
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of carbon dioxide by new mixed sandwich uranium(III) complexes incorporating cyclooctatetraenyl and pyrrolide, phospholide, or arsolide ligands†
Rachel J.
Kahan
a,
F. Geoffrey N.
Cloke
*a,
S. Mark
Roe
a and
François
Nief
b
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN1 9QJ, UK. E-mail: f.g.cloke@sussex.ac.uk
bÉcole Polytechnique, CNRS, Route de Saclay, F-91128 Palaiseau, France
First published on 22nd April 2015
Abstract
A series of uranium(III) mixed-sandwich complexes of the type [U(COTTIPS2)(CpEMe4)] (CpEMe4 = EC4Me4, E is N, P or As, and COTTIPS2 = C8H6{1,4-SiiPr3}), featuring a heterocyclic five membered ring, have been synthesised and their X-ray crystal structures determined. The redox properties of these complexes have been assessed using cyclic voltammetry and the results compared to the purely carbocyclic mixed-sandwich analogues. The reactions of [U(COTTIPS2)(CpNMe4)] and [U(COTTIPS2)(CpPMe4)] with CO2 afford the structurally characterised carbamate and phosphacarbonate complexes [U(COTTIPS2)]2(μ-O)(μ-η1:η1-O2CEC4Me4)2 (E = N and P respectively), arising from CO2 reduction and insertion.
Introduction
The use of carbocyclic aromatic ligands in organouranium chemistry has been prominent ever since the synthesis of [Cp3UCl] in 1956,1 and subsequent expansion of this area to include 6, 7 and 8 membered rings illustrates the versatility of aromatic ligands in this field. The cyclopentadienyl (Cp) ligand and its substituted derivatives are ubiquitous in organouranium chemistry,2 however aromatic heterocyclic analogues have received comparatively little attention. Of the few reported uranium complexes featuring heterocyclic 5-membered rings, we reported the only example of a homoleptic uranium complex, featuring the 1,3-di-tert-butyl-1,2,4-triphospholyl ligand,3 and more extensive studies by Ephritikhine et al. have employed the tetramethylphospholyl (CpPMe4) ligand, as this bears the most resemblance to Cp*.4,5 The latter results also demonstrated that these ligands can also bond through the pnictogen lone pair, allowing dimerisation of the complexes via η1:η5-coordination. However the CpPMe4 ligand was also observed to be more labile than its cyclopentadienyl analogues, illustrated by the ready protonation of the ligand in a mixed-sandwich complex to generate a uranium(IV) cation.6,7In recent years, the use of organometallic uranium complexes for small molecule activation, has attracted significant interest;8 in the specific case of CO2, reduction to afford uranium oxo complexes and CO has been achieved previously using U(III) complexes incorporating tripodal tris(aryloxide)9 or siloxide ligands,10 and disproportionation to CO and uranium carbonate derivatives has been described for neutral and anionic U(III) siloxide,10 and tris(aryloxide) systems.11 In recent years, we have employed uranium(III) mixed-sandwich complexes featuring substituted COT and Cp ligands for the reductive activation of CO and CO2, and comprehensive studies have determined that the steric properties of the mixed-sandwich complexes dictate the outcome of these reactions.12,13 Hence we decided to investigate the effect of changing the electronic properties of these mixed sandwich complexes, and herein we report the results obtained from incorporation of a heterocyclic ring in to the U(III) mixed sandwich motif and subsequent reactivity towards CO2.
Results and discussion
Synthesis of mixed-sandwich complexes
The three mixed-sandwich complexes [U(COTTIPS2)(CpEMe4)] (E = N (1), P (2), As (3)) were prepared by successive salt metathesis reactions of UI3 with K[CpEMe4] and K2[COTTIPS2] in low to moderate yield (Scheme 1). This ‘one-pot’ methodology is an adaptation of the synthetic route employed for the synthesis of [U(COTTIPS2)(Cp*)(THF)] and other substituted cyclopentadienyl analogues, although 1–3 are formed less cleanly and in lower yields (16–40%) than their purely carbocyclic counterparts.12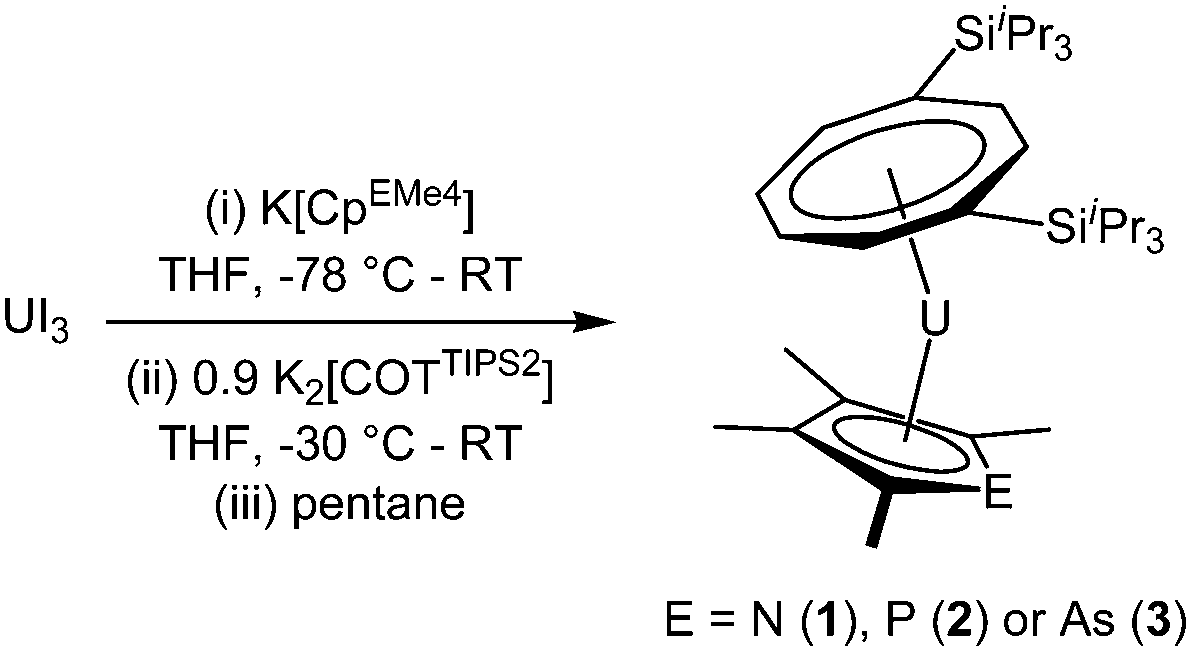 | ||
| Scheme 1 Synthetic route to uranium(III) mixed-sandwich complexes. | ||
The phospholyl and arsolyl mixed-sandwich complexes (2 and 3) displayed comparable, paramagnetically shifted 1H and 29Si{1H} NMR spectra, whereas the pyrrolyl mixed-sandwich complex 1 displayed a different pattern of proton resonances, indicative of a more complex structure in solution (vide infra). All three complexes form stable adducts with THF, 1·THF, 2·THF, and 3·THF, respectively. Mass spectrometry and microanalysis supported the formulation of 1–3, and the molecular structures were confirmed by single crystal X-ray diffraction studies on the THF complexes, and the structures are shown in Fig. 1 with selected data in Table 1.
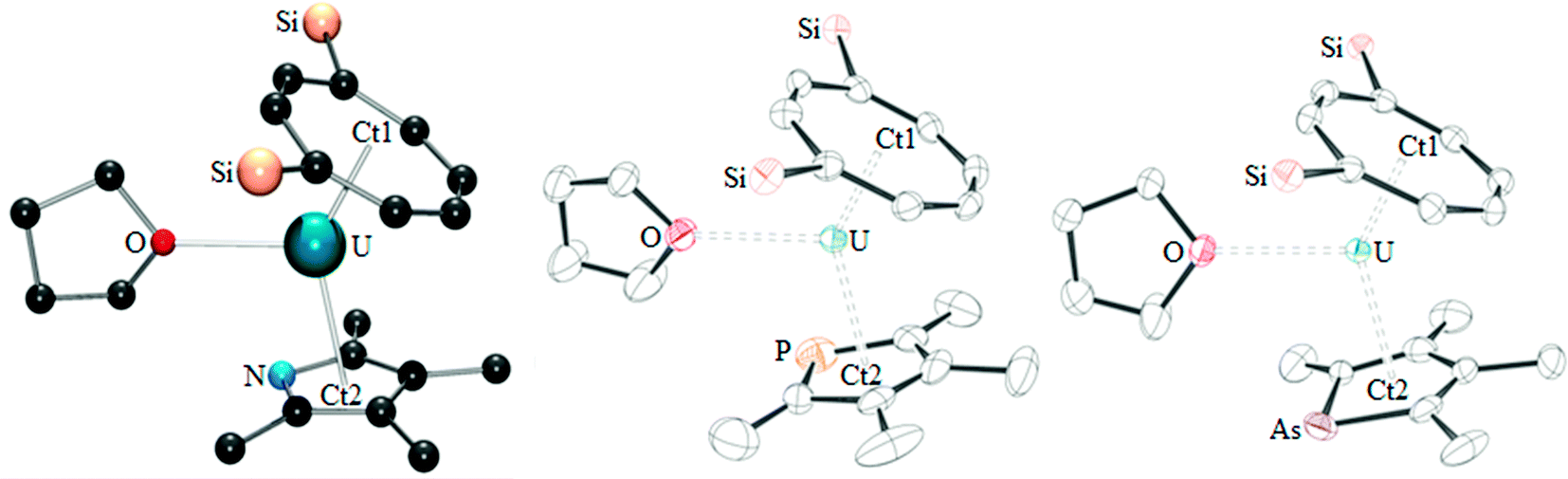 | ||
| Fig. 1 From left to right: ball and stick model of 1·THF, and ORTEP diagrams of 2·THF and 3·THF (thermal ellipsoids at 50% probability; hydrogen atoms and iPr groups have been omitted for clarity). | ||
| 2·THF | 3·THF | |
|---|---|---|
| U–Ct1 | 1.9740(4) | 1.9744(4) |
| U–Ct2 | 2.54(2) {2.59(2)} | 2.5962(4) |
| U–E | 2.9868(14) {2.776(15)} | 3.0781(7) |
| U–O | 2.716(2) | 2.726(4) |
| Ct1–U–Ct2 | 135.8(15) {142.4(14)} | 141.482(16) |
High resolution data could not be obtained for 1·THF and the molecular structure of this complex therefore only illustrates connectivity. The molecular structure of 2·THF features a phospholyl ring disordered over two positions, which has been modelled accordingly (see ESI† for full details). The three complexes are isostructural, and only small differences are observed between 2·THF and 3·THF, due to the lengthening of the U–E bond on descending the pnictogen group. These structures are similar to their carbocyclic analogue [U(COTTIPS2)(CpMe4)(THF)], demonstrating that incorporation of a pnictogen has not significantly altered the overall structural properties of the complexes. Comparison of 2·THF to the only other mixed-sandwich complex featuring a heterocyclic ligand, the U(IV) complex[U(COT)(CpPMe4)(BH4)(THF)], illustrates a similar U–Ct2 bond length (2.610(8) Å).6 However, the Ct1–U–Ct2 angle is more acute (135.6(3)°) and the U–Ct1 distance is longer (2.013(9) Å) presumably due to the presence of the BH4 group.
The molecular structure of base-free 1 was also determined by single crystal X-ray diffraction, and shows that this complex is dimeric in the solid-state (see Fig. 2). As a consequence of the dimeric structure, the Ct1–U–Ct2 angle is more acute than those in 2·THF and 3·THF, however the U–Ct1, U–Ct2 and U–O bond lengths are similar. Other heterocyclic complexes have also been reported featuring η5:η1 coordination, however only [{U(η5-CpPMe4)(μ-η5:η1-CpPMe4)(BH4)}2] is comparable to 1.4 The latter features similar U–Ct2 distances (2.56(1) and 2.54(1) Å) to 2·THF and similar U–P bond lengths (2.945(3) and 2.995(3) Å), demonstrating that η1-coordination does not affect the η5-bonding. The dimeric structure of 1 presumably persists in solution since it would account for the more complex NMR spectra observed for 1 as opposed to those for monomeric 2 and 3; unfortunately DOSY experiments on 1 were only suggestive of a dimeric structure and its low solubility in suitable solvents precluded cryoscopy.
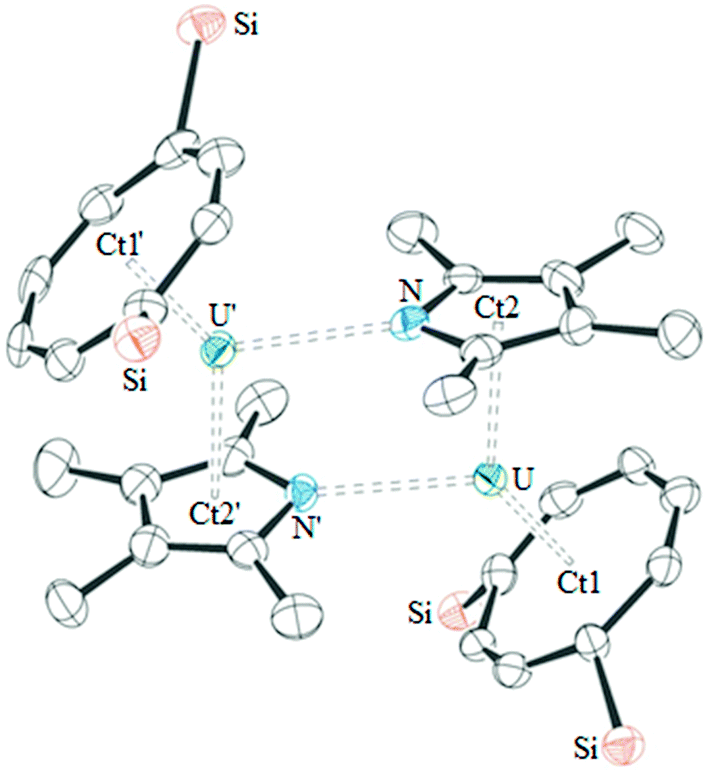 | ||
| Fig. 2 ORTEP diagram of 1 (thermal ellipsoids at 50% probability; hydrogen atoms and iPr groups have been omitted for clarity). Selected distances (Å) and angles (°): U1–Ct1 1.968(3), 1.974(3); U1–Ct2 2.548(4), 2.568(4); U1–N1 2.680(5), 2.691(6); U1–N1’ 2.598(6), 2.615(5); Ct1–U1–Ct2 138.66(11), 138.55(12). | ||
Cyclic voltammetry
In order to compare their UIV/UIII redox couples with the carbocyclic analogues, cyclic voltammetry was performed on 1–3. Complex 1 exhibits a distorted quasi-reversible wave at −1.88 V vs. FeCp2+/0, which is within the expected range for the UIV/UIII redox couple. Complexes 2 and 3 also exhibit an electrochemical event at this approximate potential. However the degree of distortion of the voltammograms becomes more pronounced descending the pnictogen group, precluding accurate determination of E1/2. Two other electrochemical events were observed for the three complexes and an additional two events were observed for 1 (see ESI†). These events could however not be unambiguously assigned and demonstrate the complex behaviour of the heteroatom containing mixed-sandwich system in the cyclic voltammetry experiment, as opposed to the more straightforward behaviour of the purely carbocyclic complexes.13The assumed E1/2 value of the UIV/UIII redox couple for 1 is slightly less negative than that for [U(COTTIPS2)(CpMe4)(THF)] (−2.08 V), demonstrating the increased thermodynamic stability of the UIII oxidation state relative to the UIV oxidation state in 1. This is in agreement with other published studies, which found the UIV/UIII redox couple is ca. 0.2 V anodically shifted for complexes featuring phospholyl ligands.14 This arises from loss of degeneracy of the five-membered ring e-symmetry orbitals, causing a decrease in the HOMO–LUMO gap, an effect which has also been observed in transition metal complexes;15 the low energy vacant orbital in the phospholyl complex 1 (and indeed the N and As analogues) thus likely stabilises the U(III) centre. Hence, whilst complexes 1–3 can still be regarded as potent reducing agents, they are somewhat less powerful than their purely carbocyclic analogues.
Reactivity with CO2
Addition of excess carbon dioxide to 1 and 2 afforded the complexes [U(COTTIPS2)]2(μ-O)(μ-O2CEC4Me4)2 (E = N (4), P (5)), which are formed by reduction of 0.5 equivalents CO2 per uranium centre to give the oxo unit. A further equivalent CO2 is inserted into the U–E bond, giving rise to the carbamate and phosphacarbonate units respectively, so that a total of 1.5 equivalents carbon dioxide are required for the transformation (Scheme 2). The reaction can be conveniently monitored by 13C NMR using 13CO2, and shows the formation of 4 and 5 by the appearance of resonances at −7.1 and −46.6 ppm corresponding to the carbamate and phosphacarbonate groups, respectively; free 13CO formed from the reduction of CO2 to form the bridging oxo unit was also observed in both cases.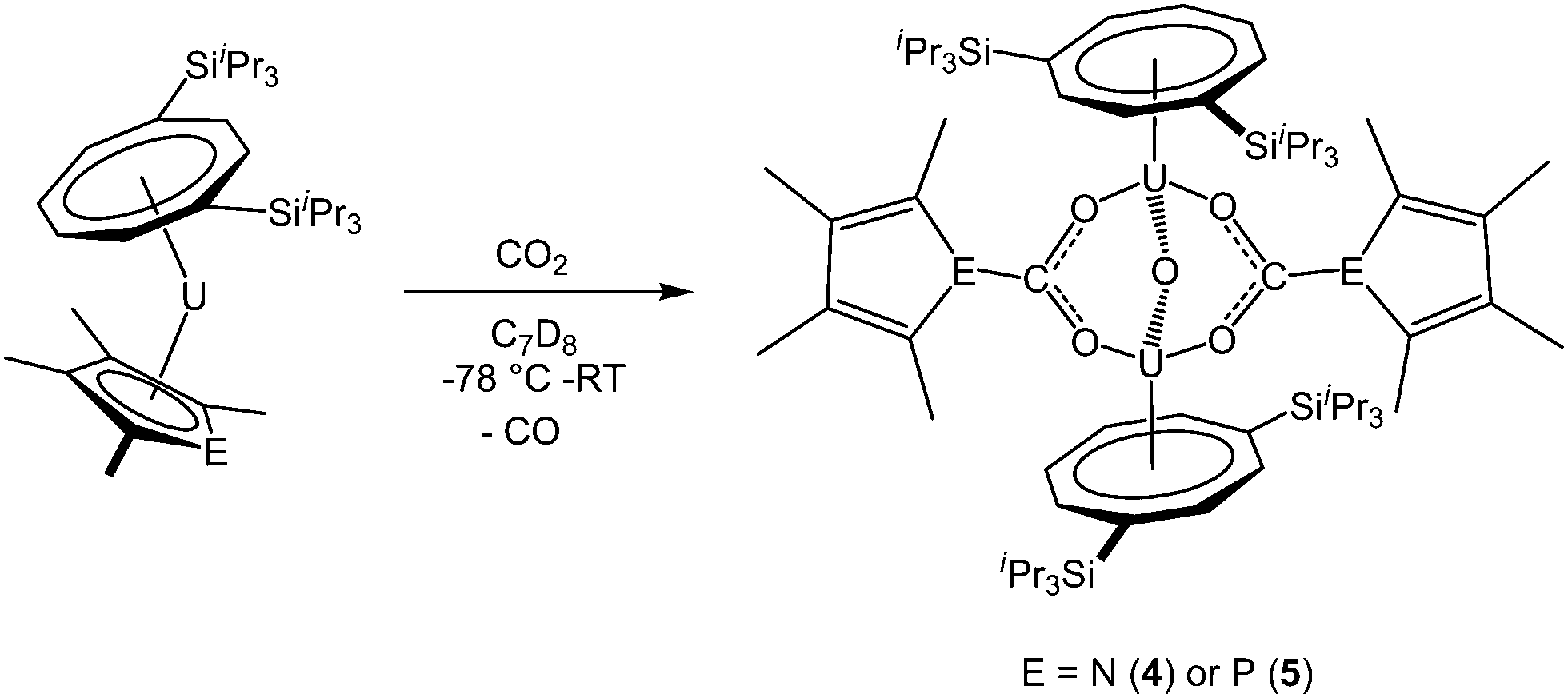 | ||
| Scheme 2 Reactions of [U(COTTIPS2)(CpEMe4)] with CO2. | ||
Monitoring of the formation of 4 in C7D8 by 1H NMR spectroscopy revealed its formation to be quantitative; however the thermal instability of this complex resulted in consistently low values of carbon by microanalysis, but 4 did display a parent ion in the mass spectrum (EI). The formation of 5 was found to proceed less cleanly and in lower yield. The 1H NMR spectrum of 5 in C7D8 at 303 K was broad and with few clearly defined resonances. The spectrum sharpened at 363 K, (possibly due to a fluxional process, the nature of which however could not be established), allowing the assignment of all but the COT ring protons. However, microanalysis and mass spectral data (EI) agreed with the proposed formulation of 5. Attempts to react 3 with carbon dioxide were unsuccessful and resulted in decomposition of the complex to form intractable products.
The proposed structures of 4 and 5 were confirmed by single crystal X-ray diffraction (see Fig. 3 and Table 2), and to the best of our knowledge, 5 represents the first example of a phosphacarbonate ligand bound to a uranium centre. Both complexes are structurally similar, and exhibit slightly shorter U–Ct1 distances than the parent mixed-sandwich complexes. The oxo unit is symmetrical in 5 but asymmetrical in 4 despite the identical U–O–U angle, but both complexes feature carboxyl fragments that are positioned off-centre between the two uranium centres. The metrics within this unit closely resemble those in [(OTtbp)2U(μ-O)(μ-O2COTtbp)2U(OTtbp)2], which also exhibits a bent oxo fragment (140.4(5)°) with identical U–O distances to 5 (2.095(3) Å), and asymmetrical bridging carbonate moieties.16
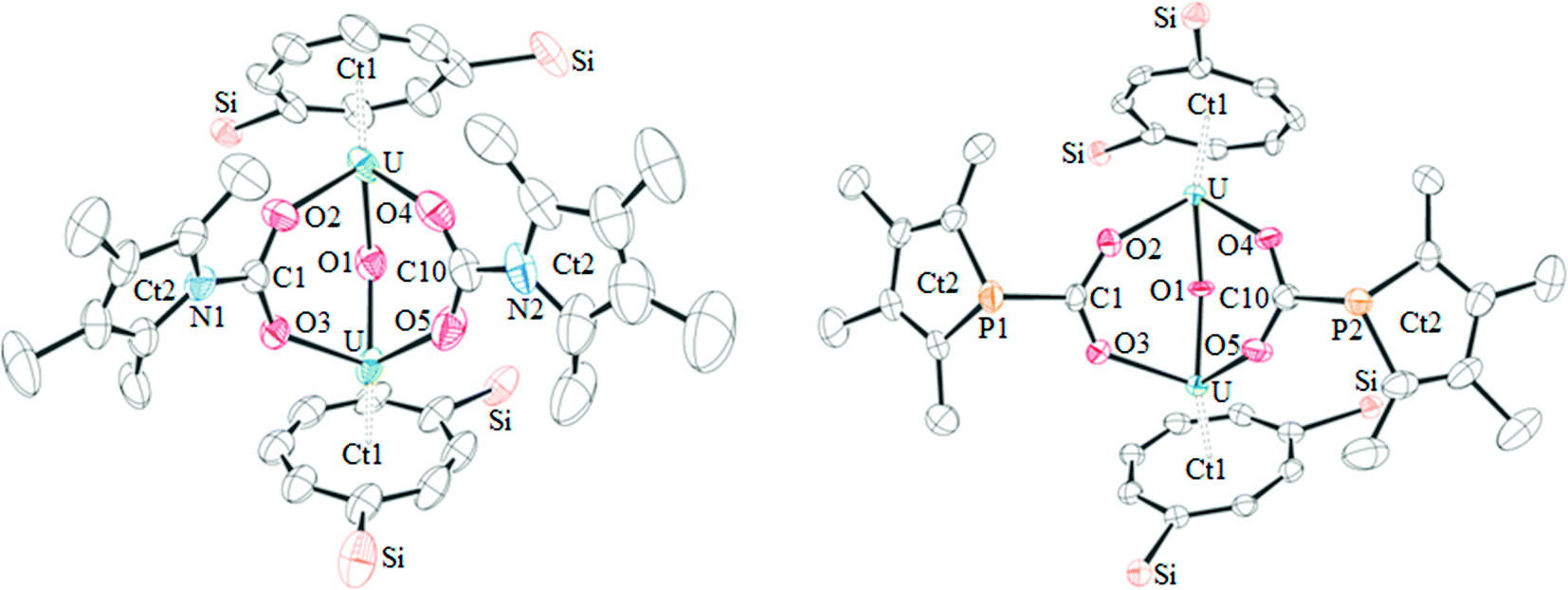 | ||
| Fig. 3 The molecular structure of 4 (left) and 5 (right, thermal ellipsoids at 50% probability. Hydrogen atoms and iPr groups have been omitted for clarity). | ||
| 4 | 5 | |
|---|---|---|
| U–Ct1 | 1.908(12), 1.921(13) | 1.9320(3), 1.9323(3) |
| U–O1 | 2.094(7), 2.074(6) | 2.096(3), 2.096(3) |
| U–O2,3,4,5 | 2.353(6), 2.350(5), 2.329(7), 2.379(7) | 2.346(4), 2.391(4), 2.362(3), 2.343(4) |
| C1,10–O2,3,4,5 | 1.262(10), 1.270(10), 1.256(12), 1.243(12) | 1.260(6), 1.262(6), 1.260(6), 1.286(6) |
| C1,10–E1,2 | 1.381(10), 1.381(14) | 1.855(6), 1.850(6) |
| U–O1–U | 139.4(3) | 139.38(19) |
However, some structural differences are observed between the carbamate and phosphacarbonate units. In 4, the nitrogen lone pair overlaps with the CO2 unit, evidenced by the short N–CO2 bonds, and with the pyrrolyl diene unit, which gives rise to near linear Ct2–N–C angles (169.1(13) and 179.6(12)°), and a delocalised carbamate moiety with an aromatic pyrrolyl ring. The phosphacarbonate fragment in 5 does not exhibit this feature, and has discrete diene and P–CO2 moieties and bent Ct2–P–C angles (116.6(2) and 116.2(3)°), with trigonal pyramidal geometry around the phosphorus atoms.
Conclusion
Three new mixed-sandwich complexes of the type [U(COTTIPS2)(CpEMe4)] (where E is N, P or As and COTTIPS2 = C8H6{1,4-SiiPr3}) have been synthesised featuring a heterocyclic alternative to the cyclopentadienyl ligand. These complexes are structurally comparable to their purely carbocyclic analogues, but feature slightly less negative UIV/UIII redox potentials as a result of the heteroatom incorporation in to the 5-membered ring. However, they are still capable of reducing CO2, but the presence of the heteroatom also results in CO2 insertion chemistry and the formation of the first uranium phosphacarbonate complex.Experimental
General considerations
All manipulations were carried out under an inert atmosphere of argon using standard Schlenk techniques or under an argon atmosphere in an MBraun glovebox. Solvents were dried over appropriate drying agents (NaK3, pentane; K, THF) prior to distillation under N2. Solvents were stored over K mirrors or 4 Å molecular sieves. Deuterated solvents were dried over K, vacuum distilled and stored over 4 Å molecular sieves under Ar. NMR spectra were recorded on a Varian VNMR spectrometer operating at 400 MHz (1H). 1H and 13C spectra were referenced internally to residual solvent signals, 28Si spectra were referenced externally to SiMe4 and 31P spectra were referenced externally to 85% H3PO4 in D2O. EI-MS was performed by Dr A. K. Abdul-Sada at the University of Sussex using a VG Autospec Fisons instrument. Elemental analyses were performed by Mikroanalytisches Labor Pascher or the University of Bristol Microanalysis Service. IR Spectra were recorded on residues between NaCl plates on a Perkin-Elmer Spectrum One FTIR instrument. The following materials were prepared according to published procedures: UI3,17 potassium tetramethylphospholyl (referred to as K[CpPMe4]),5,18 Potassium tetramethylpyrrolyl (referred to as K[CpNMe4]),19 potassium tetramethylarsolyl (referred to as K[CpAsMe4]),20 and K2[C8H6{1,4-SiiPr3}] (referred to as K2[COTTIPS2].2113CO2 (99% enrichment) was purchased from Cambridge Isotopes and transferred via a calibrated Toepler pump.Syntheses
X-ray crystallographic studies
Data for 1, 2, 3 and 5 were collected on a Enraf-Nonius CAD4 diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073) source, and data for 1·THF were collected using a Agilent Technologies Xcalibur Gemini ultra diffractometer with a Cu Kα radiation (λ = 1.54184) source at 173 K using an Oxford Cryosystems Cobra low temperature device, operating in ω scanning mode with Ψ and ω scans to fill the Ewald sphere. The programs used for control and integration were Collect,22 Scalepack and Denzo.23 Absorption corrections were based on equivalent reflections using SADABS.24 Data for 4 were collected and processed by the UK National Crystallography Service at the University of Southampton.25 The crystals were mounted on a glass fibre with silicon grease, from dried vacuum oil kept over 4 Å molecular sieves in an MBraun glovebox under Ar. All solutions and refinements were performed using the WinGX or Olex2 packages and software therein. All non-hydrogen atoms were refined with anisotropic displacement parameters and all hydrogen atoms were refined using a riding model. Disordered solvent molecules were modelled using the SQUEEZE26 function in PLATON.27 Crystal structure and refinement data are given in Table S1 of the ESI.† CCDC 1051779–1051784.Acknowledgements
We thank the European Research Council (F.G.N.C.), the EPSRC, and the Ivor Nixon Fund (R.J.K.) for financial support. We also thank Dr A. Abdul-Sada (University of Sussex) for mass spectrometry.References
- L. T. Reynolds and G. Wilkinson, J. Inorg. Nucl. Chem., 1956, 2, 246–253 CrossRef CAS.
- M. Ephritikhine, Organometallics, 2013, 32, 2464–2488 CrossRef CAS.
- G. K. B. Clentsmith, F. G. N. Cloke, M. D. Francis, J. R. Hanks, P. B. Hitchcock and J. F. Nixon, J. Organomet. Chem., 2008, 693, 2287–2292 CrossRef CAS PubMed.
- P. Gradoz, M. Ephritikhine, M. Lance, J. Vigner and M. Nierlich, J. Organomet. Chem., 1994, 481, 69–73 CrossRef CAS.
- P. Gradoz, D. Baudry, M. Ephritikhine, F. Nief and F. Mathey, J. Chem. Soc., Dalton Trans., 1992, 3047–3051 RSC; P. Gradoz, C. Boisson, D. Baudry, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1992, 1720–1721 RSC; P. Gradoz, D. Baudry, M. Ephritikhine, M. Lance, M. Nierlich and J. Vigner, J. Organomet. Chem., 1994, 466, 107–118 CrossRef CAS.
- S. M. Cendrowski-Guillaume, M. Nierlich and M. Ephritikhine, J. Organomet. Chem., 2002, 643–644, 209–213 CrossRef CAS.
- S. M. Cendrowski-guillaume, G. Le Gland, M. Nierlich and M. Ephritikhine, Eur. J. Inorg. Chem., 2003, 1388–1393 CrossRef CAS PubMed.
- For recent reviews see: P. L. Arnold, Chem. Commun., 2011, 47, 9005 RSC; T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550 RSC; M. Ephritikhine, Organometallics, 2013, 32, 2464 CrossRef CAS; B. M. Gardner and S. T. Liddle, Eur. J. Inorg. Chem., 2013, 3753–3770 CrossRef PubMed; H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303–416 CrossRef PubMed.
- I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS PubMed.
- V. Mougel, C. Camp, J. Pécaut, C. Copéret, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed., 2012, 51, 12280–12284 CrossRef CAS PubMed; O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef PubMed.
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC; A.-C. Schmidt, A. V. Nizovtsev, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2012, 48, 8634–8636 RSC.
- N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353–1362 CrossRef CAS.
- N. Tsoureas, L. Castro, A. F. R. Kilpatrick, F. G. N. Cloke and L. Maron, Chem. Sci., 2014, 5, 3777–3788 RSC.
- A. Elkechai, A. Boucekkine, L. Belkhiri, D. Hauchard, C. Clappe and M. Ephritikhine, C. R. Chim., 2010, 13, 860–869 CrossRef CAS PubMed; A. Elkechai, Y. Mani, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2012, 51, 6943–6952 CrossRef PubMed; C. Clappe, D. Leveugle, D. Hauchard and G. Durand, J. Electroanal. Chem., 1998, 448, 95–103 CrossRef.
- N. Kuhn, G. Henkel, J. Kreutzberg, S. Stubenrauch and C. Janiak, J. Organomet. Chem., 1993, 456, 97–106 CrossRef CAS; M. D. Su and S. Y. Chu, J. Phys. Chem., 1989, 93, 6043–6051 CrossRef; N. M. Kostić and R. F. Fenske, Organometallics, 1983, 2, 1008–1013 CrossRef.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef CAS PubMed.
- F. Nief, F. Mathey and L. Ricard, Organometallics, 1988, 7, 921–926 CrossRef CAS.
- C. L. Webster, J. E. Bates, M. Fang, J. W. Ziller, F. Furche and W. J. Evans, Inorg. Chem., 2013, 52, 3565–3572 CrossRef CAS PubMed.
- F. Nief, L. Ricard and F. Mathey, Polyhedron, 1993, 12, 19–26 CrossRef CAS.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602 CrossRef CAS PubMed.
- Collect, Bruker-AXS, Madison, WI, 1997–2004 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- G. M. Sheldrick, SADABS V2008/1, University of Göttingen, Göttingen, Germany Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- P. Van Der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1051779–1051784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00590f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |