
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and catalytic activity of homoleptic lanthanide-tris(cyclopropylethinyl)amidinates†‡
Farid M.
Sroor
,
Cristian G.
Hrib
,
Liane
Hilfert
,
Sabine
Busse
and
Frank T.
Edelmann
*
Chemisches Institut der Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: frank.edelmann@ovgu.de; Fax: +49 391 671-2933; Tel: +49 391 675-8327
First published on 15th April 2015
Abstract
Reactions of anhydrous lanthanide trichlorides, LnCl3 (Ln = Nd, Sm, Ho), with 3 equiv. of lithium-cyclopropylethinylamidinates, Li[c-C3H5–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C(NR)2] (1a: R = cyclohexyl (Cy), 1b: R = iPr), afforded the new homoleptic lanthanide(III) tris(cyclopropylethinylamidinate) complexes [c-C3H5–CC–C(NCy)2]3Sm (2a) and [c-C3H5–CC–C(NiPr)2]3Ln (Ln = Nd (2b), Sm (2c), Ho (2d)) as air- and moisture-sensitive crystalline solids in moderate to good isolated yields (45–79%). The formation of unsolvated, homoleptic Ln(III) tris(cyclopropylethinylamidinate) was confirmed by an X-ray diffraction study of the holmium derivative [c-C3H5–CC–C(NiPr)2]3Ho (2d). EI mass spectra of the new rare-earth metal amidinates indicated a significant volatility. An initial catalysis study revealed that these complexes catalyze the addition of terminal alkynes to carbodiimides to give propiolamidines of the type R–CC–C(
C–C(NR)2] (1a: R = cyclohexyl (Cy), 1b: R = iPr), afforded the new homoleptic lanthanide(III) tris(cyclopropylethinylamidinate) complexes [c-C3H5–CC–C(NCy)2]3Sm (2a) and [c-C3H5–CC–C(NiPr)2]3Ln (Ln = Nd (2b), Sm (2c), Ho (2d)) as air- and moisture-sensitive crystalline solids in moderate to good isolated yields (45–79%). The formation of unsolvated, homoleptic Ln(III) tris(cyclopropylethinylamidinate) was confirmed by an X-ray diffraction study of the holmium derivative [c-C3H5–CC–C(NiPr)2]3Ho (2d). EI mass spectra of the new rare-earth metal amidinates indicated a significant volatility. An initial catalysis study revealed that these complexes catalyze the addition of terminal alkynes to carbodiimides to give propiolamidines of the type R–CC–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR′)(NHR′). The molecular structure of N,N′-dicyclohexyl-phenylpropiolamidine, Ph–CC–C(NCy)(NHCy) (4), was also determined by X-ray diffraction.
NR′)(NHR′). The molecular structure of N,N′-dicyclohexyl-phenylpropiolamidine, Ph–CC–C(NCy)(NHCy) (4), was also determined by X-ray diffraction.
1. Introduction
In organolanthanide chemistry, steric saturation of the coordination sphere of the large rare-earth metal cations is generally more important than the electron count. Thus the investigation of new spectator ligands which satisfy the coordination requirements of the lanthanides continues to be of significant current interest. Anionic amidinate ligands of the type [RC(NR′)2]− (R = H, alkyl, aryl; R′ = alkyl, cycloalkyl, aryl, SiMe3) have been demonstrated to be highly useful and versatile in that respect. These readily available N-chelating ligands are generally regarded as steric cyclopentadienyl equivalents.1 In the case of rare-earth metals, mono-, di- and trisubstituted lanthanide amidinate and guanidinate complexes are all accessible, just like the mono-, di- and tricyclopentadienyl complexes. Over the past ca. 25 years, lanthanide amidinates have witnessed an impressive transformation from laboratory curiosities to highly active homogeneous catalysts as well as valuable precursors in materials science. Various rare-earth metal amidinates have been reported to be very efficient homogeneous catalysts e.g. for ring-opening polymerization reactions of lactones, the guanylation of amines or the addition of terminal alkynes to carbodiimides.2 In materials science, homoleptic alkyl-substituted lanthanide tris(amidinate) complexes are often highly volatile and can be used as promising precursors for ALD (atomic layer deposition) and MOCVD (metal–organic chemical vapor deposition) processes, e.g. for the deposition of lanthanide oxide (Ln2O3) or lanthanide nitride (LnN) thin films.3The introduction of alkinyl groups to the central carbon atom in amidines leads to alkinylamidines (or propiolamidines) of the type R–CC–C(=NR′)(NHR′). In organic synthesis, alkinylamidines have been frequently employed in the preparation of various heterocycles.4,5 More recently, alkinylamidines have attracted considerable attention due to their diverse applications in biological and pharmacological systems.6 Moreover, transition metal and lanthanide alkinylamidinate complexes have been shown to be efficient and versatile catalysts e.g. for C–C and C–N bond formation, the addition of C–H, N–H and P–H bonds to carbodiimides as well as ε-caprolactone polymerization.7 Thus far, only very few lanthanide complexes containing alkinylamidinate ligands have been described.7,8 Previously used propiolamidinate ligands include e.g. phenylethinyl derivatives [Ph–CC–C(NR)2]− (R = iPr, tBu)7a,8 and the trimethylsilylacetylene-derived anions [Me3Si–CC–C(NR)2]− (R = cyclohexyl (Cy), iPr).9
In the course of our ongoing investigation of lanthanide amidinates we recently initiated a study of alkinylamidinates derived from cyclopropylacetylene. The resulting anions [c-C3H5–CC–C(NR)2]− (R = Cy, iPr) represent a potentially useful addition to the current library of amidinate ligands. In a first contribution we described the synthesis and full characterization of the lithium-cyclopropylethinylamidinates Li[c-C3H5–CC–C(NR)2] (1a: R = cyclohexyl (Cy), 1b: R = iPr).10 These precursors are readily available on a large scale and in high yields using commercially available starting materials. In a subsequent study, their use as precursors for new lanthanide amidinates could be demonstrated by the synthesis of a series of new Ln(III) bis(cyclopropylethinylamidinates). In the case of Ce and Nd, the chloro-bridged dimers [{c-C3H5–CC–C(NR)2}2Ln(μ-Cl)(THF)]2 (Ln = Ce, Nd; R = Cy, iPr) were isolated, whereas the smaller holmium afforded the “ate” complex [c-C3H5–CC–C(NCy)2]2Ho(μ-Cl)2Li(THF)(OEt2). An initial study showed that these complexes effectively catalyze the addition of aniline derivatives to carbodiimides to give N-arylguanidines.11 Herein we report the synthesis and structural characterization of the first homoleptic Ln(III) tris(cyclopropylethinylamidinate) complexes as well as an initial study of their possible use as homogeneous catalysts for the addition of terminal alkynes to carbodiimides.
2. Results and discussion
2.1 Synthesis and structure
The starting materials used in this study, the lithium-cyclopropylethinylamidinates Li[c-C3H5–CC–C(NR)2] (1a: R = Cy, 1b: R = iPr), were prepared in a straightforward manner according to Scheme 1 by in situ-deprotonation of commercially available cyclopropylacetylene followed by treatment with either N,N′-diisopropylcarbodiimide or N,N′-dicyclohexylcarbodiimide according to the published procedure. These lithium amidinates can be isolated in the form of stable, crystalline solids as adducts with donor solvent like diethyl ether, THF or DME (1,2-dimethoxyethane).10 However, for the reactions with lanthanide trichlorides, the reagents 1a and 1b were conveniently prepared in THF solution and used in situ.
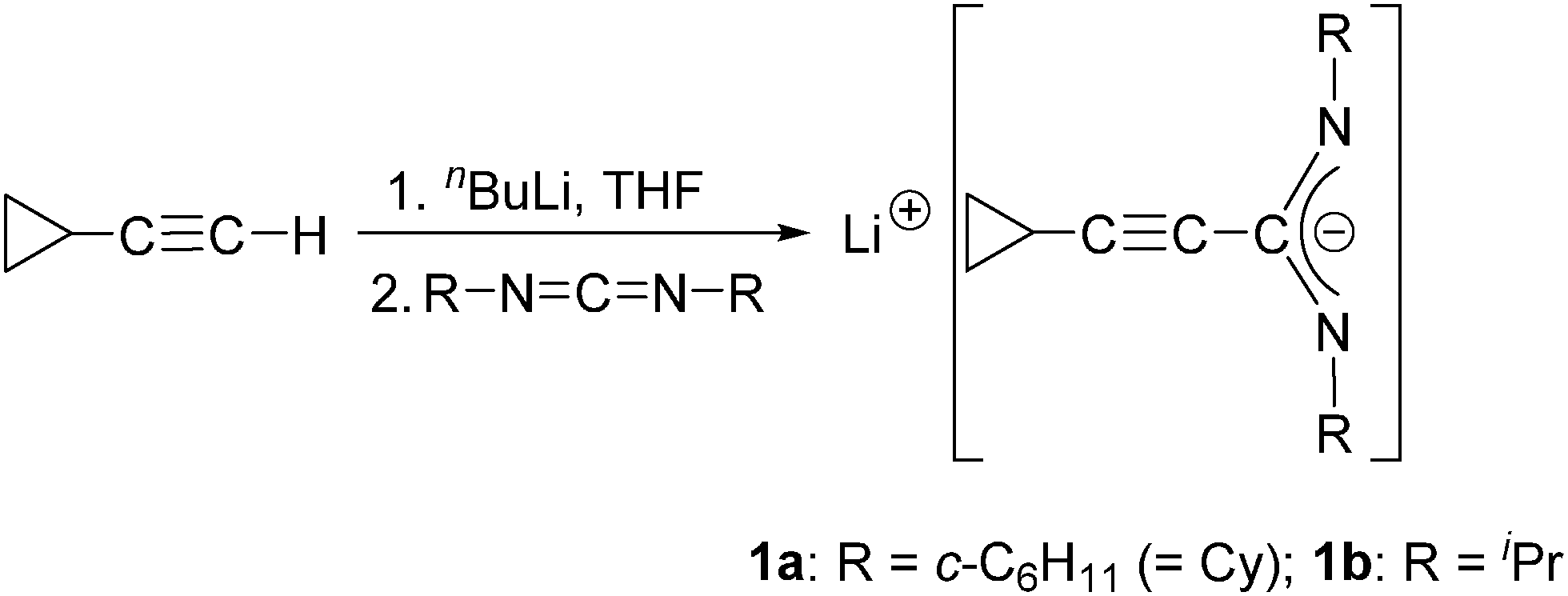 | ||
| Scheme 1 Synthesis of the lithium-cyclopropylethinylamidinates 1a and 1b. | ||
Subsequent reactions of the lithium-cyclopropylethinylamidinates 1a and 1b with anhydrous lanthanide trichlorides, LnCl3 (Ln = Nd, Sm, Ho) were carried out in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio in THF solutions according to Scheme 2. Evaporation of the volatiles and recrystallization of the crude products from n-pentane afforded the new lanthanide(III) tris(cyclopropylethinylamidinate) complexes 2a–d in moderate (2b: 54%, 2c: 45%, 2d: 55%) to good (2a: 79%) yields. The samarium and holmium derivatives 2a, 2c, and 2d were isolated as yellow, air- and moisture-sensitive crystals, while the neodymium complex 2b is a green, crystalline solid. All four compounds are highly soluble in THF, diethyl ether, toluene and n-pentane. The very high solubility even in non-polar solvents like n-pentane certainly accounts for the relatively low yields in the case of complexes 2b–d. A single-crystal X-ray diffraction study of the holmium derivative 2d (vide infra) confirmed the presence of the expected unsolvated, homoleptic lanthanide(III) tris(cyclopropylethinylamidinate) complex.
:3 molar ratio in THF solutions according to Scheme 2. Evaporation of the volatiles and recrystallization of the crude products from n-pentane afforded the new lanthanide(III) tris(cyclopropylethinylamidinate) complexes 2a–d in moderate (2b: 54%, 2c: 45%, 2d: 55%) to good (2a: 79%) yields. The samarium and holmium derivatives 2a, 2c, and 2d were isolated as yellow, air- and moisture-sensitive crystals, while the neodymium complex 2b is a green, crystalline solid. All four compounds are highly soluble in THF, diethyl ether, toluene and n-pentane. The very high solubility even in non-polar solvents like n-pentane certainly accounts for the relatively low yields in the case of complexes 2b–d. A single-crystal X-ray diffraction study of the holmium derivative 2d (vide infra) confirmed the presence of the expected unsolvated, homoleptic lanthanide(III) tris(cyclopropylethinylamidinate) complex.
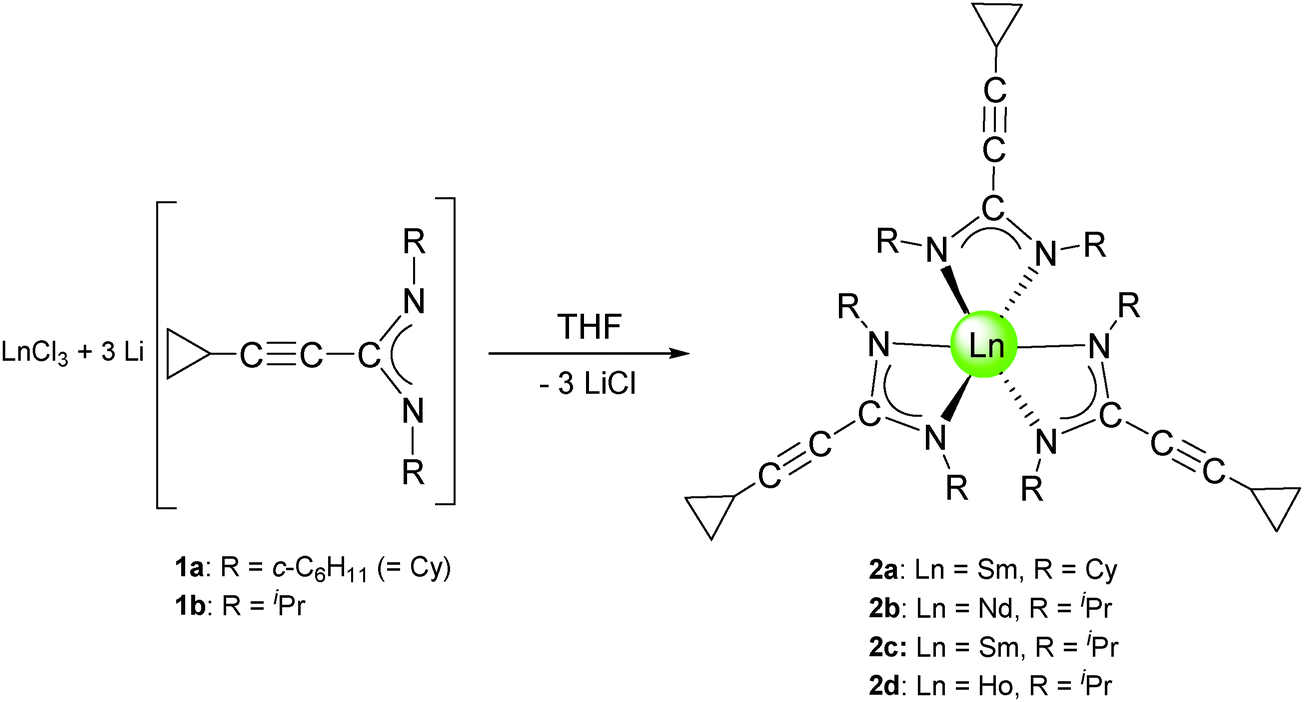 | ||
| Scheme 2 Synthesis of the Ln(III) tris(cyclopropylethinylamidinates) 2a–d. | ||
All four compounds were characterized by their NMR (1H, 13C) and IR spectra as well as elemental analyses. Despite the paramagnetic nature of the Ln3+ ions employed here, meaningful NMR spectra could be obtained for all four compounds with the exception of the 1H NMR spectrum of the Ho3+ complex 2c. The data were in good agreement with the formation of unsolvated lanthanide(III) tris(cyclopropylethinylamidinates). No signals attributable to coordinated THF could be observed. The IR spectra of 2a–c were found to be almost superimposable. IR bands resulting from the CN stretching vibrations of the N–C–N units appear at around 1606–1612 cm−1, whereas very strong bands at 2220–2227 cm−1 can be assigned to the CC vibrations. In all cases the EI mass spectra indicated good volatility of the new homoleptic lanthanide amidinates as they all showed the molecular ions in an intensity range of 20–45% relative intensity.
As a typical representative of the new homoleptic lanthanide tris(amidinates), the holmium derivative 2d was structurally authenticated through single-crystal X-ray diffraction (Fig. 1). Pale yellow, block-like single-crystals of 2d were obtained by cooling of a very concentrated solution in n-pentane to −30 °C over a prolonged period of time. Crystallographic data of 2d are listed in Table 1, while selected bond lengths and angles are summarized in the caption of Fig. 1. Compound 2d crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The crystal structure determination clearly confirmed the presence of the first unsolvated homoleptic lanthanide(III) tris(cyclopropylethinylamidinate) complex. The central Ho3+ ion is coordinated by three chelating [c-C3H5–CC–C(NiPr)2]− ligands in a highly distorted octahedral fashion. To our knowledge, only three closely related homoleptic Ln(III) tris(phenylethinylamidinate) complexes of the type [Ph–CC–C(NiPr)2]3Ln (Ln = Y,8b Ce,8a Lu8b) have been reported in the previous literature. All three complexes have also been structurally characterized by X-ray diffraction. The overall structural features of 2d are very similar to those reported for [Ph–CC–C(NiPr)2]3Ln (Ln = Y, Ce, Lu). The Ho–N distances in 2d are in the very narrow range of 2.342(2)–2.383(3) Å. As a result of the lanthanide contraction,12 these values are virtually identical with those reported for the yttrium(III)-tris(phenylethinylamidinate) complex [Ph–CC–C(NiPr)2]3Y (Y–N 2.363(4) and 2.356(4) Å). The average N–Ho–N bite angle to the chelating amidinate ligands in 2d is 57.33(9)°. This is also favorably comparable to the corresponding N–Ln–N angles found in the three phenylethinylamidinates [Ph–CC–C(NiPr)2]3Ln (Ln = Y, Ce, Lu) and in other homoleptic lanthanide tris(N,N′-dialkylamidinates).1,8 The bond lengths of the triple bonds in the cyclopropylethinyl units in 2d are 1.182(6) Å (C2–C3), 1.185(4) Å (C14–C15) and 1.184(5) Å (C22–C23).
. The crystal structure determination clearly confirmed the presence of the first unsolvated homoleptic lanthanide(III) tris(cyclopropylethinylamidinate) complex. The central Ho3+ ion is coordinated by three chelating [c-C3H5–CC–C(NiPr)2]− ligands in a highly distorted octahedral fashion. To our knowledge, only three closely related homoleptic Ln(III) tris(phenylethinylamidinate) complexes of the type [Ph–CC–C(NiPr)2]3Ln (Ln = Y,8b Ce,8a Lu8b) have been reported in the previous literature. All three complexes have also been structurally characterized by X-ray diffraction. The overall structural features of 2d are very similar to those reported for [Ph–CC–C(NiPr)2]3Ln (Ln = Y, Ce, Lu). The Ho–N distances in 2d are in the very narrow range of 2.342(2)–2.383(3) Å. As a result of the lanthanide contraction,12 these values are virtually identical with those reported for the yttrium(III)-tris(phenylethinylamidinate) complex [Ph–CC–C(NiPr)2]3Y (Y–N 2.363(4) and 2.356(4) Å). The average N–Ho–N bite angle to the chelating amidinate ligands in 2d is 57.33(9)°. This is also favorably comparable to the corresponding N–Ln–N angles found in the three phenylethinylamidinates [Ph–CC–C(NiPr)2]3Ln (Ln = Y, Ce, Lu) and in other homoleptic lanthanide tris(N,N′-dialkylamidinates).1,8 The bond lengths of the triple bonds in the cyclopropylethinyl units in 2d are 1.182(6) Å (C2–C3), 1.185(4) Å (C14–C15) and 1.184(5) Å (C22–C23).
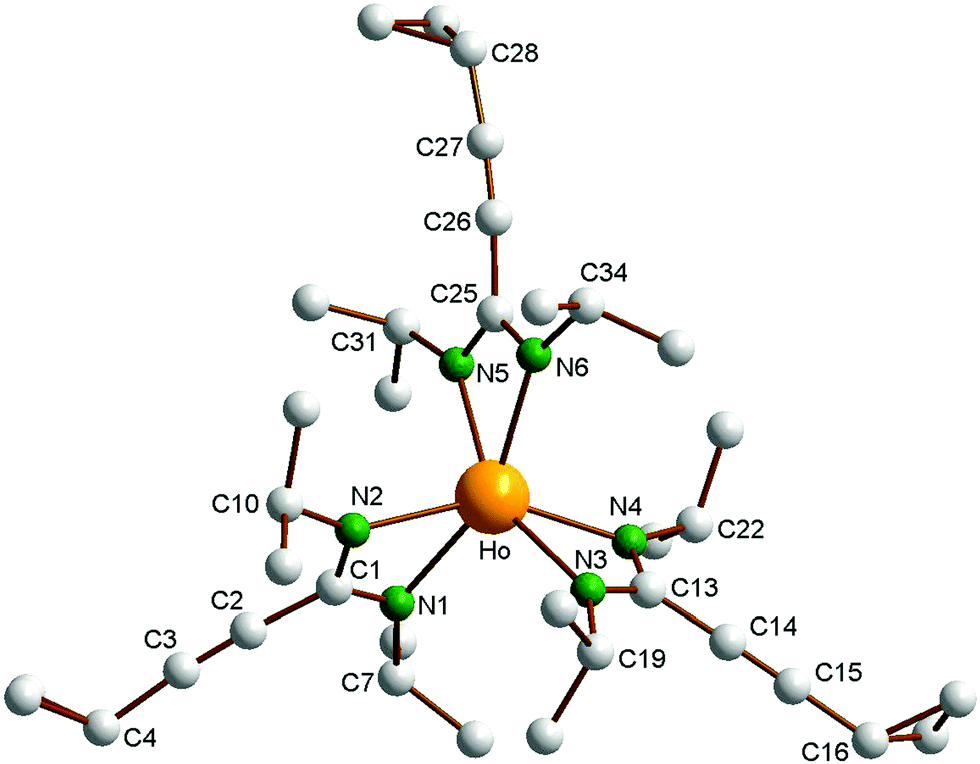 | ||
| Fig. 1 Molecular structure of complex [c-C3H5–CCC(NiPr)2]3Ho (2d). All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ho–N1 2.359(3), Ho–N2 2.351(3), Ho–N3 2.348(2), Ho–N4 2.353(3), Ho–N5 2.342(2), Ho–N6 2.383(3), C1–N1 1.312(4), C1–N2 1.327(4), C13–N3 1.333(4), C13–N4, 1.331(4), C25–N5 1.333(4), C25–N6 1.324(3), C7–N1 1.481(4), C1–C2 1.461(5), C2–C3 1.182(6), C14–C15 1.185(4), C26–C27 1.184(5), N1–Ho–N2 57.08(9), N3–Ho–N4 57.74(9), N5–Ho–N6 57.14(8), N1–Ho–N3 100.1(11), N2–Ho–N5 98.77(10), N4–Ho–N6 98.74(11), N1–C1–N2 117.0(3), N3–C13–N4 116.9(3), N5–C25–N6 116.6(3). | ||
| 2d | 4 | |
|---|---|---|
| Empirical formula | C36H57HoN6 | C21H28N2 |
| Formula weight | 738.81 | 308.45 |
| Crystal size (mm3) | 0.40 × 0.40 × 0.20 | 0.34 × 0.23 × 0.22 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P |
P |
| a (Å) | 9.776(2) | 9.7257(19) |
| b (Å) | 13.149(3) | 10.383(2) |
| c (Å) | 16.983(3) | 10.558(2) |
| α (°) | 101.28 | 70.77 |
| β (°) | 105.35 | 65.92 |
| γ (°) | 108.19 | 70.83 |
| Cell volume (Å3) | 1905.6(7) | 895.5(3) |
| Z | 2 | 2 |
| T (°C) | −120 | −120 |
| λ (Å) | 0.71703 | 0.71703 |
| D calcd (g cm−3) | 1.288 | 1.144 |
| μ (mm−1) | 2.106 | 0.067 |
| F(000) | 764 | 336 |
| Index ranges | −13 ≤ h ≤ 13 | −12 ≤ h ≤ 11 |
| −18 ≤ k ≤ 18 | −12 ≤ k ≤ 12 | |
| −19 ≤ l ≤ 23 | −13 ≤ l ≤ 12 | |
| Data/restraints/parameters | 10209/38/461 |
3625/157/267 |
| Goodness-of-fit on F2 | 1.040 | 1.071 |
| R (F0 or F02) | 0.0343 | 0.0535 |
| R w (F0 or F02) | 0.0908 | 0.1512 |
| Largest diff. peak and hole (e Å−3) | 2.465, −1.743 | 0.198, −0.223 |
2.2 Catalytic activity
For a first study of the possible catalytic activity of the new Ln(III) tris(cyclopropylethinylamidinate) we chose the catalytic addition of alkynes to carbodiimides to give substituted propiolamidines. The lanthanide-catalyzed synthesis of propiolamidines R–CC–C(=NR′)(NHR′) was first reported in 2005 by Hou et al. using rare-earth metal half-sandwich complexes as catalysts. The pre-catalysts used in this study were constrained-geometry-type complexes such as [Me2Si(C5Me4)(NPh)]Y(CH2SiMe3)(THF)2. It was found that half-sandwich complexes comprising a propiolamidinate ligand play an important role in the catalytic cycle. Upon treatment with excess acetylene, they release the propiolamidine product.7a Most recently, Zhang and Zhou et al. employed rare-earth metal alkyl complexes stabilized by the bulky pyrazolylborate ligand TpMe2 (=hydro-tris(3,5-dimethylpyrazolyl)-borate) as catalysts for the synthesis of N-aryl-substituted propiolamidines.7g
In an initial screening test, we examined the Ln-catalyzed addition of phenylacetylene to N,N′-diisopropylcarbodiimide in the presence of all four compounds 2a–d as illustrated in Scheme 3. All four new lanthanide(III)-tris(cyclopropylethinylamidinates) 2a–d were used as precatalysts, and the reactions were carried out in concentrated THF solutions at 60 °C. The results are summarized in Table 2. The isolated yields of the known compound Ph–CC–C(NiPr)(NHiPr) (3)13 varied from 27 to 85% depending of the lanthanide metal employed. Clearly the highest activity was observed for the samarium complex [c-C3H5–CC–C(NCy)2]3Sm (2a), while the lowest yields were obtained when using the holmium catalyst [c-C3H5–CC–C(NiPr)2]3Ho (2d). In a control experiment (Table 2, entry 11), an equimolar mixture of phenylacetylene and N,N′-diisopropylcarbodiimide were heated in concentrated THF solution at 60 °C for 1 h in the absence of a rare-earth metal compound. Under these conditions, no trace of Ph–CC–C(NiPr)(NHiPr) (3) could be detected in the reaction mixture.
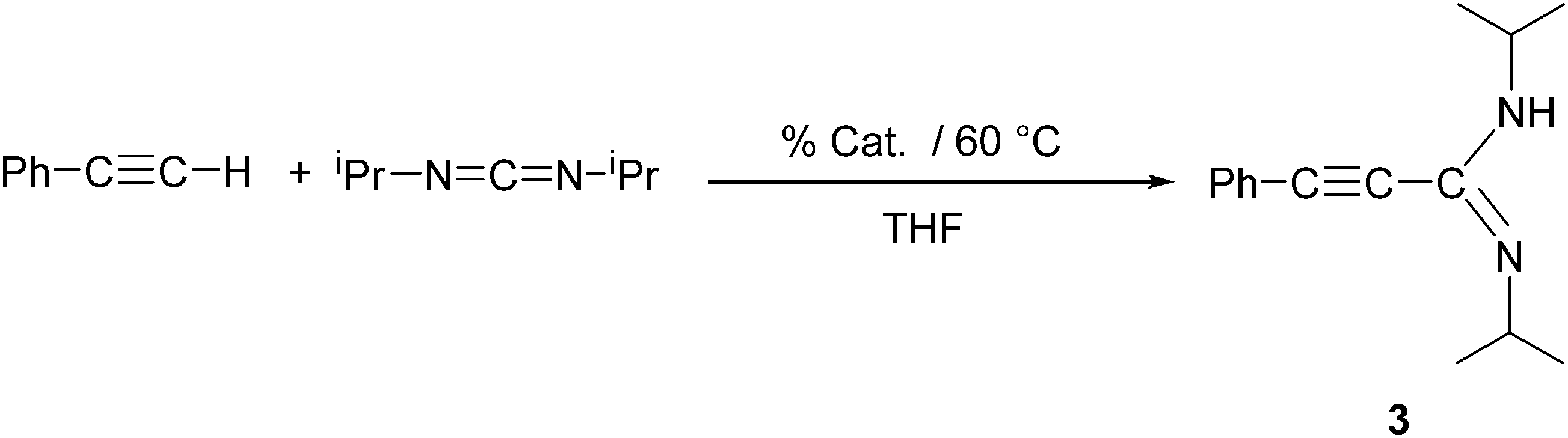 | ||
| Scheme 3 Synthesis of Ph–CC–C(NiPr)(NHiPr) (3) using 2a–d as catalyst. | ||
In a second set of experiments, the Ln-catalyzed addition of three different terminal alkynes to both N,N′-diisopropylcarbodiimide and N,N′-dicyclohexylcarbodiimide was studied. For these tests, the most active complex [c-C3H5–CC–C(NCy)2]3Sm (2a) was used as the precatalyst. The reactions were again carried out in THF at 60 °C according to Scheme 4.
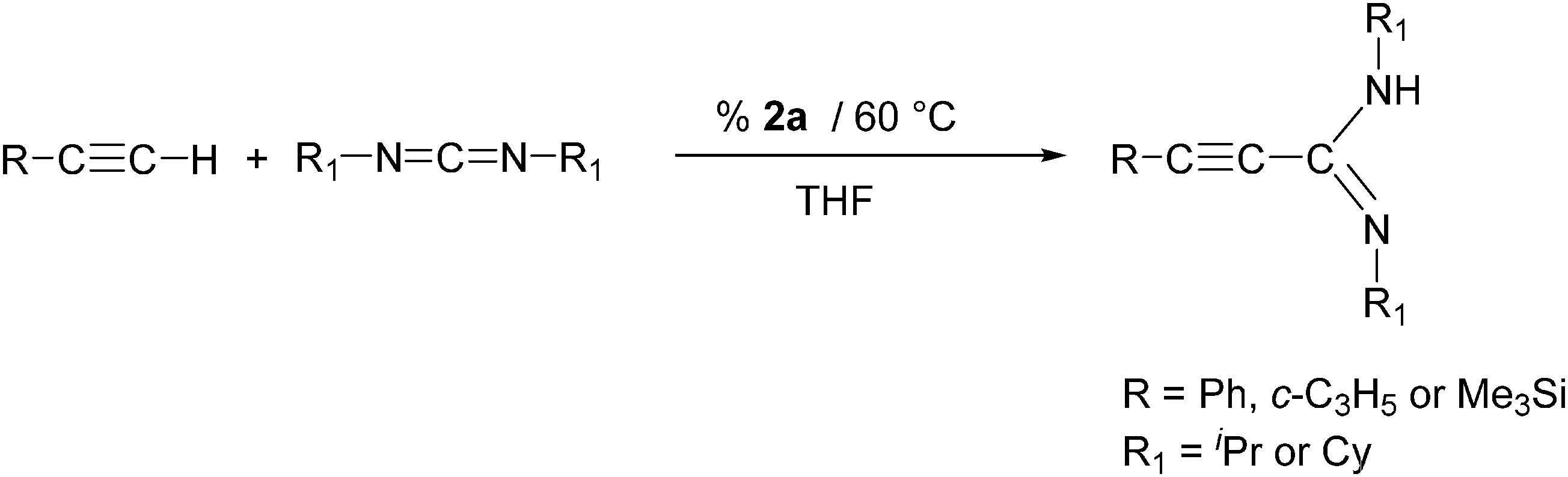 | ||
| Scheme 4 Synthesis of alkynylamidines using complex 2a as catalyst. | ||
As can be seen from the results listed in Table 3, this short screening produced mixed results. Reactions of phenylacetylene with both N,N′-diisopropylcarbodiimide and N,N′-dicyclohexylcarbodiimide gave good yields of the hydroacetylenation products 3 and 4, while cyclopropylacetylene could be added only to N,N′-dicyclohexylcarbodiimide affording a moderate yield of propiolamidine 5. In sharp contrast, virtually no reactions were observed when trimethylsilylacetylene was employed. Thus the use of the new homoleptic lanthanide(III)-tris(cyclopropylethinylamidinates) as catalysts for the addition of terminal acetylenes to carbodiimides appears to be quite limited. Obviously these amidinate complexes cannot seriously compete with previously reported rare-earth metal catalysts comprising cyclopentadienyl7a or pyrazolylborate7g ligands. These compounds all contain additional σ-alkyl groups such as –CH2Ph or –CH2SiMe3 which certainly account for the significantly higher activity of such catalysts systems.7a,g
In the course of the present study, the molecular structure of the propiolamidine 4 has been verified by single-crystal X-ray diffraction (cf.Table 1). X-Ray-quality single-crystals of 4 were grown by slowly cooling a solution in hot acetonitrile to room temperature. The molecular structure of 4 is shown in Fig. 2. Previously reported crystal structures of propiolamidines include those of 4-ClC6H4–CC–C(NiPr)(NHiPr) and 2-ClC6H4–CC–C(NiPr)(NHiPr),7a Ph–CC–C(NC6H3iPr2-2,6)(NHC6H3iPr2-2,6),14 and Ph–CC–C(NC6H3iPr2-2,6)(NHC6H3Cl2-3,4).7g The CC bond length in 4 is 1.195(3) Å, while the C1–N1 and C1–N2 distance (1.364(2) and 1.275(4) Å) correspond to standard C–N single and double bonds, respectively. As in 4-ClC6H4–CC–C(NiPr)(NHiPr) and 2-ClC6H4–CC–C(NiPr)(NHiPr),7a one cyclohexyl substituent points toward the alkynyl group and the other one away, resulting in a transoid conformation around the N–C–N unit. In contrast, a cisoid conformation (both substituents pointing toward the alkynyl group) has been reported for Ph–CC–C(NC6H3iPr2-2,6)(NHC6H3iPr2-2,6)14 and Ph–CC–C(NC6H3iPr2-2,6)(NHC6H3Cl2-3,4)7g which both contain bulky 2,6-diisopropylphenyl substituents.
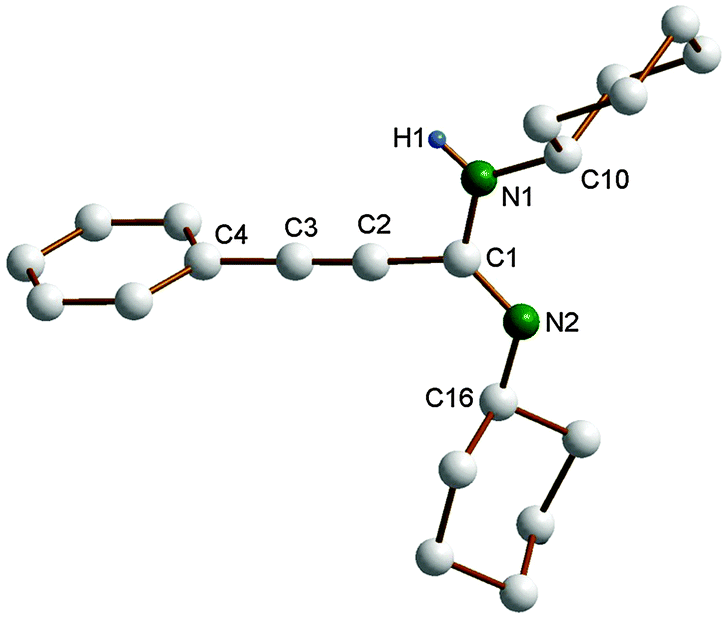 | ||
| Fig. 2 Molecular structure of complex of C6H5–CC–C(NCy)(NHCy) (4). Most of the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): C1–N1 1.364(2), C1–N2 1.275(2), C1–C2 1.451(2), C2–C3 1.195(3), C10–N1 1.451(3), N1–C1–N2 121.93(17), C10–N1–C1 125.79(15), C16–N2–C1 117.4(4). | ||
3. Conclusions
In summarizing the work reported here, we succeeded in the straightforward preparation of a series of new homoleptic lanthanide tris(cyclopropylethinylamidinate) complexes comprising neodymium, samarium, and holmium as central metals. The lithium-cyclopropylethinylamidinate precursors employed in these preparations are readily available in one step from commercially available starting materials. The new complexes 2a–d are highly soluble even in non-polar solvents such as n-pentane. The presence of unsolvated, homoleptic tris(cyclopropylethinylamidinate) complexes could be verified by an X-ray crystal structure determination of the holmium complex 2d. An initial catalysis study revealed that the new amidinates effectively catalyze the addition of phenylacetylene to N,N′-diisopropylcarbodiimide and N,N′-dicyclohexylcarbodiimide but have insufficient activity with other terminal acetylenes.4. Experimental section
4.1 General procedures
All experiments were carried out in oven-dried or flame-dried glassware under an inert atmosphere of dry argon employing standard Schlenk and glovebox techniques (<1 ppm O2, <1 ppm H2O). n-Pentane and THF were distilled from sodium/benzophenone under nitrogen atmosphere prior to use. All glassware was oven-dried at 120 °C for at least 24 h, assembled while hot, and cooled under high vacuum prior to use. The starting materials, anhydrous LnCl3 (Ln = Ce, Nd),15 and the lithium-cyclopropylethinyl-amidinate precursors 1a and 1b10 were prepared according to the literature methods. 1H-NMR (400 MHz) and 13C-NMR (100.6 MHz) were recorded in C6D6 or CDCl3 solutions on a Bruker DPX 400 spectrometer at 25 °C. Chemical shifts were referenced to TMS. Assignment of signals was made from 1H–13C HSQC 2D NMR experiments. IR spectra were recorded using KBr pellets on a Perkin Elmer FT-IR spectrometer system 2000 between 4000 cm−1 and 400 cm−1. Microanalyses of the compounds were performed using a Leco CHNS 923 apparatus.4.2 Synthesis and characterization of the Ln(III)-tris(cyclopropylethinylamidinates) 2a–d
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CC(NCy)2]3Sm (2a).
Anhydrous SmCl3 (1.0 g, 4 mmol) and 1b (3.3 g, 12 mmol) were charged in a 250 ml Schlenk flask. 100 ml of THF were added and the mixture was stirred 12 h at r.t. to give a clear yellow solution. The solvent was removed under vacuum followed by extraction with n-pentane (2 × 15 ml). The clear yellow flitrate was evaporated under vacuum affording 2a as a pale yellow solid (3.0 g, 79%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 3.34 (m, 6H, CH, Cy), 1.85 (m, 3H, CH, c-C3H5), 1.56 (br, 12H, CH2, Cy), 1.40 (m, 6H, CH2, c-C3H5), 0.97–1.32 (m, 18H, CH2, Cy), 0.87 (m, 6H, CH2, c-C3H5), 0.69 (br, 12H, CH2, Cy), −0.21 to −0.12 (q, 6H, CH2, Cy), −2.31 (br, 12H, CH2, Cy); 13C{1H} NMR (100.6 MHz, C6D6, 25 °C): δ = 201.9 (NCN), 104.1 (CC–C), 73.7 (HC–CC), 56.9 (CH, Cy), 35.8 (CH2, Cy), 25.5 (CH2, Cy), 9.8 (CH2, c-C3H5), 1.8 (CH, c-C3H5). MS (EI, M = 965.57): m/z (%) 965.7(45) [M], 695.4(70) [M–(c-C3H5–CCC(NCy)2)]+, 272.2(80) [c-C3H5–CCC(NCy)2]+, 229.1(58) [c-C3H5–CCC(NCy)2–(c-C3H5)]+, 190.1(63) [c-C3H5–CCC(NCy)2–(Cy) + 2H]+, 177(100) [c-C3H5–CCC–NCy + 2H]+. IR (KBr): 3668, 3438, 3220, 3012, 2925, 2850, 2665, 2222, 1606, 1469, 1398, 1361, 1174, 1120, 1028, 972, 888, 703, 676, 588 cm−1. Anal. calcd for C54H81N6Sm: C, 67.24; H, 8.46; N, 8.71%. Found: C, 67.22; H, 8.51; N, 8.60%.
CC(NiPr)2]3Nd (2b).
A solution of anhydrous NdCl3 (1.0 g, 4 mmol) in 30 ml of THF was added to a solution of 1a (2.3 g, 12 mmol) in 70 ml of THF. The reaction mixture was heated to 65 °C for 2 h and then stirred at r.t. for 12 h. The solution color changed to blue. Work-up using n-pentane as described for 2a afforded 2b as green crystals (1.5 g, 54%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 22.3 (m, 6H, CH–(CH3)2), 4.10 (m, 3H, CH, c-C3H5), 2.97 (m, 6H, CH2, c-C3H5), 2.02 (m, 6H, CH2, c-C3H5), −3.55 (m, 36H, CH3); 13C{1H} NMR (100.6 MHz, C6D6, 25 °C): δ = 228.6 (NCN), 108.5 (CC–C), 65.3 (CH–(CH3)2), 59.8 (HC–CC), 23.1 (CH3), 12.1 (CH2, c-C3H5), 2.4 (CH, c-C3H5). MS (EI, M = 715.37): m/z (%) 631.6(33) [M–2(iPr)]+, 396.4(20) [2(c-C3H5–CCC(NiPr)2) + CH3]+, 381.3(15) [2(c-C3H5–CCC(NiPr)2)]+, 205.2(50) [(c-C3H5–CCC(NiPr)2) + CH3]+, 177.1(34) [c-C3H5–CCC(NiPr)2–CH3]+, 149.1(17) [c-C3H5–CCC(NiPr)2–(c-C3H5)]+. IR (KBr): 3678, 3439, 3220, 3015, 2963, 2867, 2608, 2220, 1865, 1635, 1591, 1498, 1382, 1332, 1169, 811, 716, 692, 530, 445 cm−1. Anal. calcd for C36H57N6Nd: C, 60.16; H, 7.93; N, 11.69%. Found: C, 60.25; H, 7.92; N, 11.52%.
CC(NiPr)2]3Sm (2c).
A reaction of anhydrous SmCl3 (1.0 g, 4 mmol) with 1a (2.3 g, 12 mmol) following the procedure described for 2a afforded 2d as a yellow, crystalline solid (1.6 g, 55%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 3.60 (m, 6H, CH–(CH3)2), 1.81 (m, 3H, CH, c-C3H5), 1.37 (m, 6H, CH2, c-C3H5), 0.89 (m, 6H, CH2, c-C3H5), −0.47 (m, 36H, CH3); 13C{1H} NMR (100.6 MHz, C6D6, 25 °C): δ = 201.6 (NCN), 104.5 (CC–C), 73.5 (HC–CC), 48.3 (CH–(CH3)2), 25.1 (CH3), 9.7 (CH2, c-C3H5), 1.7 (CH, c-C3H5). MS (EI, M = 725.38): m/z (%) 726.4(20) [M]+, 710.5(23) [M–CH3]+, 533.3(10) [M–c-C3H5–CCC(NiPr)2 + H]+, 343.1(32) [M–2(c-C3H5–CCC(NiPr)2)], 327.1(22) [M–2(c-C3H5–CCC(NiPr)2)–CH3]+, 177.1(58) [c-C3H5–CCC(NiPr)2–CH3]+, 149.1(20) [c-C3H5–CCC(NiPr)2–(c-C3H5)]+. IR (KBr): 3653, 3440, 3096, 3015, 2963, 2866, 2608, 2221, 1612, 1466, 1330, 1263, 1210, 1185, 1052, 967, 875, 811, 707, 529, 472 cm−1. Anal. calcd for C36H57N6Sm: C, 59.70; H, 7.93; N, 11.60%. Found: C, 59.80; H, 7.83; N, 11.55%.
CC(NiPr)2]3Ho (2d).
A solution of anhydrous HoCl3 (1.0 g, 3.7 mmol) in 30 ml of THF was added to a solution of 1a (2.2 g, 11.1 mmol) in 60 ml of THF. The reaction mixture was heated to 65 °C for 3 h and then stirred at r.t. for 12 h. The solvent was removed under vacuum followed by extraction with pentane 2 × 15 ml to give a clear bright-yellow solution. The filtrate was concentrated to ca. 5 ml. Crystallization at −30 °C for three months afforded 2d as pale yellow crystals (1.2 g, 45%). Due to the strongly paramagnetic nature of the Ho3+ ion, no meaningful 1H NMR data could be obtained.13C NMR (100.6 MHz, C6D6, 25 °C): δ = 224.8 (NCN), 158.8 (CC–C), 62.7 (HC–CC), 50.4 (CH–(CH3)2), 29.8 (CH3), 26.5 (CH3), 8.7 (CH2, c-C3H5), 0.35 (CH, c-C3H5). MS (EI, M = 738.39): m/z (%) 738.5(35) [M], 723.5(50) [M–CH3]+, 695.5(32) [M–2CH3]+, 547.3(36) [M−c-C3H5–CCC(NiPr)2], 177.1(100) [c-C3H5–CCC(NiPr)2–CH3]+, 149.1(43) [c-C3H5–CCC(NiPr)2–(c-C3H5)]+. IR (KBr): 3440, 3219, 2964, 2932, 2869, 2227, 1636, 1612, 1486, 1375, 1315, 1260, 1179, 1031, 984, 879, 812, 505, 468 cm−1. Anal. calcd for C36H57HoN6: C, 58.52; H, 7.78; N, 11.38%. Found: C, 58.75; H, 7.33; N, 11.17%.
CC(NCy)2]3Sm (2a).
Anhydrous SmCl3 (1.0 g, 4 mmol) and 1b (3.3 g, 12 mmol) were charged in a 250 ml Schlenk flask. 100 ml of THF were added and the mixture was stirred 12 h at r.t. to give a clear yellow solution. The solvent was removed under vacuum followed by extraction with n-pentane (2 × 15 ml). The clear yellow flitrate was evaporated under vacuum affording 2a as a pale yellow solid (3.0 g, 79%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 3.34 (m, 6H, CH, Cy), 1.85 (m, 3H, CH, c-C3H5), 1.56 (br, 12H, CH2, Cy), 1.40 (m, 6H, CH2, c-C3H5), 0.97–1.32 (m, 18H, CH2, Cy), 0.87 (m, 6H, CH2, c-C3H5), 0.69 (br, 12H, CH2, Cy), −0.21 to −0.12 (q, 6H, CH2, Cy), −2.31 (br, 12H, CH2, Cy); 13C{1H} NMR (100.6 MHz, C6D6, 25 °C): δ = 201.9 (NCN), 104.1 (CC–C), 73.7 (HC–CC), 56.9 (CH, Cy), 35.8 (CH2, Cy), 25.5 (CH2, Cy), 9.8 (CH2, c-C3H5), 1.8 (CH, c-C3H5). MS (EI, M = 965.57): m/z (%) 965.7(45) [M], 695.4(70) [M–(c-C3H5–CCC(NCy)2)]+, 272.2(80) [c-C3H5–CCC(NCy)2]+, 229.1(58) [c-C3H5–CCC(NCy)2–(c-C3H5)]+, 190.1(63) [c-C3H5–CCC(NCy)2–(Cy) + 2H]+, 177(100) [c-C3H5–CCC–NCy + 2H]+. IR (KBr): 3668, 3438, 3220, 3012, 2925, 2850, 2665, 2222, 1606, 1469, 1398, 1361, 1174, 1120, 1028, 972, 888, 703, 676, 588 cm−1. Anal. calcd for C54H81N6Sm: C, 67.24; H, 8.46; N, 8.71%. Found: C, 67.22; H, 8.51; N, 8.60%.
CC(NiPr)2]3Nd (2b).
A solution of anhydrous NdCl3 (1.0 g, 4 mmol) in 30 ml of THF was added to a solution of 1a (2.3 g, 12 mmol) in 70 ml of THF. The reaction mixture was heated to 65 °C for 2 h and then stirred at r.t. for 12 h. The solution color changed to blue. Work-up using n-pentane as described for 2a afforded 2b as green crystals (1.5 g, 54%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 22.3 (m, 6H, CH–(CH3)2), 4.10 (m, 3H, CH, c-C3H5), 2.97 (m, 6H, CH2, c-C3H5), 2.02 (m, 6H, CH2, c-C3H5), −3.55 (m, 36H, CH3); 13C{1H} NMR (100.6 MHz, C6D6, 25 °C): δ = 228.6 (NCN), 108.5 (CC–C), 65.3 (CH–(CH3)2), 59.8 (HC–CC), 23.1 (CH3), 12.1 (CH2, c-C3H5), 2.4 (CH, c-C3H5). MS (EI, M = 715.37): m/z (%) 631.6(33) [M–2(iPr)]+, 396.4(20) [2(c-C3H5–CCC(NiPr)2) + CH3]+, 381.3(15) [2(c-C3H5–CCC(NiPr)2)]+, 205.2(50) [(c-C3H5–CCC(NiPr)2) + CH3]+, 177.1(34) [c-C3H5–CCC(NiPr)2–CH3]+, 149.1(17) [c-C3H5–CCC(NiPr)2–(c-C3H5)]+. IR (KBr): 3678, 3439, 3220, 3015, 2963, 2867, 2608, 2220, 1865, 1635, 1591, 1498, 1382, 1332, 1169, 811, 716, 692, 530, 445 cm−1. Anal. calcd for C36H57N6Nd: C, 60.16; H, 7.93; N, 11.69%. Found: C, 60.25; H, 7.92; N, 11.52%.
CC(NiPr)2]3Sm (2c).
A reaction of anhydrous SmCl3 (1.0 g, 4 mmol) with 1a (2.3 g, 12 mmol) following the procedure described for 2a afforded 2d as a yellow, crystalline solid (1.6 g, 55%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 3.60 (m, 6H, CH–(CH3)2), 1.81 (m, 3H, CH, c-C3H5), 1.37 (m, 6H, CH2, c-C3H5), 0.89 (m, 6H, CH2, c-C3H5), −0.47 (m, 36H, CH3); 13C{1H} NMR (100.6 MHz, C6D6, 25 °C): δ = 201.6 (NCN), 104.5 (CC–C), 73.5 (HC–CC), 48.3 (CH–(CH3)2), 25.1 (CH3), 9.7 (CH2, c-C3H5), 1.7 (CH, c-C3H5). MS (EI, M = 725.38): m/z (%) 726.4(20) [M]+, 710.5(23) [M–CH3]+, 533.3(10) [M–c-C3H5–CCC(NiPr)2 + H]+, 343.1(32) [M–2(c-C3H5–CCC(NiPr)2)], 327.1(22) [M–2(c-C3H5–CCC(NiPr)2)–CH3]+, 177.1(58) [c-C3H5–CCC(NiPr)2–CH3]+, 149.1(20) [c-C3H5–CCC(NiPr)2–(c-C3H5)]+. IR (KBr): 3653, 3440, 3096, 3015, 2963, 2866, 2608, 2221, 1612, 1466, 1330, 1263, 1210, 1185, 1052, 967, 875, 811, 707, 529, 472 cm−1. Anal. calcd for C36H57N6Sm: C, 59.70; H, 7.93; N, 11.60%. Found: C, 59.80; H, 7.83; N, 11.55%.
CC(NiPr)2]3Ho (2d).
A solution of anhydrous HoCl3 (1.0 g, 3.7 mmol) in 30 ml of THF was added to a solution of 1a (2.2 g, 11.1 mmol) in 60 ml of THF. The reaction mixture was heated to 65 °C for 3 h and then stirred at r.t. for 12 h. The solvent was removed under vacuum followed by extraction with pentane 2 × 15 ml to give a clear bright-yellow solution. The filtrate was concentrated to ca. 5 ml. Crystallization at −30 °C for three months afforded 2d as pale yellow crystals (1.2 g, 45%). Due to the strongly paramagnetic nature of the Ho3+ ion, no meaningful 1H NMR data could be obtained.13C NMR (100.6 MHz, C6D6, 25 °C): δ = 224.8 (NCN), 158.8 (CC–C), 62.7 (HC–CC), 50.4 (CH–(CH3)2), 29.8 (CH3), 26.5 (CH3), 8.7 (CH2, c-C3H5), 0.35 (CH, c-C3H5). MS (EI, M = 738.39): m/z (%) 738.5(35) [M], 723.5(50) [M–CH3]+, 695.5(32) [M–2CH3]+, 547.3(36) [M−c-C3H5–CCC(NiPr)2], 177.1(100) [c-C3H5–CCC(NiPr)2–CH3]+, 149.1(43) [c-C3H5–CCC(NiPr)2–(c-C3H5)]+. IR (KBr): 3440, 3219, 2964, 2932, 2869, 2227, 1636, 1612, 1486, 1375, 1315, 1260, 1179, 1031, 984, 879, 812, 505, 468 cm−1. Anal. calcd for C36H57HoN6: C, 58.52; H, 7.78; N, 11.38%. Found: C, 58.75; H, 7.33; N, 11.17%.
4.3 General procedure for the addition of phenylacetylene to N,N′-diisopropylcarbodiimide catalyzed by 2a–d.
A 100 ml Schlenk flask was charged with phenylacetylene (1.40 ml, 12.8 mmol) and N,N′-diisopropylcarbodiimide (2.0 ml, 12.8 mmol) in 20 ml of THF. To the mixture was added the catalyst (2a, 2b, 2c, or 2d) (0.5 or 1.0% mmol), dissolved in 5 ml of THF. The resulting mixture was stirred at 60 °C or at room temperature for a fixed time. The solvent was completely removed under vacuum and the product was purified by crystallization from a minimum amount of dry acetonitrile in air to give 3 in yields as shown in Table 2.4.4 General procedure for the addition of terminal alkynes to N,N′-diisopropylcarbodiimide catalyzed by 2a
A 100 ml Schlenk flask was charged with the terminal alkyne (1.0 mmol) and N,N′-diisopropylcarbodiimide (1.0 mmol) in 15 ml of THF. To the mixture was added the catalyst 2a (0.01 mmol), dissolved in 5 ml of THF. The resulting mixture was stirred at 60 °C for a fixed time, as shown in Table 2. The solvent was removed under vacuum and the product was purified by crystallization from a minimum amount of dry acetonitrile in air. The resulting propiolamidines 3–5 were identified through their 1H and 13C NMR data (cf. ESI‡).7,144.5 X-Ray crystallographic studies
The intensity data of 2d and 4 were collected on a Stoe IPDS 2T diffractometer with MoKα radiation. The data were collected with the Stoe XAREA16 program using ω-scans. The space groups were determined with the XRED3224 program. Absorption corrections were applied using the multi-scan method. The structures were solved by direct methods (SHELXS-97)17a and refined by full matrix least-squares methods on F2 using SHELXL-97.17b Data collection parameters are given in Table 1.Acknowledgements
Financial support by the Otto-von-Guericke-Universität Magdeburg is gratefully acknowledged. Farid M. Sroor is grateful to the ministry of Higher Educational Scientific Research (MHESR), Egypt, and the German Academic Exchange Service (DAAD), Germany, for a PhD scholarship within the German Egyptian Research Long-Term Scholarship (GERLS) program.References
- Recent review articles: (a) F. T. Edelmann, Adv. Organomet. Chem., 2008, 57, 183–352 CrossRef CAS; (b) M. P. Coles, Chem. Commun., 2009, 3659–3676 RSC; C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 Search PubMed; (c) A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS PubMed; (d) A. A. Mohamed, H. E. Abdou and J. P. Fackler Jr., Coord. Chem. Rev., 2010, 254, 1253–1259 CrossRef CAS PubMed; (e) S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS PubMed; (f) F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55–374 CrossRef CAS.
- (a) F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC; (b) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- A. Devi, Coord. Chem. Rev., 2013, 257, 3332–3384 CrossRef CAS PubMed , and references cited therein.
- (a) H. Fujita, R. Endo, A. Aoyama and T. Ichii, Bull. Chem. Soc. Jpn., 1972, 45, 1846–1852 CrossRef CAS; (b) G. Himbert, M. Feustel and M. Jung, Liebigs Ann. Chem., 1981, 1907–1927 CrossRef CAS PubMed; (c) G. Himbert and W. Schwickerath, Liebigs Ann. Chem., 1984, 85–97 CrossRef CAS PubMed; (d) G. F. Schmidt and G. Süss-Fink, J. Organomet. Chem., 1988, 356, 207–211 CrossRef CAS; (e) T.-G. Ong, J. S. O'Brien, I. Korobkov and D. S. Richeson, Organometallics, 2006, 25, 4728 CrossRef CAS; (f) X. Xu, J. Gao, D. Cheng, J. Li, G. Qiang and H. Guo, Adv. Synth. Catal., 2008, 350, 61–64 CrossRef CAS PubMed; (g) W. Weingärtner, W. Kantlehner and G. Maas, Synthesis, 2011, 265–272 Search PubMed; (h) W. Weingärtner and G. Maas, Eur. J. Org. Chem., 2012, 6372–6382 CrossRef PubMed.
- (a) H. Fujita, R. Endo, K. Murayama and T. Ichii, Bull. Chem. Soc. Jpn., 1972, 45, 1581 CrossRef CAS; (b) W. Ried and M. Wegwitz, Liebigs Ann. Chem., 1975, 89–94 CrossRef CAS PubMed; (c) W. Ried and R. Schweitzer, Chem. Ber., 1976, 109, 1643–1649 CrossRef CAS PubMed; (d) W. Ried and H. Winkler, Chem. Ber., 1979, 112, 384–388 CrossRef CAS PubMed.
- (a) P. Sienkiewich, K. Bielawski, A. Bielawska and J. Palka, Environ. Toxicol. Pharmacol., 2005, 20, 118–124 CrossRef PubMed; (b) T. M. Sielecki, J. Liu, S. A. Mousa, A. L. Racanelli, E. A. Hausner, R. R. Wexler and R. E. Olson, Bioorg. Med. Chem. Lett., 2001, 11, 2201–2204 CrossRef CAS; (c) C. E. Stephens, E. Tanious, S. Kim, D. W. Wilson, W. A. Schell, J. R. Perfect, S. G. Franzblau and D. W. Boykin, J. Med. Chem., 2001, 44, 1741–1748 CrossRef CAS PubMed; (d) C. N. Rowley, G. A. DiLabio and S. T. Barry, Inorg. Chem., 2005, 44, 1983–1991 CrossRef CAS PubMed.
- (a) W.-X. Zhang, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2005, 127, 16788–16789 CrossRef CAS PubMed; (b) S. Zhou, S. Wang, G. Yang, Q. Li, L. Zhang, Z. Yao, Z. Zhou and H.-B. Song, Organometallics, 2007, 26, 3755–3761 CrossRef CAS; (c) W.-X. Zhang and Z. Hou, Org. Biomol. Chem., 2008, 6, 1720–1730 RSC; (d) Z. Du, W. Li, X. Zhu, F. Xu and Q. Shen, J. Org. Chem., 2008, 73, 8966–8972 CrossRef CAS PubMed; (e) C. N. Rowley, T.-G. Ong, J. Priem, D. S. Richeson and T. K. Woo, Inorg. Chem., 2008, 47, 12024–12031 CrossRef CAS PubMed; (f) Y. Wu, S. Wang, L. Zhang, G. Yang, X. Zhu, Z. Zhou, H. Zhu and S. Wu, Eur. J. Org. Chem., 2010, 326–332 CrossRef PubMed; (g) F. Zhang, J. Zhang, Y. Zhang, J. Hong and X. Zhou, Organometallics, 2014, 33, 6186–6192 CrossRef CAS.
- (a) P. Dröse, C. G. Hrib and F. T. Edelmann, J. Organomet. Chem., 2010, 695, 1953–1956 CrossRef PubMed; (b) L. Xu, Y.-C. Wang, W.-X. Zhang and Z. Xi, Dalton Trans., 2013, 42, 16466–16469 RSC.
- W. W. Seidel, W. Dachtler and T. Pape, Z. Anorg. Allg. Chem., 2012, 638, 116–121 CrossRef CAS PubMed.
- F. M. A. Sroor, C. G. Hrib, L. Hilfert and F. T. Edelmann, Z. Anorg. Allg. Chem., 2013, 639, 2390–2394 CrossRef CAS PubMed.
- F. M. Sroor, C. G. Hrib, L. Hilfert, P. G. Jones and F. T. Edelmann, J. Organomet. Chem., 2015, 785, 1–10 CrossRef CAS PubMed.
- S. Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- G. F. Schmidt and G. Süss-Fink, J. Organomet. Chem., 1988, 356, 207–211 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill, S. L. Lomas, M. S. Heng, P. B. Hitchcock and G. Kociok-Köhn, Dalton Trans., 2014, 43, 14249–14256 RSC.
- J. H. Freeman and M. L. Smith, J. Inorg. Nucl. Chem., 1958, 7, 224 CrossRef CAS.
- Stoe, XAREA Program for Xray Crystal Data collection, (XRED32 included in XAREA), Stoe, 2002 Search PubMed.
- (a) G. M. Sheldrick, SHELXL-97 Program for Crystal Structure Refinement, Universität Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXS-97 Program for Crystal Structure Solution, Universität Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Dedicated to Professor Herbert W. Roesky on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: CIF files of the X-ray structural data for 2d and 4. CCDC 1050915 (2d) and 1050916 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00555h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |