
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConducting polymer nanofibers with controlled diameters synthesized in hexagonal mesophases†
Srabanti
Ghosh‡
a,
Laurence
Ramos
b,
Samy
Remita
ac,
Alexandre
Dazzi
a,
Ariane
Deniset-Besseau
a,
Patricia
Beaunier
de,
Fabrice
Goubard
f,
Pierre-Henri
Aubert
f and
Hynd
Remita
*ag
aLaboratoire de Chimie Physique, UMR 8000-CNRS, Université Paris-Sud, 91405 Orsay, France. E-mail: hynd.remita@u-psud.fr
bLaboratoire Charles Coulomb (L2C), UMR 5221 CNRS-Université de Montpellier, Montpellier, France
cDépartement CASER, Ecole SITI, Conservatoire National des Arts et Métiers, CNAM, 75141 Paris Cedex 03, France
dSorbonne Universités, UPMC Univ. Paris 06, UMR 7197-CNRS, Laboratoire de Réactivité de Surface, F-75005 Paris, France
eCNRS, UMR 7197, Laboratoire de Réactivité de Surface, F-75005 Paris, France
fLaboratoire de Physicochimie des Polymères et Interfaces (LPPI), Université de Cergy-Pontoise, 95031 Cergy-Pontoise Cedex, France
gCNRS, Laboratoire de Chimie Physique, UMR 8000, 91405 Orsay, France
First published on 11th August 2015
Abstract
Oil-swollen hexagonal mesophases resulting from the surfactant mediated self-assembly of a quaternary mixture of water, surfactant, co-surfactant, and oil, are versatile templates to synthesize anisotropic nanomaterials. Poly(diphenylbutadyine) (PDPB) polymer nanofibrous network structures were produced in the oil tubes of the mesophases by photo-induced radical polymerization using a chemical initiator or by gamma irradiation. The diameter of the nanofibers can be varied from 5 to 25 nm in a controlled fashion, and is directly determined by the diameter of the oil tube of the doped mesophases, proving thus a direct templating effect of the mesophase. The nanoIR technique allows chemical characterization and identification of the polymer nanostructures simultaneously with morphological characterization. Cyclic voltammetry has been used as an effective approach to evaluate both the energy level of the highest occupied molecular orbital (HOMO) as well as the energy of the lowest unoccupied molecular orbital (LUMO) and the band gap of the PDPB. The conductivity of the PDPB nanostructures obtained by gamma irradiation was estimated to be 10−1 S cm−1, which is higher than the conductivity of PDPB nanostructures previously reported in the literature. The soft template approach allows size tunable synthesis of anisotropic polymer structures with morphological homogeneity at the nanoscale with high conductivity, thus it appears to be an attractive opportunity for electronic device applications.
1. Introduction
Conducting polymers with anisotropic shape hold great promise as flexible, inexpensive materials for fundamental science as well as for various industrial applications.1,2 In recent years, one-dimensional conducting polymer nanostructures (e.g., nanofibers and nanotubes) have attracted considerable interest due to their superior optoelectronic properties. They indeed exhibit a high surface-to-volume ratio and enhanced current carrying ability, which lead to potential applications including light-emitting diodes, electrical nanodevices, solar cells, and chemosensors.3–5 In particular, poly(diacetylene)s are well known π-conjugated polymers with excellent optoelectronic properties.6,7The synthesis of poly(diphenylbutadiyne) (PDPB) usually occurs by closely packed pre-organization of the monomer, 1,4-diphenylbutadiyne (DPB), by photo-mediated or radiation-induced polymerization leading to extremely linear, aligned polymerized domains of 1,4-disubstituted polydiacetylenes.8,9 In general, unless conjugated polymers are molecularly and macroscopically assembled and aligned with a well-defined structure, their superior optoelectronic properties for device application cannot be fully realized. But the majority of these polymers are extremely brittle and difficult to process due to their conjugated backbone. The solubility and processability of the polymer can be further improved by using functionalized monomers which in turn significantly lower the conductivity.10 Nevertheless, there have been efforts to align conjugated polymers by using crystals, micelles, or Langmuir–Blodgett (LB) films.10,11 However, these techniques do not provide much flexibility in terms of controlling the size and morphology. Furthermore, a common and effective method has been used in order to create macroscopic alignment of monomers via supra molecular assemblies.12 Directed assembly and the alignment of conjugated polymers is a challenging task. Indeed, it is difficult to envisage the exact mode of assembly and the final structural parameters such as length and shape of the supramolecular architecture. On the other hand, conducting polymer nanostructures show high electrical conductivity, large surface area, and high electrochemical activity, as compared to its macrogranular structure or self-supporting films.13,14 Although the formation of assembled structures and films is widely explored, chemical based synthetic approaches for the PDPB nanostructure are still scarce in the literature.15–17 Shinkai et al. reported the preparation of porphyrin-based 1D assembly by linking the porphyrin units using the polymerization of butadiyne in the gel state.16 Morin et al. recently reported the synthesis of conjugated nanowires prepared by topochemical polymerization of butadiynes in the xerogel state.16,17 However, the production of freestanding ultrathin polymer nanostructures with high conductivity and easy processability remains a significant challenge and has not been well explored in the literature.15–17
Polymerization in self-assembled liquid crystal (LC) is an emerging and versatile technique for the preparation of polymers.18,19 Few examples have been reported for the preparation of conducting polymer nanostructures in LC as structure-directing polymerization templates.20–22 One of the significant problems in order to control polymer structure in LC template synthesis is thermodynamically driven phase separation, which in turn leads to polymers with poorly defined nanostructures.23,24 To circumvent this problem, temperature independent rapid initiation rates of photo-polymerization are required which may allow cross-linking or locking of a polymer network at a very rapid rate before phase separation or reorganization of the template.25 Photopolymerization in the LC phases has been used to generate polymer nanostructures with improved functionality, but retention of the original LC template throughout the polymerization process is again a challenging task.24–27
Swollen hexagonal mesophases made by a quaternary system water/oil/surfactant/cosurfactant and constituted of regular arrays of surfactant-stabilized tubes that can be swollen with a nonpolar solvent, can be used for the synthesis of one-, two- or three-dimensional nanostructures.28–34 We have shown that these swollen hexagonal mesophases doped with various precursors can be used as nanoreactors to synthesize various nanostructured materials both in the aqueous and in the oil phases.30–34 Here, we report the synthesis and characterization of nanofibrous network structures of tunable diameters of poly(diphenylbutadiyne) (PDPB), a polymer from the polydiacetylene family, in swollen hexagonal mesophases. The polymerization is induced by UV light (in the presence of an initiator for polymerization) or by gamma irradiation.
2. Experimental
2.1 Reagents
Sodium dodecyl sulfate (SDS), sodium chloride, cyclohexane (>99%) and pentanol (≥99%) were purchased from Sigma-Aldrich. For in situ polymerization, we used 1,4-diphenylbutadiyne (DPB) (Aldrich) as a monomer and benzoin methyl ether (BME) (Fluka) as a catalyst. All compounds were used as received. Ultrapure water (Millipore System, 18.2 MΩ cm) and ethanol (≥99% for HPLC, purchased from Sigma-Aldrich) were used as solvents.2.2 Sample preparation
2.3 Material characterization
Small-angle X-ray scattering (SAXS) was used to characterize the pure hexagonal mesophases and the mesophases doped with the monomer and with the catalyst before and after polymerization. The mesophases were inserted in glass capillaries of 1.5 mm diameter. A high brightness low power X-ray tube, coupled with aspheric multilayer optic (GeniX 3D from Xenocs) was employed, which delivered an ultralow divergent beam (0.5 mrad). Scatterless slits were used to give a clean 0.8 mm diameter X-ray spot with an estimated flux of around 35 mph s−1 at the sample position. A transmission configuration was used. The scattered intensity was collected on a two-dimensional Schneider 2D image plate detector prototype, at a distance of 1.9 m from the sample. The experimental data were corrected for the background scattering and the sample transmission. The scattering vector q can be calculated from the angle of the scattered radiation and the X-ray wavelength. The scattering pattern of a hexagonal phase consists of diffraction peaks whose positions are in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:2. The first peak position q0 allows a direct determination of the hexagonal lattice parameter a according to
:√3:2. The first peak position q0 allows a direct determination of the hexagonal lattice parameter a according to
Optical microscopy of gel samples before and after polymerization was performed using a Leica DMRX polarizing microscope.
For the structural study of polymer nanostructures synthesized by UV and gamma irradiation of doped mesophases, drops of the diluted PDPB ethanolic solutions after extraction from mesophases were deposited on carbon coated grids. Transmission electron microscopy (TEM) observations were performed on a JEOL JEM 100 CXII transmission electron microscope at an accelerating voltage of 100 kV.
The Attenuated Total Reflectance (ATR)-Fourier transformed infrared spectroscopy (FTIR) of solid DPB monomers and solid PDPB powders obtained after extraction from the mesophases as synthesized by UV or gamma irradiation were recorded using a Brüker Vertex 70 FTIR spectrometer with diamond ATR attachment (PIKEMIRACLE crystal plated diamond/ZnSe) and a MCT detector with a liquid nitrogen cooling system. Data allow the identification of the chemical structure of the as prepared PDPB nanostructure. Scanning wavelengths were varied from 4000–600 cm−1 with a 4 cm−1 spectral resolution from an average of 100 repetition scans for each spectrum. This allows the identification of the chemical structure of the as prepared PDPB nanostructures.
The morphology eventually combined with local spectrum of synthesized PDPB on the solid substrate was determined by combining the classical atomic force microscope (AFM) with tunable pulsed laser as an InfraRed (IR) source (AFMIR).36,37 For the present study, we have used a commercial setup, nanoIR (Anasys Instrument corp.) allowing us to cover the range from 3600 cm−1 to 1000 cm−1. Usually, samples are directly deposited on the upper surface of a ZnSe prism that is transparent in the mid-infrared and the tip of the AFM remains in contact with the object. When the sample absorbs the IR laser pulse, it warms via the photothermal effect, resulting in a rapid thermal expansion of the absorbing region of the sample. The thermal expansion pulse impacts the tip of the AFM cantilever and causes it to oscillate. As the amplitude of oscillations is proportional to the absorption, we are able to record infrared absorption spectra at a given point and to make chemical maps by scanning the surface at a given wavelength.38 In this study, drops of ethanolic solution of PDPB powder after extraction from mesophases were directly deposited on the upper surface of a ZnSe prism and dried at room temperature. For chemical mapping, in comparison to the topology study (0.5 mg L−1), a higher concentration of polymer solutions (3 mg mL−1) was used due to the limitation of resolution in the present nanoIR (100 nm) system.
Thermal stability analysis of PDPB solid powder was carried out by using a TGA DQ50 thermogravimetric analysis (TGA) apparatus (TA instruments, USA). The test was carried out under the nitrogen atmosphere at a heating rate of 20 °C min−1 from 50 °C to 600 °C.
Electrochemical characterization was carried out using a three-electrode single cell, with a glassy carbon (GC) disk (2 mm diameter) coated with the sample film as the working electrode, a Pt wire as the counter electrode, and a silver wire pseudo-reference electrode. Ferrocenium/ferrocene (Fc/Fc+) redox potential was measured at the end of each experiment in order to calibrate the pseudo reference electrode as recommended by IUPAC.39 Polymer samples (at a concentration of 1 mg mL−1 in tetrahydrofuran) were drop-casted onto the GC-electrode and then dried. The GC-electrode was immersed into the electrochemical cell containing acetonitrile with 0.1 M tetrabutylammoniumhexafluorophosphate (TBAPF6). The compounds were electrochemically reduced prior to being oxidized between −2.0 V and +2.0 V at a scan rate of 20 mV s−1.
Finally, spin-coated films were fabricated on glass slides at 1000 rpm for 60 s using the PDPB ethanolic solutions (1 mg mL−1) obtained after extraction from mesophases. In addition, PDPB nanostructures obtained after extraction from the mesophases were treated with nitrosonium tetrafluoborate (NOBF4), as the chemical oxidant, at a concentration of 10−2 mol L−1 in acetonitrile and were then spin-coated under the same conditions. The thickness (ca. 200–500 nm thickness) of all the films (doped or un-doped) was measured using a 3 Veeco Dektak 150 surface profiler. The electrical conductivity of the polymer films was measured using a Kelvin four-point probe technique implemented with a Keithley 2420 system. The conductivity, (S cm−1) was determined using the following equation:

3. Results and discussion
The mesophases doped with the monomer (DPB) and the initiator (BME) in the oil phase was exposed to UV-light. On the other hand, mesophases doped with the monomer without using an initiator were exposed to gamma rays for the polymerization of DPB in the oil phase. Before irradiation, both SDS-based mesophases (doped with the monomer and initiator or only with the monomer) were transparent, translucent and birefringent. After irradiation by UV light or by gamma rays, the mesophases turned yellow, but remained translucent and birefringent (see ESI,† Fig. S1). The closely packed and properly ordered, self-assembled diacetylene monomers undergo polymerization via 1,4-addition reaction to form alternating ene–yne polymer chains upon irradiation with UV light (in the presence of a precursor for polymerization) or with γ-irradiation (Scheme 1). The γ-irradiation or UV irradiation in the presence of BME can induce a free radical reaction in sequence which allows for a cascade reaction to occur throughout the distinct hydrophobic domains of the mesophases and aromatic diacetylenes were found to form stable diradicals which in turn produce oligomers.9–13 | ||
| Scheme 1 Schematic representation of polymerization of diphenylbutadiyne (DPB) by UV or gamma irradiation. | ||
The birefringence before and after irradiation revealed an anisotropic structure, which was confirmed by polarized light microscopy (Fig. 1 and see ESI,† Fig. S2). The cross section of swollen hexagonal mesophases containing the PDPB monomer is schematically shown in the inset of Fig. 1a. Polarized light micrograph patterns (Fig. S1a and b, ESI†) showed characteristic birefringence of the hexagonal liquid crystal before and after doping with the DPB monomer and the BME initiator. This indicates that the controlled addition of the monomer and the initiator in the organic confined phase did not disrupt the liquid crystalline phase. The SAXS spectra of the mesophases before and after both UV and gamma irradiation exhibited the characteristic features of a direct hexagonal phase with three Bragg peaks whose positions were in the ratio 1:√3:2:√7 (Fig. 1c).
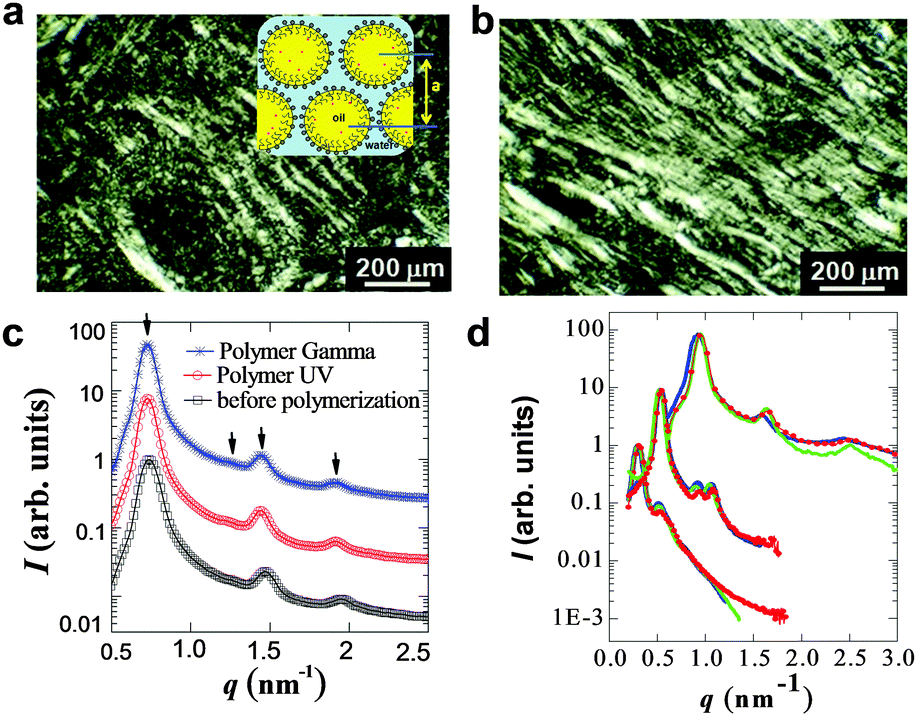 | ||
| Fig. 1 Representative polarized light micrographs of hexagonal mesophases (a) before and (b) after photopolymerization (ϕ = 2.21, Cs = 0.3 mol L−1). Inset of a: Cross section of swollen hexagonal mesophases, a is the lattice parameter of hexagonal mesophases. (c) SAXS spectra of swollen hexagonal phases before polymerization (□) and after polymerization by UV irradiation (○), gamma irradiation (*) at 0.1 M NaCl (ϕ = 0.98). The arrows specify the indexing of the Bragg peaks. (d) Representative SAXS spectra of swollen hexagonal phases with various swelling ratios and salt concentrations from top to bottom (top, ϕ = 0.22, Cs = 0 mol L−1 with less oil, middle, ϕ = 0.72 with relatively more oil, Cs = 0 mol L−1, bottom, ϕ = 2.21, Cs = 0.3 mol L−1). Before polymerization (blue line), after polymerization by UV light irradiation (red line), and gamma irradiation (green line). | ||
SAXS patterns observed before and after polymerization were found to be superimposed. Data measured for hexagonal mesophases with various swelling ratios (ϕ, from 0.22 to 2.21) and various salt concentrations (Cs, from 0 to 0.3 mol L−1), before and after polymerization both using UV and gamma-irradiation, are shown in Fig. 1d. As ϕ increased, Bragg peaks shifted to a lower wave-vector (q), showing an increase of the characteristic size of the hexagonal arrangement from 7.5 nm to 18.4 nm, as measured previously.19 For all ϕ, the SAXS profiles of the doped hexagonal mesophases following UV or γ radiation superimpose to the profiles before polymerization. Hence our results showed that the UV and γ irradiations used for in situ polymerization did not disturb the long-range order of the soft hexagonal matrix over the whole range of composition investigated for the mesophases.
Similar observations were obtained while varying the experimental conditions (various swelling ratio from 0.22 to 2.21 and salt concentration ranging from 0 to 0.3 mol L−1) indicating that a relatively large degree of liquid crystalline order was preserved when doping the oil phase of the mesophases with DPB and BME. Hence, the structure of the hexagonal mesophases remained unchanged after photo and gamma irradiation over the whole range of composition.
The monomer and the synthesized nanostructures after extraction from the mesophases were characterized by ATR-FTIR (Fig. 2).
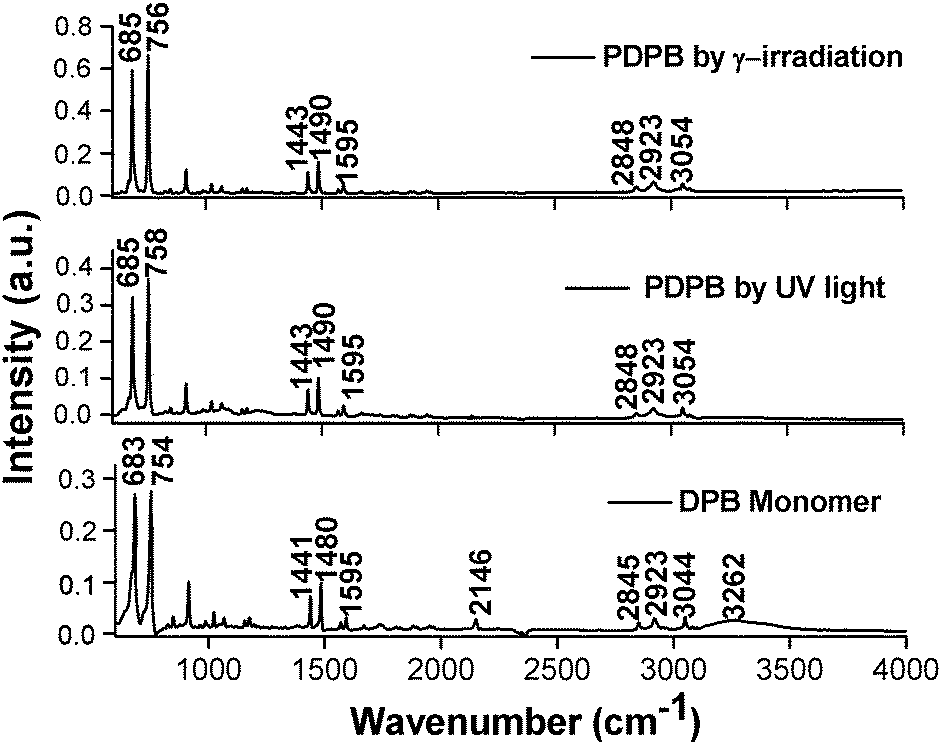 | ||
| Fig. 2 ATR-FTIR spectra of pure DPB monomer (bottom spectrum), and PDPB nanostructures synthesized either by UV light irradiation (middle spectrum), or by gamma irradiation (top spectrum) with ϕ = 2.21, Cs = 0.3 mol L−1. | ||
The IR spectrum of the DPB monomer shows characteristic peaks at 2845 and 2923 cm−1 due to the symmetrical and asymmetrical stretching modes of the C–H bond respectively.40 The typical bands observed at 683 cm−1 and 754 cm−1 are associated with the benzene ring out-of-plane deformations. Generally, the peaks in the 1400 to 1600 cm−1 region have been attributed to the formation of the π conjugated enyne unit is overlapped with the spectrum region associated with aromatic ring stretching and bending.
Before irradiation, the IR spectrum of the monomer shows triple band stretching (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) of the monomer at 2146 cm−1 as shown in Fig. 2 (bottom spectrum). Upon UV irradiation, the monomer band at 2146 cm−1 corresponding to the antisymmetric acetylene IR active band of DPB disappeared suggesting a change in the nature of the CC units. The absence of the characteristic vibration peak indicates the formation of PDPB polymer, as demonstrated in the literature.41,42 Photo-irradiation induced spectral changes of the DPB monomer were monitored in the whole region of 4000–600 cm−1. However, except the triple band stretching, the other bands of the monomer have been preserved after polymerization. The peaks at 1490 and 1443 cm−1 are due to in plane C–H bending and the band at 1595 cm−1 has been assigned to the stretching vibration of the benzene ring in PDPB polymer synthesized by photo irradiation. The large intensity change of C–H stretching modes has been associated with frequencies in the range of 3100–3000 cm−1. The band at 3054 cm−1 corresponds to the C–H vibration involving hydrogen atoms at the para and meta positions. Similar IR spectra were obtained after gamma irradiation (Fig. 2 top). This suggests a similar chemical structure formed by both photo and gamma irradiation, and these two spectra are in good agreement with the spectrum of PDPB obtained in the literature.41,42
C) of the monomer at 2146 cm−1 as shown in Fig. 2 (bottom spectrum). Upon UV irradiation, the monomer band at 2146 cm−1 corresponding to the antisymmetric acetylene IR active band of DPB disappeared suggesting a change in the nature of the CC units. The absence of the characteristic vibration peak indicates the formation of PDPB polymer, as demonstrated in the literature.41,42 Photo-irradiation induced spectral changes of the DPB monomer were monitored in the whole region of 4000–600 cm−1. However, except the triple band stretching, the other bands of the monomer have been preserved after polymerization. The peaks at 1490 and 1443 cm−1 are due to in plane C–H bending and the band at 1595 cm−1 has been assigned to the stretching vibration of the benzene ring in PDPB polymer synthesized by photo irradiation. The large intensity change of C–H stretching modes has been associated with frequencies in the range of 3100–3000 cm−1. The band at 3054 cm−1 corresponds to the C–H vibration involving hydrogen atoms at the para and meta positions. Similar IR spectra were obtained after gamma irradiation (Fig. 2 top). This suggests a similar chemical structure formed by both photo and gamma irradiation, and these two spectra are in good agreement with the spectrum of PDPB obtained in the literature.41,42
The polymer nanostructures were also characterized by the AFMIR technique using the nanoIR instrument. The nanoIR can quickly survey the polymer regions via AFM topography imaging and then rapidly acquire high-resolution local chemical spectra at selected regions on the sample.43 The surface topography of the photo-induced PDPB nanostructures showed a dendritic arrangement formed during deposition on the ZnSe substrates as shown in Fig. 3a. The dendritic nanostructures correspond to the self assembly of polymer nanostructures which interact by π-stacking. The combination of a nanoscale probe from an atomic force microscope with a tunable IR source provides simultaneous measurements of nanoscale morphology along with the chemical composition mapping (Fig. 3(b–d)).36,37
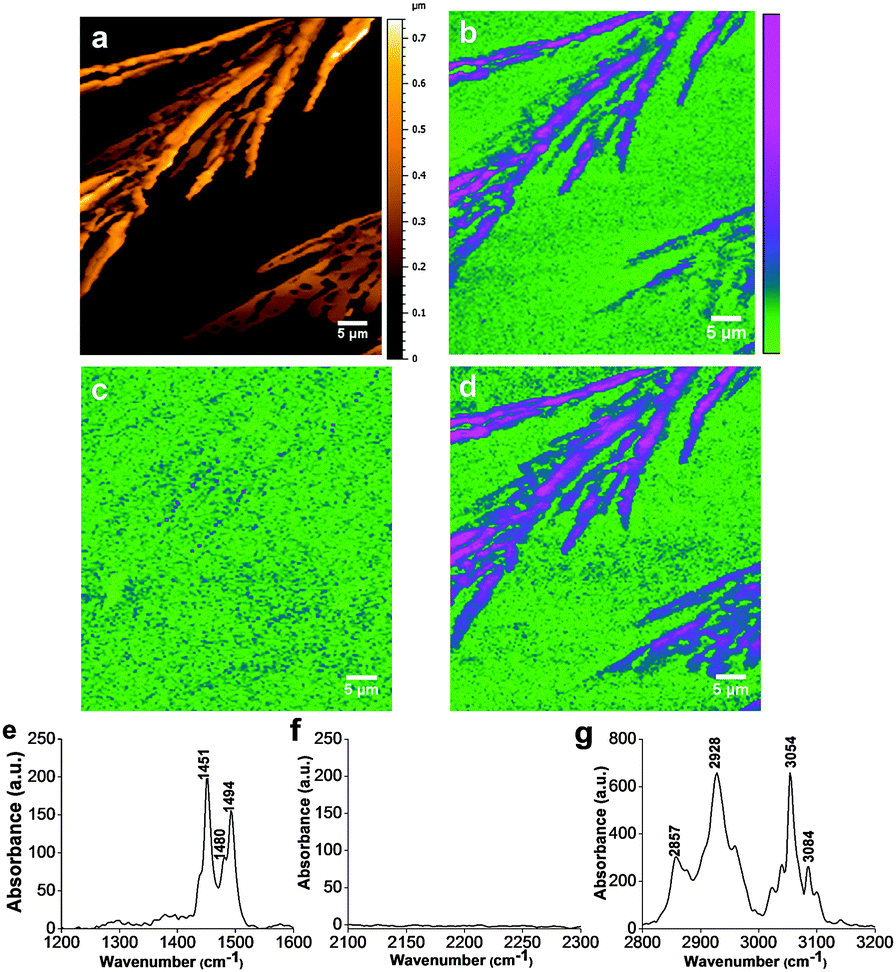 | ||
| Fig. 3 (a) Topographic image of photosynthesized PDPB by conventional AFM. AFM-IR mappings of the photo-induced PDPB polymer nanostructures synthesized in a swollen hexagonal phase with ϕ = 2.21, Cs = 0.3 mol L−1 measured at different fixed wavenumbers: 1490 cm−1 (b), 2146 cm−1 (c) and 3054 cm−1 (d). (e–g) nanoIR spectra recorded at three different spectral regions of the PDPB polymer. | ||
Fig. 3(e) shows the nanoIR spectrum of PDPB in the 1200–1600 cm−1 region, which is quite similar to the FTIR spectrum of PDPB, generally associated with the formation of the enyne unit. The other peaks around 1451, 1480 and 1494 cm−1 corresponds to in plane C–H bending. The band at 1451 cm−1 is larger in relative intensity in the nanoIR spectrum compared to that observed in the FTIR spectrum of PDPB (Fig. 2). Additionally, the spectral region at 2300–2000 cm−1 of the triple bond stretching was thoroughly analyzed in order to identify the presence of the monomer DPB band at 2146 cm−1 as shown in Fig. 3(f). The disappearance of the monomer band at 2146 cm−1 after UV irradiation demonstrates that the polymerization is complete. Fig. 3(g) shows the IR spectra measured at a fixed point in each domain in the wavenumber range of 2800–3200 cm−1. The benzene ring C–H stretching leads to characteristic peaks at 2857, 2928 and 3023, 3054, and 3084 cm−1.44 On the other hand, the intense absorption bands at 2857, 1451, and 1494 cm−1 are due to deformational vibrations of C–H in the polymer spectra. The spectral changes in the 3100–3000 cm−1 region are noticeable in nanoIR spectra (100 nm) due to the presence of significant steric hindrances between neighboring phenyl groups within the large oligomers as shown in Fig. 3(g). As nanoIR is accomplished with high spatial resolution compared to classical FTIR measurement, we can identify even lower intensity bands present in the polymer structure by photo-irradiation. Furthermore, the strength of the benzene ring stretching measured by nanoIR around 2928 and 3054, and 3084 cm−1 is much more intense than the band measured by FTIR. In contrast to classical FTIR measurement with average data of several polymers, nanoIR provides the local spectra of the polymer molecule. Hence, each nanoIR spectrum is compared with the corresponding FTIR spectrum and importantly the major absorption band occurs at substantially the same positions for both techniques. In order to have a comparative study for the photosynthesized PDPB, gamma radiation induced PDPB have also been characterized by the AFMIR technique (see ESI,† Fig. S4). The IR absorption spectrum of gamma induced-synthesized PDPB (see ESI,† Fig. S4) was similar to that of the photosynthesized polymer (Fig. 3). It displays intense absorption bands at 1440, 1451 and 1480, 1494 cm−1 in addition to the band at 3054 cm−1 which corresponds to the C–H vibration. Note that once again the monomer band at 2146 cm−1 is absent indicating the formation of PDPB by gamma radiolysis. To conclude, the nanoIR system provides local absorption spectra of the nanoscale region of polymer and demonstrates good correlation with the FTIR measurements.
Contrary to our observation with the polymer structure synthesized upon UV-irradiation, the topographic image of PDPB polymer nanostructures synthesized by gamma irradiation shows well-dispersed fibers on ZnSe substrate (see ESI,† Fig. S3). Interestingly, we identified a strong signal at 1490 cm−1 and 3054 cm−1 characteristic of the polymer structure with a lack of signal at 2146 cm−1 indicating the absence of the DPB monomer. This observation also supports effective polymerization of the DPB monomer within the mesophase by gamma irradiation.
The polymer nanostructures synthesized by both UV and gamma irradiation were further characterized by transmission electron microscopy (TEM). In order to see the effect of the confinement of the polymer nanostructures within oil-swollen tubes during polymerization, the size of the oil-swollen tubes was controlled by varying the amount of oil (cyclohexane) and the salt concentration whose compositions are given in Table S1 of the ESI.† Irradiations carried out in doped mesophases with different swelling ratios and hence different oil tube diameters have been investigated for the polymers produced by UV and gamma irardiation. In all cases, connected polymer nanofibers are observed. Interestingly, we found that the average nanofiber diameter varies as the oil-swollen tubes of the hexagonal phases varied. Fig. 4 shows TEM images of polymers synthesized upon UV-irradiation.
 | ||
| Fig. 4 Transmission electron micrographs of PDPB nanostructures prepared by UV-irradiation in mesophases with (a and b) ϕ =2.21 and Cs = 0.3 M NaCl, (c and d) ϕ =0.98 and Cs = 0.1 M NaCl, and (e and f) ϕ =0.72 and Cs = 0 M NaCl. | ||
PDPB nanofibers with uniform diameters of ∼19 nm and a few micrometers long are obtained in mesophases with ϕ = 2.21 and comprising oil tubes of 18.4 nm (Table 1, Fig. 4a and b). Nanofibers of average diameter 12 nm are synthesized in mesophases with ϕ = 0.98 and comprising oil tubes of 7.4 nm (Table 1, Fig. 4c and d). Even thinner nanofibers (average diameter 5 nm) are obtained for mesophases with ϕ = 0.72 and comprising oil tubes of 5.2 nm (Table 1, Fig. 4e and f). Interestingly, we therefore find that the average diameter of the polymer nanofibers increases with the diameter of the oil cylinder of the soft template from 5 to 19 nm (Fig. 4). Thus, the diameter of the as prepared PDPB nanostructures can be varied by ∼4-fold by tuning the diameter of the confining oil tubes, which can be simply swelled by simultaneous variation of the water/oil ratio (ϕ) and the salt concentration in the aqueous phase.
| C s (mol L−1) | ϕ | a (nm) | D (nm) | Diameter of PDPB fibers (nm) | Conductivity (S cm−1) | ||
|---|---|---|---|---|---|---|---|
| UV | Gamma | UV | Gamma | ||||
| 0 | 0.72 | 6.68 | 5.2 | 5 ± 1 | 10 ± 0.5 | 0.035 ± 0.003 | 0.130 ± 0.008 |
| 0.1 | 0.98 | 11.8 | 11.1 | 12 ± 1 | 14 ± 2 | 0.020 ± 0.006 | 0.100 ± 0.009 |
| 0.3 | 2.21 | 20.9 | 21.6 | 19 ± 2 | 25 ± 3 | 0.012 ± 0.005 | 0.092 ± 0.005 |
Similar fiber morphology was obtained with polymers prepared by gamma-irradiation. In the case of gamma induced polymerization, the diameter of the polymer nanofibrous network structures varied in the range of 12–25 nm as shown in Fig. 5. The diameter of the fibers increases as the diameter of the oil tubes increases, and the diameter of fibers are slightly larger with gamma irradiation compared to UV irradiation. The anisotropic shape of the templated polymer nanostructure presumably reflects the geometry of the hydrophobic domains of the hexagonal mesophases.
 | ||
| Fig. 5 Transmission electron micrographs of the PDPB nanostructure prepared by gamma irradiation in mesophases with (a) ϕ = 2.21 and Cs = 0.3 M NaCl, (b) ϕ = 0.98 and Cs = 0.1 M NaCl and (c) ϕ = 0.72 and Cs = 0 M NaCl. | ||
For both UV and gamma polymerization, the diameter of the nanofibers depends on the swelling ratio of the template hexagonal phase, which sets the diameter of the oil tubes. Remarkably, both set of experimental data matched well with the evolution of the oil tube diameters of the hexagonal phase (Fig. 6 and Table 1), suggesting a direct templating effect of the mesophases on the formation of the polymer nanofibers in the confined oil phase.
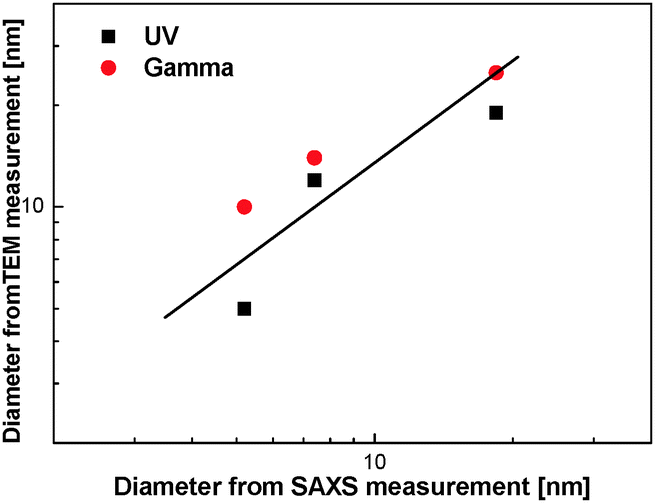 | ||
| Fig. 6 Composition of the diameter of the oil tubes of hexagonal mesophases and the diameter of PDPB nanostructures by both UV and gamma-irradiation with various swelling ratios (ϕ) and structural parameters, as determined from SAXS and TEM measurements. The continuous line has a slope of 1. | ||
In addition, control experiments showed that only micron-sized spherical particles were obtained when polymer was synthesized by UV irradiation directly from surfactant-free DPB and BME in cyclohexane. However, no such fiber structure has been formed without using mesophases (see ESI,† Fig. S5), indicating that the hydrophobic region of the mesophases accommodates DPB monomer within the nonpolar solvent filled tubes to align them in a one-dimensional packing mode suitable for polymerization.15 Since the morphologies of poly(diacetylene)s are strikingly dependent on the superstructure of the monomer assemblies before UV-irradiation, pre-organization of the corresponding monomers into well-designed fibrous architectures is a prerequisite to obtain poly(diacetylene) nanofibers.45,46
Thermogravimetric analysis (TGA) can provide information relating to the thermal stability of the as-prepared polymer nanostructures. The TGA graph of PDPB polymer powder after extraction from the mesophases showed an onset of decomposition at about 200 °C until a major decomposition happens around 540 °C (see ESI,† Fig. S6). However, it has been observed that at about 260 °C, the PDPB weight decreased significantly by ∼41%. Furthermore, at higher temperatures, PDPB displayed a decomposition pattern up to 590 °C with a weight loss of about 95 wt%. Hence, the thermal stability of PDPB polymer nanofibrous structures synthesized in hexagonal mesophases is consistent with the other conductive polymers as described in the literature.16,47
Wide-angle X-ray scattering (WAXD) measurement has been conducted for the detection of crystallinity of the PDPB nanofibers (as shown in Fig. S7, ESI†). The XRD pattern of the PDPB nanofibers shows sharp features, revealing highly crystalline nature and a well-organized material which is well consistent with the literature report (see Fig. S7 in ESI†).16
The molar mass of polymer has been found to be 1625 g mol−1 as determined by gel permeation chromatography (GPC) and corresponds to oligomers of degree of polymerization 8. The polymers are relatively short chains presumably because they are induced by polymerization in the oil confined domains. The PDPB nanofibers are probably formed by π-stacking of these oligomers in the oil tubes of the mesophases. The polymerization process happened within a confined geometry of the liquid crystal which may restrict the growth of polymerization of poly(diphenylbutadyine) which is consistent with the literature reports.15–17
The oxidation and reduction potentials of PDPB have been determined under the similar experimental conditions to estimate both the energy level of the highest occupied molecular orbital (HOMO) and the energy of the lowest unoccupied molecular orbital (LUMO) from the ionization potential and the electronic affinity, respectively and the band gap using cyclic voltammetry (CV) measurement. For PDPB, we found that the main p-doping (oxidation) and n-doping (reduction) were irreversible processes, and the values of the peak potentials vs. Ag pseudo-reference were: +1.91 V (oxidation) and −1.71 V (reduction) yielding an energy gap of ∼3.62 eV as a first approximation as shown in Fig. 7a. Furthermore, to explain the visible light absorption of PDPB, having a careful analysis of the CV of polymer structures (zoom at lower current, Fig. 7b) reveals onsets of oxidation (+1.35 V) which means a HOMO level at −5.52 eV, and reduction (−0.45 V) processes occurring at lower potentials leading to a HOMO LUMO level calculated at −3.72 eV. From the CV curve, the bandgap is estimated to be 1.80 eV, in agreement with the bandgap estimated from the onset of optical absorption (1.77 eV) which is remarkably narrow for a polymer. Additionally, these results are consistent with the calculated value of the PDPB band gap, which is 1.95 eV on the basis of DFT (considering oligomeric PDPB structures comprising various numbers of units from 1 to 8).48 The low bandgap implies the possibility of efficient injection of carriers, useful for potential application in optoelectronics or as support in electrocatalysis.48–50
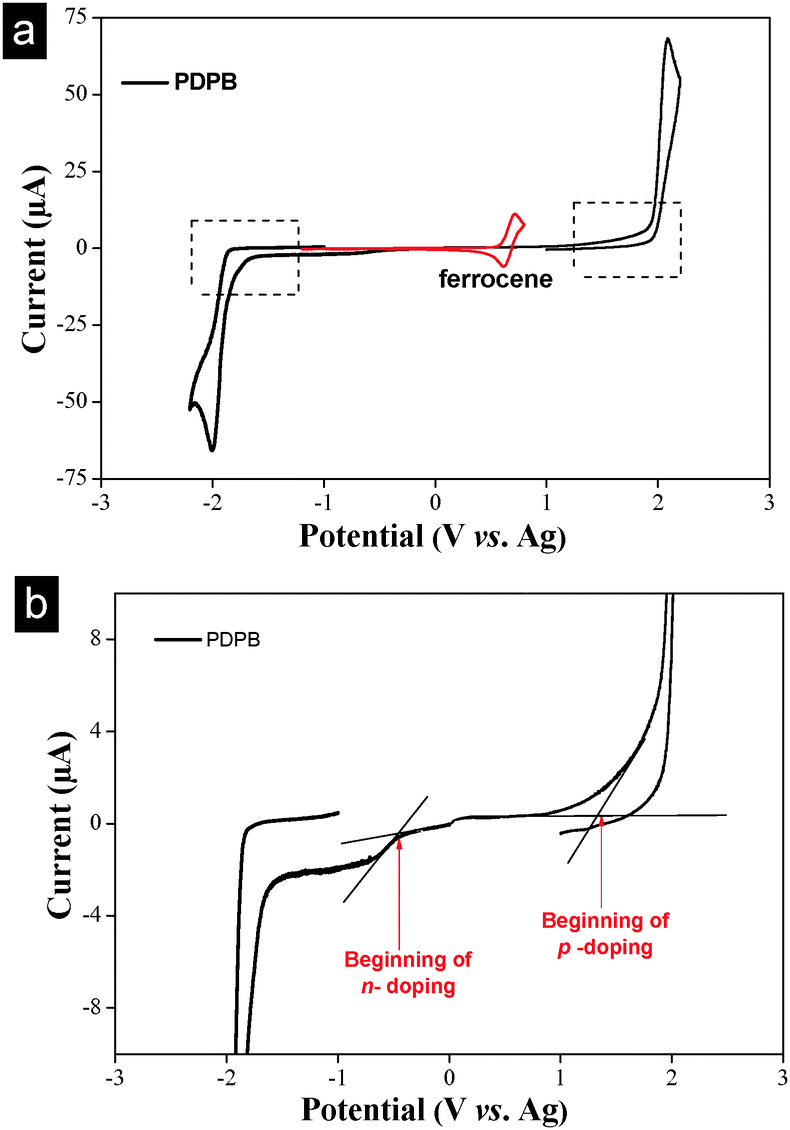 | ||
| Fig. 7 Cyclic voltammograms of the PDPB nanostructure prepared by UV-irradiation in mesophases with ϕ = 2.21 and Cs = 0.3 M NaCl. (a) Cyclic voltammogram obtained at 20 mV s−1 in acetonitrile and 0.1 M tetrabutylammonium perchlorate. Ferrocenium/ferrocene (Fc/Fc+) redox potential has been measured at the end of each experiment in order to calibrate the pseudo reference electrode (0.63 V vs. Ag in the present study). (b) Zoom of the voltammogram as shown in (a). The energy levels of the PDPB nanostructure are determined as follows: EHOMO (eV) from ionization potential = −4.8e (Eox_onset − 0.63) and ELUMO (eV) from electronic affinity = −4.8e (Ered_onset − 0.63). | ||
Importantly, polydiacetylene (PDA) has a quasi one dimensional π-conjugated backbone which posses interesting electrical and optical properties.51 Conducting polymers are able to conduct electricity through their conjugated backbone when properly doped, through either chemical or electrochemical processes.52
Oxidative doping of a conjugated polymer makes it conductive because of the extraction of electrons from the polymer chain which creates free carriers. After extraction from the mesophases, the electrical conductivity of NOBF4-doped thin films of PDPB (thickness of ca. 200–500 nm) was evaluated by using a four-point probe device. Table 1 shows the conductivities of doped PDPB nanofibers obtained by UV and gamma induced polymerization techniques. The electrical conductivity of the photo-induced PDPB nanostructures at room temperature is about 10−2 S cm−1. This value is comparable with conductivity values already reported in the literature for PDPB.52–54 In contrast, the electrical conductivity of the PDPB nanostructures obtained by gamma irradiation is 0.13 S cm−1, which is one order of magnitude higher than that of the UV-induced polymer nanostructure. It has to be noted that the conductivity increases slightly with the decrease of the diameter of the nanofibers (induced by UV- or gamma irradiation). Polyacetylene has been considered as an insulator without doping.52 However, the conductivity of bulk polyacetylene was found to be 10−11 S cm−1 by NOBF4 doping, which is consistent with that previously reported in the literature for doped polyacetylene.52 The conductivity of polydiacetylene thin films has been evaluated in the region below 20 μm using a newly constructed independently driven double-tip scanning tunneling microscope, and was found to be (3–5) × 10−6 S cm−1.53 Baba et al., reported highest conductivity ca. (1.3 ± 0.8) × 10−2 S cm−1 of poly(diacetylene) nanocrystals after chemical doping.54 In contrast, the as prepared PDPB nanostructures have higher conductivity in the range of 10−1 S cm−1, which can be used as semiconducting thin layers for organic electronic devices. A comparative conductivity data of PDPB has been tabulated in S1 (ESI†). Additionally, these one-dimensional PDPB nanostructures were readily dispersed in alcohol and form transparent continuous films by simple drop casting on a substrate and subsequent drying, which may be used for electronic device applications.
4. Conclusions
Hexagonal mesophases composed of oil-swollen surfactant stabilized tubes arranged on a triangular lattice in water and doped with monomers were used as soft templates for the synthesis of polymer nanostructures upon UV and gamma irradiations. Micrometers long nanofibers of conducting poly(diphenylbutadiyne) (PDPB) were synthesized in the oil tubes of the hexagonal mesophases. Catalyst-free γ-ray initiated polymerization reactions in mesophases are particularly interesting in order to have a pure final product without using additional photo initiators. The diameter of the nanofibers can be varied from 5 to 25 nm in a controlled fashion, and is found to be directly determined by the diameter of the oil tube of the doped mesophases, thus proving a direct templating effect of the mesophase. The polymer nanofibers can be easily extracted from the mesophases by a simple washing process, and this synthetic approach is fast, simple, and reproducible. We demonstrated that the nano-IR technique can improve chemical characterization and identification of the polymer nanostructures when compared with traditional FTIR spectra. In addition, we found that the polymer nanofibers produced by radiolysis exhibit relatively high conductivity and may find applications in electronic devices or solar light harvesting.Acknowledgements
S. G. acknowledges Marie Curie Cofund, RBUCE-UP (Research Based University Chairs of Excellence of Paris) and PRES UniverSud Paris for a postdoctoral fellowship. The authors gratefully acknowledge the C'Nano Ile de France and the Université Paris-Sud (ERM project) for financial support for the Cobalt-60 panoramic gamma source.Notes and references
- A. N. Aleshin, Adv. Mater., 2006, 18, 17–27 CrossRef CAS PubMed.
- N. R. Chiou, C. M. Lui, J. J. Guan, L. J. Lee and A. J. Epstein, Nat. Nanotechnol., 2007, 2, 354–357 CrossRef CAS PubMed.
- M. Granström, M. Berggren and O. Inganäs, Science, 1995, 267, 1479–1481 CrossRef PubMed.
- H. Sirringhaus, T. Kawase, R. H. Friend, T. Shimoda, M. Inbasekaran, W. Wu and E. P. Woo, Science, 2000, 290, 2123–2126 CrossRef CAS.
- E. Pardieu, H. Cheap, C. Vedrine, M. Lazerges, Y. Lattach, F. Garnier, C. Pernelle and S. Remita, Anal. Chim. Acta, 2009, 649, 236–245 CrossRef CAS PubMed.
- D. H. Charych, J. O. Nagy, W. Spevak and M. D. Bednarski, Science, 1993, 261, 585–588 CAS.
- R. Jelinek and M. Ritenberga, RSC Adv., 2013, 3, 21192–21201 RSC.
- R. W. Carpick, D. Y. Sasaki, M. S. Marcus, M. A. Eriksson and A. R. Burns, J. Phys.: Condens. Matter, 2004, 16, R679–R697 CrossRef CAS.
- M. F. Beristain, T. Ogawa, G. Gomez-Sosa, E. Muñoz, Y. Maekawa, F. Halim, F. Smith, A. Walser and R. Dorsinville, Mol. Cryst. Liq. Cryst., 2010, 521, 237–245 CrossRef CAS PubMed.
- A. Sakamoto, K. Mori, K. Imura and H. Okamoto, J. Phys. Chem. C, 2011, 115, 6190–6194 CAS.
- N. Mackiewicz, E. Gravel, A. Garofalakis, J. Ogier, J. John, D. M. Dupont, K. Gombert, B. Tavitian, E. Doris and F. Ducongé, Small, 2011, 7, 2786–2792 CrossRef CAS PubMed.
- S. Rondeau-Gagné, J. R. Neabo, M. Desroches, J. Larouche, J. Brisson and J.-F. Morin, J. Am. Chem. Soc., 2013, 135, 110–113 CrossRef PubMed.
- K. Ramanathan, M. A. Bangar, M. H. Yun, W. Chen, A. Mulchandani and N. V. Myung, Nano Lett., 2004, 4, 1237–1239 CrossRef CAS.
- H. D. Tran, D. Li and R. B. Kaner, Adv. Mater., 2009, 21, 1487–1499 CrossRef CAS PubMed.
- N. R. Chiou, C. M. Lui, J. J. Guan, L. J. Lee and A. J. Epstein, Nat. Nanotechnol., 2007, 2, 354–357 CrossRef CAS PubMed.
- M. Shirakawa, N. Fujita and S. A. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164–4165 CrossRef CAS PubMed.
- J. R. Néabo, S. Rondeau-Gagné, C. Vigier-Carrière and J. F. Morin, Langmuir, 2013, 29, 3446–3452 CrossRef PubMed.
- J. R. Néabo, K. I. S. Tohoundjona and J. F. Morin, Org. Lett., 2011, 13, 1358–1361 CrossRef PubMed.
- C. Tschierske, Chem. Soc. Rev., 2007, 36, 1930–1970 RSC.
- B. S. Forney, C. Baguenard and C. A. Guymon, Chem. Mater., 2013, 25, 2950–2960 CrossRef CAS.
- E. Pena dos Santos, M. S. Tokumoto, G. Surendran, H. Remita, C. Bourgaux, P. Dieudonné, P. Prouzet and L. Ramos, Langmuir, 2005, 21, 4362–4369 CrossRef.
- J. F. Hulvat and S. I. Stupp, Angew. Chem., Int. Ed., 2003, 42, 778–781 CrossRef CAS PubMed.
- J. F. Hulvat and S. I. Stupp, Adv. Mater., 2004, 16, 589–592 CrossRef CAS PubMed.
- B.-G. Kim, E. J. Jeong, J. W. Chung, S. Seo and B. K. J. Kim, Nat. Mater., 2013, 12, 659–664 CrossRef CAS PubMed.
- M. Antonietti, R. Basten and S. Lohmann, Macromol. Chem. Phys., 1995, 196, 441–466 CrossRef CAS PubMed.
- M. Antonietti, R. A. Caruso, C. G. Göltner and M. C. Weissenberger, Macromolecules, 1999, 32, 1383–1389 CrossRef CAS.
- B. S. Forney and C. A. Guymon, Macromolecules, 2010, 43, 8502–8510 CrossRef CAS.
- C. L. Lester, S. M. Smith, W. L. Jarrett and C. A. Guymon, Langmuir, 2003, 19, 9466–9472 CrossRef CAS.
- M. A. DePierro, K. G. Carpenter and C. A. Guymon, Chem. Mater., 2006, 18, 5609–5617 CrossRef CAS.
- O. Stamatoiu, J. Mirzaei, X. Feng and T. Hegmann, Top. Curr. Chem., 2012, 318, 331–393 CrossRef CAS.
- S. Dutt, P. F. Siril, V. Sharma and S. Periasamy, New J. Chem., 2015, 39, 902–908 RSC.
- A. Lehoux, L. Ramos, P. Beaunier, D. B. Uribe, P. Dieudonné, F. Audonnet, A. Etcheberry, M. José-Yacaman and H. Remita, Adv. Funct. Mater., 2012, 22, 4900–4908 CrossRef CAS PubMed.
- G. Surendran, G. Apostolescu, M. Tokumoto, E. Prouzet, L. Ramos, P. Beaunier, P. J. Kooyman, A. Etcheberry and H. Remita, Small, 2005, 1, 964–967 CrossRef CAS PubMed.
- P. F. Siril, L. Ramos, P. Beaunier, P. Archirel, A. Etcheberry and H. Remita, Chem. Mater., 2009, 21, 5170–5175 CrossRef CAS.
- G. Surendran, M. S. Tokumoto, E. Pena dos Santos, H. Remita, L. Ramos, P. J. Kooyman, C. V. Santilli, C. Bourgaux, P. Dieudonné and E. Prouzet, Chem. Mater., 2005, 17, 1505–1514 CrossRef CAS.
- A. Dazzi, R. Prazeres, F. Glotin and J. M. Ortega, Opt. Lett., 2005, 30, 2388–2390 CrossRef CAS.
- A. Dazzi, C. B. Prater, Q. Hu, D. B. Chase, J. F. Rabolt and C. Marcott, Appl. Spectrosc., 2012, 66, 1365–1384 CrossRef CAS PubMed.
- A. Dazzi, F. Glotin and R. Carminati, Appl. Phys., 2010, 107, 124519 Search PubMed.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 CrossRef.
- J.-M. Kim, J.-S. Lee, H. Choi, D. Sohn and D. J. Ahn, Macromolecules, 2005, 38, 9366–9376 CrossRef CAS.
- B. Zimmermann and G. Baranović, Vib. Spectrosc., 2006, 41, 126–135 CrossRef CAS PubMed.
- T. Hasegawa, S. Haraguchi, M. Numata, T. Fujisawa, C. Li, K. Kaneko, K. Sakurai and S. Shinkai, Chem. Lett., 2005, 34, 40–41 CrossRef CAS.
- J. Felts, H. Cho, M.-F. Yu, L. Bergman, A. Vakakis and W. King, Rev. Sci. Instrum., 2013, 84, 023709 CrossRef PubMed.
- H. Cho, J. R. Felts, M.-F. Yu, L. A. Bergman, A. F. Vakakis and W. P. King, Nanotechnology, 2013, 24, 444007 CrossRef PubMed.
- A. Matsumoto, in Control of stereochemistry of polymers in free radical polymerization, ed. K. Matyjaszewski and T. Davis, Handbook of Radical Polymerization, John Wiley, Hoboken, 2002 Search PubMed.
- D. J. Ahn, S. Lee and J.-M. Kim, Adv. Funct. Mater., 2009, 19, 1483–1496 CrossRef CAS PubMed.
- S. Ghosh, H. Remita, L. Ramos, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and S. Remita, New J. Chem., 2014, 38, 1106–1115 RSC.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Conducting Polymer Nanostructures as Promising Photocatalysts under Visible-Light, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- S. Ghosh, A.-L. Teillout, D. Floresyona, P. d. Oliveira, A. Hagège and H. Remita, Int. J. Hydrogen Energy, 2015, 40, 4951–4959 CrossRef CAS PubMed.
- A. Sarkar, S. Okada, H. Matsuzawa, H. Matsuda and H. Nakanishi, J. Mater. Chem., 2000, 10, 819–828 RSC.
- H. Nakanishi, H. Matsuda and M. Kato, Mol. Cryst. Liq. Cryst., 1984, 105, 77–88 CrossRef CAS PubMed.
- D. Day and J. B. Lando, Macromolecules, 1980, 13, 1478–1483 CrossRef CAS.
- K. Takami, J. Mizuno, M. Akai-kasaya, A. Saito, M. Aono and Y. Kuwahara, J. Phys. Chem. B, 2004, 108, 16353–16356 CrossRef CAS.
- K. Baba, H. Kasai, Y. Shinohara, S. Okada, H. Oikawa, H. Matsuda and H. Nakanishi, Jpn. J. Appl. Phys., 2008, 47, 3769–3780 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information, polarized light micrographs, AFMIR spectra, AFM and SEM images. See DOI: 10.1039/c5nj00826c |
| ‡ Present address: Department of Chemical, Biological and Macromolecular Sciences, S. N. Bose National Centre for Basic Sciences, Block JD, Sector III, Salt Lake, Kolkata 700 098, India. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |