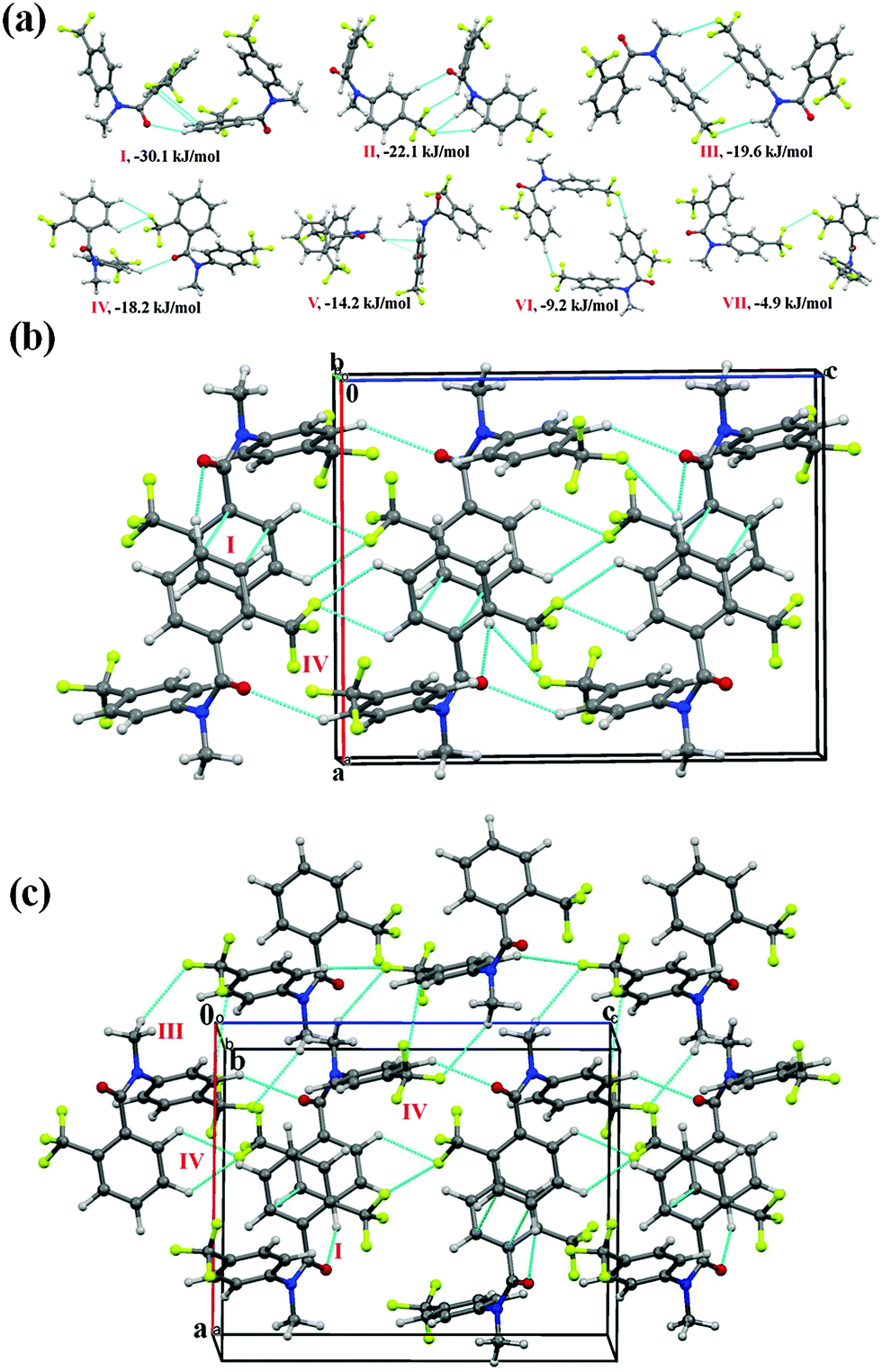 | ||
| Fig. 11 (a) Molecular pairs extracted from the crystal packing of NM31 along with their interaction energies. (b) Packing of molecules down the ab plane in NM31via weak C(sp2)–H⋯OC, C–H⋯F–C(sp3) hydrogen bonds and π⋯π interactions. (c) Packing of molecules down the ac plane with the utilization of weak C(sp2)–H⋯OC, C–H⋯F–C(sp3) hydrogen bonds and π⋯π interactions in NM31. | ||
N-Methyl-4-(trifluoromethyl)-N-(4-(trifluoromethyl)phenyl)benzamide (NM33)
Compound NM33 crystallizes in the centrosymmetric triclinic space group P with Z = 4 (Z′ = 2). Selected molecular motifs, which contribute towards the stabilization of the crystal packing, are presented in Fig. 12(a). The two molecules in the asymmetric unit were observed to be connected via motif IV (I.E = −28.2 kJ mol−1), which involved the presence of a bifurcated, short and directional C(sp2)–H⋯OC (2.30 Å, 169°; 2.42 Å, 136°) hydrogen bond with the stabilization energy having a substantial electrostatic contribution of 59% (Table 5). There are three more stabilized molecular pairs (motif I, II, III) other than motif IV, which were recognized in the crystal packing. The arrangements of the first four molecular motifs in the crystal packing of NM33 are depicted in Fig. 12(b) down the crystallographic bc plane. The highest stabilized molecular motif I (I.E = −37.8 kJ mol−1; %Eelec of 40%) consists of a short and highly directional dimeric C(sp2)–H⋯OC (2.49 Å, 173°) hydrogen bond. The stabilization of motif I is significantly higher than motif IV, although both possess similar interactions. The reason for this may be the presence of some long-range dispersion interactions in motif I, as the net contribution from the dispersion energy in motif I were observed to be almost double than that in motif IV (Table 5). In motif II (I.E = −35.3 kJ mol−1), the molecules were found to be linked via weak C–H⋯π and π⋯π interactions with the contribution from dispersion being significantly high (75%), whereas three weak C(sp2)–H⋯F–C(sp3) hydrogen bonds along with π⋯π interactions were recognized to link the molecules in motif III with the %Edisp contribution being reduced to 66%. Furthermore, motif V (I.E = −21.9 kJ mol−1) and VI (I.E = −21.9 kJ mol−1) were observed to provide similar stabilization to the crystal packing; however, the involved interactions were recognized to be significantly different. A weak C(sp3)–H⋯OC hydrogen bond along with a short C(sp3)–H⋯π (2.63 Å, 138°) and π⋯π interactions were found to stabilize motif V, whereas it was mainly the latter which linked the molecules in motif VI. The differences associated in the nature of interactions in the two motifs V and VI are clearly reflected in the dispersion energy contribution, as this is 67% in the case of the former, but 78% in the latter. A very short C(sp2)–H⋯OC (2.20 Å, 148°) hydrogen bond, involving acidic hydrogen, along with a weak C(sp3)–H⋯F bond at higher distance (2.81 Å, 125°) were observed to stabilize the crystal packing (motif VII, I.E = −15.0 kJ mol−1), with a substantial electrostatic contribution (65%). A weak C(sp3)–F⋯CO interaction and C(sp3)–H⋯F hydrogen bond (motif VIII, I.E is −14.0 kJ mol−1) were found to direct the molecular chain of molecule 2 along the crystallographic a-axis [Fig. 12(c)]. Such chains were observed to be linked with adjacent molecular chains, formed with the utilization of a weak bifurcated C(sp2)/(sp3)–H⋯F hydrogen bond along the a-axis (motif IX; I.E = −10 kJ mol−1) via the presence of the different intermolecular interactions involved in motifs II, IV, V and VII [Fig. 12(c)].
 | ||
| Fig. 12 (a) Displaying selected molecular motifs connected with different intermolecular interactions in the crystal packing of NM33. (b) Part of the crystal packing down the bc plane in NM33, depicting the presence of weak C(sp2)–H⋯OC, C–H⋯π and C(sp2)–H⋯F–C(sp3) hydrogen bonds along with π⋯π interactions. (c) Packing of molecules in NM33via the network of weak C–H⋯OC, C–H⋯π and C–H⋯F–C(sp3) hydrogen bonds along with π⋯π and C(sp3)–F⋯CO interactions. | ||
It can be noted that weakly stabilized molecular motifs possessing interactions involving organic fluorine were recognized in the crystal packing of NM33, with stabilization energies in the range from 10 kJ mol−1 to 1.2 kJ mol−1 [motif IX–XIV, Fig. 12(a)]. Motifs IX, X and XI were observed to provide similar stabilization (−10 kJ mol−1) but involve interactions of a different nature and geometry. Motif IX was found to involve bifurcated C(sp2)/(sp3)–H⋯F hydrogen bonds (with one at a short distance of 2.43 Å, 148°), whereas a dimeric C(sp2)–H⋯F bond and C(sp3)–F⋯F–C(sp3) bond were observed in motif X. Furthermore, in the case of motif XI, bifurcated C(sp3)–H⋯F hydrogen bonds (with one being short and directional; 2.48 Å, 160°) was recognized. Unlike motif IX, it involves a bifurcated acceptor, wherein two fluorine atoms of one CF3 group are involved in the formation of the hydrogen bond with a hydrogen atom of the CH3 group. Moreover, motifs XII and XIII were observed to consist of weak C(sp2)–H⋯F–C(sp3) hydrogen bonds, providing similar stabilization (8.0 and 7.2 kJ mol−1, respectively). A dimeric C(sp3)–F⋯F–C(sp3) interaction (with one contact, Type I geometry: 2.889(1) Å/101(1)°/101(1)°) was recognized in the formation of the molecular motif XIV [Fig. 11(a)], which provided the least stabilization (I.E = −1.2 kJ mol−1) to the crystal packing. The partition of the interaction energy into different contributions indicated a positive coulombic contribution, with the net stabilization originating mainly from the dispersive contribution (96%, Table 5).
Comparison of the crystal structures
From the analysis of the crystal structures of 11 compounds (ten derivatives of N-methyl-N-phenylbenzamide plus one unsubstituted compound) in this study, it was observed that seven molecules crystallized in the monoclinic space group P21/c (including NM03 and NM30 with Z′ = 2 and NM11, wherein the molecule preferred the trans geometry) and none of them appeared to be isostructural.72 This also included compounds NM10 and NM00, which crystallized in the same space group: orthorhombic centrosymmetric Pbca. Furthermore, except for NM11, all the compounds in this series appeared to have a similar molecular conformation (cis-geometry) [Fig. 1(c)]. Hence, it was of interest to compare these crystal structures to gain insights into the similarities and dissimilarities associated with the crystal packing. Therefore, XPac 2.073,74 was used to analyze the crystal packing of these structures, excluding NM11. The details of this analysis are presented in Section S2 in the ESI.† XPac identified the similar packing arrangements in the two crystal structures, termed as ‘supramolecular constructs (SC)’. It can be 3D (exactly similar arrangement or isostructural), 2D (layers of molecules are similar), 1D (a row of molecules is similar) or 0D similarity (isolated unit-like dimers are identical in the packing). The measure of the extent with which the two crystal structures deviate from the perfect geometrical similarity is defined as the ‘dissimilarity index (X)’.75 The lower the value of X, the better is the structural match. The analysis of the ten crystal structures (Table S2, ESI†) revealed that the arrangement of the molecules match (regarding the presence of 2D SCs) in the case of NM02 (packing of molecule 1) and NM03 (packing of molecule 2) with X = 6.7 (labeled as ‘C1’ Fig. 13 and Fig. S7, Table S2, ESI†). There was also the presence of 1D SCs [the presence of a molecular chain (6 types, B1 to B6), Fig. 13 and Fig. S8, Table S2, ESI†] observed in the case of the pairs NM02_2/NM03_1; NM02_2/NM10; NM03_1/NM10; NM12/NM22; NM22/NM23 and NM31/NM33_2. There were 6 different types (A1 to A6, Fig. 13 and Fig. S9, ESI†) of similar molecular dimers (the presence of a 0D SC) also recognized in the different pairs of the crystal structure (Table S2, ESI†). | ||
| Fig. 13 Relationship of all the crystal structures from the XPac analysis (Section S2, ESI†). The compounds NM02, NM03, NM30 and NM33 have two symmetry independent molecules, represented by the number in the circle. | ||
It was of further interest to compare all the present crystal structures with related crystal structures reported in CSD [Fig. 1(d)]. Comparisons with structures with a cis-geometry (CSD ref. code: YEGJEY, YEGKEA, YEGKIE, YEGKOK and YEGLAX) revealed no similarity with the unsubstituted compound, NM00 (ref. code: JAZJOJ10) [Table S2, ESI†]. There was also the presence of a similar molecular chain (1D SCs) on comparison of NM03_1, NM10, NM12 with YEGLAX [Fig. S10(a) and Table S2, ESI†], which is analogous with the chain ‘B2’ [in pair NM03_1/NM10; Fig. S8(b), ESI†]. In addition, the existence of 1D SC (similar chain) was also recognized for NM22/YEGLAX. Moreover, the pairs NM03_1/YEGKEA, NM10/YEGKEA, NM10/YEGKOK, NM12/YEGKEA and NM23/YEGKEA display the presence of a similar molecular robust dimer (equivalent with the dimer ‘A1’; 0D SCs) in their crystal packing [Fig. S11(a), ESI†]. Furthermore, the presence of 0D SCs (similar molecular pairs) was also observed for the pairs NM02_1/YEGKOK, NM02_1/YEGLAX NM22/YEGKOK, NM31/YEGLAX and NM22/YEGKOK [Fig. S11(b)–(d), ESI†]. Furthermore, the comparison of the crystal structure of NM11 with the structure reported in CSD with the trans geometry (ref. code: YEGJEY, DIBGIF and DIBGAX) indicates the presence of similar chains in the case of the pairs, NM11/DIBGAX_1 and NM11/DIBGAX_4, and surprisingly, no similarity was observed for NM11/YEGJEY (having four methyl substitution at ortho positions of both the phenyl rings in the molecule). Hence, from the overall comparison of the crystal structures, it can be observed that although none of these structures are isostructural, the presence of similar structural motifs can be realized in their crystal packing.
Conclusions
The complete quantitative analysis of the molecular and crystal structure of ten out of the fifteen newly synthesized trifluoromethyl-substituted N-methyl-N-phenylbenzamides revealed the significance of weak interactions in stabilizing the molecular and crystal structure in the absence of any strong donor atom. Unlike the N-phenylbenzamides, the derivatives of N-methyl-N-phenylbenzamide prefer to possess the cis-conformation, wherein the molecular structure is stabilized by the presence of a weak C(sp3)–H⋯OC hydrogen bond. The steric crowd at the ortho position of both the phenyl rings may change the conformation to the trans geometry, similar to as observed in N-phenylbenzamide.
The computational procedures, which involve calculation of the lattice energy and the evaluation of the interaction energies for different intermolecular interactions, provided detailed insights into the nature of the weak intermolecular interactions present in the crystal packing of this series of compounds. In the absence of a strong donor, the crystal packing was observed to be stabilized by the cooperative interplay in the presence of weak intermolecular interactions, such as C–H⋯OC and C–H⋯π hydrogen bonds, along with other weak interactions such as π⋯π stacking. There are short C–H⋯OC hydrogen bonds observed in the crystal packing of these compounds with a substantially high electrostatic (coulombic + polarization) contribution. The interactions involving organic fluorine, namely, C–H⋯F–C, C–F⋯F–C, and C–F⋯F–C, are ubiquitous and provide stabilization, albeit less, to the crystal packing, and are observed to be involved in the formation of different unique structural motifs. The detailed and comparative analysis of the nature of the different interactions involved in the different molecular motifs in the crystal packing with detailed inputs from energy calculations using the PIXEL method brings out the following observations: (i) the interaction energy in the decreasing order of weak hydrogen bonds was as follow: C–H⋯OC > C–H⋯π > C–H⋯F–C (ii) the contribution from dispersion energy towards the total stabilization follows the order: C–H⋯OC < C–H⋯F–C < C–H⋯π (the contribution from the electrostatic follows the opposite order). (iii) There is an increase in the electrostatic contribution observed at short distances, and directional hydrogen bonds are present in the molecular motif. In future studies, it would be of interest to extend this study to the investigation of interactions involving organic fluorine in different electronic and chemical environments.
Acknowledgements
PP thanks UGC-India for research scholarship. We acknowledge the IISER Bhopal for research facilities and infrastructure. The authors also thank Prof. T. N. Guru Row for SCXRD and PXRD data collection on the CCD facility at IISc, Bangalore under the IRHPA-DST Scheme. DC thanks DST-Fast Track Scheme for research funding.References
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- J. D. Dunitz and A. Gavezzotti, Chem. Soc. Rev., 2009, 38, 2622–2633 RSC.
- A. Nangia and G. R. Desiraju, in Design of Organic Solids, ed. E. Weber, Springer-Verlag, Berlin, 1998 Search PubMed.
- D. Philip and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154–1196 CrossRef PubMed.
- P. Panini, K. N. Venugopala, B. Odhav and D. Chopra, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 681–696 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaeergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS PubMed.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- (a) K. Reichenbacher, H. I. Suss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC; (b) P. Panini and D. Chopra, Hydrogen Bonded Supramolecular Structures, In Lect. Notes Chem., ed. Z. Li and L. Wu, Springer-Verlag, Berlin, Heidelberg, 2015, vol. 87, pp. 37–67 Search PubMed.
- (a) H.-J. Schneider, Chem. Sci., 2012, 3, 1381–1394 RSC; (b) R. Shukla and D. Chopra, CrystEngComm, 2015, 17, 3596–3609 RSC.
- M. Egli, Acc. Chem. Res., 2012, 45, 1237–1246 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
- H. J. Bohm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Muller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. D. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- J. D. Dunitz, ChemBioChem, 2004, 5, 614–621 CrossRef CAS PubMed.
- P. Panini and D. Chopra, CrystEngComm, 2013, 15, 3711–3733 RSC.
- (a) P. Panini and D. Chopra, CrystEngComm, 2012, 14, 1972–1989 RSC; (b) P. Panini and D. Chopra, Cryst. Growth Des., 2014, 14, 3155–3168 CrossRef CAS.
- M. Perez-Torralba, M. A. García, C. Lopez, M. C. Torralba, M. R. Torres, R. M. Claramunt and J. Elguer, Cryst. Growth Des., 2014, 14, 3499–3509 CAS.
- S. Terada, K. Katagiri, H. Masu, H. Danjo, Y. Sei, M. Kawahata, M. Tominaga, K. Yamaguchi and I. Azumaya, Cryst. Growth Des., 2012, 12, 2908–2916 CAS.
- A. Abad, C. Agulló, A. C. Cuñat, C. Vilanova, d. Ramírez and M. C. Arellano, Cryst. Growth Des., 2006, 6, 46–57 CAS.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- D. Chopra, Cryst. Growth Des., 2012, 12, 541–546 CAS.
- D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 2175–2186 RSC.
- A. R. Choudhury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47–52 CAS.
- D. Chopra and T. N. Guru Row, CrystEngComm, 2008, 10, 54–67 RSC.
- G. Kaur and A. R. Choudhury, Cryst. Growth Des., 2014, 14, 1600–1616 CAS.
- G. Kaur, P. Panini, D. Chopra and A. R. Choudhury, Cryst. Growth Des., 2012, 12, 5096–5110 CAS.
- M. Karanam and A. R. Choudhury, Cryst. Growth Des., 2012, 13, 4803 Search PubMed.
- V. Vasylyeva and K. Merz, Cryst. Growth Des., 2010, 10, 4250–4255 CAS.
- A. G. Dikundwar, R. Sathishkumar, T. N. Guru Row and G. R. Desiraju, Cryst. Growth Des., 2011, 11, 3954–3963 CAS.
- V. R. Hathwar, D. Chopra, P. Panini and T. N. Guru Row, Cryst. Growth Des., 2014, 14, 5366–5369 CAS.
- O. Jeannin and M. Fourmigue, Chem. – Eur. J., 2006, 12, 2994–3005 CrossRef CAS PubMed.
- C. Jackel, M. Salwiczek and B. Koksch, Angew. Chem., Int. Ed., 2006, 45, 4198–4203 CrossRef PubMed.
- M. Fioroni, K. Burger, A. E. Mark and D. Roccatano, J. Phys. Chem. B, 2003, 107, 4855–4861 CrossRef CAS.
- J. L. Kgokong, P. P. Smith and G. M. Matsabisa, Bioorg. Med. Chem., 2005, 13, 2935–2942 CrossRef CAS PubMed.
- J. A. K. Howard and H. A. Sparkes, CrystEngComm, 2008, 10, 502–506 RSC.
- (a) D. E. Braun, T. Gelbrich, V. Kahlenberg, G. Laus, J. Wieser and U. J. Griesser, New J. Chem., 2008, 32, 1677–1685 RSC; (b) M. O. BaniKhaled, J. D. Mottishaw and H. Sun, Cryst. Growth Des., 2015, 15, 2235–2242 CrossRef CAS.
- I. Azumaya, K. Yamaguchi, H. Kagechika, S. Saito, A. Itai and K. Shudo, J. Pharm. Soc. Japan, 1994, 114, 414–430 CAS.
- I. Azumaya, H. Kagechika, K. Yamaguchi and K. Shudo, Tetrahedron, 1995, 51, 5277–5290 CrossRef CAS.
- R. Yamasaki, A. Tanatani, I. Azumaya, H. Masu, K. Yamaguchi and H. Kagechika, Cryst. Growth Des., 2006, 6, 2007–2010 CAS.
- I. Azumaya, H. Kagechika, Y. Fujiwara, M. Itoh, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1991, 113, 2833–2838 CrossRef CAS.
- F. D. Lewis and W. Liu, J. Phys. Chem. A, 2002, 106, 1976–1984 CrossRef CAS.
- T. Hirano, T. Osaki, S. Fujii, D. Komatsu, I. Azumaya, A. Tanatani and H. Kagechika, Tetrahedron Lett., 2009, 50, 488–491 CrossRef CAS PubMed.
- (a) L. Maschio, B. Civalleri, P. Ugliengo and A. Gavezzotti, J. Phys. Chem. A, 2011, 115, 11179–11186 CrossRef CAS PubMed; (b) A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- (a) J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2005, 5, 2180–2189 CrossRef CAS; (b) P. Panini, K. N. Venugopala, B. Odhav and D. Chopra, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 681–696 CAS.
- J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2012, 12, 5873–5877 CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- TURBOMOLE V6.2 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- M. J. Frisch, et al., GAUSSIAN09, Revision A. 02, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, Chem. Phys., 2010, 132, 154104 Search PubMed.
- W. Hujo and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 13942–13950 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS PubMed.
- S. L Cockroft, J. Perkin, C. Zonta, H. Adams, S. E. Spey, C. M. R. Low, J. G. Vinter, K. R. Lawson, C. J. Urch and C. A. Hunter, Org. Biomol. Chem., 2007, 5, 1062–1080 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. Mukherjee and G. R. Desiraju, IUCrJ, 2014, 1, 49–60 CAS.
- P. Metrangolo and G. Resnati, IUCrJ, 2014, 1, 5–7 CrossRef CAS PubMed.
- E. D'Oria and J. J. Novoa, CrystEngComm, 2008, 10, 423–436 RSC.
- S. Kawahara, S. Tsuzuki and T. Uchimaru, J. Phys. Chem. A, 2004, 108, 6744–6749 CrossRef CAS.
- R. M. Osuna, V. Hernàndez, J. T. L. Navarrete, E. D'Oria and J. J. Novoa, Theor. Chem. Acc., 2011, 128, 541–553 CrossRef CAS.
- A. Kàlmàn, L. Pàrnkànyi and G. Argay, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324–336 RSC.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 448–460 RSC.
- T. Gelbrich, T. L. Threlfall and M. B. Hursthouse, CrystEngComm, 2012, 14, 5454–5464 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1025677–1025685 and 1027431. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01670c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |