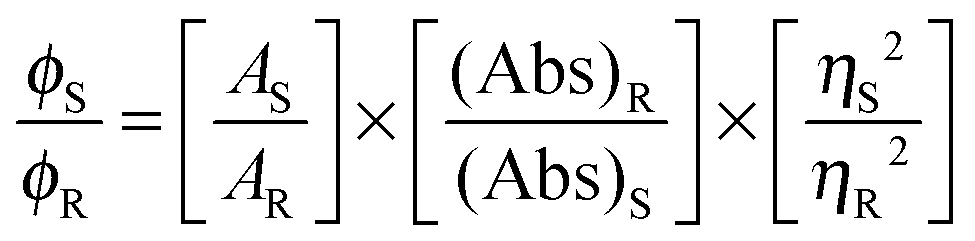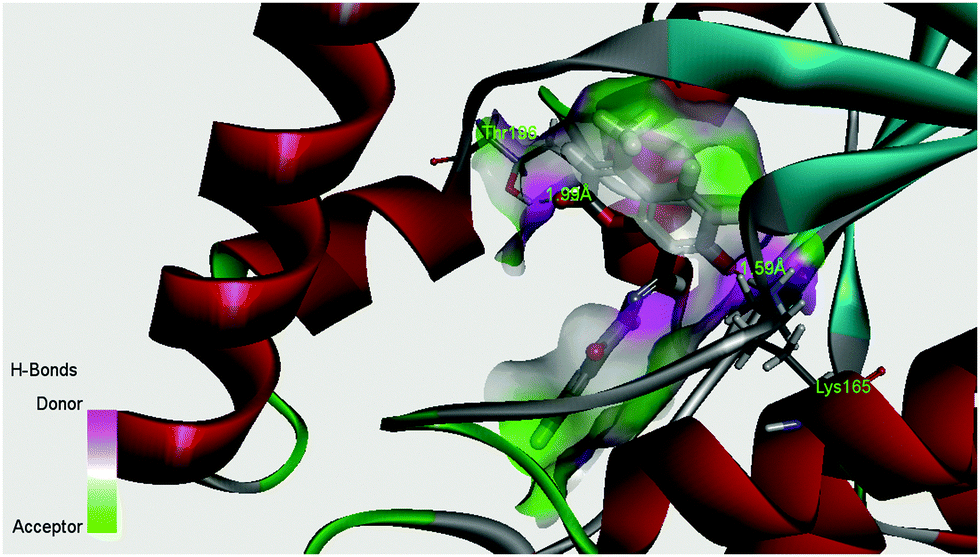DOI:
10.1039/C5NJ00789E
(Paper)
New J. Chem., 2015,
39, 7309-7321
An enolato-bridged dinuclear Cu(II) complex with a coumarin-assisted precursor: a spectral, magnetic and biological study†
Received
(in Victoria, Australia)
30th March 2015
, Accepted 16th July 2015
First published on 16th July 2015
Abstract
A new, phenoxo-bridged CuII dinuclear complex Cu2[(L)2(DMF)2] (1) has been obtained by employing the coumarin-assisted tridentate precursor, H2L, [benzoic acid(7-hydroxy-4-methyl-2-oxo-2H-chromen-8-ylmethylene)-hydrazide]. Complex 1 has been systematically characterized by FTIR, UV-Vis, fluorescence and EPR spectrometry. The single crystal X-ray diffraction analysis of 1 shows that the geometry around each copper ion is square pyramidal, comprising two enolato oxygen atoms belonging to different ligands (which assemble the dimer bridging the two metal centers), one imine-N and one phenolic-O atoms of the Schiff base and one oxygen atom from the DMF molecule. The temperature dependent magnetic interpretation agrees with the existence of weak ferromagnetic interactions between the bridging dinuclear Cu(II) ions. Both the ligand and complex 1 exhibit anti-mycobacterial activity and considerable efficacy towards M. tuberculosis H37Rv ATCC 27294 and M. tuberculosis H37Ra ATCC 25177 strains. The cytotoxicity study on human adenocarcinoma cell lines (MCF7) suggests that the ligand and complex 1 have potential anticancer properties. Molecular docking of H2L with the enoyl acyl carrier protein reductase of M. tuberculosis H37Rv (PDB ID: 4U0K) is examined and the best docked pose of H2L shows one hydrogen bond with Thr196 (1.99 Å).
Introduction
Recent research trends reveal that there has been considerable interest among researchers in studying the chemistry of coumarin functionalised compounds due to their broad spectrum of activities ranging from photophysical or photochemical properties1 to biological properties.2 Many products that contain a coumarin subunit exhibit diverse biological activities such as molluscicidal3a and insecticidal activities,3b cytotoxicity,3c and anti-HIV activity;3d some serve as antioxidant3c and anticoagulant agents.4 Basically, the coumarin nucleus is one of the most interesting chromophores with high fluorescence efficiency,5 which opens up a new way for recognition and determination of metal cations or anions.6 There are several coumarin appended fluorescent sensors reported in the literature which are potentially used to detect cations e.g. Ca2+,6a Zn2+,6b and Cu2+6c as well as anions e.g. fluorides,6d cyanides, etc. It is well established that the biological activity associated with hydrazone compounds is attributed to the presence of the active pharmacophore (–CONH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–),7 and many hydrazone compounds containing this active moiety showed good anticancer, antituberculosis and antibacterial activities.8 Additionally, the azomethine (–CONH–NC–) group in hydrazone derivatives possesses strong binding ability due to the formation of stable chelates with metal ions. So, coumarin containing hydrazone derivatives are not only expected to have enhanced biological activities but also show high sensitivity towards metal ions. Indeed, chelation studies involving metal ions coordinated to coumarin derivatives at different positions have been reported in the literature.9 8-Substituted-4-methyl-7-hydroxycoumarin and 6-substituted-4-methyl-7-hydroxycoumarin have been investigated due to their complexation ability and exhibited anticoagulant and plant growth regulating properties.10 Some of us previously reported one Cu(II) derivative mediated by N-[(2-pyridyl)methyliden]-6-coumarin.11
C–),7 and many hydrazone compounds containing this active moiety showed good anticancer, antituberculosis and antibacterial activities.8 Additionally, the azomethine (–CONH–NC–) group in hydrazone derivatives possesses strong binding ability due to the formation of stable chelates with metal ions. So, coumarin containing hydrazone derivatives are not only expected to have enhanced biological activities but also show high sensitivity towards metal ions. Indeed, chelation studies involving metal ions coordinated to coumarin derivatives at different positions have been reported in the literature.9 8-Substituted-4-methyl-7-hydroxycoumarin and 6-substituted-4-methyl-7-hydroxycoumarin have been investigated due to their complexation ability and exhibited anticoagulant and plant growth regulating properties.10 Some of us previously reported one Cu(II) derivative mediated by N-[(2-pyridyl)methyliden]-6-coumarin.11
Dinuclear transition metal complexes have gained significant interest over the years due to their diverse properties and reactivity, originating from the magnetic exchange occurring between neighbouring metal centres and potential applications in the field of magnetism12 and biochemistry.13 So far, several copper ions coordinated to oxygen and/or nitrogen atoms as structural and functional models in magneto-chemistry and metalloenzymes have been reported in the literature.14 Copper is the third most abundant transition metal found in living systems,15 and it plays an essential role in biological systems. The nature (antiferromagnetic or ferromagnetic) and magnitude of a magnetic exchange interaction between two metal ions are reasonably well understood in terms of the energetics and overlap of 'magnetic’ orbitals.16 In the last few years, the success in understanding magnetic exchange interactions in metal complexes has prompted efforts to see if these complexes can be used as molecular building blocks for materials exhibiting interesting properties. The magnetic spin exchange in dinuclear copper(II) complexes in relation to the participation of donor atoms of the corresponding ligand is of continuing interest.17 The design of ligands capable of binding two metal ions in close proximity is, therefore, an important subject in this field. In order to gain an insight into the correlation between the structure and magnetism of dinuclear copper(II) complexes, detailed investigations based on crystal structure analyses are needed. Tuberculosis, which is caused primarily by Mycobacterium tuberculosis and rarely by M. bovis and M. africanum, is one of the oldest disease in the world affecting lungs and spreading to the other organs.18 At present, a million children per year have died from this disease.19 Mycobacteria resist to many chemicals, disinfectant antibiotics and chemotherapeutical agents.20 Mycobacteria show a strong resistance to most of the antibiotics and chemicals due to the low permeability of the mycobacterial cell wall. Porins of the thickness and low permeability of this bilayer membrane allow inefficient permeability of the substances.20b The prospect of developing new and effective drugs is central to the global control of tuberculosis.
In the present contribution, the ligand [benzoic acid(7-hydroxy-4-methyl-2-oxo-2H-chromen-8-ylmethylene)-hydrazide] (H2L), a tridentate ONO donor,21 was used in order to synthesize the Cu(II) dinuclear complex Cu2[(L)2(DMF)2] (1). Besides studying the systematic spectral and magnetic properties, we have focused our attention on the anti-mycobacterial activity of human tuberculosis cells like M. tuberculosis H37Rv ATCC 27294 and M. tuberculosis H37Ra ATCC 25177 strains. We also have examined the cytotoxicity study on human adenocarcinoma cell lines (MCF7). The anti-microbacterial efficiency of H2L is examined by molecular docking with the enoyl acyl carrier protein reductase of M. tuberculosis H37Rv (PDB ID: 4U0K).
Experimental
Materials
All chemicals and solvents were of reagent grade and used as received. Cu(NO3)2·3H2O was purchased from Merck, India. Benzohydrazide and 7-hydroxy-4-methyl-coumarin were purchased from Aldrich Chemicals, India.
Physical measurements
Microanalytical data (C, H, and N) were collected on a Perkin-Elmer 2400 CHNS/O elemental analyzer. FTIR spectra were recorded on a Perkin-Elmer RX-1 spectrophotometer in the range 4000–400 cm−1 as KBr pellets. Electronic spectra were measured on a Lambda 25 (UV-vis–NIR) spectrophotometer. Emission spectra were recorded using an LS 55 Perkin-Elmer spectrofluorometer at room temperature (298 K) in different solutions under degassed conditions. The fluorescence quantum yield of the complexes was determined using carbazole as a reference with a known ϕR of 0.42 in benzene.22 The complex and the reference dye were excited at the same wavelength, maintaining a nearly equal absorbance (∼0.1), and the emission spectra were recorded. The area of the emission spectrum was integrated using the software available in the instrument and the quantum yield was calculated according to the following equation:
here, ϕS and ϕR are the fluorescence quantum yield of the sample and the reference, respectively. AS and AR are the area under the fluorescence spectra of the sample and the reference, respectively, (Abs)S and (Abs)R are the respective optical densities of the sample and the reference solution at the wavelength of excitation, and ηS and ηR are the values of the refractive index of the respective solvent used for the sample and the reference. Fluorescence lifetimes were measured using a time-resolved spectrofluorometer from IBH, UK. The instrument uses a picosecond diode laser (NanoLed-03, 370 nm) as the excitation source and works on the principle of time-correlated single photon counting.23 The goodness of fit was evaluated by the χ2 criterion and the visual inspection of the residuals of the fitted function to the data. EPR spectra were recorded from 0 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Gauss in the temperature range 77–298 K using an X-band (9.4 GHz) Bruker EMX spectrometer. EPR parameters reported in the text were obtained by simulating the spectra using the computer program Bruker WinEPR SimFonia.24 Electronic absorption spectra were recorded using a Perkin-Elmer Lambda 35 spectrophotometer. Magnetic properties were investigated using a Quantum Design MPMS-XL superconducting quantum interference device magnetometer (SQUID) at an applied field 0.5 T in a temperature range 5–300 K. The diamagnetic correction was carried out by using Pascal constants.
000 Gauss in the temperature range 77–298 K using an X-band (9.4 GHz) Bruker EMX spectrometer. EPR parameters reported in the text were obtained by simulating the spectra using the computer program Bruker WinEPR SimFonia.24 Electronic absorption spectra were recorded using a Perkin-Elmer Lambda 35 spectrophotometer. Magnetic properties were investigated using a Quantum Design MPMS-XL superconducting quantum interference device magnetometer (SQUID) at an applied field 0.5 T in a temperature range 5–300 K. The diamagnetic correction was carried out by using Pascal constants.
Synthesis of the ligand and the complex
Synthesis of the ligand (H2L).
8-Formyl-7-hydroxy-4-methyl-coumarin was prepared from 7-hydroxy-4-methyl-coumarin using the reported procedure.25 Ligand H2L was synthesized by refluxing 8-formyl-7-hydroxy-4-methyl-coumarin (0.408 g, 2 mmol) and benzohydrazide (0.272 g, 2 mmol) for a period of 2 h in methanol to obtain the yellow colored compound. It was filtered, washed with excess of methanol and recrystallized from dioxane (see Scheme ESI†). Yield: 0.231 g (72%). Anal. calc. for C18H14N2O4: C, 66.72; H, 4.91; N, 8.45. Found: C, 67.07; H, 4.38; N, 8.69%. 1H NMR (300 MHz, CDCl3): δ 2.4 (s, 3H, C18-CH3), 6.25 (s, 1H, C3-H), 6.96–6.99 (d, 1H, C6-H, J = 9 Hz), 7.53–7.63 (m, 3H, C-14, C-15, C-16), 7.71–7.74 (d, 1H, C5-H, J = 9 Hz), 7.98–7.96 (d, 2H, C-13,C-17, J = 6.9 Hz), 8.70 (s, 1H, HC = N), 12.44 (s, 1H, NH), 12.87 (s, 1H, OH).
Synthesis of Cu2[(L)2(DMF)2] (1).
To a methanol solution (10 mL) of Cu(NO3)2·3H2O (0.482 g, 2 mmol), the ligand H2L (0.644 g, 2 mmol) in 15 mL of DMF was added upon constant stirring. The resulting green solution was kept undisturbed at room temperature. Dark-green square-shaped single crystals of 1 were generated after one week. These were separated by filtration and air-dried before X-ray diffraction analysis. Yield: 0.69 g. Anal. calc. for C42H38Cu2N6O10: C, 55.20; H, 4.20; N, 9.19. Found: C, 55.373; H, 3.87; N, 9.58%.
X-ray crystallography
The crystal structure of 1 was determined by X-ray diffraction methods. Crystal data and experimental details of data collection and structure refinement are reported in Table 1. Intensity data and cell parameters were recorded at 293(2) K on a Bruker Breeze (MoKα radiation λ = 0.71069 Å) equipped with a CCD area detector and a graphite monochromator. The ω:2θ scan technique was applied within a θ range of 1.91−30.63°. No significant crystal decay was observed. A total of 31462 reflections were collected of which 4850 were independent [R(int) = 0.0358] reflections. The raw frame data were processed using SAINT and SADABS to yield the reflection data file.26 The structure was solved by Direct Methods using the SIR97 program27 and refined on Fo2 by full-matrix least-squares procedures, using the SHELXL-97 program28 in the WinGX suite v.1.80.05.29 All non-hydrogen atoms were refined by anisotropic atomic displacements. The hydrogen atoms were included in the refinement at idealized geometry (C–H 0.95 Å) and refined “riding” on the corresponding parent atoms. The weighting scheme used in the last cycle of refinement was w = 1/ [σ2Fo2 + (0.0583P)2 + 0.4073P], where P = (Fo2 + 2Fc2)/3. CCDC-938644 for 1.
Table 1 Crystallographic data of 1
| Empirical formula |
C42H38Cu2N6O10 |
|
Goodness-of-fit S = [∑w(Fo2 − Fc2)2/(n − p)]1/2, where n is the number of reflections and p is the number of parameters.
R
1 = ∑∥Fo∣ − ∣Fc∥/∑∣Fo∣; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2.
|
| Formula weight |
913.86 |
| Temperature |
293(2) |
| Wavelength (Å) |
0.71069 |
| Crystal system |
Monoclinic |
| Space group |
P21/n |
|
a (Å) |
8.2580(10) |
|
b (Å) |
11.221(2) |
|
c (Å) |
21.508(3) |
|
β (deg) |
97.190(2) |
| Volume (Å3) |
1977.3(5) |
|
Z
|
2 |
|
D
calc (mg m−3) |
1.535 |
|
μ (MoKα) (mm−1) |
1.144 |
|
F(000) |
940 |
| Crystal size, mm−3 |
0.15 × 0.12 × 0.09 |
|
θ range for data collection |
1.91–30.63 |
| Reflections collected/unique |
31462/6071 [R(int) = 0.0358] |
| Observed reflections [Fo > 4σ(Fo)] |
4850 |
| Data/restraints/parameters |
6071/0/314 |
| Final R indices [Fo > 4σ(Fo)]b |
R
1 = 0.0358, wR2 = 0.0963 |
|
R indices (all data) |
R
1 = 0.0483, wR2 = 0.1040 |
| Goodness-of-fit on F2a |
1.046 |
| Largest diff. peak and hole, e Å−3 |
0.616 and −0.251 |
Theoretical calculation
Full geometry optimization of 1 was carried out using density functional theory (DFT) at the B3LYP level.30 All calculations were performed using the Gaussian 03 program package31 with the aid of the Gauss View visualization program.32 For C, H, N, O, and Cl the 6-31G(d) basis set was assigned, while for Cu and Ni the LanL2DZ basis set with effective core potential was employed.33 The vibrational frequency calculations were performed to ensure that the optimized geometries represent the local minima and there are only positive eigen values. Vertical electronic excitations based on B3LYP optimized geometries were computed by the time-dependent density functional theory (TD-DFT) formalism34–36 in acetonitrile using the conductor-like polarizable continuum model (CPCM).37 The Gauss sum was used to calculate the fractional contributions of various groups to each molecular orbital.38
Anti-mycobacterial activity
Medium.
In the assays, Mycobacteria Growth Indicator Tubes (MGITs) and their supplements, BBL MGIT OADC enrichment and BBL MGIT PANTA were purchased from BD. The MGIT (Mycobacteria Growth Indicator Tube) contains 4 mL of modified Middlebrook 7H9 Broth base.
Inoculum preparation.
For the cultivation of mycobacteria, the MGIT (Mycobacteria Growth Indicator Tube), a fluorescent compound, is embedded in silicone at the bottom, and then 4 mL of modified Middle brook 7H9 Broth base are added to the mixture. After that 0.5 mL of OADC (oleic acid, albumin, dextrose and catalase) enrichment, and the PANTA antibiotic mixture used to prevent the growth of any non-mycobacteria (0.1 mL) are added to this medium. Oleic acid plays an important role in the metabolism of mycobacteria; albumin acts as a protective agent; dextrose is an energy source; catalase destroys toxic peroxides. Tubes are incubated at 37 °C. For positive control, MGIT tubes are prepared by inoculating bacteria. An un-inoculated MGIT tube is used as a negative control. Blood agar is used for checking the growth of other bacteria. Daily tube reading starts on the second day of incubation using a Micro MGIT fluorescence reader which has long wave UV light.39 To prepare inoculums from a positive BACTEC MGIT tube, the positive tubes (day 1 or day 2 positive) are used directly as inoculums. The positive tubes between day 3 and day 5 are diluted to 1:4 ratio using sterile saline. Inoculums, prepared from a Day 1 to Day 5 MGIT 7 mL positive tube, range between 0.8 × 105 and 3.2 × 105 CFU mL−1. Each assay is performed according to the MGIT manual fluorometric susceptibility test procedure recommended by the manufacturer.39,40
Antimycobacterial susceptibility assay.
The activity of the ligand, H2L, and complex 1 against M. tuberculosis strains was tested using the Microplate Presto Blue Assay (MPBA) by the method described by Collins & Franzblau41 and modified by Jimenez-Arellanes et al.42 128 μL of compound 1 and 72 μL of 7H9 broth were transferred into the first column, and then 100 μL of 7H9 broth was transferred from column 2 to column 8. Column 9 and 10 were the positive and the negative control, respectively. 100 μL of a mixture of broth and the compound were transferred from column 1 to column 2 and then mixed using pipettes three times; the procedure was repeated to provide serial 1:2 dilutions. 100 μL of the excess medium was discarded from the wells in column 8. Afterwards, 20 μL of M. tuberculosis inoculum was added to the wells of column 1 to 8 and to the positive control columns. Negative control columns were not inoculated with bacteria. Positive control columns included 7H9 broth and bacteria, while negative control columns contained 7H9 broth and compound 1. Final-test concentration ranges were 423.68–6.62 μmol L−1 in the mixture. Microplates were inoculated with the bacterial suspension (20 μL per well) except for the negative control and incubated at 37 °C for 6 days. Presto blue (15 μL, Life Technologies) was then added to the bacterial growth control wells (without compound 1) and the microplates were incubated at 37 °C for an additional 24 h. If the dye turned from blue to pink (indicating positive bacterial growth), the Presto blue solution was added to the other wells to determine the MIC values. All tests were performed in triplicate. The minimal inhibitory concentration (MIC) was defined as the lowest concentration of the sample that prevents a color change to pink. The minimal bactericidal concentration (MBC) corresponded to the minimum compound concentration which does not cause a color change in the cultures when re-incubated in fresh medium.43 Streptomycin (STR) from Sigma and isoniazid (INH) from Fluka were used as standard drugs.
Cytotoxicity assay
Cell lines and culture conditions.
MCF7, a human breast adenocarcinoma cell line, was obtained from the American Type Culture Collection (ATCC, Bethesda, MD, USA). Cells were grown in DMEM (Invitrogen, USA) containing 10% heat inactivated fetal bovine serum (FBS) (GIBCO BRL, USA), at 37 °C in a humidified atmosphere containing 5% CO2.
Cell viability test by MTT assay.
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay was performed to determine the effect of compound 1 and the ligand (0.4, 0.8, 1.6, 3.12, 6.25, 12.50, 25, 50, and 100 μM) on the proliferation of MCF7 cells by their utilization and conversion to formazan according to the method of Mosmann.44 MCF7 cells were plated into 96-well microtiter plates at a density of 2.5 × 104 cells per well. After 24 h, the culture medium was replaced by 200 μL (0–100 μM) of compound, and the cells were incubated for 24 and 48 h. DMSO treated cells were used as controls. After treatment, the media were replaced with serum free DMEM media containing 1 mg mL−1 of MTT and incubated at 37 °C in a 5% CO2 incubator for additional 5 h. After 5 h, the unreacted dye was removed, and the insoluble formazan crystals were dissolved in 100 μL per well DMSO and measured spectrophotometrically in a VersaMax tunable microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 570 nm. The relative cell viability (%) related to control wells containing cell culture medium without samples was calculated by A570nm[sample]/A570nm[control] × 100.
Docking studies
Molecular docking is used to predict how a protein interacts with small molecules. The crystal structure of enoyl acyl carrier protein reductase of M. tuberculosis H37Rv was downloaded from the RCSB protein data bank (http://www.pdb.org) and used for docking. The protein (PDB id: 4U0K) was co-crystallized with (3S)-N-(5-chloro-2-methylphenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide and nicotinamide-adenine-dinucleotide. In silico docking studies were performed using the CDOCKER module of the Receptor–Ligand interaction protocol section of Discovery Studio client 3.5.45 Initially, there was a pretreatment process for the protein and the ligand. The structure of the ligand was drawn using Chemdraw 5.0, saved in .mol file and finally the .mol file was imported to the Discovery studio 4.0 platform. Ligand preparation was done using the Prepare Ligand module in the Receptor–Ligand interaction tool of Discovery studio 4.0 and the prepared ligand was used for docking. Protein preparation was done under the Prepare Protein module of the Receptor–Ligand interaction tool of Discovery Studio 4.0 and that was used for docking. Then we have defined the protein as the total receptor and the active site was selected based on the ligand binding domain of (3S)-N-(5-chloro-2-methylphenyl)-1-cyclohexyl-5-oxopyrrolidine-3-carboxamide and nicotinamide-adenine-dinucleotide. Then the pre-existing ligand was removed and the prepared ligand was placed. The most favorable docked pose was selected according to the minimum free energy of the protein–ligand complex and analyzed to investigate the interaction.
ADMET prediction
Absorption, distribution, metabolism, excretion and toxicity (ADMET) predictions were done in the ADMET descriptor module of the Small molecule protocol of Discovery studio client 4.0. The druglikeness of the compounds was also checked following Lipinski’s rule of five.46,47
Results and discussion
Synthesis and formulation
The ligand, [benzoic acid(7-hydroxy-4-methyl-2-oxo-2H-chromen-8-yl-methylene)-hydrazide] (H2L), was prepared by the condensation of 8-formyl-7-hydroxy-4-methyl-coumarin with benzohydrazide (1:1 mole proportion) in methanol. H2L was then reacted with Cu(NO3)2·3H2O in methanol to yield the complex Cu2[(L)2(DMF)2] (1), which was crystallized by slow evaporation of the solvent. The IR spectra of 1 show a ν(OC–O) band at 1720 cm−1 that represents the stretching frequency of the lactone (the corresponding peak appears at 1707 cm−1 in H2L). The band at 1618 cm−1 is assigned to coordinated ν(CN),48 whereas in the free ligand, the band appears at a lower wavenumber (1613 cm−1). A high intensity band in the region 1303–1269 cm−1 is assigned to the phenolic ν(C–O) vibration, whereas the shifting of this band at 1370–1400 cm−1 supports the coordination of the phenolic oxygen to the metal ion via deprotonation. The ν(M–N) and ν(M–O) stretching frequencies are observed at 712 and 462 cm−1.
Crystal structure
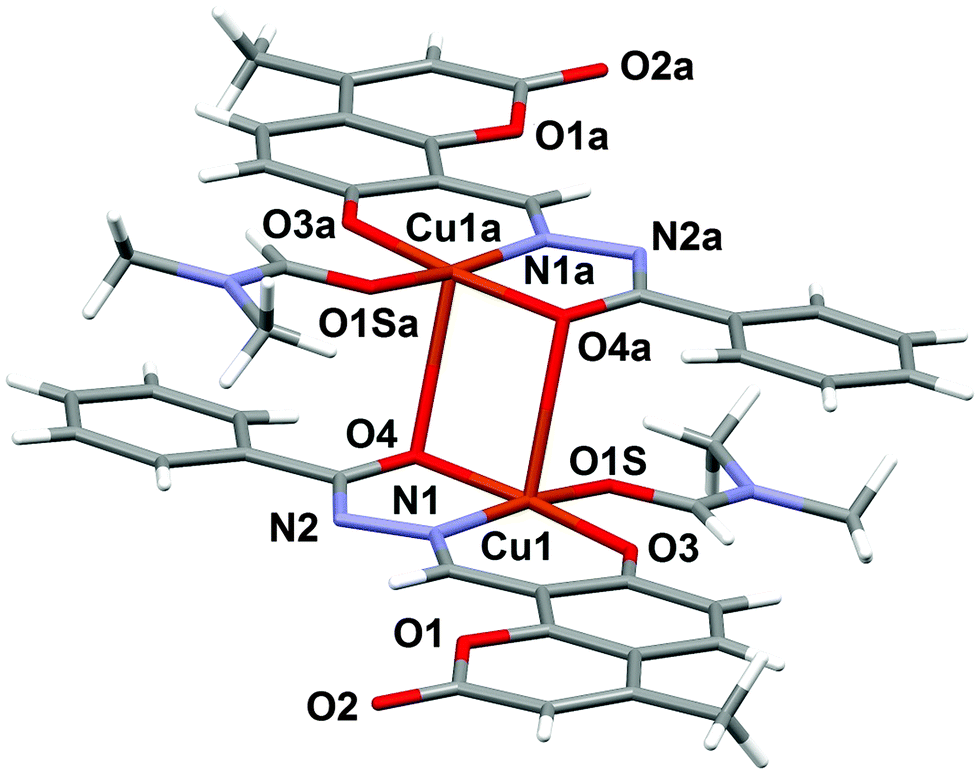
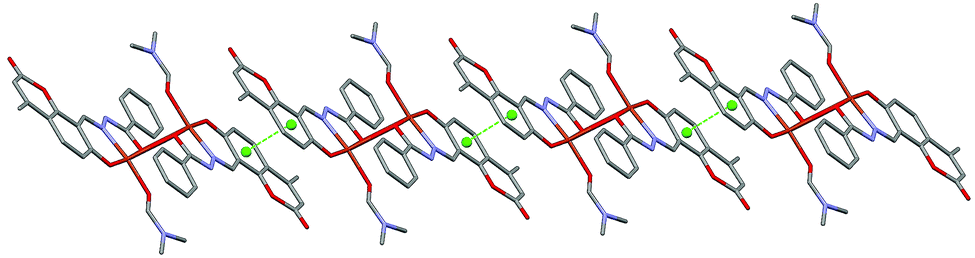
The molecular structure of the dinuclear copper compound is shown in Fig. 1. Relevant bond lengths and angles are given in Table 2. The geometric centre of the molecule lies on a crystallographic inversion centre. The geometry of the NO4 environment around each copper ion is square pyramidal, as evidenced by the τ value49 of 0.08. The dimer is assembled via the μ2-enolato oxygen atoms O4 and its centrosymmetric analogue O4a, belonging to two distinct deprotonated ligand L2−. Each of the oxygen atoms bridge two adjacent copper ions, thus occupying both the apical position of the pyramid around one metal center, and the equatorial position of its centrosymmetric counterpart. The coordination around copper is completed by the imine-N(1) and phenolic-O(3) atoms of the Schiff base and by the O1S atom of the DMF molecule. This arrangement gives rise to a Cu2O2 distorted square and to a pair of five and six-membered chelate rings. The Cu1–Cu1a distance is 3.429(6) Å, which is in good agreement with previously reported doubly phenolato-bridged copper complexes,50,51 where the Cu–Cu separation is in the range 2.901–3.345 Å. The Cu1–O4 distance is 1.943(1) Å, which is comparable with similar bond distances [1.922(4) and 1.941(4) Å] reported in the literature.50a On the other hand, the Cu1–O4–Cu1a angle of 95.33(4)° is relatively small if compared with literature data,50a,d,52 where the angular values span from 95.7 to 107.5°.53 In the lattice, the crystal structure is stabilized by weak π–π stacking interactions involving the aromatic rings of the ligand. Fig. 2 shows the packing along the b axis of the unit cell, with centroid⋯centroid distances of 3.849(5) Å.
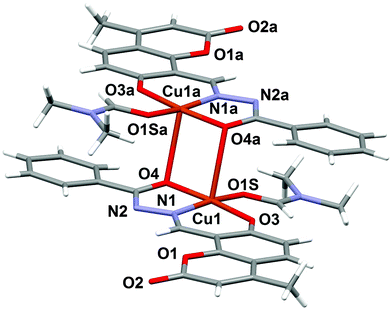 |
| | Fig. 1 Molecular structure of complex 1 with the partial labeling scheme. | |
Table 2 Selected bond lengths [Å] and angles [°] for 1
|
−x, −y + 1, −z.
|
| Cu1–O3 |
1.892(1) |
O4–Cu1–O3 |
174.34(5) |
| Cu1–O4 |
1.943(1) |
O3–Cu1–N1 |
93.08(5) |
| Cu1–O4a |
2.651(1) |
O3–Cu1–O1S |
93.12(5) |
| Cu1–N1 |
1.919(1) |
O4–Cu1–O1S |
92.03(5) |
| Cu1–O1S |
1.971(1) |
N1–Cu1–O4 |
81.42(5) |
|
|
|
N1–Cu1–O1S |
167.52(5) |
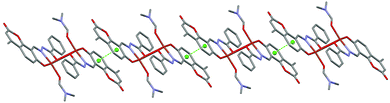 |
| | Fig. 2 π–π stacking interactions present in the crystal structure of 1, shown along the b axis of the unit cell. Centroids are represented as green spheres. | |
Electronic spectra
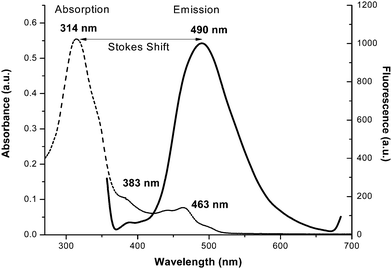
The electronic spectra for H2L and complex 1 were recorded in HPLC grade dimethyl formamide and methanol, respectively. For H2L, the absorption bands at 314 (ε = 201600 M−1 cm−1), 347 (ε = 115818 M−1 cm−1), 383 (ε = 38218 M−1 cm−1) and 463 nm (ε = 28072 M−1 cm−1) are attributed to π → π* and n → π* transitions.54 Upon complexation, the π → π* band shifts from 314 to 337 nm (ε = 38750 M−1 cm−1), while the n → π* band moves to 392 (ε = 21583 M−1 cm−1) and 408 nm (ε = 20541 M−1 cm−1). Furthermore for the complex, a pure d–d transition appears at 650 nm (ε = 1008 M−1 cm−1).55
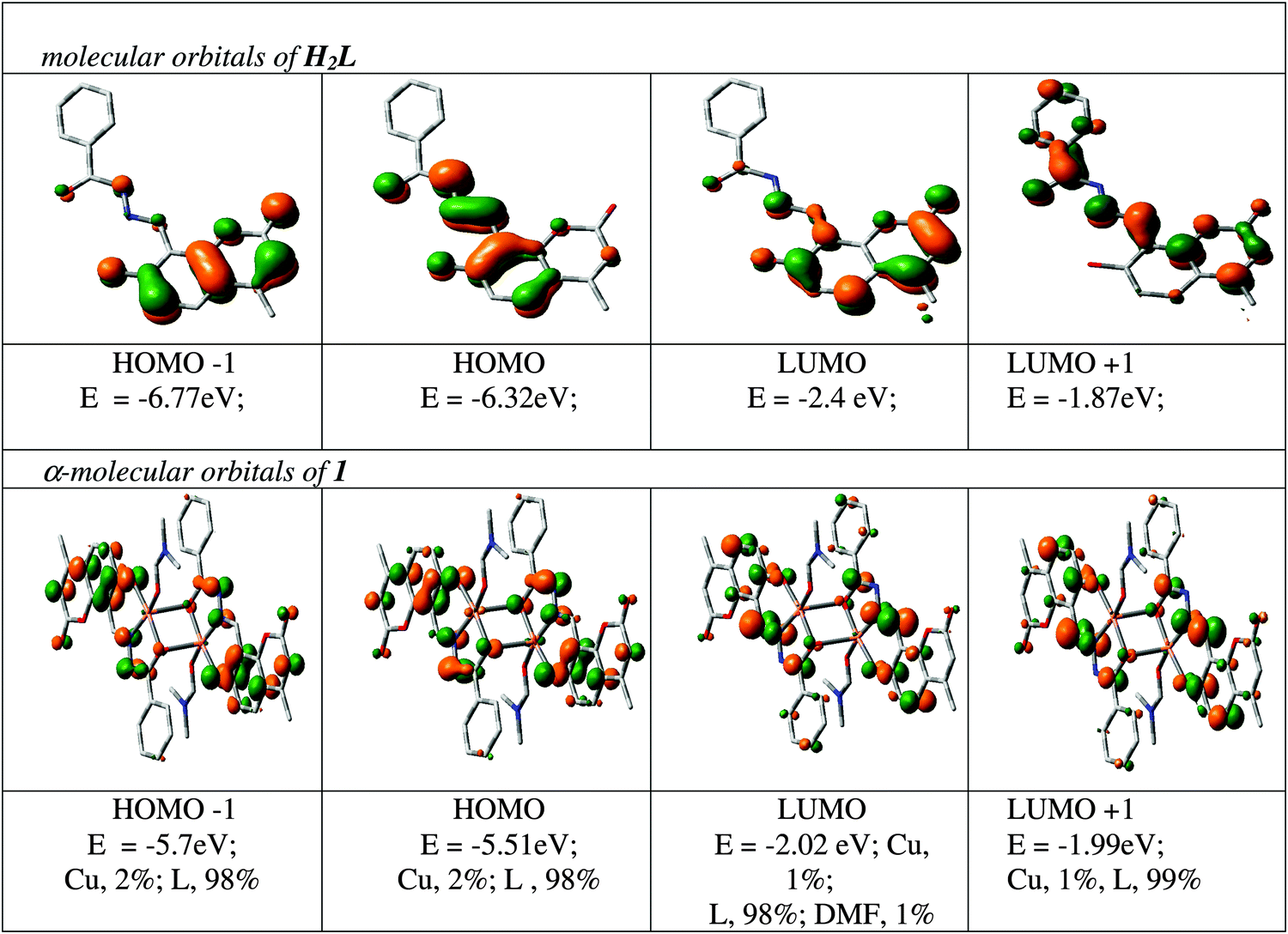
The spectral characteristics are explained with the help of a DFT computation of the optimized structure of 1. The orbital energies along with the contributions to some MOs from the ligands and the metal for a few MOs are given in Fig. 3, while the orbital energies of the ligands and the complex are given in Table S1 (ESI†). As complex 1 contains two unpaired electrons as s = 1/2, it shows two spin states designated as α and β. Both filled and vacant α-MOs from HOMO−5 to LUMO+5 constitute mainly of ligand orbitals (L2−). The metal and DMF contributions are insignificant in both filled and vacant MOs; only HOMO−5 and HOMO−4 carry 15% and 14% Cu functions whereas LUMO+4 and LUMO+5 carry 9% and 12% DMF functions in the α state, respectively. Thus, the ligand L2− controls the molecular functions and hence the electronic properties of the complexes. Thus, in the α state all the transition bands are basically considered as ILCT in nature. The intensity of these transitions has been assessed by the oscillator strength (f). HOMO → LUMO is considered as L(π) → L(π*) with f = 0.5762 at 405.8 nm; HOMO−1 → LUMO+3 is found at 335.3 nm with f = 0.3549 which is also represented as ILCT (see Table S2, ESI†). In a similar way, the orbital energy of the β state of 1 was also calculated (see Table S3, ESI†) and the calculated transitions are grouped. Here also the contribution of the ligand L2− in both filled and vacant MOs is >80% except for LUMO and LUMO+1 where the contribution is: 34% L; 62% Cu; 4% DMF; and 36% L; 60% Cu; 4% DMF, respectively. In this case the transition bands inferior to 400 nm are ILCT in nature except for the HOMO−9 → LUMO+1 transition at 360 nm (f = 0.0201) which is a mixture of LMCT and ILCT. Additionally, absorption bands at 434 (f = 0.0025) and 525 (f = 0.0046) nm are also considered as the mixture of LMCT and ILCT which are due to HOMO−3 → LUMO+1 and HOMO → LUMO+1 transitions.
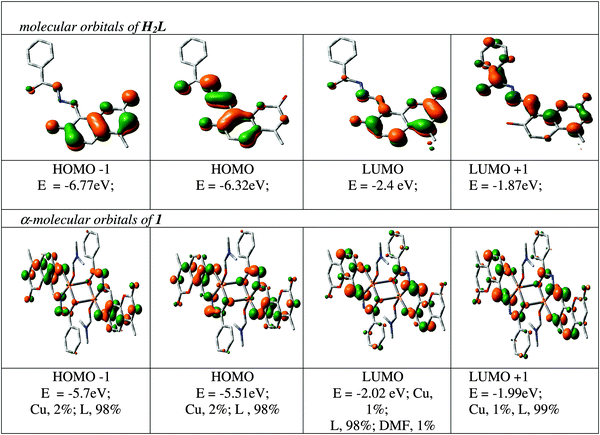 |
| | Fig. 3 Contour plots of some selected MOs of H2L and Cu2[(L)2(DMF)2] (1). | |
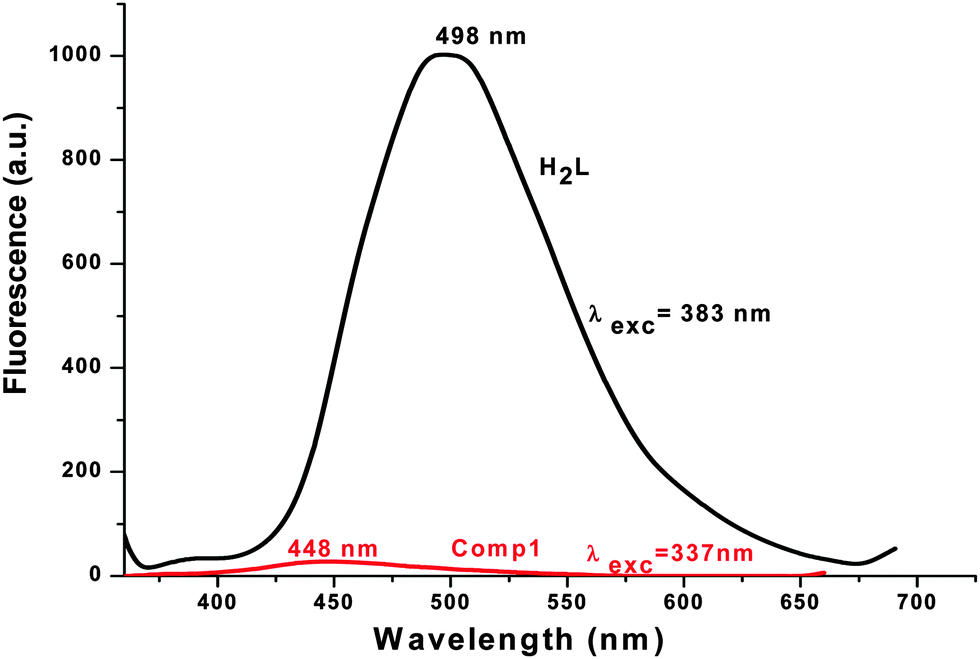
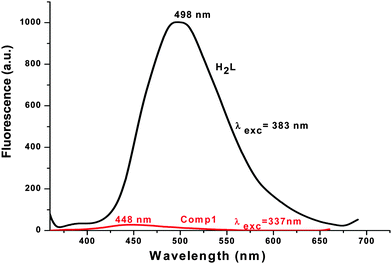
Emission spectra
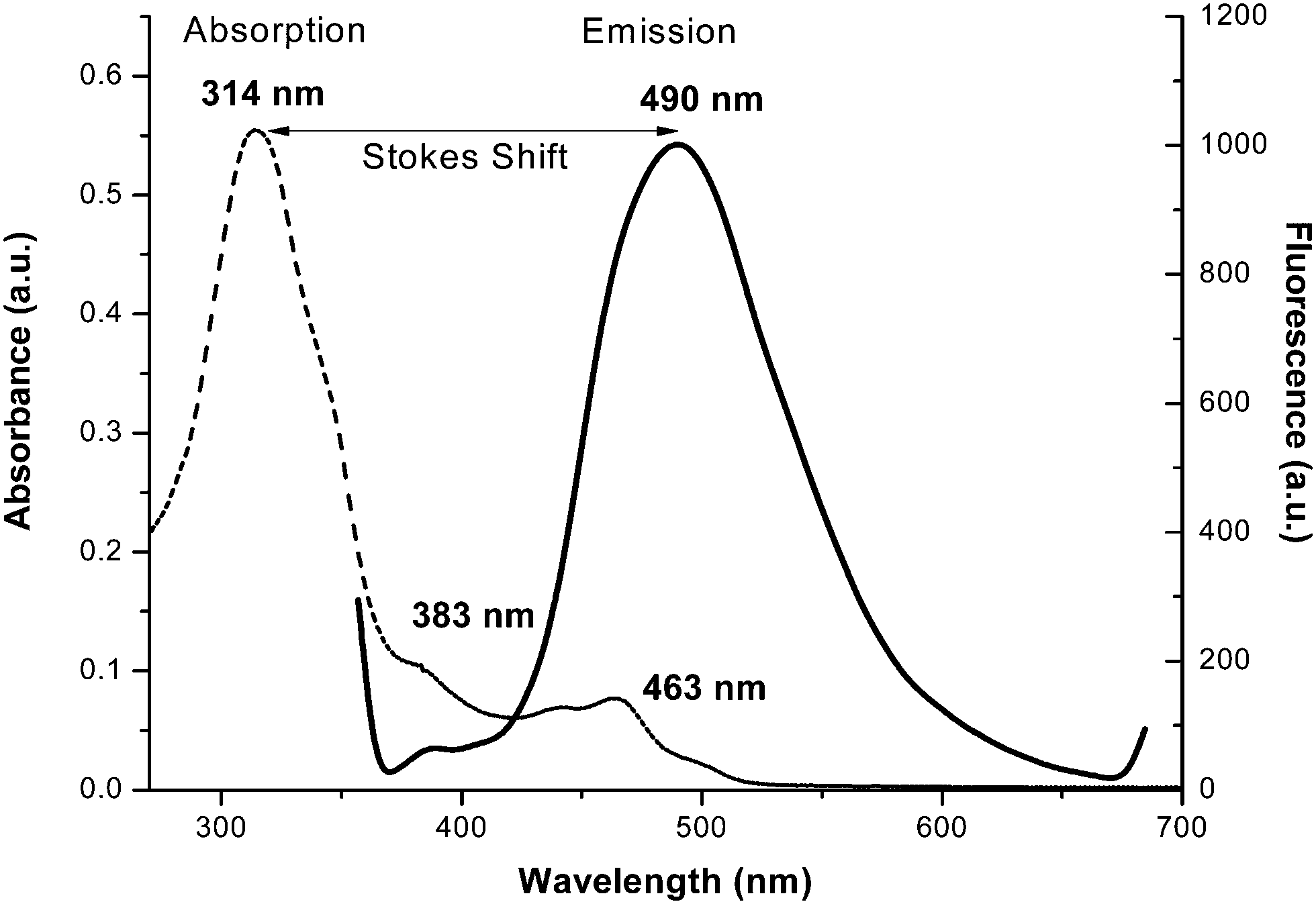
The emission spectra of H2L and 1 were carried out in DMF and methanol, respectively, at room temperature. They show that H2L fluoresces strongly when it’s π → π* transition band is excited, whereas complex 1 is a weak emitter (Fig. 4). For H2L, a strong emission band appears at 498 nm upon excitation at 314, 347 or 383 nm (which are π → π* bands). This means that H2L shows a high Stokes shift value of 184 nm (Δλ′ = λemission − λabsorption) (Fig. 5) which corresponds to the Stokes shift parameter of 11767 cm−1. The Stokes shift parameter indicates the difference in properties and structure between the ground state S0 and the first exited state S1,56 and has been estimated according to the following equation:57
| Δυ′ = υabs′ − υem′ = (1/λabs − 1/λem) × 107 cm−1 |
where: λabs and λem = absorbance and emission wavelength (nm), υabs′ and υem′ = wavenumbers for the absorption and emission maxima (cm−1).
 |
| | Fig. 4 Emission spectra of H2L (black curve, in DMF) and of the Cu(II) complex (1) (red curve, in MeOH). | |
 |
| | Fig. 5 Emission spectra with the Stokes shift value. | |
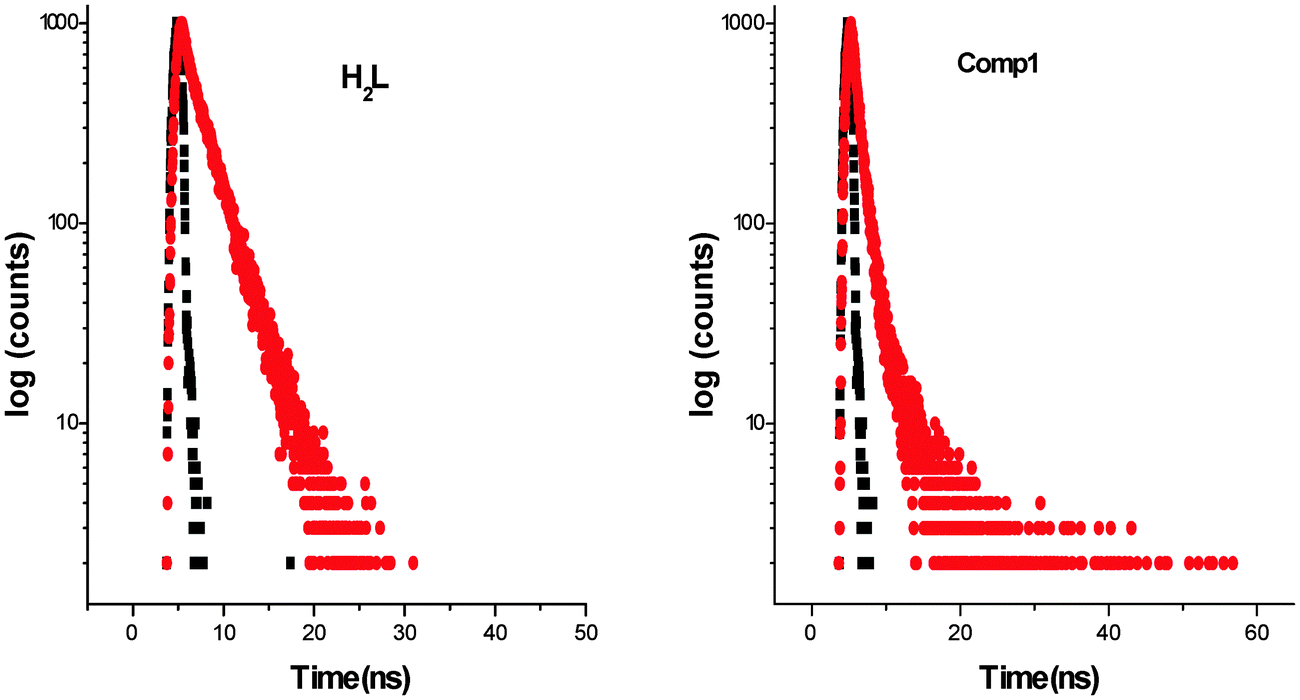
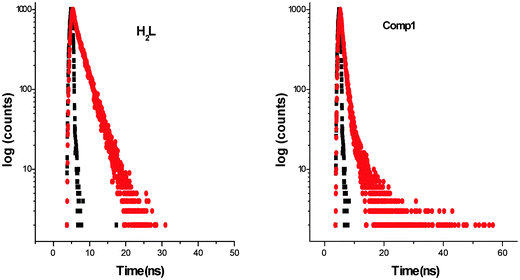
Complex 1 fluoresces very weakly at 448 nm when it is excited at 337 nm. Longer wavelengths of excitation (MLCT/d–d bands) do not produce any emission. Though the Stokes shift parameter of the ligand is comparatively higher with respect to the well-known coumarin dye, coumarin-1 (C1; 7-NEt2-4-CH3-1,2-benzopyrone, Δυ′= 4259 cm−1 in acetonitrile), the fluorescence quantum yield of the ligand (ϕH2L: 0.17) is much less than C1(ϕC1: 0.62). This is basically due to the presence of an OH oscillator, which acts as an efficient quencher enhancing the intersystem crossing process. In addition to that, the stretching vibrations of the CH, NH and CO oscillators also have an effect on quenching of luminescence.58 In the case of 1, the fluorescence quantum yield (ϕ1: 0.008) decreases by ∼20 times due to the high paramagnetic quenching of the Cu(II) metal (d9).59 The decay studies of the free ligand and the complex have been performed in DMF and methanol respectively, upon excitation at 370 nm (Fig. 6, left). The decay profile of the ligand fits with a mono exponential decay whereas in the case of the complex, the nature of the decay is bi-exponential (Fig. 6, right) The mean lifetime (τf = a1τ1 + a2τ2, where a1 and a2 are relative amplitudes of the decay process for bi-exponential) has been calculated to compare the excited state stability of the complex (1 ns) and the free ligand (3 ns). This indicates that the excited states of 1 are unstable compared to those of H2L. Radiative and non-radiative rate constant values have also been calculated showing, as usual, knr values [3.09 × 108 (H2L) and 9.8 × 108 (1)] higher than the relative kr values [6.32 × 107 (H2L) and 7.97 × 106 (1)]. In the complex, the rate constant of non-radiative decay is much higher (∼100 times) than the radiative decay, evidencing that 1 is a very weaker emitter than the ligand.
 |
| | Fig. 6 Exponential decay profile (red) of H2L and complex 1. | |
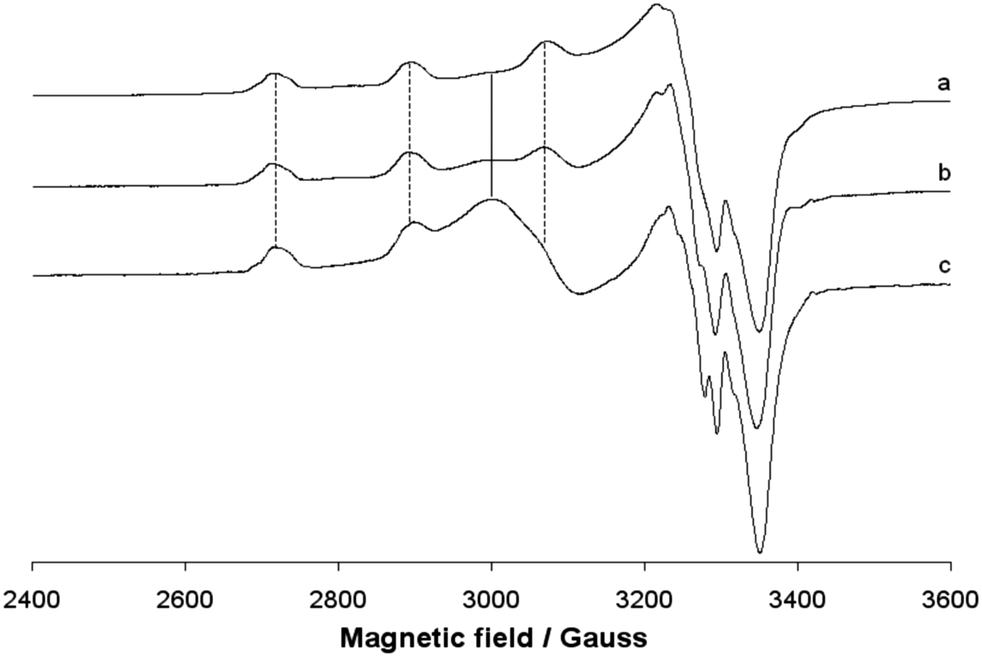
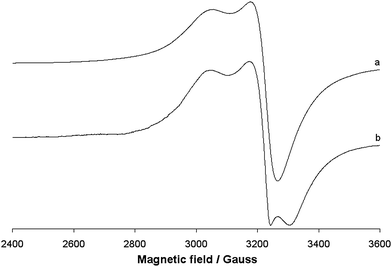
EPR spectra
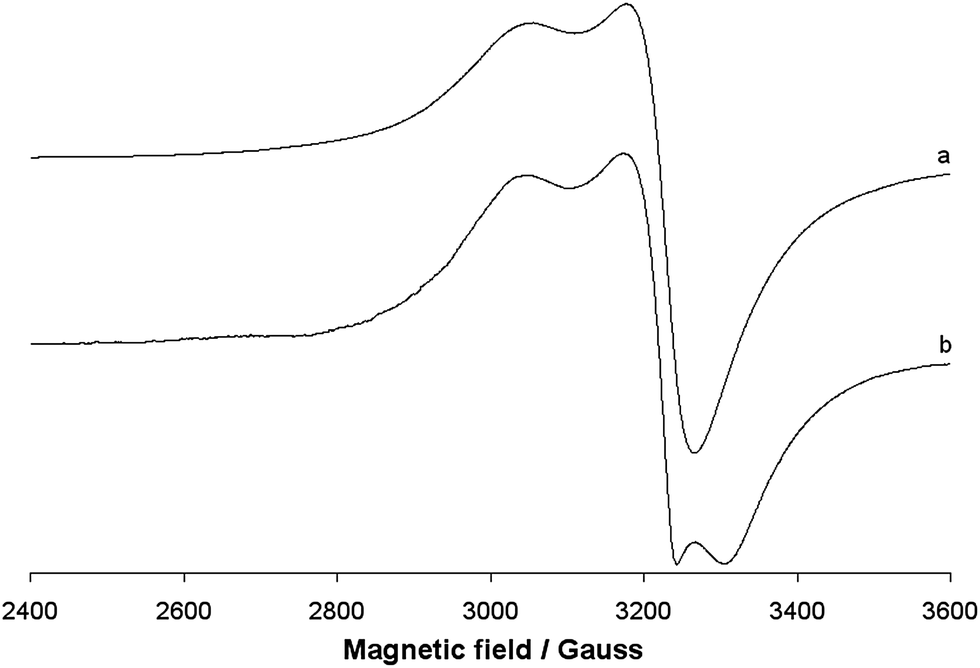
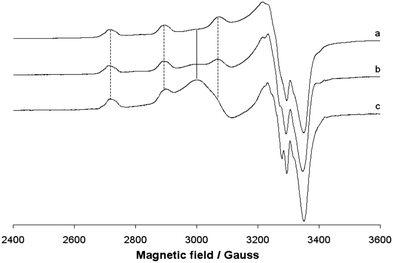
The EPR spectra of the polycrystalline solid complex 1 were recorded at 298 and 77 K. The two spectra are shown in Fig. 7. No resonances below 2800 and above 3400 Gauss and no half-field transition ΔM = ±2, attributable to a magnetic coupling between two 3d9 Cu(II) ions, were detected. This indicates that the two ions, separated by 3.429 Å, are not interacting, in agreement with the EPR behaviour of other Cu dimeric complexes reported in the literature.60 EPR spectra are ‘tetragonal’ at RT, with two g values, and ‘rhombic’ at 77 K, with three g values. This suggests a geometric distortion when lowering the temperature, which determines a slight x, y anisotropy. At RT, the order gz ≫ gx = gy > ge (Table 3) is characteristic of a ground state Ψ based on the Cu-dx2−y2 orbital and it is compatible with the square pyramidal environment of each copper ion. At 77 K, the order gz ≫ gy > gy > ge (Table 3) indicates a ground state Ψ which can be described as a linear combination of Cu-dx2−y2 and Cu-dz2 orbitals, Ψ = c1|dx2−y2〉 + c2|dz2〉,61 the parameter R (R = (gy − gx)/(gz − gy), with gz > gy > gx) being indicative of the predominance of the Cu-dx2−y2 orbital (c1 > c2) or Cu-dz2·(c2 > c1).62 If R <1, the greater contribution to the ground state arises from the Cu-dz2 orbital, while if R > 1 this arises from Cu-dx2−y2 orbital.61b For 1, R is 2.7 and this means that c1 > c2 and that a slight difference between the x and the y axis occurs at low temperatures, but the structure remains close to the square pyramidal geometry.61b Therefore, variations in temperature cause slight structural changes in bond lengths and angles in the xy plane, which results in a mixing between the Cu-dx2−y2 and Cu-dz2 orbitals in the singly occupied molecular orbital that bears the unpaired electron. Similar effects induced by the temperature were observed from some other Cu(II) compounds.63 When 1 is dissolved in an organic solvent (DMSO, DMF or MeOH) at 298 K, the dinuclear Cu(II) structure is destroyed; the solvent breaks the long Cu–O4# bond, replaces the alkoxide oxygen in the axial position and mononuclear Cu(II) species are formed (Fig. 8). This has been observed for other polynuclear Cu(II) structures.64 In all the three solvents, the spectra are slightly ‘rhombic’ with gz ≫ gy > gy > ge, similarly to what were observed in the solid state (Table 3). Therefore, the ground state is based on the Cu-dx2−y2 orbital and the geometry around copper is a distorted square pyramid. The values of gz and Az are compatible with an equatorial NO3 coordination and a charge close to the neutrality around the metal ion.65
 |
| | Fig. 7 X-band EPR spectra of the polycrystalline solid complex 1 at (a) 298 and (b) 77 K. | |
Table 3 EPR parameters of complex 1 in the solid state and in some organic solventsa
| |
g
x
|
g
y
|
g
z
|
A
x
|
A
y
|
A
z
|
R
|
Ground state |
|
A
x
, Ay and Az values reported in 10−4 cm−1.
|
| Solid at 298 K |
2.080 |
2.080 |
2.205 |
— |
— |
— |
∞ |
Cu-dx2−y2 |
| Solid at 77 K |
2.038 |
2.084 |
2.208 |
— |
— |
— |
2.7 |
Cu-dx2−y2 |
| Solid dissolved in DMSO |
2.040 |
2.069 |
2.253 |
13.0 |
15.0 |
186.9 |
6.3 |
Cu-dx2−y2 |
| Solid dissolved in MeOH |
2.041 |
2.069 |
2.254 |
12.0 |
14.0 |
185.4 |
6.6 |
Cu-dx2−y2 |
| Solid dissolved in DMF |
2.040 |
2.068 |
2.251 |
13.0 |
15.5 |
187.3 |
6.5 |
Cu-dx2−y2 |
 |
| | Fig. 8 Anisotropic X-band EPR spectra of the solid complex 1 dissolved in (a) DMSO, (b) MeOH and (c) DMF. The dotted lines indicate the first three parallel resonances due to the coupling between the unpaired electron and the63,65 Cu nucleus, while the full line shows the position of the isotropic band due to the aggregate observed in MeOH and DMF. | |
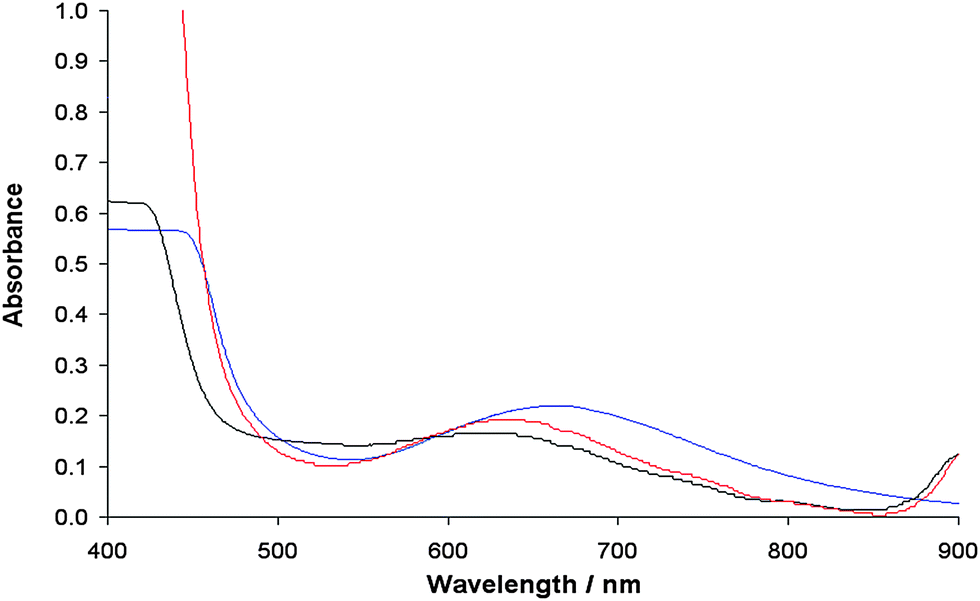
Interestingly, in the spectra recorded in MeOH and DMF, an EPR resonance centered at g ∼ 2.24 is detected. The intensity of this band increases from MeOH to DMF and cannot be ascribed to a residual presence of the dimeric structure because no other absorptions (nor at lower and higher magnetic fields) are observed. The broad and unresolved band can be assigned to an aggregation processes originated in MeOH and DMF, probably favored from the formation of π stacking interactions. Owing to such processes, the distance between the Cu centers decreases significantly and an isotropic band due to the magnetic interaction of the S = 1/2 paramagnetic metal ions appears. These aggregations have been observed for other paramagnetic species and depend on the solvent used; the effect can be detected using EPR spectroscopy but not using UV-vis spectrophotometry.66
The appearance of this band, characteristic of the solid samples, is not accompanied by the formation of a precipitate, ruled out by the electronic absorption spectra (Fig. 9). Therefore, this state can be interpreted as the intermediate between the solution and the solid state, in which the molecules undergo aggregation through π stacking favored by the presence of the two aromatic rings in the ligand structure.
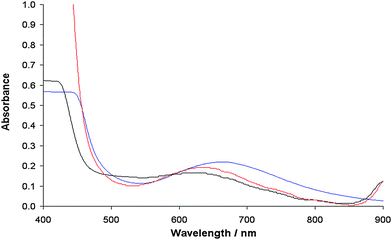 |
| | Fig. 9 Electronic absorption spectra of the solid complex 1 dissolved in DMSO (blue), DMF (red) and MeOH (black). The complex concentration was 1 × 10−3 M. | |
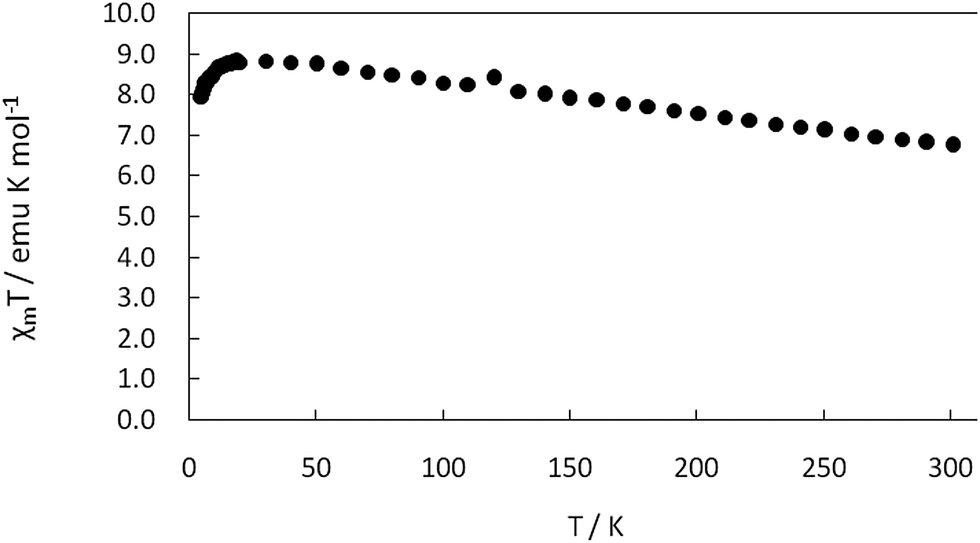
Magnetic properties
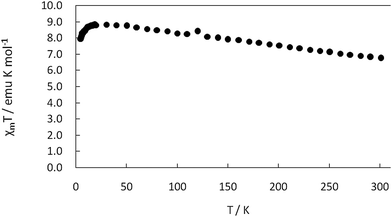
The variable-temperature magnetic properties (in the temperature range from 5 to 300 K under an external field of 10000 Oe) of complex 1 in the form of χMT vs. T (χMvs. T inset) plots are illustrated in Fig. 10 (χM is the molar susceptibility for two Cu(II)). At room temperature, the χMT value is 6.74 emu K mol−1. When lowering the temperature, the χMT value increases smoothly to a maximum value of 8.83 emu K mol−1 at 30 K. It then decreases sharply and reaches a value of 7.94 emu K mol−1 at 5 K. The behavior suggests that there is weak ferromagnetic interactions between the bridging dinuclear Cu(II) ions. Furthermore, the effective magnetic moment at room temperature is 2.07 B.M., which is less than the spin only value of Cu(II), 2.45 B.M., calculated for two uncoupled spin 1/2 centers.
 |
| | Fig. 10 The χMT vs. T plots for complex 1 in the temperature range 5–300 K. | |
Antimycobacterial activity
In the anti-mycobacterial assay, H2L and complex 1 were tested against M. tuberculosis H37Rv ATCC 27294 and M. tuberculosis H37Ra ATCC 25177 strains as well as against two clinical strains (strain 1 and strain 2). The results are shown in Table S4 (ESI†). M. tuberculosis H37Rv and M. tuberculosis H37R, well-known indicators, were used for the drug sensitivity tests.20 The results showed that the ligand H2L and complex 1 both exhibited anti-mycobacterial activity against all the tested M. tuberculosis strains, with MIC values of 52.96 μmol L−1 and MBC values in the range of 52.96–423.68 μmol L−1. Both H2L and complex 1 showed the bactericidal activity and killed the bacteria (Table S4 and Fig. S4, ESI†). Trias et al.67 showed that Mycobacterium chelonae developed a pore-forming protein as a mycobacterial porin. In the study of permeability of Mycobacterium smegmatis, Trias and Benz68 reported that the mycobacterial porin formed an important hydrophilic structure where small molecules pass through the pores and diffuse into the cell. In this study, we evidence that all compounds showed a considerable efficacy on the Mycobacterial strains. The mycobacterial cell wall includes a large amount of complex lipids, lipopolysaccharides and mycolic acids. This constitution makes the cell wall a hydrophobic strong barrier against antimicrobial agents.69 These results are well comparable with previous literature.70 The MIC (Minimal Inhibitory Concentration) and MBC (Minimal Bactericidal Concentration) values are higher in the present case. The concerned data respond well while comparing with known drugs like streptomycin and isoniazid.
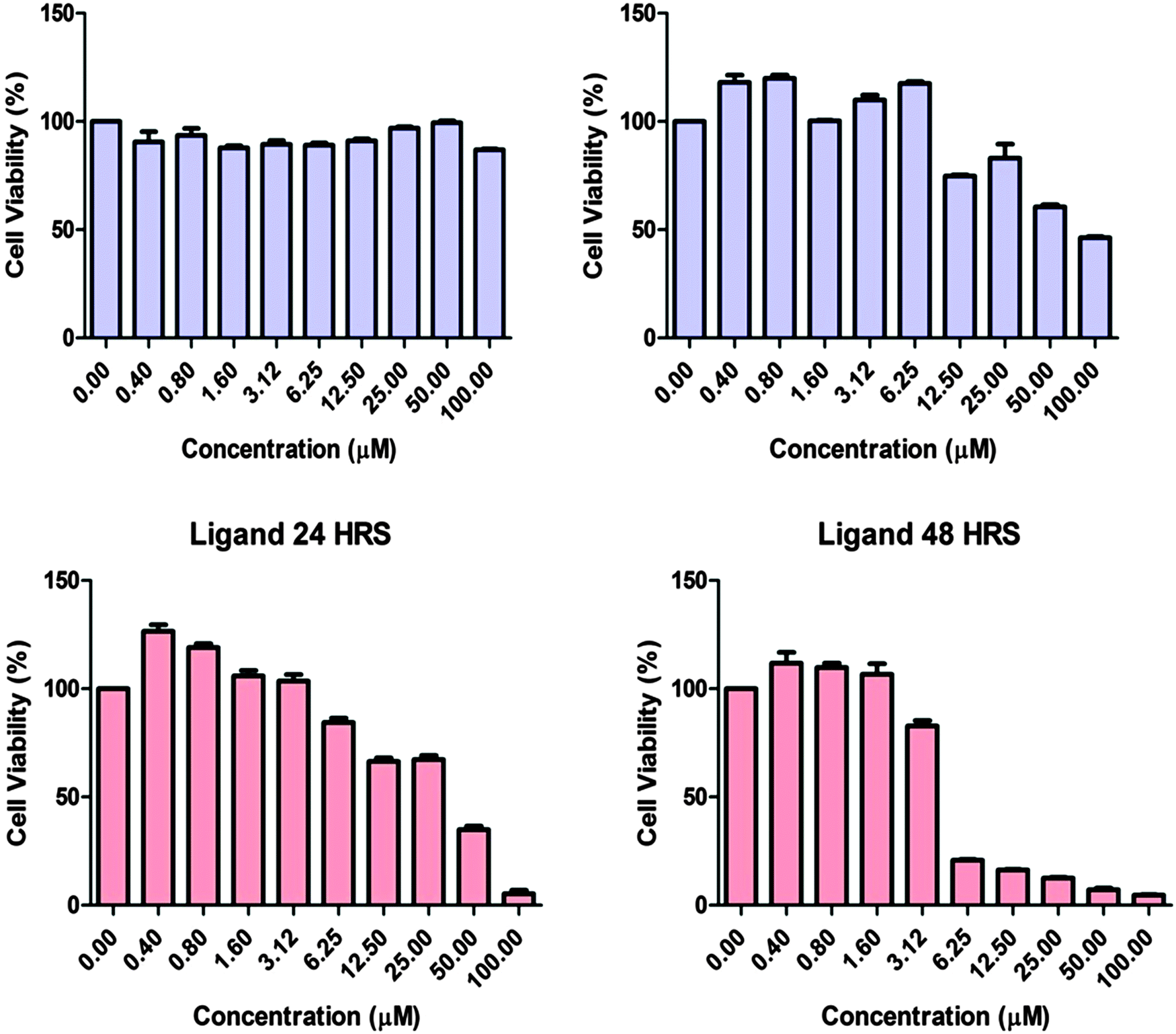
Cytotoxicity study
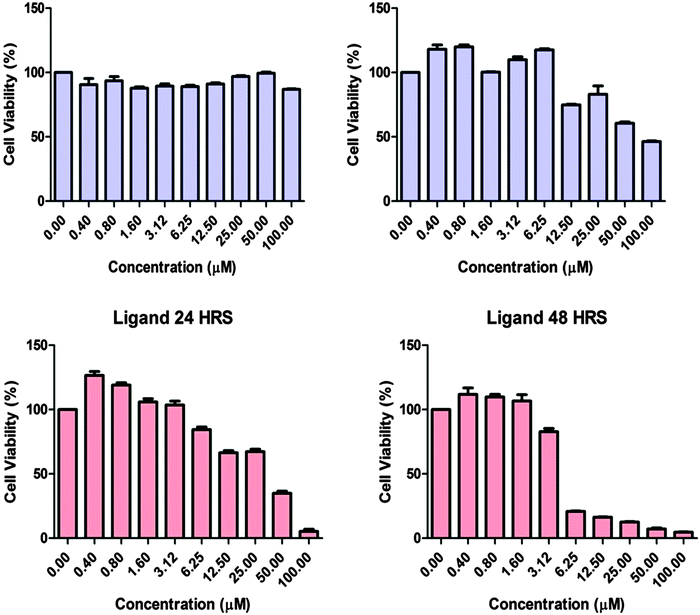
Fig. 11 shows the effects of complex 1 and ligand H2L in human adenocarcinoma cells. In order to determine the cytotoxic effect of complex 1 and ligand H2L, the adenocarcinoma cells were treated with or without the compounds and the cell viability was tested after 24 and 48 hours by MTT Assay. The results showed that the cell population was significantly decreased after 48 hours of treatment with H2L. However complex 1 produced the cytotoxic effect only after 48 hours of incubation. Our results suggest that both complex 1 and the ligand are able to induce cytotoxicity in MCF7 cells with an IC50 value of 100 μM and 5 μM. Hence the ligand and complex 1 can have potential anticancer properties.
 |
| | Fig. 11 Evaluation of the cytotoxic effect on the adenocarcinoma cell line. MCF7 cells were seeded (2.5 × 104 cells per mL) and treated with the compounds (0.4, 0.8, 1.6, 3.12, 6.25, 12.50, 25, 50 and 100 μM) and cytotoxicity was determined after 24 and 48 hours by MTT assay. | |
Docking study
The enoyl acyl carrier protein reductase (ENR) from M. tuberculosis is one of the key enzymes involved in the mycobacterial fatty acid elongation cycle and has been validated as an effective antimicrobial target. In the current study we have docked our ligand inside the binding pocket of enoyl acyl carrier protein reductase of M. tuberculosis to get an idea about the mode of action. The crystallographic structure of enoyl acyl carrier protein reductase of M. tuberculosis H37Rv was downloaded from the RCSB protein data bank (PDB ID: 4U0K), which was resolved at 1.09 Å by X-ray diffraction. Energy minimized structure of the ligand (H2L) was used for in silico protein ligand docking studies in the cavity of the enzyme. A total of 16 amino acids (Ile21, Gly96, Phe97, Met98, Met103, Met147, Asp148, Phe149, lys165, Ala191, Gly192, Pro193, Ile194, Thr196, and Met199) were present in the cavity sites and involved in the binding process within 3.5 Å radius of the ligand. The ligand, H2L, interacts with the protein by ionic interaction with Lys165 (1.59 Å, Fig. S5 and S6, ESI†) and hydrogen bond interaction with Thr196 (1.99 Å, Fig. S5 and S6, ESI†). The relevant data of the docking study are given in Tables S5 and S6 (ESI†). The surface area of the active site cavity (with respect to the H-bond donor/acceptor) is depicted in Fig. 12; the carbonyl group and the –OH group of the coumarin moiety is oriented towards the H-bond donor rich surface of the protein.
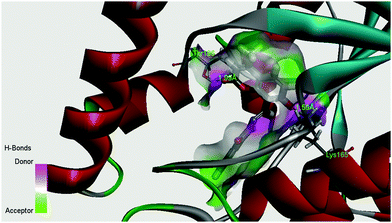 |
| | Fig. 12 Surface area (with respect to H-bond donors & acceptors) of the binding pocket in the best docked pose of the ligand and the protein. | |
Druglikeness and ADMET prediction
The druglikeness of H2L was checked following Lipinski's rule of five. The ADMET modules of Discovery Studio were used to check the ADMET (absorption, distribution, metabolism, excretion and toxicity) properties of the compounds. Toxicity prediction, aqueous solubility, blood brain barrier penetration, human intestinal absorption, and ames mutagenicity were predicted under the Calculate Molecular Property module of the Small molecule tool of Discovery Studio client 4.0. The predicted data are summarized in Table 4. According to these predictions, the ligand, H2L, is well absorbed and non-mutagenic.
Table 4 ADMET prediction of H2L
| Compounds |
Molecular weight |
ADMET solubility (aqueous) |
ADMET solubility level |
ADMET absorption levela |
ADMET_A logP98 |
No. of H-bond acceptor |
No. of H-bond donor |
Lipniski's filter |
Druglikeness inference |
Ames prediction |
|
ADMET absorption level: 0, good; 1, moderate; 2, low.
|
| H2L |
322.3 |
−4.039 |
2 |
0 (good) |
2.957 |
5 |
2 |
Yes |
Yes, low |
Non-mutagen |
Conclusions
The coumarin-assisted tridentate ligand, [benzoic acid(7-hydroxy-4-methyl-2-oxo-2H-chromen-8-ylmethylene)-hydrazide], has been used to synthesize a phenoxo-bridged Cu(II) dinuclear complex, Cu2[(L)2(DMF)2] (1). The solid state structure of 1 shows that the Cu atoms are present in a square-pyramidal arrangement. The variable temperature EPR spectra suggest that a geometrical distortion takes place upon lowering the temperature and that the dinuclear structure is destroyed if the complex is dissolved in coordinating solvents. The temperature dependent magnetic moment supports the presence of weak ferromagnetic interactions between the bridging dinuclear Cu(II) ions. Both the ligand H2L and complex 1 exhibit anti-mycobacterial activity and considerable efficacy on M. tuberculosis H37Rv ATCC 27294 and M. tuberculosis H37Ra ATCC 25177 strains. Our cytotoxicity study on human adenocarcinoma cells suggests that both complex 1 and the ligand H2L are able to induce cytotoxicity in MCF7 cells. In summary, the studies here discussed have evidenced the activity in the biomedical field of a new copper complex; further investigations in this area with other transition metal ions and chemo-sensor activities of the ligand for selective detection of metal ions are currently being carried out in our laboratories.
Acknowledgements
KD and CS would like to thank DST (Department of Science and Technology, New Delhi, India) for the grant (SR/S1/IC-31/2008) to carry out the present study. AD would like to thank the National Science Council, Taiwan for financial assistance.
References
- L. Taneja, A. K. Sharma and R. D. Singh, J. Lumin., 1995, 63, 203–214 CrossRef CAS.
-
(a) L. G. Samsonova, R. M. Gadirov, V. V. Ishchenko and O. V. Khilya, Opt. Spectrosc., 2007, 102, 878–884 CrossRef CAS;
(b) A. Prakash and B. K. Singh, Int. J. Pure Appl. Chem., 2010, 5, 291–297 Search PubMed;
(c) A. Prakash, B. K. Singh, N. Bhojak and D. Adhikari, Spectrochim. Acta, Part A, 2010, 76, 356–362 CrossRef PubMed.
-
(a) A. Schonberg and N. Latif, J. Am. Chem. Soc., 1954, 76, 6208 CrossRef CAS;
(b) A. K. Mitra, S. K. Misra and A. Patra, Synth. Commun., 1980, 10, 915–919 CrossRef CAS PubMed;
(c) R. N. Gacche and S. G. Jadhav, J. Exp. Clin. Med., 2012, 4, 165–169 CrossRef CAS PubMed;
(d) H. Zhao, N. Neamati, H. Hong and A. Mazumder, J. Med. Chem., 1997, 40, 242–249 CrossRef CAS PubMed.
-
(a) L. A. Singer and N. P. Kong, J. Am. Chem. Soc., 1966, 88, 5213–5219 CrossRef CAS;
(b) S. Weigt, N. Huebler, R. Strecker, T. Braunbeck and T. H. Broschard, Reprod. Toxicol., 2012, 33, 133–141 CrossRef CAS PubMed.
- G. Signore, R. Nifosì, L. Albertazzi and R. Bizzarri, J. Biomed. Nanotechnol., 2009, 5, 722–729 CrossRef CAS PubMed.
-
(a) J.-M. An, Z.-Y. Yang, M.-H. Yan and T.-R. Li, J. Lumin., 2013, 139, 79–83 CrossRef CAS PubMed;
(b) Z. Su, K. Chen, Y. Guo, H. Qi, X.-F. Yang and M. Zhao, J. Fluoresc., 2010, 20, 851–856 CrossRef CAS PubMed;
(c) F. Chen, G. Liu, Y. Shi, P. Xi, J. Cheng, J. Hong, R. Shen, X. Yao, D. Bai and Z. Zeng, Talanta, 2014, 124, 139–145 CrossRef CAS PubMed;
(d) S. W. Hong, N. J. Jeon, J. Jeong, J. Hong and K. C. Nam, Bull. Korean Chem. Soc., 2013, 34, 1530–1532 CrossRef CAS.
-
(a) G. Verma, A. Marella, M. Shaquiquzzaman, M. Akhtar, M. R. ahmat Ali and M. M. Alam, J. Pharm. BioAllied Sci., 2014, 6, 69–80 CrossRef PubMed;
(b) E. Macková, K. Hrušková, P. Bendová, A. Vávrová, H. Jansová, P. Hašková, P. Kovaříková, K. Vávrová and T. Šimunek, Chem.-Biol. Interact., 2012, 197, 69–79 CrossRef PubMed;
(c) L. V. de Freitas, C. C. P. da Silva, J. Ellena, L. Antônio Sodré Costa and N. A. Rey, Spectrochim. Acta, Part A, 2013, 116, 41–48 CrossRef PubMed.
-
(a) L. Jin, J. Chen, B. Song, Z. Chen, S. Yang, Q. Li, D. Hu and R. Xu, Bioorg. Med. Chem. Lett., 2006, 16, 5036–5040 CrossRef CAS PubMed;
(b) A. C. González-Baró, P. Diez, B. S. Parajón-Costa and N. A. Rey, J. Mol. Struct., 2012, 1007, 95–101 CrossRef PubMed;
(c) P. G. Avaj, C. H. V. Kumar, S. A. Patil, K. N. Shivananda and C. Nagaraju, Eur. J. Med. Chem., 2009, 44, 3552–3559 CrossRef PubMed.
-
(a) A. U. Mandakmare and M. L. Navwade, Orient. J. Chem., 1997, 13, 155–158 CAS;
(b) E. M. Marshall, M. Ryles, K. Butler and L. Weiss, J. Cancer Res. Clin. Oncol., 1994, 120, 535–538 Search PubMed.
- R. D. Thornes, L. Daly and G. Lynch, J. Cancer Res. Clin. Oncol., 1994, 120, S32–S34 CrossRef.
-
(a) S. Roy, T. K. Mondal, P. Mitra, E. L. Torres and C. Sinha, Polyhedron, 2011, 30, 913–922 CrossRef CAS PubMed;
(b) S. Roy, T. K. Mondal, P. Mitra and C. Sinha, Polyhedron, 2013, 51, 27–40 CrossRef CAS PubMed.
-
(a) M. Zhao, C. Stern, A. G. M. Barrett and B. M. Hoffman, Angew. Chem., Int. Ed., 2003, 42, 462–464 CrossRef CAS PubMed;
(b) H. Zheng, W. Zhe-Ming, G. Song and Y. Chun-Hua, Inorg. Chem., 2006, 45, 6694–6705 CrossRef PubMed;
(c) J. S. Miller, Dalton Trans., 2006, 2742–2749 RSC;
(d) C. Cadiou, M. Murrie, C. Paulsen, V. Villar, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2001, 2666–2667 RSC.
-
(a) P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75–86 RSC;
(b) T. Niittymaki and H. Lonnberg, Org. Biomol. Chem., 2006, 4, 15–25 RSC;
(c) Q.-X. Xiang, J. Zhang, P.-Y. Liu, C.-Q. Xia, Z.-Y. Zhou, R.-G. Xie and X.-Q. Yu, J. Inorg. Biochem., 2005, 99, 1661–1669 CrossRef CAS PubMed.
-
(a) D. Mastropaolo, D. A. Powers, J. A. Potenza and H. J. Schugar, Inorg. Chem., 1976, 15, 1444–1451 CrossRef CAS;
(b) A. Drzewiecka, A. E. Koziol, M. Lowczak and T. Lis, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2339 CAS;
(c) G. Zhang, G. Yang and J. S. Ma, Cryst. Growth Des., 2006, 6, 375–381 CrossRef CAS.
-
Bioinorganic Chemistry, ed. I. Bertini, H. B. Gray, S. J. Lippard and J. S. Valentine, University Science Books, Mill Valley, CA, 1994 Search PubMed.
-
Magneto-Structural Correlation in Exchange-Coupled Systems, ed. R. D. Willett, D. Gatteschi and O. Kahn, NATO AS1 Series C, 140, D. Reidel Publishing Co, Dordrecht, The Netherlands, 1985 Search PubMed.
- D. J. Hodgson, Prog. Inorg. Chem., 1975, 19, 173–178 CrossRef CAS PubMed.
- A. L. Okunade and M. P. F. Elvin-Lewis, Phytochemistry, 2004, 65, 1017–1022 CrossRef CAS PubMed.
- N. Lall, M. D. Sarma, B. Hazra and J. J. Meyer, J. Antimicrob. Chemother., 2003, 51, 435–438 CrossRef CAS PubMed.
-
(a) E. Banfi, M. G. Mamolo, D. Zampieri, L. Vio and C. M. Bragadin, J. Antimicrob. Chemother., 2001, 48, 705–711 CrossRef CAS PubMed;
(b) V. Jarlier and H. Nikaido, FEMS Microbiol. Lett., 1994, 123, 11–18 CrossRef CAS PubMed.
- K. H. K. Naik, S. Selvaraj and N. Naik, Spectrochim. Acta, Part A, 2014, 131, 599–605 CrossRef PubMed.
- D. F. Eaten, Pure Appl. Chem., 1988, 60, 1107–1114 Search PubMed.
- S. R. Stoyanov, J. M. Villegas and D. P. Rillema, Inorg. Chem., 2002, 41, 2941–2945 CrossRef CAS PubMed.
-
WINEPR SimFonia, version 1.25 aass, Bruker Analytische Messtechnik GmbH, Karlsruhe, 1996 Search PubMed.
- G. B. Bagihalli, P. G. Avaji, S. A. Patil and P. S. Badam, Eur. J. Med. Chem., 2008, 43, 2639–2649 CrossRef CAS PubMed.
-
(a) SADABS Bruker AXS, Madison, Wisconsin, USA, 2004; SAINT, Software Users Guide, Version 6.0; Bruker Analytical X-ray Systems, Madison, WI, 1999;
(b)
G. M. Sheldrick, SADABS v2.03: Area-Detector Absorption Correction, University of Göttingen, Germany, 1999 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 Revision D 01, Gaussian Inc., Wallingford, CT, 2004 Search PubMed.
-
GaussView3.0, Gaussian, Pittsburgh, PA Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. K. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- N. M. O’Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- B. D. Becton, Dickinson and Company Newsletter BD Bactec MGIT 960 SIRE kit now FDA-cleared for susceptibility testing of Mycobacterium tuberculosis, Microbiology News & Ideas, 2002, 13, 4 Search PubMed.
- NCCLS, (2003). National Committee for Clinical Laboratory Standards (NCCLS). Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes; Approved Standard. NCCLS document M24-A [ISBN 1-56238-500-3]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003.
- L. Collins and S. G. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004–1009 CAS.
- A. Jimenez-Arellanes, M. Meckes, R. Ramirez, J. Torres and J. Luna-Herrera, Phytother. Res., 2003, 17, 903–908 CrossRef PubMed.
-
(a) G. R. Battu and B. M. Kumar, Pharmacogn. J., 2010, 2, 456–463 CrossRef;
(b) P. Bontempo, V. Carafa, R. Grassi, A. Basile, G. C. Tenore, C. Formisano, D. Rigano and L. Altucci, Food Chem. Toxicol., 2013, 304–312 CrossRef CAS PubMed.
- T. Mosman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- Discovery Studio 4.0 is a product of Accelrys Inc, San Diego, CA, USA.
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Parts A and B, John Wiley, New York, 5th edn, 1997 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
-
(a) M. Sakamoto, S. Itose, T. Ishimori, N. Matsumoto, H. Ōkawa and S. Kida, Dalton Trans., 1989, 2083–2088 RSC;
(b) Y. Shiping, C. Peng, L. Daizheng, J. Zonghui, W. Genglin, W. Honggen and Y. Xinkan, Polyhedron, 1992, 11, 879–883 CrossRef;
(c) P. Cheng, D. Liao, S. Yan, J. Cui, Z. Jiang, G. Wang, X. Yao and H. Wang, Helv. Chim. Acta, 1997, 80, 213–219 CrossRef CAS PubMed;
(d) K. Bertoncello, G. D. Fallon, J. H. Hodgkin and K. S. Murray, Inorg. Chem., 1988, 27, 4750–4758 CrossRef CAS.
- R. Gupta, S. Mukherjee and R. Mukherjee, J. Chem. Soc., Dalton Trans., 1999, 4025–4030 RSC.
- Y. Shiping, C. Peng, L. Daizheng, J. Zonghui, W. Genglin, W. Honggen and Y. Xinkan, Polyhedron, 1992, 11, 879–883 CrossRef.
-
(a) S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. Le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526–3536 CrossRef CAS;
(b) S. K. Dutta, U. Florke, S. Mohanta and K. Nag, Inorg. Chem., 1998, 37, 5029–5032 CrossRef CAS.
-
A. B. P. Lever, Inorganic Electronic Spectroscopy, 2nd edn, Elsevier, New York, 1984 Search PubMed.
- U. Ray, K. K. Sarker, G. Mostafa, T.-H. Lu, M. S. El Fallah and C. Sinha, Polyhedron, 2006, 25, 2764–2772 CrossRef CAS PubMed.
- M. S. Refat, Spectrochim. Acta, Part A, 2007, 68, 1393–1405 CrossRef PubMed.
- A. E. H. Machado, D. Severino, J. Ribeiro, R. De Paula, M. H. Gehlen, H. P. M. de Oliveira, M. D. S. Matos and J. A. de Miranda, Photochem. Photobiol. Sci., 2004, 3, 79–86 CAS.
-
(a) J. L. Kropp and M. W. Windsor, J. Chem. Phys., 1963, 39, 2769–2770 CrossRef CAS PubMed;
(b) J. L. Kropp and M. W. Windsor, J. Chem. Phys., 1965, 42, 1599–1608 CrossRef CAS PubMed.
- J. H. Chang, Y. M. Choi and Y.-K. Shin, Bull. Korean Chem. Soc., 2001, 22, 527–533 CAS.
- S. Thakurta, P. Roy, G. Rosair, C. J. Gómez-García, E. Garribba and S. Mitra, Polyhedron, 2009, 28, 695–702 CrossRef CAS PubMed.
-
(a) B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS;
(b) E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229–1235 CrossRef CAS.
- R. J. Fereday, P. Hodgson, S. Tyagi and B. J. Hathaway, J. Chem. Soc., Dalton Trans., 1981, 2070–2077 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784–1795 RSC.
-
(a) J. C. Jeffery, J. P. Maher, C. A. Otter, P. Thornton and M. D. Ward, J. Chem. Soc., Dalton Trans., 1995, 819–824 RSC;
(b) G. van Albada, I. Mutikainen, W. J. J. Smeets, A. L. Spek, U. Turpeinen and J. Reedijk, Inorg. Chim. Acta, 2002, 327, 134–139 CrossRef CAS;
(c) W. A. Alves, R. H. de Almeida Santos, A. Paduan-Filho, C. C. Becerra, A. C. Borin and A. M. Da Costa Ferriera, Inorg. Chim. Acta, 2004, 357, 2269–2278 CrossRef CAS PubMed;
(d) M. A. Ali, A. H. Mirza, R. J. Fereday, R. J. Butcher, J. M. Fuller, S. C. Drew, L. R. Gahan, G. R. Hanson, B. Moubaraki and K. S. Murray, Inorg. Chim. Acta, 2005, 358, 3937–3948 CrossRef CAS PubMed;
(e) I. A. Koval, M. Sgobba, M. Huisman, M. Lüken, E. Saint-Aman, P. Gamez, B. Krebs and J. Reedijk, Inorg. Chim. Acta, 2006, 359, 4071–4078 CrossRef CAS PubMed.
- J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS.
- A. Stasko, V. Brezova, S. Biskupic and V. Misik, Free Radical Res., 2007, 41, 379–383 CrossRef CAS PubMed.
- J. Trias, V. Jarlier and R. Benz, Science, 1992, 258, 1479–1481 CAS.
- J. Trias and R. Benz, Mol. Microbiol., 1994, 14, 283–286 CrossRef CAS PubMed.
-
(a)
D. E. Minnikin and M. Goodfellow, in Microbiological classification and identification, ed. R. G. Board, Academic, London, 1980, p. 189 Search PubMed;
(b)
D. E. Minnikin, Lipids, complex lipids, their chemistry, biosynthesis and roles, in The biology of mycobacteria, ed. C. Ratledge and J. Stanford, Academic Press, Inc, London, 1982, p. 95 Search PubMed.
-
(a) K. Das, S. Nandi, S. Mondal, T. Askun, Z. Canturk, P. Celikboyun, C. Massera, E. Garribba, A. Datta, C. Sinha and T. Akitsu, New J. Chem., 2015, 39, 1101–1114 RSC;
(b) K. Das, A. Datta, S. Nandi, S. B. Mane, S. Mondal, C. Massera, C. Sinha, C.-H. Hung, T. Askun, P. Celikboyun, Z. Cantürk, E. Garribba and T. Akitsu, Inorg. Chem. Front., 2015 10.1039/C5QI00060B.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 938644. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00789e |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence