
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dioxidomolybdenum(VI) complexes with isoniazid-related hydrazones: solution-based, mechanochemical and UV-light assisted deprotonation†
Višnja
Vrdoljak
*a,
Biserka
Prugovečki
a,
Ivana
Pulić
a,
Marko
Cigler
a,
Dora
Sviben
a,
Jelena
Parlov Vuković
b,
Predrag
Novak
a,
Dubravka
Matković-Čalogović
a and
Marina
Cindrić
a
aUniversity of Zagreb, Faculty of Science, Department of Chemistry, Horvatovac, 102a, 10000 Zagreb, Croatia. E-mail: visnja.vrdoljak@chem.pmf.hr; Fax: +385-1-4606341; Tel: +385-1-4606353
bINA-Industrija nafte d.d., Refining & marketing business division, Product development department, Lovinčićeva bb, 10002 Zagreb, Croatia
First published on 15th July 2015
Abstract
Synthesis of the dioxidomolybdenum(VI) complexes [MoO2(HLR)(MeOH)]Cl (1–3) was carried out by using MoO2Cl2 and the corresponding ONO aroylhydrazone ligand H2LR (ligand H2LR is salicylaldehyde isonicotinoylhydrazone (H2LSIH), 2-hydroxy-naphthaldehyde isonicotinoylhydrazone (H2LNIH), or p-(N,N′-diethylamino)salicylaldehyde isonicotinoylhydrazone (H2LEt2NSIH) in methanol. Compounds [MoO2(HLR)(H2O)]Cl (1a–3a) were obtained upon exposure of the corresponding mononuclear complexes 1–3 to moisture. Deprotonation of the mononuclear complexes 1–3 was performed by using Et3N as a base (by the conventional solution based-method and by the mechanochemical approach) as well as by UV-light assisted reactions yielding [MoO2(LSIH)(MeOH)] (4), [MoO2(LNIH)(MeOH)] (5) and [MoO2(LEt2NSIH)]n (6), respectively. Crystal and molecular structures of all complexes were determined by the single crystal X-ray diffraction method. The complexes were further characterized by elemental analysis, IR spectroscopy, TG analysis, one- and two-dimensional NMR spectroscopy and powder X-ray diffraction.
Introduction
The chemistry of hydrazones is continuing to be an interesting area of research because of their modularity, easiness of synthesis and stability towards hydrolysis.1 It is known that aroylhydrazones can exist in solution as configurational isomers (in E or Z forms) or in tautomeric forms (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH–(CO)– or N–N(C–OH)–) that occur in equilibrium. In most cases they have an acidic proton and their coordination to transition metals often leads to proton displacement.1 Depending on the proton-accepting ability, metal complexes with neutral (H2L), singly- (HL−) and doubly-deprotonated ligands (L2−) can be obtained.2 In such a way different dimensionalities of the hydrogen-bonded networks can be formed. The protonation state of these ligands in metal complexes plays an important role since it offers fine-tuning of properties such as electrochemical, photophysical or catalytic.3 Recent attention has been paid to the configurational switching mechanism based on coordination-coupled deprotonation which explains the role of hydrazone deprotonation in activating the molecular switch.4
N–NH–(CO)– or N–N(C–OH)–) that occur in equilibrium. In most cases they have an acidic proton and their coordination to transition metals often leads to proton displacement.1 Depending on the proton-accepting ability, metal complexes with neutral (H2L), singly- (HL−) and doubly-deprotonated ligands (L2−) can be obtained.2 In such a way different dimensionalities of the hydrogen-bonded networks can be formed. The protonation state of these ligands in metal complexes plays an important role since it offers fine-tuning of properties such as electrochemical, photophysical or catalytic.3 Recent attention has been paid to the configurational switching mechanism based on coordination-coupled deprotonation which explains the role of hydrazone deprotonation in activating the molecular switch.4
Isoniazid-related aroylhydrazones and their coordination compounds are of great interest owing to their biological activities5 and their structural diversity. Thus, depending on the reaction conditions, cis-{MoO2}2+ structural units can form interesting supramolecular assemblies [MoO2(L)]x (x = 4, 6 or n), where the nitrogen atom of the isonicotinyl moiety coordinates an additional neighboring metal atom.6–9 Otherwise, the sixth coordination site is occupied by the oxygen atom from the solvent D thus forming mononuclear complexes [MoO2(L)D]. In these compounds, the hydrazone ligand is in the doubly-deprotonated (L2−) form. Surprisingly, chloride salts of charged dioxidomolybdenum(VI) complexes with tridentate ONO-donor ligands are very rare. Only two such structures have been published to date.10
A typical method employed in the preparation of some fully deprotonated metal complexes involves addition of a base during the complexation reaction, commonly in organic solvents.1 To the best of our knowledge, alternative methods towards deprotonation of cis-dioxidomolybdenum(VI) complexes (including grinding or UV irradiation as the method of activation) were not investigated. With these possibilities in mind, we set out to explore the influence of UV light on singly protonated isonicotinoyl hydrazone complexes to see if deprotonation of the pyN–H+ moiety could proceed without a photoinduced E-to-Z configurational switch about the hydrazone double bond. Deprotonation has been investigated also by using Et3N as a base (by a conventional solution-based method and a mechanochemical approach). To achieve this aim we have prepared and characterized dioxidomolybdenum(VI) complexes [MoO2(HLR)(MeOH)]Cl (1–3) with three ligands, H2LR, salicylaldehyde isonicotinoylhydrazone (H2LSIH), 2-hydroxy-naphthaldehyde isonicotinoylhydrazone (H2LNIH), and p-(N,N′-diethylamino)salicylaldehyde isonicotinoylhydrazone (H2LEt2NSIH), Scheme 1. We were also interested to investigate the importance of nonbonding interactions in the structures as well as the ability of these complexes to form different hydrogen-bonded networks depending on the protonation state of the complexes.
 | ||
| Scheme 1 The isoniazid-based hydrazones H2LR. | ||
Results and discussion
Synthesis of dioxidomolybdenum(VI) complexes
Synthesis of [MoO2(HLR)(MeOH)]Cl was carried out in dry methanol using MoO2Cl2 and the corresponding aroylhydrazone H2LR (R = SIH, NIH or Et2NSIH), Scheme 2. In all of the investigated compounds formed after chelation, the ligands are found to be in the singly-deprotonated form (HLR)− coordinated to the cis-{MoO2}2+ core via the ONO donor atoms. The cis-{MoO2}2+ core is additionally coordinated by a solvent molecule resulting in the formation of [MoO2(HLR)(MeOH)]Cl, where R = SIH (1), NIH (2) or Et2NSIH (3), respectively. If the reaction was not performed in dry methanol a mixture of the corresponding [MoO2(HLR)(MeOH)]Cl and [MoO2(HLR)(H2O)]Cl complexes was obtained. All of the isolated compounds 1–3 are moisture-sensitive crystalline solids (Fig. S1, see ESI†). Exposure of samples 1–3 to water vapour resulted in crystalline products distinct from the starting compounds. In all cases they were identified to be [MoO2(HLR)(H2O)]Cl (1a–3a) where the coordination sphere around the molybdenum atom is completed by coordination of a water molecule. Crystals of 1a suitable for single crystal X-ray diffraction were obtained from wet methanol, whereas those of 2a and 3a·H2O were obtained from wet acetonitrile.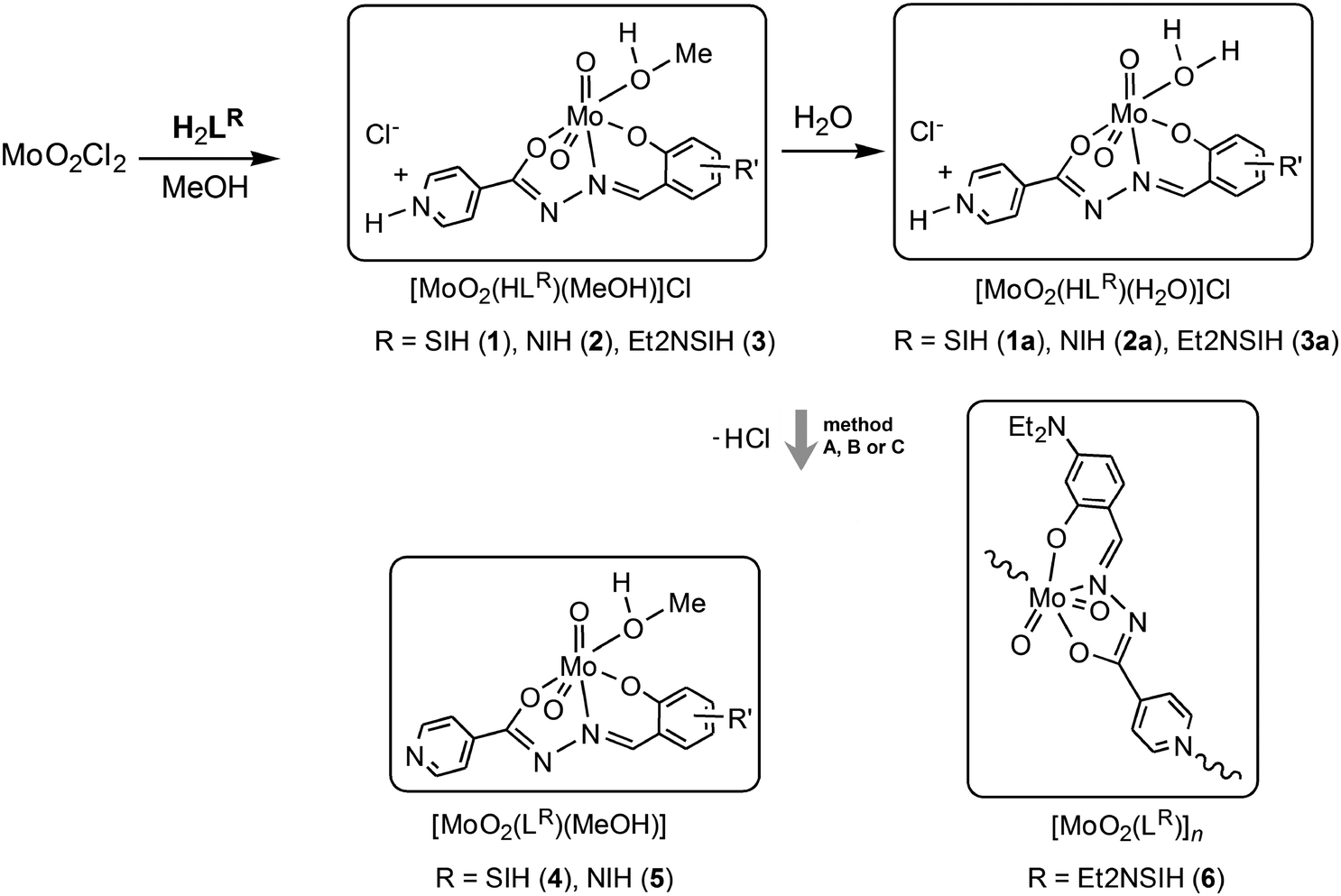 | ||
| Scheme 2 Synthesis of the complexes [MoO2(HLR)(MeOH)]Cl (1–3) and [MoO2(HLR)(H2O)]Cl (1a–3a) with singly-deprotonated ligands and synthesis of the doubly-deprotonated complexes [MoO2(LR)(MeOH)] (4 and 5) and [MoO2(LEt2NSIH)]n (6) by using Et3N as a base (by the conventional solution-based method (A), by the mechanochemical approach (B)), and by the UV-light assisted method (C). | ||
Deprotonation reactions
Treatment of [MoO2(HLR)(MeOH)]Cl (1 and 2) with a stoichiometric amount of Et3N in methanol (method A) at room temperature afforded Et3NCl and the mononuclear complexes [MoO2(LSIH)(MeOH)] (4) and [MoO2(LNIH)(MeOH)] (5), respectively. The reaction of 3 with Et3N proceeds differently and the coordination polymer [MoO2(LEt2NSIH)]n (6) was obtained without isolation of the corresponding mononuclear complex. It is expected that the deprotonation reaction leads to the formation of a labile intermediate complex with the coordinated methanol molecule, which is then displaced by the isonicotinoyl part of the neighbouring molecule. Compounds 4–6 were synthesised more easily by liquid-assisted grinding (LAG) of the complexes 1–3 and Et3N in the presence of a small amount of methanol (method B). Triethylammonium chloride impurities can be removed by rinsing the products with water affording 4–6 in a pure, chloride-free form. Compounds 4–6 can be prepared alternatively by photoassisted deprotonation of the corresponding complexes 1–3 (method C). To achieve deprotonation, mononuclear complexes were exposed to UV radiation (254 nm) in dry methanol. Absence of chloride in the prepared compounds was proven by a negative test reaction with aqueous AgNO3.In complexes 4–6, the ligands are coordinated tridentately in the doubly-deprotonated form (LR)2− to the molybdenum centre. The remaining sixth coordination site is occupied by the oxygen atom of the solvent methanol molecule (in 4 and 5) or by the nitrogen atom of the bridging isonicotinyl moiety of the neighboring complex (in 6). This suggests that the obtained complexes are inert towards further photoisomerization. To the best of our knowledge, deprotonation of cis-dioxidomolybdenum(VI) complexes leading to formation of the mononuclear complexes or polynuclear assemblies through a UV-light assisted reaction or by mechanochemical synthesis has not been reported so far.
Crystal and molecular structures of 5 and 6 were determined by the single crystal X-ray diffraction method. The crystals of 4 are identical to those known from the literature.11 The products obtained by methods A, B and C (Scheme 2) were examined also by PXRD, Fig. 1 (Fig. S2 and S3, see ESI†). Although grinding of 1–5 during sample preparation for PXRD experiments resulted in a partial release of the coordinated solvent molecule (about 1% according to TG measurements) this change was not significant and PXRD patterns could be used for comparison with those calculated from the structures obtained by the single crystal X-ray diffraction method.
 | ||
| Fig. 1 PXRD patterns of 3 (a and b); 6 (c–f). The colored lines indicate patterns obtained by powder diffraction, Cu Kα radiation ((c) sample obtained by method A, (d) sample obtained by method B and (e) sample obtained by method C)), while the black lines indicate patterns calculated from the X-ray single-crystal structures of the corresponding compounds. | ||
Thermogravimetric analyses
Crystals of all samples were used for TG analysis without grinding in the atmosphere of pure oxygen (at a heating rate of 5 °C min−1). TG study of the [MoO2(HLR)(MeOH)]Cl complexes revealed that three main processes occurred: desolvation, loss of a HCl molecule and decomposition. In the case of the mononuclear complexes 1 and 2 the loss of MeOH and HCl molecules proceeded in a stepwise fashion: in the range 174–204 °C and 223–276 °C for 1, 113–151 °C and 172–206 °C for 2. However, desolvation of 3 was accompanied by the release of a HCl molecule without formation of a stable intermediate product (in the range 120–194 °C). Upon further heating, the non-solvated products decomposed. During decomposition a significant mass loss occurred in the range 288–507 °C (1); 317–546 °C (2); 251–503 °C (3) and afforded MoO3 as the final residue.The thermal analysis data for the mononuclear complexes [MO2(LR)(H2O)]Cl showed a similar thermal behaviour. The mass loss for complexes 1a, 2a and 3a·H2O corresponded to the loss of H2O and HCl molecules (in the range 120–142 °C and 208–249 °C for 1a, 50–105 °C and 128–246 °C for 2a, and 59–96 °C and 201–250 °C for 3a·H2O), whereas decomposition of these complexes occurred in the range 323–512 °C for 1a, 278–494 °C for 1b and 286–501 °C for 3a·H2O. Complexes 4 and 5 exhibited a first mass loss (in the range 158–181 °C for 4, and 161–182 °C for 5) corresponding to the release of the coordinated methanol molecule, whereas decomposition of the complexes occurred in the range 349–477 °C for 4, and 304–500 °C for 5. For the polynuclear complex, the mass loss was observed only in the temperature range 254–474 °C (6).
Spectroscopic characterisation
All complexes were characterized also by IR spectral data. Compounds were identified by the appearance of the stretching frequencies characteristic for νasym(MoO2) (found at about 945–935 cm−1) and antisymmetric combination of MoO and Mo–OEtOH stretchings (found at ca. 910 cm−1 for 1–5). The corresponding symmetric stretching bands νsym(MoO2) having significantly lower intensities appear in the same region but they either overlap with the asymmetric ones or appear as shoulders.12 The bands found in the IR spectra of H2LR, characteristic for the CO vibration at ca. 1680 cm−1 and N–H vibration (at 3180 cm−1 (H2LSIH), 3223 cm−1 (H2LNIH), 3215 cm−1 (H2LEt2NSIH)), are absent in the IR spectra of 1–3, suggesting the hydrazone tautomerism (N–NH–(CO)– → N–N(C–OH)–), deprotonation and coordination through the oxygen atom. This is also supported by the presence of a new band at ca. 1330 cm−1 assigned to the C–O group of the hydrazone moiety.13 The bands typical for CNimine and C–Ophenolic appear at ca. 1610 cm−1 and 1550 cm−1, respectively. A band at around 1050 cm−1 seen in the IR spectra of 1–5 is assigned to the C–O stretching vibration of the coordinated MeOH molecule.14 This band is absent in the spectra of 1a–3a and 6. Formation of the Mo–Nisonicotinyl bond is additionally supported by the presence of a band at 907 cm−1 assigned to νasym(OMo–Nisonicotinyl) and absence of a broad band at ≈850 cm−1 characteristic for an intermolecular MoO⋯Mo interaction.15
The NMR analysis has confirmed chemical structures of all compounds in solution. The proton and carbon chemical shifts (Tables S1–S3, see ESI†) were assigned by using one (1H and APT) and two-dimensional NMR experiments (COSY, HSQC and HMBC). The proton spectra of the ligand molecules displayed signals resonating at ∼11 ppm and ∼12 ppm which were assigned to OH and NNH protons, respectively (Fig. 2). The signals were somewhat broadened indicating their involvement in hydrogen bonding interactions.
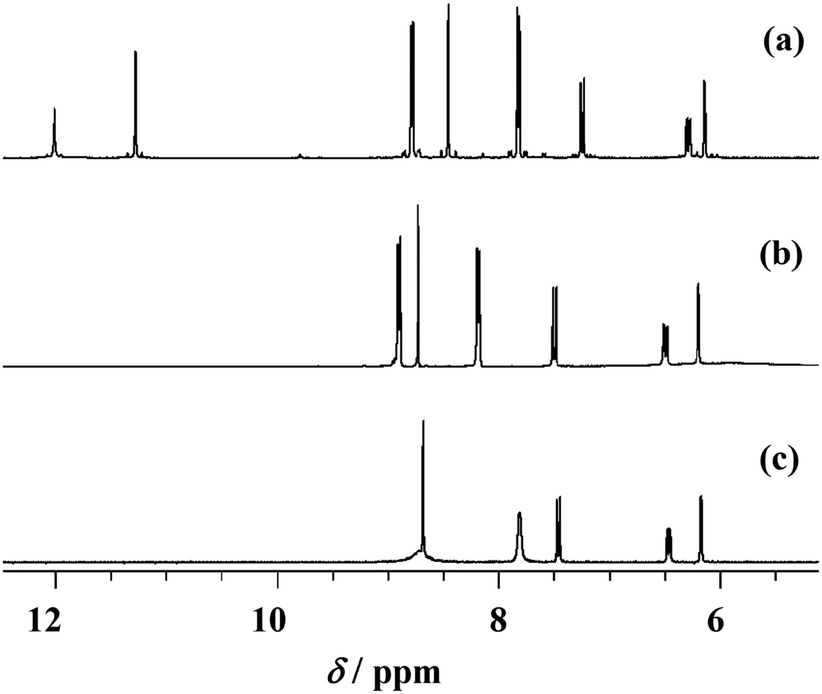 | ||
| Fig. 2 1H NMR spectra of H2LEt2NSIH (a), 3 (b) and 6 (c) in DMSO-d6. | ||
In the compounds [MoO2(HLR)(MeOH)]Cl (1–3) these signals disappeared indicating their deprotonation upon coordination of the ligand to molybdenum. Simultaneously, the pyridine nitrogen became protonated which is supported by the appearance of new signals at 3.17–4.66 ppm (Tables S1–S3, see ESI†). As a consequence of these reactions the neighboring atoms were also affected resulting in changes in their chemical shifts. In compounds [MoO2(LSIH)(MeOH)] (4), [MoO2(LNIH)(MeOH)] (5) and [MoO2(LEt2NSIH)]n (6) the signal belonging to the pyN–H+ moiety is absent indicating full deprotonation and formation of new molybdenum complexes. That was confirmed by up- and down-field shifts observed for neighboring hydrogen and carbon atoms (as can be noticed in Fig. 2), as a result of electron redistribution upon complexation. The largest effects were detected in compound 6 for carbons C-5 and C-7,9 amounting to 5.42 ppm and −5.15 ppm, respectively. The atoms C-6,10, C-4 and C-1 were also affected but to a smaller extent. These findings are in accordance with the crystal structures obtained for these compounds.
Crystallographic studies
The ligand coordinates the metal centre of the cis-MoO22+ core tridentately via the phenolic-oxygen, azomethine-nitrogen and isonicotinic-oxygen forming five and six membered chelate rings in all complexes. The sixth coordination molybdenum site is occupied by the oxygen atom from the solvent (methanol or water) thus forming mononuclear complexes 1–3 (Fig. 3), 1a, 2a, 3a·H2O (Fig. 4) and 5 (Fig. 5(a)). An exception is the coordination polymer 6 (Fig. 5(b)) where the coordination sphere is completed by the nitrogen atom of the isonicotinyl moiety of a neighboring molybdenum complex. This moiety acts as a linker between the molybdenum centers and enables formation of a larger structural assembly, a 1D zig-zag coordination polymer. | ||
| Fig. 3 ORTEP drawings of the ions in 1 (a), 2 (b) and 3 (c) with the atom-labeling scheme (displacement ellipsoids of non-hydrogen atoms are drawn at the 50% probability level). | ||
 | ||
| Fig. 4 ORTEP drawings of the ions in 1a (a), 2a (b) and 3a·H2O (c) with the atom-labeling scheme (displacement ellipsoids of non-hydrogen atoms are drawn at the 50% probability level). | ||
 | ||
| Fig. 5 ORTEP drawings of the molecules of 5 (a) and 6 (b) with the atom-labeling scheme (displacement ellipsoids of non-hydrogen atoms are drawn at the 50% probability level). | ||
In all reported complexes the coordination sphere of molybdenum is a distorted octahedron (Tables 1 and 2). The smallest cis-angle at the Mo atom is that of O1–Mo–N1 being in the range from 71.63(5)° in 1 to 72.42(8)° in 2a, while the largest one involves the oxo-oxygen atoms O3–Mo–O4 being in the range from 105.21(7)° in 1 to 105.99(9)° in 5.
| 1 | 2 | 3 | 5 | 6 | 1a | 2a | 3a·H2O | |
|---|---|---|---|---|---|---|---|---|
| Mo–O1 | 2.0046(13) | 1.9976(15) | 2.021(2) | 2.0083(17) | 2.0218(17) | 2.0141(18) | 2.0156(19) | 2.0309(12) |
| Mo–O2 | 1.9221(14) | 1.9275(16) | 1.911(2) | 1.9439(18) | 1.9399(19) | 1.9183(17) | 1.9539(17) | 1.9316(11) |
| Mo–O3 | 1.6965(15) | 1.6847(16) | 1.685(2) | 1.6951(18) | 1.689(2) | 1.687(2) | 1.746(2) | 1.6949(17) |
| Mo–O4 | 1.7039(13) | 1.6921(17) | 1.709(2) | 1.7044(18) | 1.7069(19) | 1.6969(18) | 1.7026(18) | 1.7134(13) |
| Mo–O5 | 2.3363(15) | 2.3828(16) | 2.400(3) | 2.3538(18) | — | 2.381(2) | 2.288(2) | 2.3503(17) |
| Mo–N1 | 2.2485(15) | 2.2399(19) | 2.213(2) | 2.228(2) | 2.211(2) | 2.2431(19) | 2.232(3) | 2.2224(13) |
| Mo–N3 | — | — | — | — | 2.531(2) | — | — | — |
| C1–N1 | 1.291(2) | 1.297(3) | 1.303(4) | 1.290(3) | 1.298(3) | 1.288(3) | 1.293(4) | 1.297(2) |
| N1–N2 | 1.405(2) | 1.397(3) | 1.388(3) | 1.398(3) | 1.395(3) | 1.395(3) | 1.396(3) | 1.3955(19) |
| N2–C2 | 1.293(2) | 1.289(3) | 1.297(4) | 1.294(3) | 1.289(3) | 1.292(3) | 1.294(4) | 1.294(2) |
| 1 | 2 | 3 | 5 | 6 | 1a | 2a | 3a·H2O | |
|---|---|---|---|---|---|---|---|---|
| O1–Mo–O2 | 151.42(5) | 148.37(6) | 151.43(8) | 147.62(7) | 149.65(8) | 149.24(8) | 148.88(7) | 149.96(5) |
| O1–Mo–O3 | 96.27(6) | 97.39(8) | 94.46(9) | 99.39(8) | 98.67(9) | 97.07(9) | 97.77(8) | 97.68(7) |
| O1–Mo–O4 | 95.91(6) | 97.79(7) | 95.31(9) | 96.56(8) | 95.90(8) | 95.92(8) | 97.96(8) | 94.97(6) |
| O1–Mo–O5 | 81.73(5) | 80.88(6) | 80.62(9) | 78.06(7) | — | 79.27(7) | 80.68(8) | 80.44(6) |
| O1–Mo–N1 | 71.63(5) | 71.78(6) | 71.85(8) | 72.41(7) | 72.15(7) | 71.87(7) | 72.42(8) | 71.76(5) |
| O1–Mo–N3 | — | — | — | — | 81.34(7) | — | — | — |
| O2–Mo–O3 | 97.82(7) | 99.55(8) | 100.61(10) | 99.42(8) | 98.54(9) | 99.65(9) | 97.45(8) | 99.37(7) |
| O2–Mo–O4 | 104.16(6) | 102.92(8) | 103.78(9) | 103.25(8) | 103.38(9) | 103.98(8) | 103.83(8) | 104.09(5) |
| O2–Mo–O5 | 80.93(6) | 77.52(6) | 82.06(9) | 79.10(7) | — | 80.35(8) | 80.36(8) | 79.12(5) |
| O2–Mo–N1 | 81.90(5) | 80.67(7) | 81.66(8) | 79.88(7) | 81.58(8) | 80.78(7) | 79.71(8) | 82.52(5) |
| O2–Mo–N3 | — | — | — | — | 79.10(7) | — | — | — |
| O3–Mo–O4 | 105.21(7) | 105.94(8) | 105.34(10) | 105.99(9) | 105.35(9) | 105.92(10) | 105.61(9) | 105.28(8) |
| O3–Mo–O5 | 172.16(6) | 169.25(8) | 173.27(10) | 170.63(8) | — | 171.20(8) | 171.71(9) | 171.68(6) |
| O3–Mo–N1 | 97.70(6) | 93.48(8) | 100.02(9) | 95.07(8) | 95.10(9) | 96.17(9) | 93.45(8) | 93.96(6) |
| O3–Mo–N3 | — | — | 174.27(8) | — | — | — |
The distance from the molybdenum atom to the O atom from the solvent molecule (1–3, 5, 1a, 2a, 3a·H2O) or the N atom of the isonicotinyl moiety (6) represents the largest bond length within the octahedron. The ligand is singly deprotonated in compounds 1–3 and 1a, 2a and 3a·H2O which are all chloride salts, whereas in compounds 5 and 6 it is doubly deprotonated resulting in neutral complexes. The ligands are not planar, the smallest and largest deviation from planarity is that between the isonicotinyl and phenyl/naphthaldehyde moieties in compounds 6 (3.85(12)°) and 2, respectively (8.47(9)°, Table S4, see ESI†), and between the five- and six-membered chelate rings in compounds 3 (5.20(10)°) and 5 (8.32(9)°, Table S4, ESI†). The C1–N1 bond in the complexes is not significantly different from that in the free ligands.16,17 However, the bond length N2−C2 is shortened, whereas that of N1−N2 is lengthened in the complexes in comparison to the free ligands H2LNIH (1.357(3) Å and 1.370(2) Å, respectively)16 and H2LSIH (1.3558(17) Å and 1.3699(15) Å, respectively)17 due to the electron delocalization.
Complexes 1, 2 and 3 (Fig. 6) have two hydrogen bond donors, the methanol hydroxyl group and the protonated nitrogen atom of the isonicotinyl moiety. They are both involved in hydrogen bonding to the chloride ion (Table S5, see ESI†). Hydrogen bonds of the type N−H⋯Cl are in the range from 2.988(3) Å in 3 to 3.0199(16) Å in 1, and are shorter than those involving the hydroxyl group, O−H⋯Cl which are from 3.057(3) Å in 3 to 3.0936(16) Å in 1.
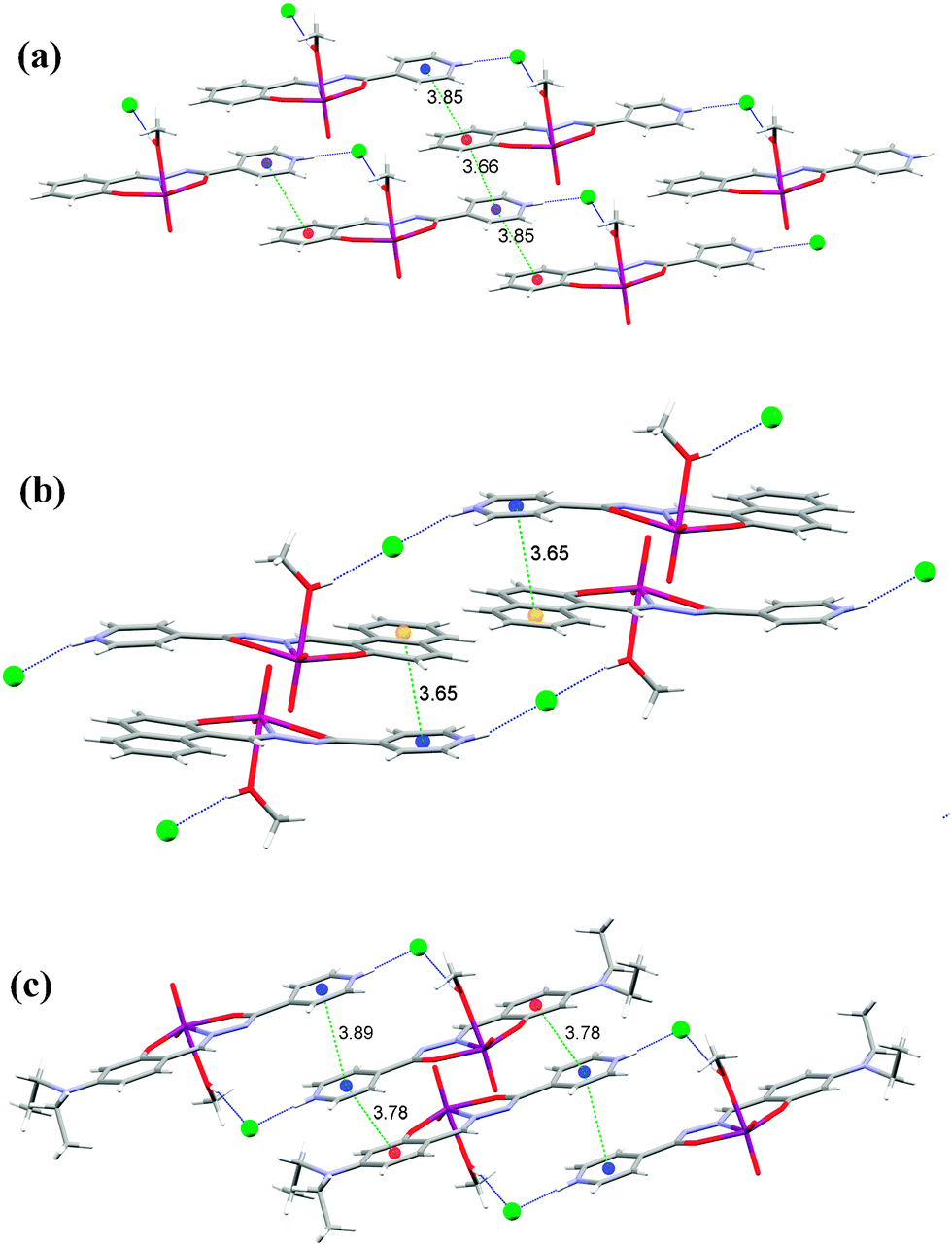 | ||
| Fig. 6 Nonbonding interactions connecting the ions into supramolecular motifs in 1 (a), 2 (b) and 3 (c). Hydrogen bonds are shown by blue dotted lines. The distance between centroids (green dashed lines) is in Å. | ||
The longer hydrogen bonds are formed in 1 and 2 where the hydrogen bonds connect the complex molecules into infinite one-dimensional chains (C12(11)) parallel to the c-axis, in contrast to 3 which is built up of centrosymmetrical dimers (R24(22)). Therefore, in these three complexes the typical hydrogen bonding connection is N−H⋯Cl⋯H−O, which is either bent (1 and 3) or linear (2). It seems that the linear connection is more favorable for a shorter distance between the π systems (Fig. 6).
In 1 there are π⋯π interactions Cg3⋯Cg4[x, y, 1 + z] of 3.6597(11) Å and Cg3⋯Cg4[1 + x, y, 1 + z] of 3.8484(11) Å between the isonicotinyl (Cg3 is the centroid of the ring N3, C3−C7) and the phenyl moieties (Cg4 is the centroid of the ring C8−C13) (Fig. 6(a) and Table S6 see ESI†). This centroid labeling is valid for all complexes. In 2 the chains are held together by π⋯π interactions between the isonicotinyl and the naphthaldehyde moieties, Cg3⋯Cg4[2 − x,1 − y, z] of 3.7770(14) Å and Cg3⋯Cg5[2 − x,1 − y, z] of 3.6530(15) Å. Cg5 is the centroid of the outer phenyl ring of the naphthaldehyde moiety (C10, C11, C14−C17), which forms a shorter contact and is shown in Fig. 6(b) (Table S6, ESI†). Complex 3 has the shortest hydrogen bonds among complexes 1–3 but the weakest π⋯π interactions due to the bulky diethylamino substituent on the salicylaldehyde moiety. These contacts amount to Cg3⋯Cg4[1 − x, −y, 1 − z] of 3.7879(19) Å and Cg3⋯Cg3[1 − x, −y, −z] of 3.8898(18) Å (Fig. 6(c) and Table S6, ESI†).
Complexes 1a, 2a and 3a·H2O (Fig. 7) have a water molecule coordinating the molybdenum atom and so there is one more hydrogen atom donor than in 1–3 thus forming a more extensive hydrogen bonding network. Indeed, both hydrogen atoms are involved as donors, in 1a and 2a toward chloride ions, while in 3a·H2O one is toward a chloride ion and the other toward the solvent water molecule. The presence of the solvent water molecules increases the number of hydrogen bonds that are found in the structure 3a·H2O. The chloride ion is an acceptor of three (1a and 3a·H2O) or four (2a) hydrogen bonds.
 | ||
| Fig. 7 Nonbonding interactions connecting the ions into supramolecular motifs in 1a (a), 2a (b) and 3a·H2O (c). Hydrogen bonds are shown by blue dotted lines. The distance between centroids (green dashed lines) is in Å. | ||
The O−H⋯Cl bonds are in the range from 3.044(3) Å in 2a to 3.1729(18) Å in 3a·H2O, similarly as in 1–3. Hydrogen bonds of the type N−H⋯Cl are longer than in 1–3 and range from 3.053(3) Å in 1a to 3.394(2) Å in 2a. The supramolecular motifs that are formed are layers parallel to (100) made up of interconnected rings R24(22) in 1a (Fig. 7(a)), a three-dimensional network in 2a (Fig. 7(b)), and double-layers parallel to (100) in 3a·H2O (Fig. 7(c)). Inside the double layer is the hydrophilic part while the bulky diethylamino substituents on the salicylaldehyde moiety extend into the hydrophobic part. π⋯π interactions are present only in 2a Cg5⋯Cg3[1 − x, 1 − y, 2 − z] of 3.5529(16) Å and Cg4⋯Cg3[1 − x, 1 − y, 2 − z] of 3.5855(15) Å, again with better stacking of Cg5⋯Cg3 as in 2 (Table S6, ESI†).
Interestingly, only two ionic chloride structures of molybdenum complexes with ONO-donor ligands were found in the Cambridge Structural Database:18cis-dioxido-methanol-(N-salicylidene-N′-(pyridinium)ethylidenehydrazone)-molybdenum(VI) chloride,10b and (3-(salicylideneiminato)-2-(2-hydroxyphenyl)piperidinum-3-carboxylato-N,O,O′)-methanol-dioxido-molybdenum(VI) chloride methanol solvate.10a
Complexes 5 and 6 are deprotonated and chloride free 2 and 3, respectively. In 5 the molecules are connected by hydrogen bonds into helical chains around the 41 axis involving the methanol hydroxyl group and the deprotonated isonicotinyl nitrogen O−H⋯N of 2.709(3) Å. The chains are connected by π⋯π interactions between the isonicotinyl and naphthaldehyde moieties with the strongest interactions between centroids Cg3⋯Cg5[1 − x, −y, −z] of 3.7563(15) Å suggesting that π⋯π interactions are weaker than in 2 (Fig. 8a and Table S6, ESI†). The only complex with no classical hydrogen bonds within this study is 6. The polymeric zig-zag chains are parallel to the b-axis (Fig. 8(b)). There are only van der Waals interactions between the chains. Coordination polymers with such linking through the nitrogen atom are very rare. A methanol solvate of 6 has been recently reported.9 It is also polymeric without hydrogen bonds between the solvent methanol molecules and the complex molecules. Interestingly, the two diethyl substituents are in a trans position whereas in all our structures they are cis to each other. Two other coordination polymers with the linker being the isonicotinyl moiety have been described previously.6,7 Packing diagrams of all complexes are given in Fig. S4 to S11, see ESI.†
 | ||
| Fig. 8 (a) Nonbonding interactions connecting the molecules into a 3D-network in 5. Hydrogen bonds are shown by blue dotted lines. The distance between centroids is in Å; (b) two polymeric chains in 6. | ||
Conclusions
Substitution of the chloride ligands in MoO2Cl2 by the corresponding aroylhydrazone ligand H2LR in methanol gives rise to formation of the mononuclear complexes [MO2(HLR)(MeOH)]Cl (1–3). In these compounds, the ligands (HLR)− are coordinated to the cis-{MoO2}2+ core via the ONO donor atoms. We have shown that mononuclear complexes 1–3 can be readily deprotonated into the mononuclear [MoO2(HLR)(MeOH)] (4 and 5) and/or polynuclear [MoO2(LR)]n (6) complexes by using Et3N as a base, either by a conventional solution-based method or by a mechanochemical approach. Compounds 4–6 can be prepared alternatively without using a base by photoassisted deprotonation of the corresponding complexes 1–3. The introduction of UV light enables deprotonation without altering the tridentate presentation of donor atoms characteristic of this class of chelating agent. In the polynuclear complex 6, the isoniazid ligand is coordinated instead of the solvent molecule.A diversity of supramolecular achitectures are formed in the complexes by non-bonding interactions, especially hydrogen bonds and π⋯π interactions. In the singly-deprotonated ionic complexes 1–3 and 1a–3a·H2O the protonated nitrogen atom of the isonicotinyl moiety and the coordinated solvent molecules are the hydrogen bond donors while the chloride ion is the main hydrogen bond acceptor (of two hydrogen bonds in 1–3, three in 1a and 3a·H2O, and four in 2a). π⋯π interactions are found in all of these complexes except 1a and 3a·H2O. The main motifs formed are either layers (1) or chains (2, 3), whereas in 1a, 2a and 3a·H2O which has an extra hydrogen bond donor (coordinated water instead of methanol) there are either layers (1a and 3a·H2O) or a 3D-network (2a). Quite different is the doubly-deprotonated molecular complex 5 where the molecules are connected by hydrogen bonds into helical chains around the 41 axis involving the methanol hydroxyl group and the deprotonated isonicotinyl nitrogen. Weak π⋯π interactions interconnect the chains into a 3D-network. As expected, the complexes with coordinated solvent molecules have low thermal stability. In the structure of the heteronuclear complex 6 there are only van der Waals interactions between the chains. This is also the thermally most stable complex since it is polymeric without coordinated solvent molecules.
Experimental section
Preparative part. Ligands H2LSIH, H2LNIH, and H2LEt2NSIH were prepared by the reaction of isonicotinyl hydrazine (“isoniazid”) with an appropriate aldehyde according to the procedures described in the literature.16 The starting MoO2Cl2 was prepared as described in the literature.19 Methanol was dried using magnesium turnings and iodine and then distilled.Synthesis of 1–3
MoO2Cl2 (0.06 g, 0.30 mmol) was added to a solution of H2LR (0.30 mmol) in dry methanol (25 mL) at room temperature and was stirred for four hours. The solution was left at room temperature for a few days and the resulting crystalline substance was filtered, rinsed with cold methanol and dried in a desiccator up to the constant mass. The crystals lose methanol molecules upon prolonged standing at room temperature. Therefore after filtration, the crystals were transferred into a desiccator and then placed in a freezer (at −15 °C).N)py, 1609 (CN), 1545 (C–Ophenolate), 1342 (C–Oisonicotinic), 1037 (C–OMeOH), 944 (MoO2), 918, 906 (OMo–OMeOH).
N)py, 1616 (CN), 1555 (C–Ophenolic), 1334 (C–Oisonicotinic), 1028 (C–OMeOH), 945, 925 (MoO2), 907 (OMo–OMeOH).
N)py, 1608 (CN), 1547 (C–Ophenolate), 1327 (C–Oisonicotinic), 1027 (C–OMeOH), 944, 929 (MoO2), 907 (OMo–OMeOH).
Conversion of 1–3 to 1a–3a
Samples of 1–3 were exposed to water vapour under ambient conditions.N)py, 1605(CN), 1549 (C–Ophenolate), 1347 (C–Oisonicotinic), 943 (MoO2), 910 (OMo–O).
N)py, 1600 (CN), 1547 (C–Ophenolate), 1332 (C–Oisonicotinic), 935 (MoO2), 907 (OMo–O).
N)py, 1613 (CN), 1549 (C–Ophenolate), 1327 (C–Oisonicotinic), 938 (MoO2), 906 (OMo–O).
Deprotonation of 1–3
N)py, 1601 (CN), 1553 (C–Ophenolate), 1338 (C–Oisonicotinic), 1062 (C–OMeOH), 938, 931 (MoO2), 912, 902 (OMo–OMeOH).
N)py, 1600 (CN), 1550 (C–Ophenolate), 1334 (C–Oisonicotinic), 1062 (C–OMeOH), 938, 927 (MoO2), 910 (OMo–OMeOH).
N), 1564 (C–Ophenolate), 1333 (C–Oisonicotinyl), 938 (MoO2), 907 (OMo–N).
All the 1H NMR spectra were recorded on a Bruker Avance 300 NMR spectrometer operating at 300 MHz for 1H and 75 MHz for 13C using a C/H dual 5 mm probe. DMSO-d6 was used as the solvent and TMS as the internal standard. Proton spectra with a spectral width of 6200 Hz and a digital resolution of 0.09 Hz per point were measured with 16-32 scans. APT spectra with spectral widths of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 030 Hz were collected with 256–1024 scans. The digital resolution was 0.11 Hz per point. In the gCOSY experiment 2046 points in the f2 dimension and 256 increments in the f1 dimension were used. For each increment 4 scans and a spectral width of 3086 Hz were applied. The digital resolution was 3.03 Hz and 24.39 Hz per point in f2 and f1 dimensions, respectively. Typical spectral conditions for gHSQC and gHMBC spectra were as follows. The spectral width was 2994 Hz in f2 and 16605 Hz in f1 dimension for both experiments. 2k data points were applied in the time domains and for each data set 256 and 128 increments were collected for gHSQC and gHMBC spectra, respectively. The resulting digital resolution was 2.94 Hz per point in f2 dimension and 125.1 Hz and 250.0 Hz per point in f1 dimension in gHSQC and gHMBC spectra, respectively. 1H and 13C chemical shifts of 1–6 are given in Tables S1–S3 (see ESI†).
030 Hz were collected with 256–1024 scans. The digital resolution was 0.11 Hz per point. In the gCOSY experiment 2046 points in the f2 dimension and 256 increments in the f1 dimension were used. For each increment 4 scans and a spectral width of 3086 Hz were applied. The digital resolution was 3.03 Hz and 24.39 Hz per point in f2 and f1 dimensions, respectively. Typical spectral conditions for gHSQC and gHMBC spectra were as follows. The spectral width was 2994 Hz in f2 and 16605 Hz in f1 dimension for both experiments. 2k data points were applied in the time domains and for each data set 256 and 128 increments were collected for gHSQC and gHMBC spectra, respectively. The resulting digital resolution was 2.94 Hz per point in f2 dimension and 125.1 Hz and 250.0 Hz per point in f1 dimension in gHSQC and gHMBC spectra, respectively. 1H and 13C chemical shifts of 1–6 are given in Tables S1–S3 (see ESI†).
X-Ray crystallography
| 1 | 2 | 3 | 5 | 6 | 1a | 2a | 3a·H2O | |
|---|---|---|---|---|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|. b wR = [∑(Fo2 − Fc2)2/∑w(Fo2)2]1/2. c S = ∑[w(Fo2 − Fc2)2/(Nobs − Nparam)]1/2. | ||||||||
| Chemical formula | C14H14ClMoN3O5 | C18H16ClMoN3O5 | C18H23ClMoN4O5 | C18H15MoN3O5 | C17H18MoN4O4 | C13H12ClMoN3O5 | C17H14ClMoN3O5 | C17H23ClMoN4O6 |
| M r | 435.67 | 485.73 | 506.83 | 449.27 | 438.29 | 421.65 | 471.70 | 510.78 |
| Crystal colour, habit | Red, prism | Red, plate | Purple-red, plate | Red, plate | Dark red, plate | Yellow, plate | Red, prism | Dark green, plate |
| Crystal size (mm3) | 0.13 × 0.30 × 0.16 | 0.46 × 0.38 × 0.20 | 0.38 × 0.26 × 0.18 | 0.34 × 0.24 × 0.10 | 0.42 × 0.28 × 0.12 | 0.04 × 0.15 × 0.39 | 0.26 × 0.09 × 0.12 | 0.07 × 0.23 × 0.09 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Tetragonal | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
I41/a | P21/n | P21/c | P21/n | P21/c |
| Unit cell parameters | ||||||||
| a (Å) | 7.48269(13) | 7.5013(5) | 9.2614(4) | 25.2633(3) | 6.59351(14) | 13.3090(5) | 8.08986(18) | 15.9580(4) |
| b (Å) | 25.6417(5) | 9.4538(9) | 9.9979(6) | 25.2633(3) | 13.0715(3) | 9.40511(19) | 17.0004(3) | 11.4043(2) |
| c (Å) | 8.45241(14) | 13.3244(7) | 11.8619(5) | 11.2412(2) | 20.2077(4) | 13.7393(4) | 13.1601(3) | 12.2325(3) |
| α (°) | 90 | 96.890(6) | 82.827(4) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 94.5659(15) | 90.248(5) | 72.104(4) | 90 | 96.718(2) | 114.964(4) | 105.619(2) | 109.225(3) |
| γ (°) | 90 | 97.558(7) | 89.482(4) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 1616.61(5) | 929.74(12) | 1036.52(9) | 7174.5(2) | 1729.68(6) | 1559.11(10) | 1743.09(7) | 2102.04(8) |
| Z | 4 | 2 | 2 | 16 | 4 | 4 | 4 | 4 |
| D calc (g cm−3) | 1.790 | 1.735 | 1.624 | 1.664 | 1.683 | 1.796 | 1.798 | 1.614 |
| Temperature (K) | 150 | 295 | 295 | 295 | 295 | 295 | 150 | 295 |
| μ (mm−1) | 1.007 | 0.885 | 0.799 | 0.767 | 0.790 | 1.041 | 0.942 | 0.792 |
| F(000) | 872 | 488 | 516 | 3616 | 888 | 840 | 944 | 1040 |
| Number of unique data | 4232 | 6216 | 5519 | 4133 | 4372 | 4535 | 4001 | 5580 |
| Number of data [Fo ≥ 4σ(Fo)] | 3972 | 4762 | 4217 | 3522 | 3592 | 3558 | 3265 | 4947 |
| Number of parameters | 222 | 289 | 321 | 248 | 267 | 220 | 252 | 280 |
| R 1 , [Fo ≥ 4σ(Fo)] | 0.0255 | 0.0336 | 0.0435 | 0.0343 | 0.0321 | 0.0337 | 0.0357 | 0.0248 |
| wR2b | 0.0635 | 0.0921 | 0.1101 | 0.0668 | 0.0851 | 0.0730 | 0.0780 | 0.0709 |
| Goodness of fit on F2, Sc | 1.17 | 1.05 | 0.93 | 1.09 | 0.94 | 1.05 | 1.05 | 0.95 |
| Min. and max. electron density (e Å−3) | −0.74, 0.34 | −0.78, 0.64 | −0.99, 1.10 | −0.36, 0.35 | −0.50, 0.64 | −0.55, 0.40 | −0.62, 0.68 | −0.36, 0.42 |
Acknowledgements
Financial support for this research was provided by Ministry of Science and Technology of the Republic of Croatia.Notes and references
- (a) S. Banerjee, A. Ray, S. Sen, S. Mitra, D. L. Hughes, R. J. Butcher, S. R. Batten and D. R. Turner, Inorg. Chim. Acta, 2008, 361, 2692 CrossRef CAS PubMed; (b) J. G. Vos and M. T. Pryce, Coord. Chem. Rev., 2010, 254, 2519 CrossRef CAS PubMed; (c) A. Kobayashi, D. Yamamoto, H. Horiki, K. Sawaguchi, T. Matsumoto, K. Nakajima, H.-C. Chang and M. Kato, Inorg. Chem., 2014, 53, 2573 CrossRef CAS PubMed.
- M. Chang, A. Kobayashi, K. Nakajima, H.-C. Chang and M. Kato, Inorg. Chem., 2011, 50, 8308 CrossRef CAS PubMed.
- (a) S. Taktak, W. Ye, A. M. Herrera and E. V. Rybak-Akimova, Inorg. Chem., 2007, 46, 2929 CrossRef CAS PubMed; (b) M. Haga, M. Ali, S. Koseki, K. Fujimoto, A. Yoshimura, K. Nozaki, T. Ohno, K. Nakajima and D. Stufkens, Inorg. Chem., 1996, 35, 3335 CrossRef CAS PubMed; (c) S. Klein, W. G. Dougherty, W. S. Kassel, T. J. Dudley and J. J. Paul, Inorg. Chem., 2011, 50, 2754 CrossRef CAS PubMed.
- (a) S. M. Landge and I. Aprahamian, J. Am. Chem. Soc., 2009, 131, 18269 CrossRef CAS PubMed; (b) X. Su, T. F. Robbins and I. Aprahamian, Angew. Chem., Int. Ed., 2011, 50, 1841 CrossRef CAS PubMed.
- (a) M. C. Rodríguez-Argüelles, S. Mosquera-Vázquez, P. Tourón-Touceda, J. Sanmartín-Matalobos, A. M. García-Deibe, M. Belicchi Ferrari, G. Pelosi, C. Pelizzi and F. Zani, J. Inorg. Biochem., 2007, 101, 138 CrossRef PubMed; (b) D. S. Kalinowski, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2008, 51, 331 CrossRef CAS PubMed.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, R. Dreos, P. Siega and C. Tavagnacco, Cryst. Growth Des., 2010, 10, 1373 CAS.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, J. Pisk, R. Dreos and P. Siega, Cryst. Growth Des., 2011, 11, 1244 CAS.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, T. Hrenar, R. Dreos and P. Siega, Cryst. Growth Des., 2013, 13, 3773 CAS.
- W. X. Xu and W. H. Li, Koord. Khim., 2012, 38, 98 Search PubMed.
- (a) M. Cindrić, G. Galin, D. Matković-Čalogović, P. Novak, T. Hrenar, I. Ljubić and T. K. Novak, Polyhedron, 2009, 28, 562 CrossRef PubMed; (b) H. X. Liu and X. M. Wang, Polyhedron, 1994, 13, 441 CrossRef CAS.
- (a) S. Gao, L.-H. Huo, H. Zhao and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1757 CAS; (b) Y.-L. Zhai, X.-X. Xu and X. Wang, Polyhedron, 1992, 11, 415 CrossRef CAS; (c) H. D. Yin, M. Hong, G. Li and D. Q. Wang, J. Org. Chem., 2005, 690, 3714 CrossRef CAS PubMed; (d) I. I. Seifullina, N. V. Shmatkova and Z. A. Starikova, Russ. J. Inorg. Chem., 2001, 46, 1150 Search PubMed.
- O. A. Rajan and A. Chakratvorty, Inorg. Chem., 1981, 20, 660 CrossRef CAS.
- M. R. Maurya, S. Agarwal, C. Bader and D. Rehder, Eur. J. Inorg. Chem., 2005, 147 CrossRef CAS PubMed.
- M. R. Maurya, S. Gopinathan, C. Gopinathan and R. C. Maurya, Polyhedron, 1993, 12, 159 CrossRef CAS.
- (a) K. Nakajima, K. Yokoyama, T. Kano and M. Kojima, Inorg. Chim. Acta, 1998, 282, 209 CrossRef CAS; (b) J. M. Berg and R. H. Holm, Inorg. Chem., 1983, 22, 1768 CrossRef CAS.
- D. R. Richardson and P. V. Bernhardt, J. Biol. Inorg. Chem., 1999, 4, 266 CrossRef CAS.
- Y.-J. Xu, S. Zhao and S. Bi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o4633 CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- R. L. Graham and L. G. Helper, J. Phys. Chem., 1959, 63, 723 CrossRef CAS.
- X'Pert Software Suite, Version 1.3e, Panalytical B. V. Almelo, The Netherlands, 2001 Search PubMed.
- CrysAlisPro, Version 1.171.37.33, Agilent Technologies, (release 27-03-2014 CrysAlis171.NET) Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- G. M. Sheldrick, SHELXS97, Program for the Solution of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, M. Towler, J. Van der Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) XRPD patterns, (2) spectral data, (3) tables and (4) figures for compounds. CCDC 1062693–1062700. See DOI: 10.1039/c5nj01567g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |