
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A structural investigation of heteroleptic lanthanide substituted cyclopentadienyl complexes†‡
Fabrizio
Ortu
,
Jonathan M.
Fowler
,
Matthew
Burton
,
Alasdair
Formanuik
and
David P.
Mills
*
School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.mills@manchester.ac.uk
First published on 12th May 2015
Abstract
The substituted cyclopentadienyl group 1 transfer agents KCp′′, KCp′′′ and KCptt (Cp′′ = {C5H3(SiMe3)2-1,3}−; Cp′′′ = {C5H2(SiMe3)3-1,2,4}−; Cptt = {C5H3(tBu)2-1,3}−) were prepared by modification of established procedures and the structure of [K(Cp′′)(THF)]∞·THF (1) was obtained. KCp′′ and KCptt were reacted variously with [Ln(I)3(THF)4] (Ln = La, Ce) in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometries to afford monomeric [La(Cp′′)2(I)(THF)] (2a·THF) and the dimeric complexes [La(Cp′′)2(μ-I)]2 (2a), [Ce(Cp′′)2(μ-I)]2 (2b) and [Ce(Cptt)2(μ-I)]2 (3). KCp′′′ was reacted with [Ce(I)3(THF)4] to afford the mono-ring complex [Ce(Cp′′′)(I)2(THF)2] (4), regardless of the stoichiometric ratio of the reagents. Complex 4 was reacted with [KN(SiMe3)2] to yield [Ce(Cp′′′)2(I)(THF)] (5), [Ce(Cp′′′){N(SiMe3)2}2] (6) and [Ce{N(SiMe3)2}3] by ligand scrambling. Complexes 1–6 have all been structurally authenticated and are variously characterised by other physical methods.
:1 stoichiometries to afford monomeric [La(Cp′′)2(I)(THF)] (2a·THF) and the dimeric complexes [La(Cp′′)2(μ-I)]2 (2a), [Ce(Cp′′)2(μ-I)]2 (2b) and [Ce(Cptt)2(μ-I)]2 (3). KCp′′′ was reacted with [Ce(I)3(THF)4] to afford the mono-ring complex [Ce(Cp′′′)(I)2(THF)2] (4), regardless of the stoichiometric ratio of the reagents. Complex 4 was reacted with [KN(SiMe3)2] to yield [Ce(Cp′′′)2(I)(THF)] (5), [Ce(Cp′′′){N(SiMe3)2}2] (6) and [Ce{N(SiMe3)2}3] by ligand scrambling. Complexes 1–6 have all been structurally authenticated and are variously characterised by other physical methods.
Introduction
Since the first reported synthesis of lanthanide (Ln) cyclopentadienyl (Cp) complexes in the 1950s,1 substituted Cp ligands, CpR, in which up to five ring protons have been replaced by various R-groups, have been employed ubiquitously in f-element organometallic chemistry.2 CpR ligands typically occupy three coordination sites and their steric demands are readily tunable. Bulky R-groups may be used to block undesired ligand scrambling and oligomerisation decomposition pathways. This, together with the electronically stabilising multihapto-donor properties of CpR ligands, has been exploited in the stabilisation of Ln CpR complexes that exhibit unusual Ln oxidation states and bonding modes.3 CpR ligands have been shown to be particularly effective in stabilising heterobimetallic systems that contain Ln–transition metal (TM) bonds, in which the two fragments are supported by the metal–metal interaction.4,5 There are currently very few examples of structurally characterised Ln–TM bonds,6 and given that recent landmark metal–metal bonds have provided step changes in our understanding of chemical bonding7 it would be considered prudent to explore novel Ln–TM systems.A well-established synthetic route for the synthesis of heterobimetallic Ln–TM complexes is alkane elimination.4c However, alkali metal salt elimination pathways provide a useful synthetic alternative that can be less sluggish and produce fewer byproducts over alkane elimination in some cases.4b,5b It follows that novel heteroleptic [Ln(CpR)2(X)] (X = halide) complexes that offer significant kinetic stabilisation and are robust with respect to ligand exchange could be useful precursors for supporting novel Ln-element bonding motifs in the future. For the larger lanthanides (La, Ce, Pr, Nd) there is a surprising lack of reports on structurally authenticated [Ln(CpR)2(X)] complexes,4b,8 and there are very few examples that contain bromide or iodide.9 The employment of the heavier halides is particularly advantageous in salt elimination reactions, where they are less prone to ate complex formation and can offer facile reaction work-ups due to the insolubility of salts such as KI in most organic solvents.5a,b
To target the synthesis of novel [Ln(CpR)2(I)] complexes of the larger Ln, we focused our attention on a family of established CpR ligands: 1,3-bis(trimethylsilyl)cyclopentadienyl (Cp′′, {C5H3(SiMe3)2-1,3}−), 1,2,4-tris(trimethylsilyl)cyclopenta-dienyl (Cp′′′, {C5H2(SiMe3)3-1,2,4}−) and 1,3-bis(tert-butyl)cyclo-pentadienyl (Cptt, {C5H3(tBu)2-1,3}−). These ligands have been previously utilised to prepare a variety of homoleptic and heteroleptic La and Ce complexes.3d,8b–d,10 Herein we report the reactions of the group 1 ligand transfer agents KCp′′, KCp′′′ and KCptt in salt metathesis reactions with selected Ln triiodides, leading to the stabilisation and structural authentication of novel monomeric and dimeric [Ln(CpR)2(I)] (Ln = La, Ce) complexes that are robust towards ligand exchange side-reactions over typical experimental timescales.
Results and discussion
KCp′′,11 KCp′′′11 and KCptt12 (Cp′′ = {C5H3(SiMe3)2-1,3}−; Cp′′′ = {C5H2(SiMe3)3-1,2,4}−; Cptt = {C5H3(tBu)2-1,3}−) were prepared by slight modifications of published procedures by deprotonation of the pro-ligands with KH or [KN(SiMe3)2]. On one occasion, crystals of [K(Cp′′)(THF)]∞·THF (1) were obtained, and these were subjected to a single crystal XRD study. The crystals were of poor quality and weakly diffracting, leading to a poor quality dataset. As a result no discussion is given of the metrical parameters although the connectivity is clear-cut (see ESI‡). KCp′′ and KCptt were reacted separately in 2:1 stoichiometric ratios with [La(I)3(THF)4] and [Ce(I)3(THF)4] to afford [La(Cp′′)2(μ-I)]2 (2a), [Ce(Cp′′)2(μ-I)]2 (2b) and [Ce(Cptt)2(μ-I)]2 (3) (Scheme 1). The synthesis of [La(Cptt)2(μ-I)]2 was not attempted. Complexes 2a and 2b were obtained in fair crystalline yields but the yield of 3 was significantly lower. It is noteworthy that the syntheses of 2b and 3 from CeI3 were discussed previously by Andersen, although no characterisation data were reported.10h
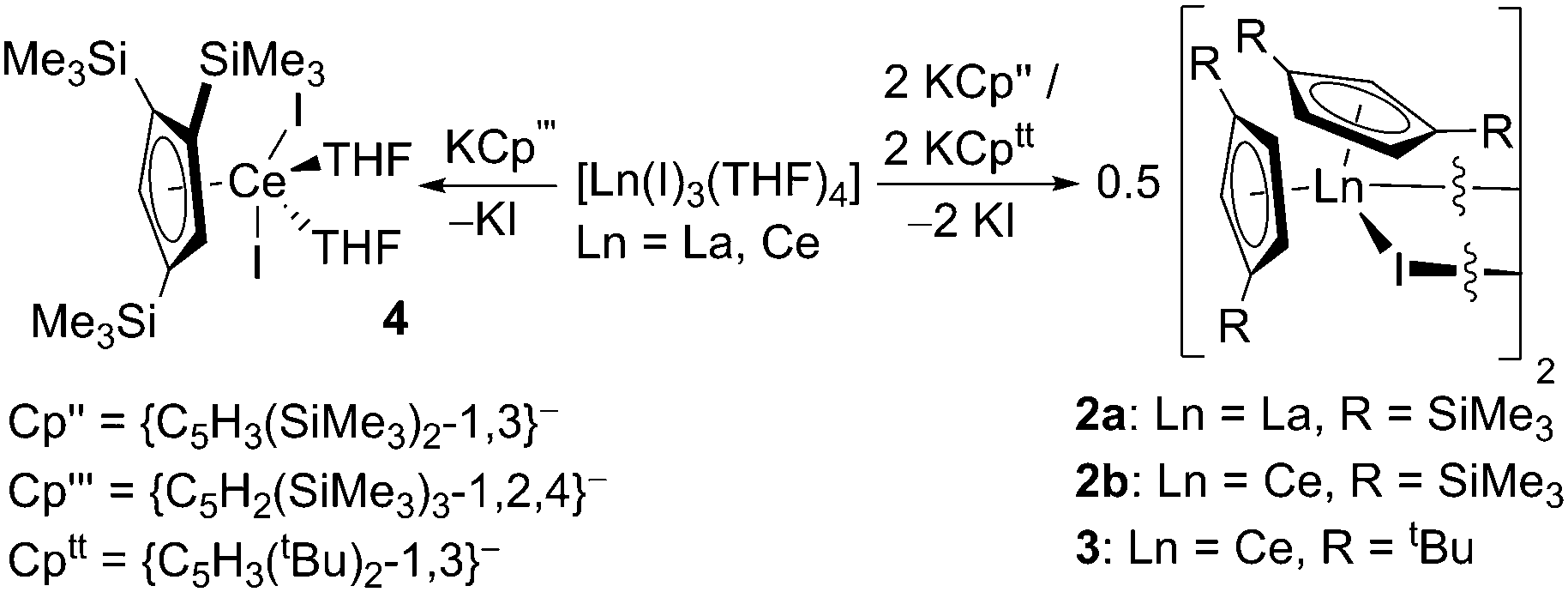 | ||
| Scheme 1 Synthesis of complexes 2–4. | ||
The 1H NMR spectra of 2a and 2b in d6-benzene exhibit resonances for the SiMe3 protons (2a: δ = 0.49 ppm; 2b: δ = −3.23 and 0.29 ppm) and three signals for the Cp′′ ring protons (2a: δ = 7.16, 7.22 and 7.27 ppm; 2b: δ = −7.30, −6.88 and −0.04 ppm). The reason for the difference in the number of SiMe3 group resonances in solution for 2a and 2b is unknown as they have similar solid state structures (see below). A single resonance was observed in the 29Si{1H} NMR spectrum of 2a (δ = −9.69 ppm), but no resonance was observed in the 29Si{1H} NMR spectrum of 2b due to paramagnetic broadening. The 1H NMR spectrum of 3 displays one broad signal for the tert-butyl protons (δ = −1.71 ppm) and three resonances (δ = −5.86 ppm, 0.29 and 1.13 ppm) for the Cptt ring protons. Elemental analyses of all three complexes are in good agreement with donor solvent-free [Ln(CpR)2(μ-I)]2 formulations.
Crystals suitable for X-ray diffraction studies were obtained for 2a, 2b and 3, and their solid state structures were determined (Fig. 1 and 2; see ESI‡). Complexes 2a and 2b are structurally analogous, featuring the same open metallocene-type array, so only the structure of 2b is depicted here (for a picture of 2a see ESI‡). The two compounds display very similar metrical parameters, such as the Ln⋯Cpcentroid [2a: La⋯Cpcentroid = 2.519(7) Å mean; 2b: Ce⋯Cpcentroid = 2.505(3) Å mean] and Ln–I distances [2a: La–I = 3.2128(16) Å mean; 2b: Ce–I = 3.1958(6) Å mean]. These values may be compared to [La(Cp′′)2(μ-F)]2,8c the only other structurally authenticated heteroleptic Cp′′ La or Ce halide-bridged dimer, to the best of our knowledge. In [La(Cp′′)2(μ-F)]2 the La⋯Cpcentroid distances [2.548(9) and 2.561(8) Å] are longer than the corresponding distances in 2a and 2b; also the Cpcentroid–Ln–Cpcentroid angle in [La(Cp′′)2(μ-F)]2 [129.42(19)°] is smaller than the analogous angles in 2a [132.8(3)°] and 2b [132.19(10)°]. Such variations are consistent with previous observations on the flexibility of coordinated CpR ligands in Ln chemistry, as they are capable of interlocking with each other in various conformations in order to best accommodate the space required by co-ligands. This behaviour was reported for a series of heteroleptic Cptt cerium complexes, which exhibited three different Cptt ring orientations in the solid state (Fig. 3A–C).8d,10i Complexes 2a and 2b adopt orientation C in the solid state and the presence of two SiMe3 resonances in the 1H NMR spectra of 2a and 2b (see above) indicates that a low-symmetry orientation is maintained in solution.
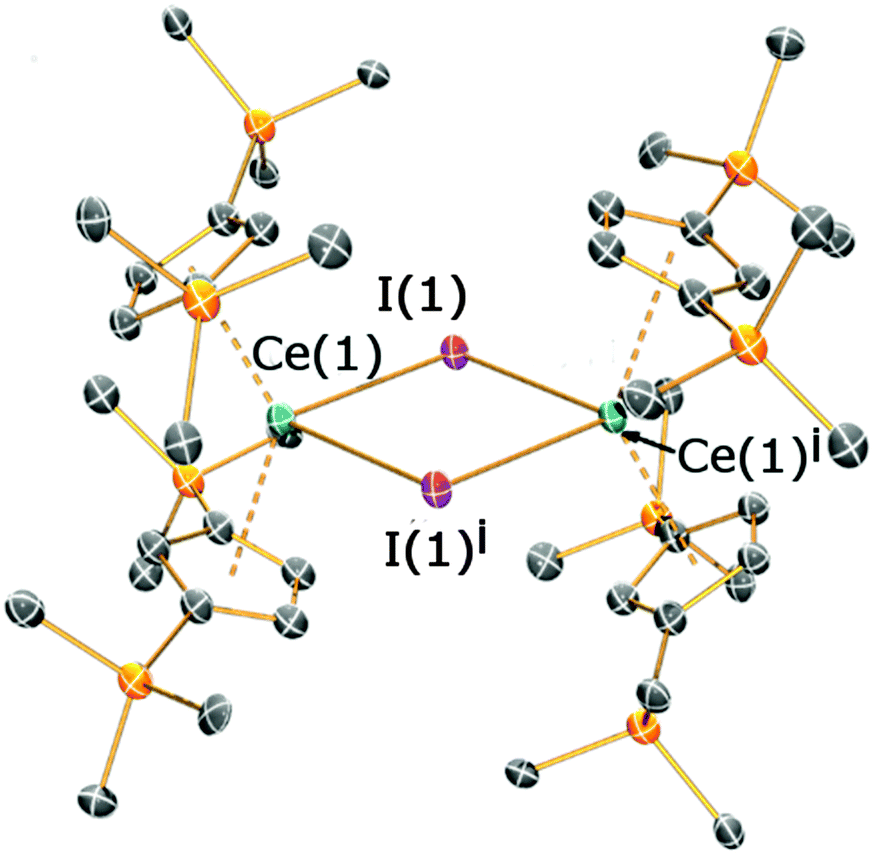 | ||
| Fig. 1 Molecular structure of 2b with selective atom labelling, with displacement ellipsoids set at the 50% probability level; hydrogen atoms have been omitted for clarity. Symmetry operation to generate equivalent atoms: i = 2 − x, −y, −z. Selected bond distances [Å] and angles [°] for 2b: Ce(1)–I(1) 3.1943(6), Ce(1)–I(1i) 3.1973(6), Ce(1)⋯Cpcentroid(1) 2.511(3), Ce(1)⋯Cpcentroid(2) 2.499(3), Ce(1)⋯Ce(1i) 4.7342(9), I(1)–Ce(1)–I(1i) 84.422(15), Ce(1)–I(1)–Ce(1i) 95.578(15). | ||
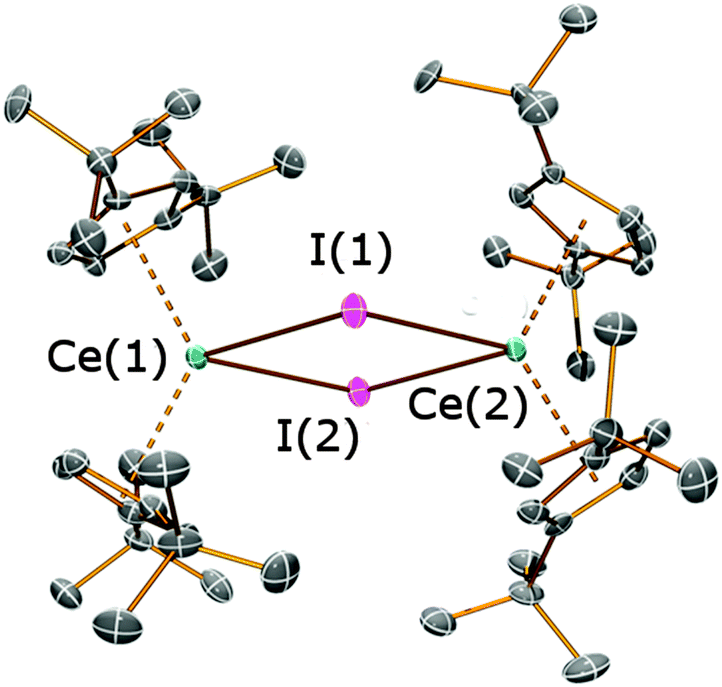 | ||
| Fig. 2 Molecular structure of 3 with selective atom labelling, with displacement ellipsoids set at the 50% probability level; hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°] for 3: Ce(1)–I(1) 3.2238(5), Ce(1)–I(2) 3.2410(4), Ce(2)–I(1) 3.2157(4), Ce(2)–I(2) 3.2286(5), Ce(1)⋯Cpcentroid(1) 2.510(2), Ce(1)⋯Cpcentroid(2) 2.516(2), Ce(1)⋯Ce(2) 5.0121(4), I(1)–Ce(1)–I(2) 77.963(11), I(1)–Ce(2)–I(2) 78.259(11), Ce(1)–I(1)–Ce(2) 102.218(12), Ce(1)–I(2)–Ce(2) 101.560(12). | ||
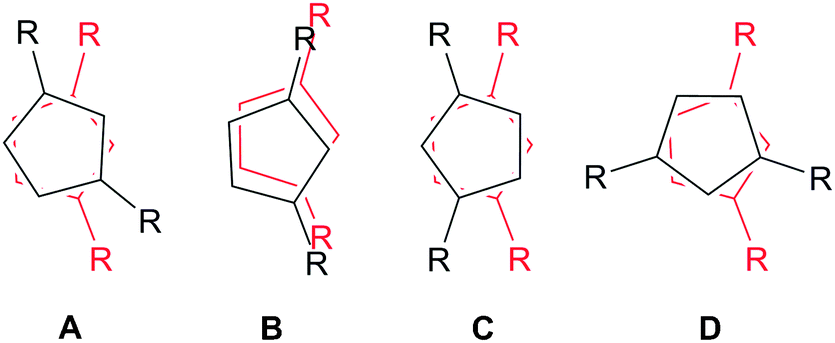 | ||
| Fig. 3 Different conformations of La and Ce open metallocenes supported by 1,3-di-substituted CpR ligands.8d,10i | ||
On one occasion in an attempted synthesis of 2a we were able to determine the molecular structure of the monomeric THF adduct [La(Cp′′)2(I)(THF)], 2a·THF (Fig. 4; see ESI‡), which resulted from incomplete removal of THF in vacuo prior to recrystallization from toluene. Complex 2a·THF displays similar La⋯Cpcentroid distances to its dimeric counterpart [La(1)⋯Cpcentroid = 2.539(4) Å mean], however the Cpcentroid–La–Cpcentroid angle in 2a·THF [127.97(12)°] is smaller. This is due to the absence of a relatively rigid La2I2 rhomboid in 2a·THF, which enables the two Cp′′ rings to interlock in a staggered conformation (Fig. 3D). To the best of our knowledge, this has not previously been observed for heteroleptic Cp′′ and Cptt lanthanum and cerium complexes, but it has been reported in Cp′′ and Cptt U(IV) chemistry.13
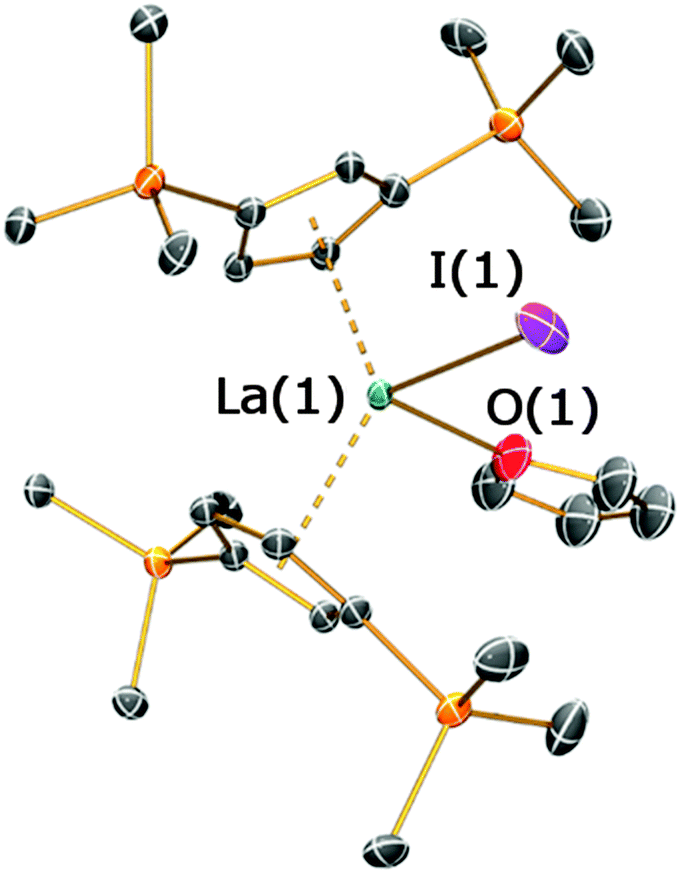 | ||
| Fig. 4 Molecular structure of 2a·THF with selective atom labelling, with displacement ellipsoids set at the 50% probability level; hydrogen atoms and disorder components have been omitted for clarity. Selected bond distances [Å] and angles [°] for 2a·THF: La(1)–I(1) 3.094(16), La(1)–O(1) 2.51(3), La(1)⋯Cpcentroid(1) 2.538(4), La(1)⋯Cpcentroid(2) 2.539(4), I(1)–La(1)–O(1) 93.0(7), Cpcentroid(1)–La(1)–Cpcentroid(2) 127.97(12). | ||
In the solid state 3 exhibits a dimeric structure, with Ce⋯Cpcentroid distances ranging between 2.510(2) and 2.528(2) Å. These distances are comparable to those observed for [Ce(Cptt)2(μ-Cl)]2 [2.523(10) Å mean] and [Ce(Cptt)2(μ-H)]2 [2.538(4) Å mean]8d,10i but are longer than those observed in [Ce(Cpt)2(μ-I)]2 [range Ce⋯Cpcentroid 2.492(5)–2.510(4) Å].9a Furthermore, the Cpcentroid–Ce–Cpcentroid angles in 3 are relatively small [124.49(7)° and 125.29(7)°], bringing the Cptt rings closer to each other than the Cp′′ rings in 2a and 2b. These angles are even smaller than those observed in 2a·THF, in which a staggered conformation of the Cptt rings was observed in the solid state, yet 3 exhibits a pseudo-eclipsed configuration of the Cptt rings (Fig. 3B). It is noteworthy that the 1H NMR spectrum of 3 exhibits only one signal for the tert-butyl protons (see above), indicating that this high symmetry environment is maintained in solution. In [Ce(Cptt)2(μ-Cl)]2 the two Cptt rings deviate even further from the ideal parallel sandwich configuration [Cpcentroid–Ce–Cpcentroid = 121.0(2)°],8d highlighting the potentially large effect of crystal packing interactions on these configurations.14
An increase in the number of sterically demanding R-groups around a CpR ring can have significant effects on its coordination to metal centres. It has previously been shown that salt metathesis reactions between group 1 salts of the more sterically encumbered Cp′′′ ligand with LaI3 afford the mono-ring complex [La(Cp′′′)(I)2(py)3] as the main product,15 with the bis-Cp′′′ derivative [La(Cp′′′)2(I)(py)] isolated in poor yields.8b Germane to this, the mono-ring complex [Ce(C5Me5)(I)2(THF)3] was synthesised from [CeI3(THF)x] and KC5Me5, whilst [Li(Et2O)2][Ce(C5Me5)2(Cl)2] was isolated when CeCl3 and LiC5Me5 were employed as starting materials.16 Similarly, the reaction of KCp′′′ with [Ce(I)3(THF)4] yielded [Ce(Cp′′′)(I)2(THF)2] (4), regardless of the stoichiometric ratio employed. The 1H NMR spectrum of 4 exhibits two SiMe3 proton resonances (δ = −3.74 and 0.27 ppm) and two signals are observed for the Cp′′′ ring protons (δ = −0.03 and 0.76 ppm). The elemental analysis of 4 was consistent with its formulation.
The solid state structure of 4 is depicted in Fig. 5 (see ESI‡). Complex 4 exhibits a four-legged piano stool coordination motif, with the two iodides and THF molecules mutually trans-with respect to each other [I(1)–Ce(1)–I(2) = 125.53(2)°; O(1)–Ce(1)–O(2) = 149.48(18)°]. The Cp′′′ ring is closer to the cerium centre [Ce(1)⋯Cpcentroid = 2.495(4) Å] than the CpR rings in 2b and 3 and this parameter is shorter than the corresponding distance in [La(Cp′′′)(I)2(py)3] (La⋯Cpcentroid = 2.557 Å).15 As a mono-ring complex typically has reduced steric crowding around the metal centre compared with the corresponding open metallocene, the Ce–I bond lengths of 4 [3.0859(7) and 3.1209(6) Å] are shorter than those observed in 2b and 3. These distances are also shorter than those exhibited by [Ce(C5Me5)(I)2(THF)3] [3.2269(12) and 3.1733(12) Å], although this complex contains an additional molecule of THF.16
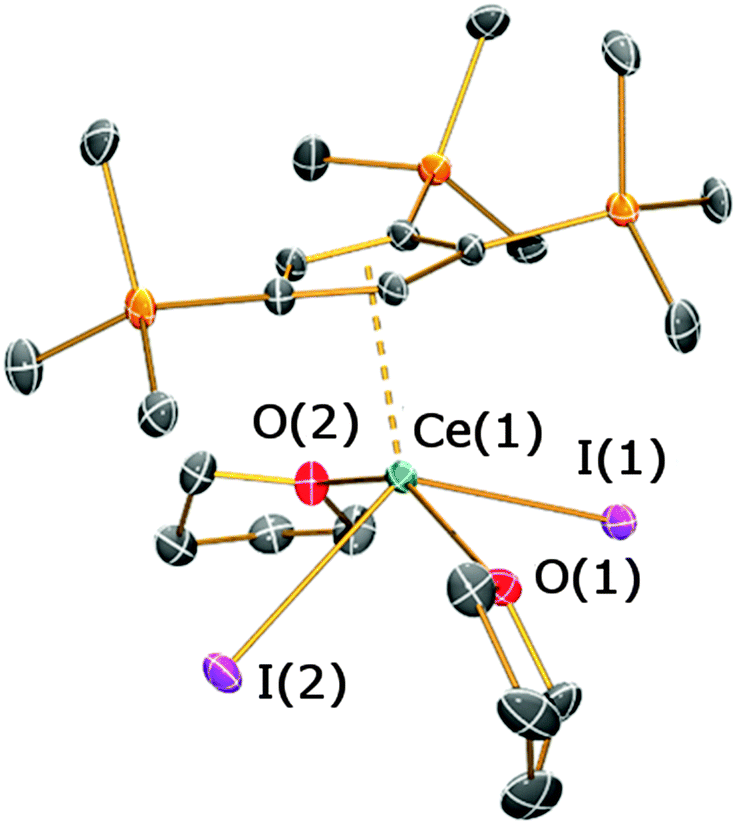 | ||
| Fig. 5 Molecular structure of 4 with selective atom labelling, with displacement ellipsoids set at the 50% probability level; hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°] for 4: Ce(1)–I(1) 3.0859(7), Ce(1)–I(2) 3.1209(6), Ce(1)–O(1) 2.495(5), Ce(1)–O(2) 2.505(5), Ce(1)⋯Cpcentroid(1) 2.495(4), I(1)–Ce(1)–I(2) 125.53(2), O(1)–Ce(1)–O(2) 149.48(18), I(1)–Ce(1)–O(1) 81.88(13), I(1)–Ce(1)–O(2) 89.39(13). | ||
In an attempt to prepare a heteroleptic Cp′′′-silylamide cerium complex, “[Ce(Cp′′′)(I){N(SiMe3)2}]”, complex 4 was reacted with one equivalent of [KN(SiMe3)2] (Scheme 2). An orange crystalline product was obtained from the reaction mixture, allowing the structural identification of [Ce(Cp′′′)2(I)(THF)] (5), which has formed by ligand scrambling (Fig. 6; see ESI‡). Complex 5 is monomeric in the solid state, analogous to the related La Cp′′′ complex [La(Cp′′′)2(I)(py)].8b The Ce–O distance in 5 [Ce(1)–O(1) = 2.490(3) Å] is consistent with those measured for 4 (see above) and the Ce⋯Cpcentroid distances of 5 [2.534(2) Å mean] are longer than those in the mono-ring complex 4, as would be expected from the increased steric demands of two Cp′′′ ligands in 5. The Cpcentroid–Ce–Cpcentroid angle in 5 [133.77(6)°] is comparable to the Cpcentroid–La–Cpcentroid angle observed for [La(Cp′′′)2(I)(py)] [134.50(7)°],8b though it is noteworthy that [Ce(CpR)2(X)]n (X = anionic ligand) complexes with larger Cpcentroid–Ce–Cpcentroid angles have been previously reported in the literature.8a,9b,c
 | ||
| Scheme 2 Synthesis of 5–6. | ||
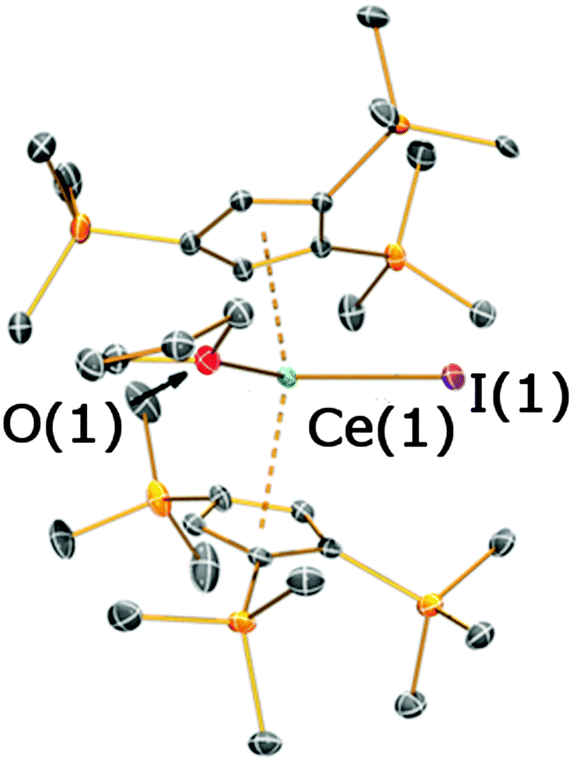 | ||
| Fig. 6 Molecular structure of 5 with selective atom labelling, with displacement ellipsoids set at the 50% probability level; hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°] for 5: Ce(1)–I(1) 3.1000(4), Ce(1)–O(1) 2.490(3), Ce(1)⋯Cpcentroid(1) 2.5416(16), Ce(1)⋯Cpcentroid(2) 2.5259(19), I(1)–Ce(1)–O(1) 89.01(7), Cpcentroid(1)–Ce(1)–Cpcentroid(2) 133.77(6). | ||
Further evidence of the steric repulsion in 5 is given by the SiMe3 substituents, which are pushed away from the cerium centre in comparison to 4 [range of Cpcentroid–C–Si angles, 5: 157.4(3)–169.4(4)°; 4: 168.1(6)–168.9(5)°] and are similar to the corresponding angles observed for [La(Cp′′′)2(I)(py)] [156.5(4)–167.4(4)°].8b Complex 5 was further characterised by 1H NMR spectroscopy, giving a simple spectrum with two resonances for the SiMe3 protons (δ = 0.30 and 0.90 ppm) and two signals for the Cp′′′ protons (δ = −5.75 and −5.34 ppm). The elemental analysis of 5 was in good agreement with the proposed formulation.
In order to better understand the ligand scrambling process which led to the formation of 5, several crystals obtained from the third crop of 5 were screened by single crystal XRD. Two additional crystal types were observed: orange needles and dark orange blocks. The needles were identified as [Ce{N(SiMe3)2}3] and the blocks were found to be the heteroleptic complex [Ce(Cp′′′){N(SiMe3)2}2] (6) (Fig. 7; see ESI‡). The structure of 6 is based on a two-legged piano stool motif and is comparable to [Ce(C5Me5){N(SiMe3)2}2],17 which exhibits similar Ce–N distances [2.353(7) Å mean] to 6 [2.353(8) Å mean]. The Ce⋯Cpcentroid distance in 6 [2.551(5) Å] is comparable to that observed for 4 and all other metrical parameters of 6 are unremarkable. It was not possible to fractionally crystallise analytically pure 6 under the conditions employed due to the similar solubility of [Ce{N(SiMe3)2}3] in hexanes/toluene, therefore no further analytical data were obtained. The global yield of 5 from this reaction was 50%, however, reaction conditions were not optimised as such ligand scrambling mechanisms can be difficult to control. It has been reported that mono-ring C5Me5 cerium complexes can undergo ligand scrambling in solution (Scheme 3),17 and if similar processes are occurring during the synthesis of 5 this would affect the yields of all the products.
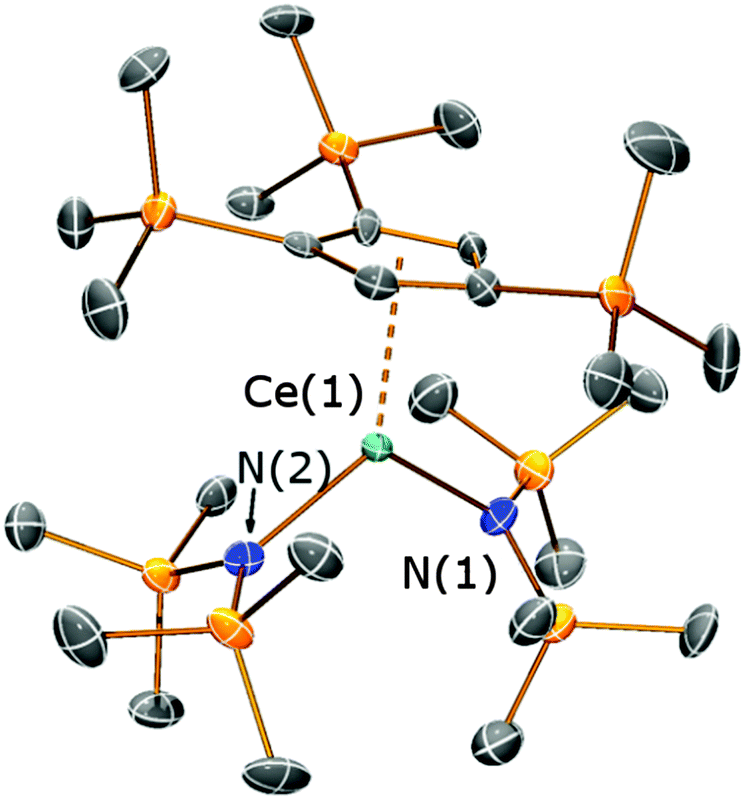 | ||
| Fig. 7 Molecular structure of 6 with selective atom labelling, with displacement ellipsoids set at the 50% probability level; hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°] for 6: Ce(1)–N(1) 2.355(8), Ce(1)–N(2) 2.350(7), Ce(1)⋯Cpcentroid(1) 2.551(5), N(1)–Ce(1)–N(2) 119.0(3). | ||
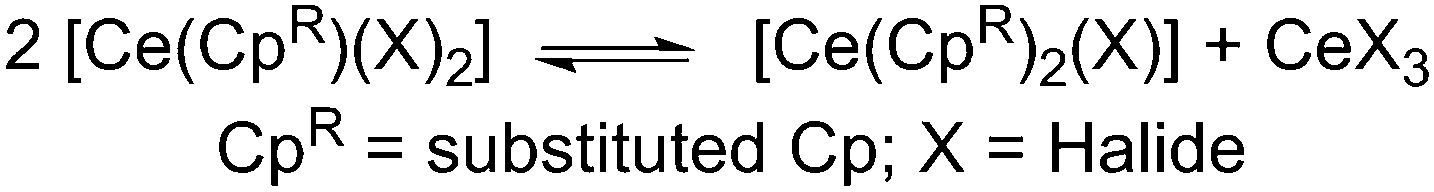 | ||
| Scheme 3 Ligand scrambling of heteroleptic mono-ring Ce complexes.17 | ||
The Evans method was employed to determine the room temperature solution magnetic moments of all isolated cerium complexes.18 The values obtained for 2b (μeff = 2.79 μB), 3 (2.23 μB), 4 (2.41 μB) and 5 (2.22 μB) are in good agreement with the predicted magnetic moment for a Ce(III) 4f1 2F5/2 ground state (2.54 μB)19 and previously reported magnetic moments of Ce(III) complexes in the literature (range 1.88–2.75 μB).20
Conclusions
We have reported the synthesis and structural characterisation of a novel series of heteroleptic substituted Cp Ln complexes using salt metathesis methodologies. These include bis-substituted CpR Ln dimeric complexes with bridging iodides (2–3) and for the bulky Cp′′′ ligand we were able to isolate a mono-ring Ce complex 4, as disubstitution was not possible for this ligand under the conditions employed. The monomeric Ce complex 5 was prepared by the reaction of 4 with [KN(SiMe3)2], triggering a ligand scrambling process which gave 5 in fair yield together with the mixed substituted Cp′′′ silylamide Ce complex 6 and [Ce{N(SiMe3)2}3]. Although the yield of 5 is only moderate, it is considerable when compared to the previously reported low-yielding synthesis of the closely related La complex, [La(Cp′′′)2(I)(py)].8b Solid state characterisation of 2–6 provides in-depth knowledge of the structural features of these systems and this will help us to predict their reactivity profile. In our hands, complexes 2a–b and 5 have not shown any tendency to ligand scramble in both coordinating and non-coordinating solvents over experimental timescales. As such, we envisage that they will be useful precursors for the preparation and stabilisation of novel La/Ce-element bonding motifs, including the preparation of heterobimetallic species, by salt metathesis methodologies.Experimental
Materials and methods
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of dry argon. Solvents were dried by refluxing over potassium and were degassed before use. All solvents were stored over potassium mirrors (with the exception of THF, which was stored over activated 4 Å molecular sieves). Deuterated solvents were distilled from potassium, degassed by three freeze-pump-thaw cycles, and stored under argon. KCp′′,11 KCp′′′,11 KCptt,12 [Ln(I)3(THF)4] (Ln = La, Ce)21 and [KN(SiMe3)2]22 were prepared according to published procedures. KH was obtained as a suspension in mineral oil and was washed three times with hexane and dried in vacuo. 1H, 13C{1H} and 29Si{1H} NMR spectra were recorded on a spectrometer operating at 400.2, 100.6 and 79.5 MHz respectively; chemical shifts are quoted in ppm and are relative to TMS. FTIR spectra were recorded as Nujol mulls in KBr discs on a PerkinElmer Spectrum RX1 spectrometer. Elemental microanalyses were carried out by Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK.Synthetic procedures
Acknowledgements
We thank the EPSRC and The University of Manchester for generously supporting this work. This work was funded by the Engineering and Physical Sciences Research Council (grant numbers EP/K039547/1 and EP/L014416/1).Notes and references
- G. Wilkinson and J. M. Birmingham, J. Am. Chem. Soc., 1954, 76, 6210 CrossRef CAS.
- (a) S. Arndt and J. Okuda, Chem. Rev., 2002, 102, 1953 CrossRef CAS PubMed; (b) F. T. Edelmann and V. Lorenz, Coord. Chem. Rev., 2000, 209, 99 CrossRef CAS.
- (a) M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857 CrossRef CAS PubMed; (b) W. J. Evans, J. Alloys Compd., 2009, 488, 493 CrossRef CAS PubMed; (c) W. J. Evans, Inorg. Chem., 2007, 46, 3435 CrossRef CAS PubMed; (d) M. C. Cassani, D. J. Duncalf and M. F. Lappert, J. Am. Chem. Soc., 1998, 120, 12958 CrossRef CAS; (e) W. J. Evans, S. L. Gonzales and J. W. Ziller, J. Am. Chem. Soc., 1991, 113, 7423 CrossRef CAS.
- (a) M. V. Butovskii, B. Oelkers, T. Bauer, J. M. Bakker, V. Bezugly, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2014, 20, 2804 CrossRef CAS PubMed; (b) M. V. Butovskii, C. Döring, V. Bezugly, F. R. Wagner, Y. Grin and R. Kempe, Nat. Chem., 2010, 2, 741 CrossRef CAS PubMed; (c) M. V. Butovskii, O. L. Tok, F. R. Wagner and R. Kempe, Angew. Chem., Int. Ed., 2008, 47, 6469 CrossRef CAS PubMed; (d) I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, N. I. Kirillova, A. K. Shestakova, I. N. Parshina, A. I. Gusev and G. K. I. Magomedov, J. Am. Chem. Soc., 1993, 115, 3156 CrossRef CAS.
- For other ligand systems see: (a) M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2013, 49, 3315 RSC; (b) P. L. Arnold, J. Mc Master and S. T. Liddle, Chem. Commun., 2009, 818 RSC; (c) S. T. Liddle, Proc. R. Soc. A, 2009, 465, 1673 CrossRef CAS.
- (a) D. Patel and S. T. Liddle, Rev. Inorg. Chem., 2012, 32, 1 CrossRef CAS; (b) S. T. Liddle and D. P. Mills, Dalton Trans., 2009, 5592 RSC.
- See, for example: (a) S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 844 CrossRef PubMed; (b) T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 1754 CrossRef PubMed; (c) I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136 CrossRef CAS PubMed.
- (a) L. Maron, E. L. Werkema, L. Perrin, O. Esienstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 2792 CrossRef PubMed; (b) G. R. Giesbrecht, G. E. Collis, J. C. Gordon, D. L. Clark, B. L. Scott and N. J. Hardman, J. Organomet. Chem., 2004, 689, 2177 CrossRef CAS PubMed; (c) Z. Xie, K. Chui, Q. Yang, T. C. W. Mak and J. Sun, Organometallics, 1998, 17, 3937 CrossRef CAS; (d) E. B. Lobkovsky, Yu. K. Gun'ko, B. M. Bulychev, V. K. Belsky, G. L. Soloveichik and M. Yu. Antipin, J. Organomet. Chem., 1991, 406, 343 CrossRef; (e) W. J. Evans, J. M. Olofson, H. Zhang and J. L. Atwood, Organometallics, 1988, 7, 629 CrossRef CAS.
- (a) T. Mehdoui, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Dalton Trans., 2005, 1263 RSC; (b) T. Mehdoui, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Chem. Commun., 2005, 2860 RSC; (c) P. N. Hazin, C. Lakshminarayan, L. S. Brinen, J. L. Knee, J. W. Bruno, W. E. Streib and K. Folting, Inorg. Chem., 1988, 27, 1393 CrossRef CAS; (d) M. D. Walter, D. Bentz, F. Weber, O. Schmitt, G. Wolmershäuser and H. Sitzmann, New J. Chem., 2007, 31, 305 RSC.
- (a) M. P. Coles, P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, Organometallics, 2012, 31, 2682 CrossRef CAS; (b) P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed., 2008, 47, 1488 CrossRef CAS PubMed; (c) Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, Organometallics, 2000, 19, 2832 CrossRef; (d) M. C. Cassani, Y. K. Gun'ko, P. B. Hitchcock, M. F. Lappert and F. Laschi, Organometallics, 1999, 18, 5539 CrossRef CAS; (e) Z. Xie, K. Chui, Z. Liu, F. Xue, Z. Zhang, T. C. W. Mak and J. Sun, J. Organomet. Chem., 1997, 16, 239 CrossRef; (f) M. C. Cassani, Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, Chem. Commun., 1996, 1987 RSC; (g) Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, J. Organomet. Chem., 1995, 499, 213 CrossRef; (h) C. D. Sofield and R. A. Andersen, J. Organomet. Chem., 1995, 501, 271 CrossRef CAS; (i) Y. K. Gun'ko, B. M. Bulychev, G. L. Soloveichik and V. K. Belsky, J. Organomet. Chem., 1992, 424, 289 CrossRef; (j) S. D. Stults, R. A. Andersen and A. Zalkin, Organometallics, 1990, 9, 115 CrossRef CAS; (k) P. N. Hazin, J. W. Bruno and G. K. Schulte, Organometallics, 1990, 9, 416 CrossRef CAS.
- M. J. Harvey, T. P. Hanusa and M. Pink, J. Chem. Soc., Dalton Trans., 2001, 1128 RSC.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 3552 CrossRef CAS.
- W. W. Lukens Jr., S. M. Beshouri, L. L. Blosch, A. L. Stuart and R. A. Andersen, Organometallics, 1999, 18, 1235 CrossRef.
- A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 201 CrossRef.
- G. R. Giesbrecht, D. L. Clark, J. C. Gordon and B. L. Scott, Appl. Organomet. Chem., 2003, 17, 473 CrossRef CAS PubMed.
- (a) P. N. Hazin, J. C. Huffman and J. W. Bruno, Organometallics, 1987, 6, 23 CrossRef CAS; (b) M. D. Rausch, K. J. Moriarty, J. L. Atwood, J. A. Weeks, W. E. Hunter and H. G. Brittain, Organometallics, 1986, 5, 1281 CrossRef CAS.
- H. J. Heeres, A. Meetsma, J. H. Teuben and R. D. Rogers, Organometallics, 1989, 8, 2637 CrossRef CAS.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003 RSC; (b) S. K. Sur, J. Magn. Reson., 1989, 82, 169 CAS; (c) D. H. Grant, J. Chem. Educ., 1995, 72, 39 CrossRef CAS.
- J. H. van Vleck, Theory of Electric and Magnetic Susceptibilities, Oxford University Press, Oxford, UK, 1932 Search PubMed.
- See, for example: (a) A. J. Wooles, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 39, 500 RSC; (b) G. Marshall, A. J. Wooles, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Inorganics, 2013, 1, 46 CrossRef PubMed.
- K. Izod, S. T. Liddle and W. Clegg, Inorg. Chem., 2004, 43, 214 CrossRef CAS PubMed.
- K. F. Tesh, T. P. Hanusa and J. C. Huffman, Inorg. Chem., 1990, 29, 1584 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Frontiers of Organo-f-element Chemistry’ themed issue. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1056158–1056165 for 1, 2a–b, 2a·THF and 3–6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00761e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |