
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinyl sulfone building blocks in covalently reversible reactions with thiols†
Thomas H.
Schneider
a,
Max
Rieger
b,
Kay
Ansorg
b,
Alexandre N.
Sobolev
c,
Tanja
Schirmeister
a,
Bernd
Engels
b and
Simon
Grabowsky
*cd
aUniversity of Mainz, Institute of Pharmacy and Biochemistry, Staudingerweg 5, 55128 Mainz, Germany
bUniversity of Würzburg, Institute of Physical and Theoretical Chemistry, Emil-Fischer-Str. 42, 97074 Würzburg, Germany
cThe University of Western Australia, School of Chemistry and Biochemistry, M310, 35 Stirling Highway, Perth, WA 6009, Australia
dUniversität Bremen, Fachbereich 2 – Biologie/Chemie, Leobener Str. NW2, 28359 Bremen, Germany. E-mail: simon.grabowsky@uni-bremen.de; Tel: +49 421 218 63152
First published on 17th June 2015
Abstract
In the present study we use quantum-chemical calculations to investigate how the reactivity of vinyl sulfone-based compounds can be modified from an irreversible to a reversible reaction with thiols. Based on the predictions from theory, an array of nine different vinyl sulfones with systematically varying substitution pattern was synthesized and their crystal structures were determined. Subsequent Hirshfeld surface analyses employing the principle of electrostatic complementarity aid the understanding of the crystal packing of the synthesized compounds. Reactivity studies against the nucleophile 2-phenylethanethiol mirror the properties predicted by the quantum-chemical computations in solution.
Introduction
In drug design vinyl sulfones are widely used as potent irreversible inhibitors of cysteine proteases.1 The most promising vinyl sulfone might be K11777, a potent inhibitor of parasitic and mammalian proteases in preclinical studies (see Fig. 1a), for which Palmer et al. reported inhibition data and synthesis.2,3 K11777 also targets cruzain, which is the major cysteine protease of the protozoa Trypanosoma cruzi causing Chagas disease.4–8 K11777 and related vinyl sulfones inactivate their target enzymes by the addition of the cysteinate residue of the active site at the double bond. A subsequent protonation of the emerging carbanion drives the reaction towards the thermodynamically stable alkylated enzyme (Fig. 1b).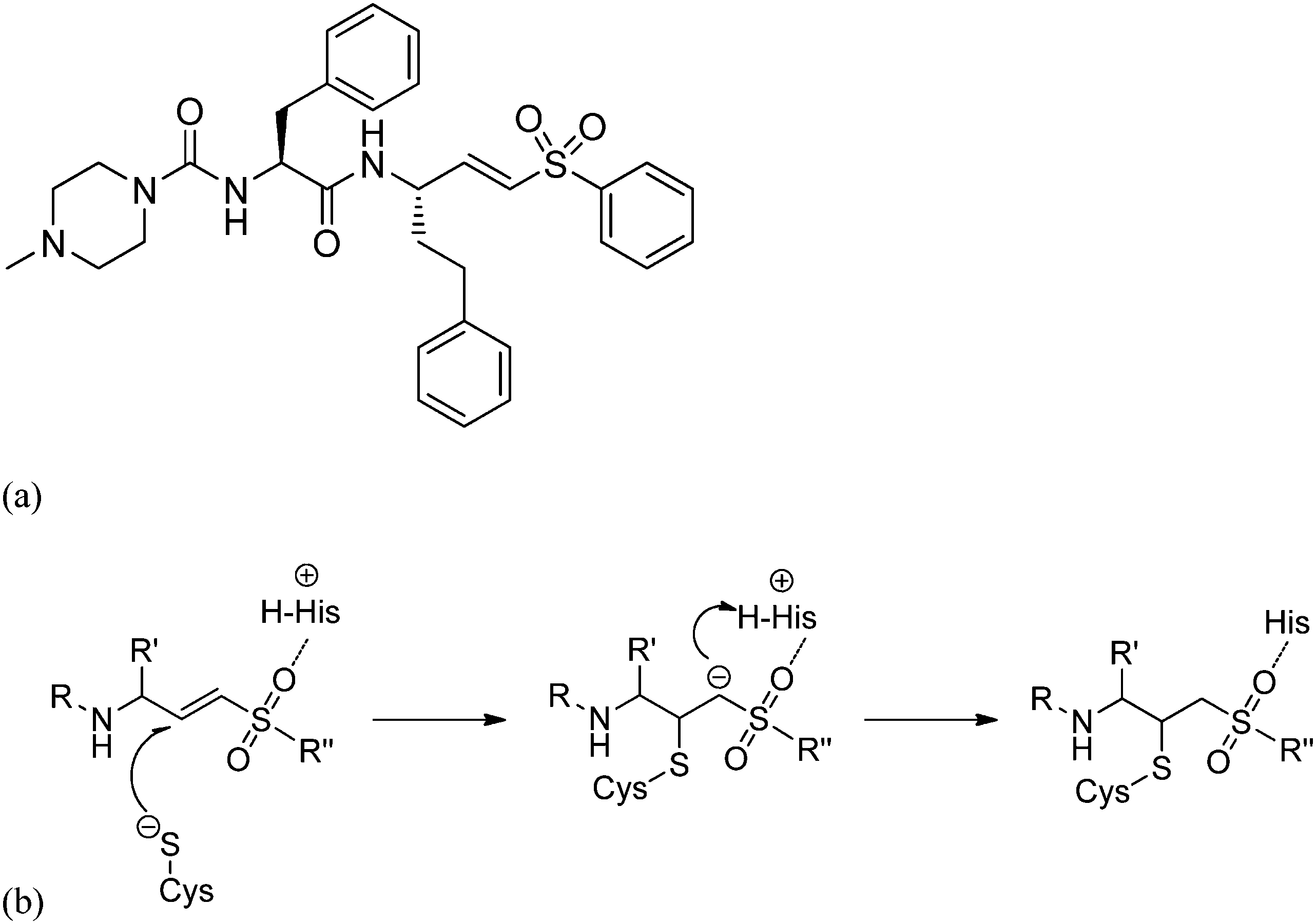 | ||
| Fig. 1 (a) Molecular structure of the vinyl sulfone K11777 and (b) reaction mechanism of the irreversible inactivation of cysteine proteases. | ||
The irreversible reaction of a drug with a biologically relevant thiol can be a source for undesired side reactions, e.g. toxicity due to reaction with glutathione and depletion of the glutathione pool, or alkylation of membrane proteins. On the other hand, a covalent reaction with a target enzyme strongly enhances binding affinity and prolongs the resting time at the enzyme.9 This time is strongly influenced by the free binding energy of the corresponding enzyme–inhibitor complex. Longer resting times can be achieved e.g. by a covalently interacting inhibitor9–12 (Fig. 1b and 2) which leads to an enzyme–inhibitor complex connected via a covalent bond between enzyme E and inhibitor I (E–I). A possibility to exploit the advantage of a covalently interacting inhibitor while avoiding the possible disadvantages of an irreversible reaction with biologically relevant thiols is offered by modulation of the free reaction energy of the underlying thiol reaction ΔG0R (Fig. 2). The free reaction energy strongly influences the half-life time of the covalently bonded adduct E–I. For strongly exergonic reactions (i.e. ΔG0R ≪ 0) the half-life time becomes infinite, i.e. the reaction is irreversible. For slightly exergonic reactions, however, the half-life time is still considerably longer than for non-covalently interacting compounds, but the reaction remains reversible. This strategy to address a Cys residue by an electrophilic moiety which covalently, but still reversibly reacts with the target nucleophilic center was applied recently to kinase inhibitors targeting a non-catalytic, allosteric Cys residue.13
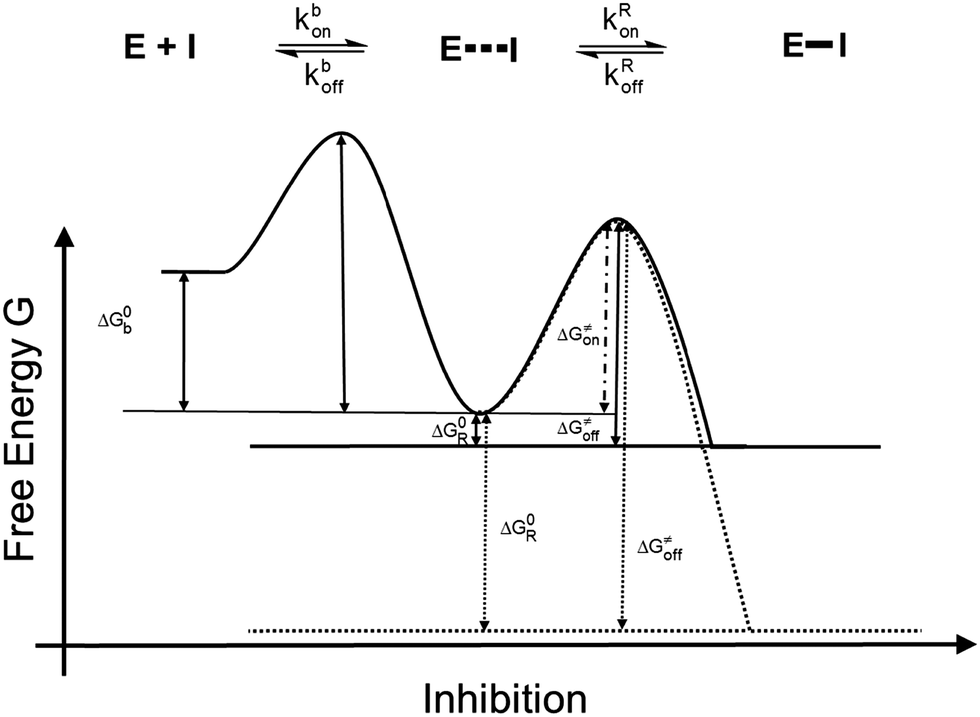 | ||
| Fig. 2 Energy diagrams of the inhibition mechanisms of covalently interacting inhibitors. Solid line: reversibly interacting inhibitors; dotted line: irreversibly interacting inhibitors. | ||
In a first step of an ongoing study with the aim to rationally modulate the thiol reactivity of vinyl sulfones toward a mechanism which is covalent, but still reversible, we present a systematic and comprehensive study on substituted vinyl sulfones. We use vinyl sulfones containing S-Me as substituent at β-position. In our study we started from a quantum-chemical computational screening of the thermodynamics of the reactions of various substituted vinyl sulfones with thiolate, followed by syntheses of model compounds, and structural elucidation by X-ray crystallography. The determination of the crystal structures of the compounds synthesized allowed careful study of intra- and intermolecular geometrical phenomena, and how they depend on variations in the substitution pattern. The importance of steric and electrostatic complementarity on crystal packing was investigated by way of Hirshfeld-surface14 and Politzer-statistics analyses.15 Finally, determination of products, rates and equilibrium constants of the reactions with a low-molecular weight thiol in solution by means of LC/MS, quantitative NMR, and IR spectroscopy was performed.
Results and discussion
Quantum-chemical calculations
Fig. 3 summarizes all possible reactions between the vinyl sulfones and a thiol, e.g. methylthiol (Me'SH). These reactions include the addition at the α- or β-carbon (reactions A and B), as well as the vinylic substitution SNV of the vinyl sulfone moiety (VS, reaction E), of the substituent at α-position X (reaction C) or the substituent S-Me located at the β-position (reaction D). Works of Rappoport, Bernasconi and coworkers revealed that through the right choice of the substituents at the double bond such SNV reactions become slightly exothermic while additional reaction paths are suppressed.16,17 For (RS)PhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh(NO2), the exothermicity of SNV reactions with thiolates was proven experimentally.17 The same was found for vinyl sulfones with X = Br (Fig. 3).18 These data indicate that vinyl sulfones are suitable for the development of compounds which react only slightly exothermic with thiols.
CPh(NO2), the exothermicity of SNV reactions with thiolates was proven experimentally.17 The same was found for vinyl sulfones with X = Br (Fig. 3).18 These data indicate that vinyl sulfones are suitable for the development of compounds which react only slightly exothermic with thiols.
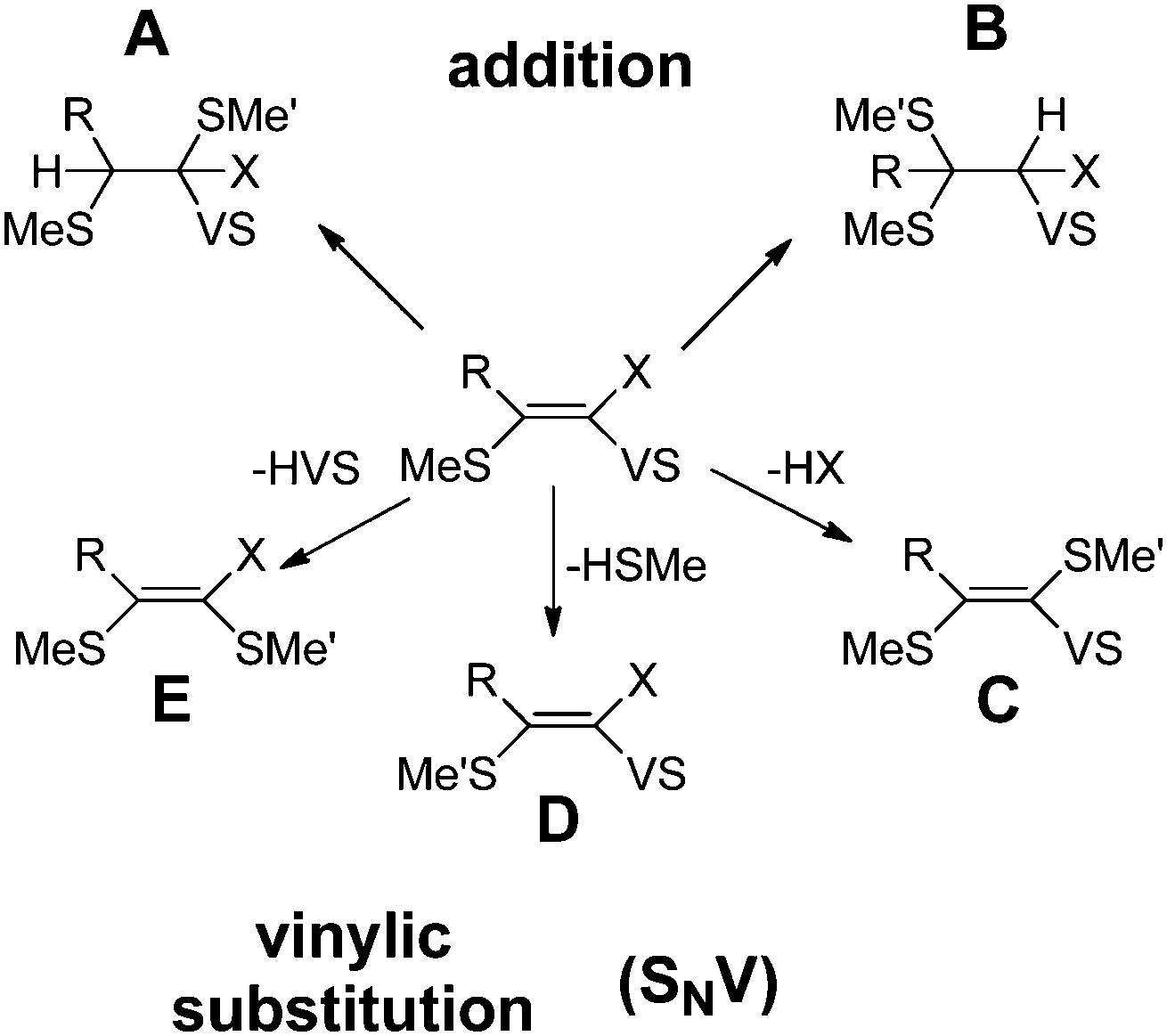 | ||
| Fig. 3 Possible reactions of vinyl sulfones (MeS)RCCX(SO2Ph) with methyl thiol HSMe′. VS abbreviates the sulfone substituent SO2Ph. The reaction energy of reaction D is 0 kcal mol−1 by definition. | ||
In the reaction of a vinyl sulfone with a thiol the substitution of a S–R substitutent (R = alkyl group) (reaction D in Fig. 3) should be almost thermoneutral since the attacking thiolate group replaces an alkyl-thiolate entity. Nevertheless, to be a selective reversibly reacting electrophile which reacts nearly thermoneutrally with the attacking thiol all other possible reactions (addition to α- or β-position (A, B), substitution of the X group (C), substitution of the VS group (E), Fig. 3) must possess positive reaction energies. To obtain information about promising substitution patterns we computed the corresponding reaction energies of various VS derivatives (MeS)RCCX(SO2Ph) with Me′SH. The results are summarized in Table 1. As indicated in Fig. 2 the free energy decides if a reaction takes place or not. However, because in this screening step only a rough estimate is necessary Table 1 gives the computed energy differences between reactants and products without considering vibrational effects or even free energies.
CX(SO2Ph) with Me′SH. See Fig. 3 for abbreviations. The reaction energy for reaction D is 0 kcal mol−1 by definition
| Substituents | Configurationa | Reaction energy ΔER for the product | ||||
|---|---|---|---|---|---|---|
| R | X | A | B | C | E | |
| a Configuration with respect to the positions of MeS and SO2Ph. | ||||||
| N(H)Me | CN | E | 19 | 20 | 16 | 4 |
| = compound 1 | ||||||
| N(H)Me | CN | Z | 6 | 3 | 1 | −10 |
| N(H)Me | Br | E | −6 | −8 | 10 | −2 |
| tert-Butyl | CN | E | 2 | 13 | 12 | −3 |
| tert-Butyl | CN | Z | −10 | −4 | 5 | −33 |
| tert-Butyl | Br | E | −1 | 6 | −8 | −4 |
| Phenyl | CN | E | 4 | 7 | 8 | −2 |
| Phenyl | CN | Z | −6 | −1 | 5 | −32 |
| Phenyl | Br | E | 1 | 3 | −8 | −1 |
The reaction energies for the substitution pattern R = N(H)Me and X = CN in case of the E configured isomer are most promising. All reactions (A, B, C, E) except the vinylic substitution of MeS (reaction D) are not expected to take place due to their positive reaction energies (4–20 kcal mol−1). For the corresponding Z isomer, reaction E becomes strongly exothermic (−10 kcal mol−1), i.e. it will be preferred with respect to the other SNV reactions (C and D) and the addition reactions. Furthermore, the small difference in reaction energies between pathways B, C and D should lead to side reactions. The difference partly results because the Z isomers of the reactants are always higher in energy than the E isomers (R = H, 8 kcal mol−1; R = N(H)Me, 5 kcal mol−1; R = tert-butyl, 1 kcal mol−1; R = phenyl, 4 kcal mol−1) due to steric interactions. Derivatives with X = Br seem to be also less promising since side reactions are predicted to take place due to only small differences between the reaction energies of the possible pathways. The same holds for R = tert-butyl or phenyl.
Syntheses
Based on the quantum-chemical studies, a first series of functionalized vinyl sulfone building blocks with various substituents at the nitrogen atom were synthesized. Besides their promising reaction energies another advantage of such nitrogen containing vinyl sulfones is the facile possibility to connect them to amino acids and peptides which may improve the interactions with binding pockets of a target enzyme. Besides the compound identified as most promising in the computational studies bearing a N(H)Me substituent (first entry of Table 1; compound 1 in Fig. 4) we also included the ethyl derivative (2) and analogs with an aromatic residue (3–9) which influences electronic and steric properties of the vinyl sulfone system.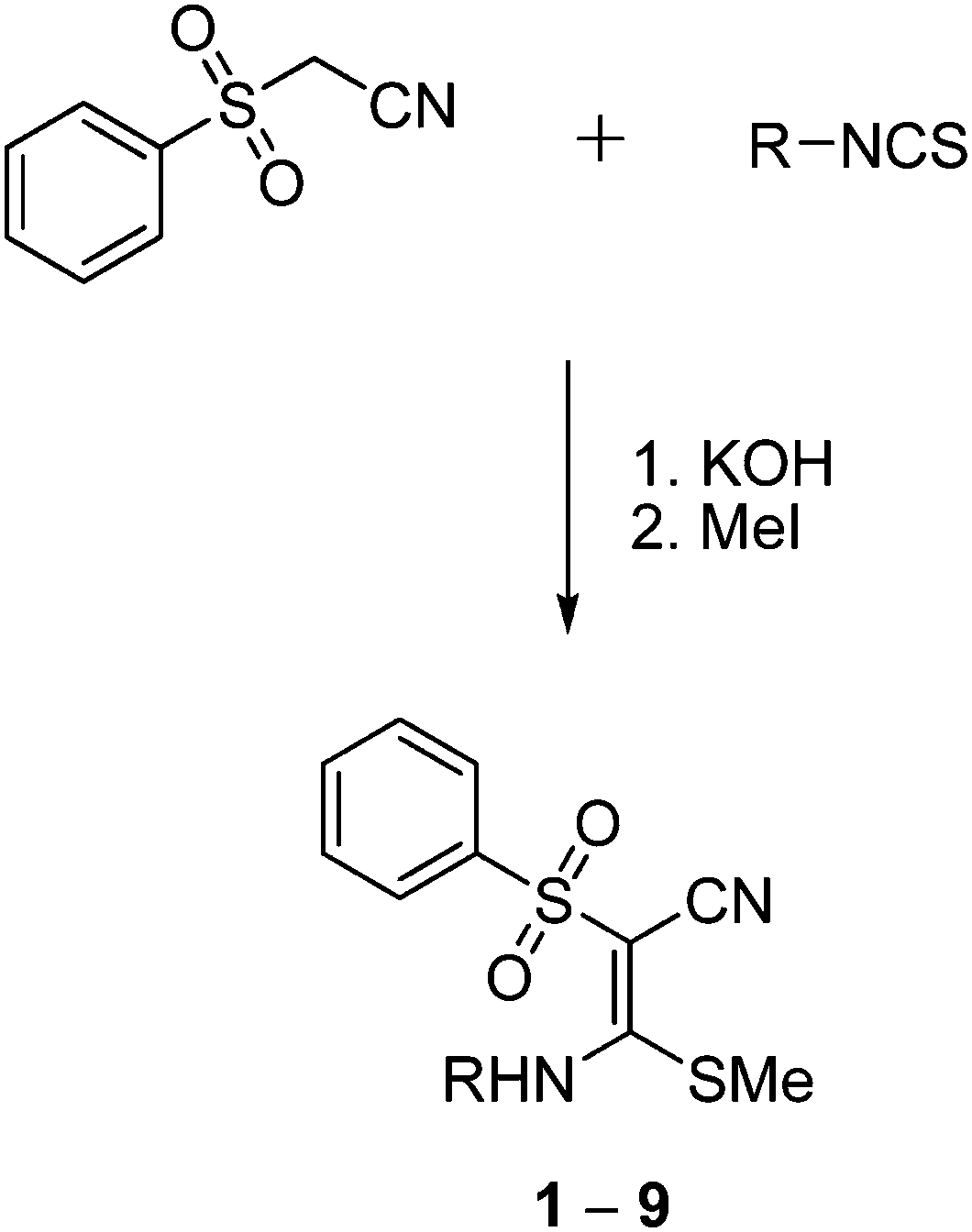 | ||
| Fig. 4 Syntheses of compounds 1 (R = methyl), yield = 36%; 2 (R = ethyl), 59%, 3 (R = 4-methylphenyl), 51%; 4 (R = benzyl), 32%; 5 (R = 4-fluorophenyl), 84%; 6 (R = 4-nitrophenyl), 70%; 7 (R = 4-methoxyphenyl), 53%; 8 (R = 4-iodophenyl), 68%; 9 (R = phenyl), 45%. | ||
Compounds 1–9 were synthesized (Fig. 4) by treatment of (phenylsulfonyl)acetonitrile with potassium hydroxide (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and the respective isothiocyanates in dioxane at 0 °C and subsequent addition of methyl iodide (molar ratio 1:1) to the resulting suspension at 20 °C.19,20 Compounds 1, 3, 7, 9 are known,18,21 but crystal structures of 3, 7, and 9 have not been published before. The formation of the compounds occurred stereospecifically yielding only the E-configured isomers. This stereospecificity results because the E isomer is more stable than the Z isomer (see above) and is supported by the hydrogen bonding between N–H and SO as shown below in the crystal structure analysis.
:2) and the respective isothiocyanates in dioxane at 0 °C and subsequent addition of methyl iodide (molar ratio 1:1) to the resulting suspension at 20 °C.19,20 Compounds 1, 3, 7, 9 are known,18,21 but crystal structures of 3, 7, and 9 have not been published before. The formation of the compounds occurred stereospecifically yielding only the E-configured isomers. This stereospecificity results because the E isomer is more stable than the Z isomer (see above) and is supported by the hydrogen bonding between N–H and SO as shown below in the crystal structure analysis.
Crystal structure and Hirshfeld surface analyses
The structures of all nine investigated compounds were determined by means of single-crystal X-ray diffraction. The data sets measured are of high quality, in fact, the data set of compound 1 is suited for an experimental electron-density investigation, which we published recently.21 It is reconsidered here in the framework of the systematic comparison of the present array of compounds, all of the other eight crystal structures being new determinations. For details on the crystallographic procedure and refinement, see the Experimental section.In principle the synthesis would also allow Z-configuration but as mentioned above, all compounds exhibit E-configuration at the double bond. This is in agreement with our quantum chemical computations which predict the E-configuration to be more stable. In parts the higher stability results because the E-configured isomer can form an intramolecular N2–H2⋯O1 hydrogen bond and a concomitant six-membered ring (see Fig. 5). As the nitrogen atom, the C–C double bond and the S–O bond are in resonance, the hydrogen bond can be described as a resonance assisted hydrogen bond (RAHB), which is a well-studied phenomenon.23 The fact that the double bond in all compounds (range 1.388 to 1.411 Å) is significantly longer than a reference isolated double bond (1.340 Å)24 shows the extent of resonance in the system which in turn explains the electron deficient character of the double bond C1–C2 which makes it attackable by nucleophiles. Hence, the closure of the six-membered ring by the hydrogen bond is energetically very favorable, which yields a rigid fragment around the reactive center measured in terms of the C1–C2, N2⋯O1, H2⋯O1 distances and the N2–H2⋯O1 angle α, cf.Table 2.
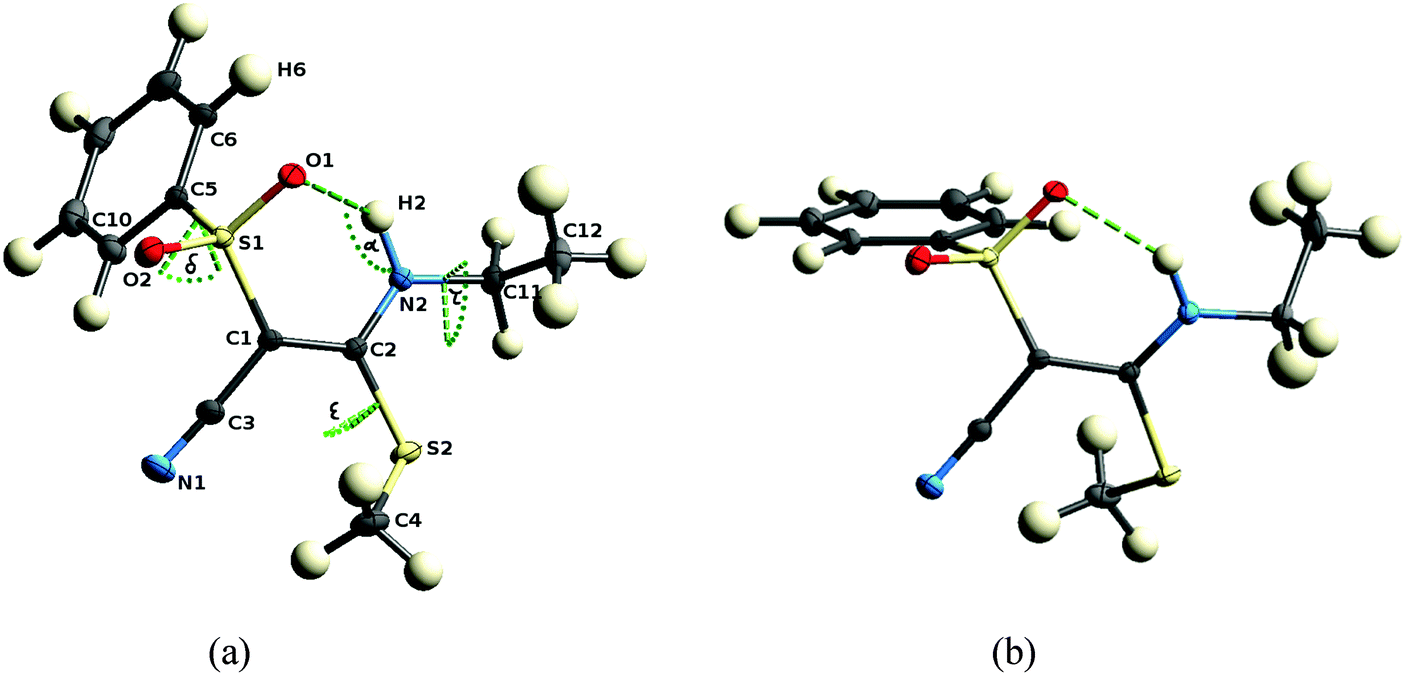 | ||
| Fig. 5 Molecular structures of both independent molecules in the asymmetric unit of 2 with global labeling scheme of atoms and parameters. α = N2–H2⋯O1 bond angle, δ = C1–S1–C5–C10, ε = C1–C2–S2–C4, and τ = C2–N2–C11–C12 torsion angles. | ||
| C1–C2 | N2⋯O1 | H2⋯O1 | α | δ | ε | τ | θ | φ | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.409(1) | 2.787(1) | 2.13 | 121.4 | 119.4(1) | 45.7(1) | N/A | 53.9(1) | 2.4(1) |
| 2(A) | 1.402(1) | 2.877(1) | 2.19 | 123.8 | 58.8 (1) | 32.8(1) | −92.9(1) | −7.1(1) | −56.0(1) |
| 2(B) | 1.411(1) | 2.762(1) | 2.06 | 125.1 | 110.7(1) | −52.5(1) | 138.3(1) | 43.0(1) | −5.6(1) |
| 3 | 1.403(2) | 2.778(2) | 1.98 | 134.2 | 110.0(1) | 144.7(1) | −122.5(1) | 47.2(1) | −5.0(1) |
| 4 | 1.395(1) | 2.783(2) | 2.11 | 122.7 | 109.7(1) | −50.4(1) | −99.8(1) | 43.2(1) | −7.6(1) |
| 5 | 1.396(2) | 2.810(2) | 2.03 | 132.7 | 87.9(1) | 144.9(1) | −132.6(2) | 25.7 (1) | −25.7(1) |
| 6 | 1.400(1) | 2.758(2) | 2.12 | 119.7 | 96.6(1) | −148.1(1) | −141.9(1) | 165.9(1) | −147.8(1) |
| 7 | 1.398(2) | 2.796(2) | 2.01 | 132.5 | 96.3(1) | 151.7(1) | −138.8(1) | 35.1(1) | −17.9(1) |
| 8 | 1.388(2) | 2.726(2) | 1.93 | 133.3 | 104.0(2) | −53.9(2) | −92.9(2) | −12.4(1) | 104.0(1) |
| 9(A) | 1.397(2) | 2.793(2) | 1.98 | 136.0 | 93.7(2) | 147.9(1) | −126.5(2) | 30.1(1) | −20.9(1) |
| 9(B) | 1.396(2) | 2.766(2) | 1.96 | 134.6 | 104.6(1) | 144.0(1) | −142.0(2) | 41.4(1) | −10.4(1) |
Conformational flexibility is only given outside the active region with respect to a rotation of the phenyl group at S1, the methyl group at S2, and the substituent at N2, measured in terms of the torsion angles C1–S1–C5–C10 (δ), C1–C2–S2–C4 (ε), and C2–N2–C11–C12 (τ) (whereby C11 and C12 could be alkylic or phenylic in the various compounds). These torsion angles are thus a proxy of the outer molecular shape of the molecules. A comparison of the two independent molecules in the asymmetric unit of 2 (Fig. 5) is a good example for the central part being only slightly changed, but the peripheral functional groups being significantly rotated. Values for the above defined parameters are listed in Table 2 for all eleven molecules across the nine different compounds.
Different arrangements exist for the orientation of the phenyl group at S1 with respect to the S–O bonds. Fig. 5(a) and (b) are good examples facilitating understanding of this phenomenon. If the phenyl ring is coplanar with the bond S1–O1 (Fig. 5(a), torsion angle θ in Table 2) there is some interaction between H6 and O1 and consequent on this the hydrogen bond H2⋯O1 is lengthened. If the phenyl ring is coplanar with the bond S1–O2 (Fig. 5(b), torsion angle φ in Table 2) not influencing the hydrogen bond, it is shorter. Therefore, indirectly the change of substituents at N2 influences the geometry of the hydrogen bond, because a different substituent leads to a different crystal packing, which in turn leads to a different orientation of the phenyl group.
It is important to study the availability of the groups in the reactive region for intermolecular interactions because the binding of a low-molecular weight ligand to the pocket of an enzyme in general involves the formation of hydrogen bonds.25Fig. 6 exemplifies that for compound 1 both sulfonyl oxygen atoms (O1 and O2) are involved in two intermolecular hydrogen bonds each, regardless of an intramolecular hydrogen bond existing for oxygen O1 (N2–H2⋯O1). Furthermore, H2 forms an intermolecular hydrogen bond with a symmetry-generated N1 nitrogen atom. N1 is furthermore a potent acceptor of hydrogen bonds. In all the investigated compounds, these atoms are the major protagonists of the crystal packing scheme, in each case they are involved in at least one hydrogen bond (compare complete lists of hydrogen bonds in the ESI†).
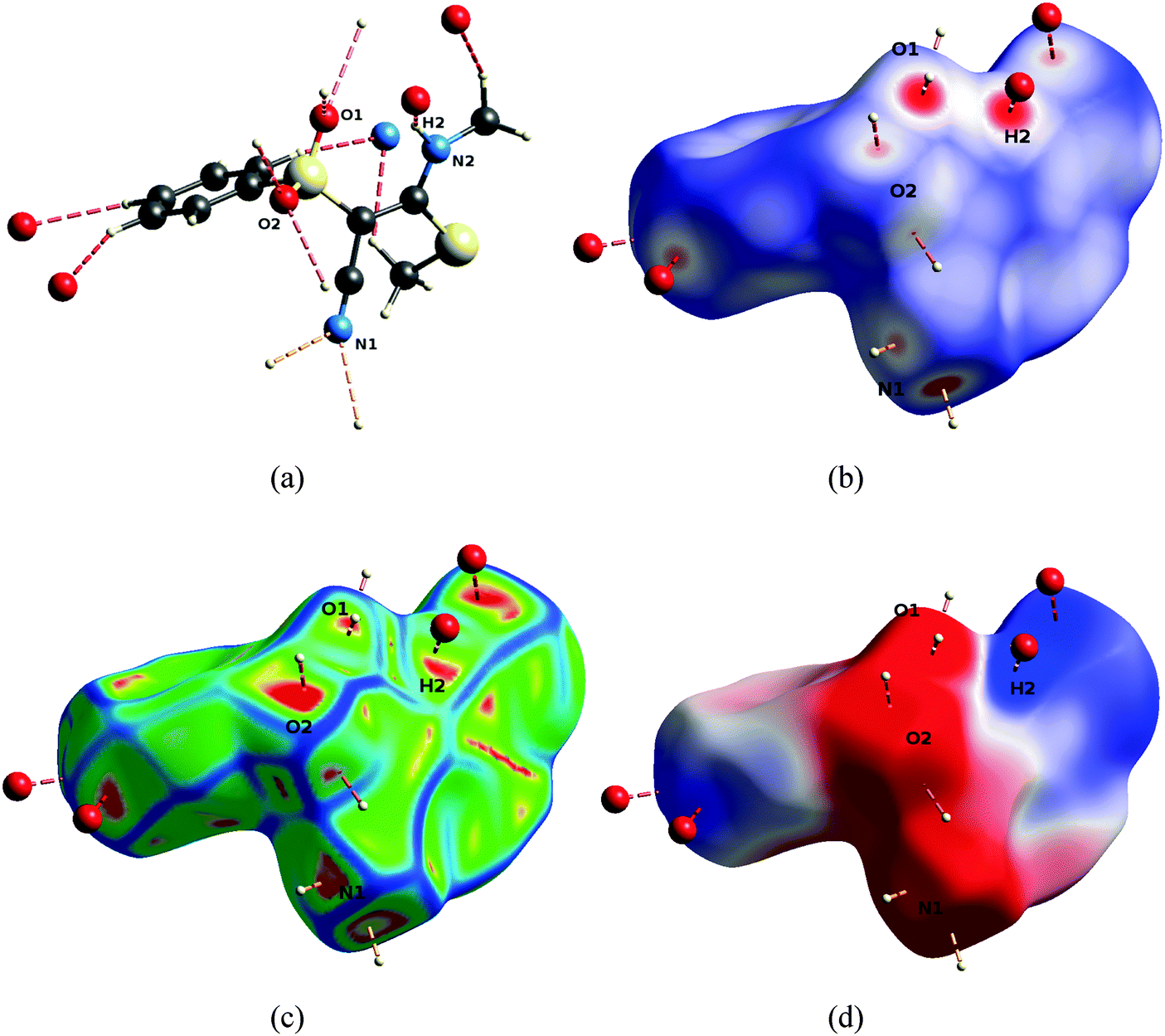 | ||
| Fig. 6 Hydrogen bonding pattern of 1, orientation as defined in Fig. 5. Pictures were generated with the program CrystalExplorer.22 (a) Hydrogen-bonding geometries. (b) Descriptor dnorm mapped onto the Hirshfeld Surface (HS), scale −0.7 (red) to 0 (white) to 1.0 (blue). (c) Descriptor curvedness mapped onto the HS, arbitrary colour scale, red regions indicate highest flatness of the surface. (d) Electrostatic potential (ESP) mapped onto the HS, colour scale −0.05 (red) to 0 (white) to 0.05 a.u. (blue). | ||
An analysis of properties mediated by the shapes of the molecular surfaces of the compounds is a global way to compare interaction networks and packing capacities. Here, we use Hirshfeld surface properties. A Hirshfeld Surface (HS)14 is the outer contour of the space which a molecule or an atom consumes in a crystalline environment. It can be compared to the van der Waals envelope which other molecules or atoms come into contact with when interactions are present, for example in the biological recognition process. The directions and strengths of these interactions can be mapped onto the HSs using various descriptors such as dnorm26 and the curvedness of the surface.27 The former is a ratio encompassing the distances of any surface point to the nearby interior and exterior atoms and the van der Waals radii of the atoms.26 Red colored regions in Fig. 6(b) (i.e. negative values of dnorm) as well as flattened regions of the HS itself (called contact patches, red color for flattest regions, Fig. 6(c)) locally indicate the existence of hydrogen bonds. The centers of the colored regions approximately agree with the intersections of the bond axes of the hydrogen bonds with the surface. Therefore, the locations of interactions of the molecule with its environment can be identified very easily.
The electrostatic potential (ESP) calculated for an isolated molecule at the experimental geometry of that molecule inside the crystal was mapped onto the HSs. It gives complementary information that is less local, more general, and not only based on geometric descriptors. Fig. 6(d) shows that the hydrogen bonds are located in the regions of highest positive or negative ESP. Therefore the ESP mapped onto HSs can be understood as a proxy for the complete intermolecular interactions network. But it furthermore allows a direct appreciation of electrostatic complementarity, which is the guiding force of crystal packing beyond local atom–atom interactions as well as a guiding principle in biological recognition.28,29
The way that electrostatic complementarity works in the crystal packings of the investigated array of compounds is shown in Fig. 7 using 1 as an example. Hirshfeld surfaces are used as a vehicle for displaying the inherent electrostatic nature of the molecules. Therefore, they are not influenced by polarization effects due to neighbouring molecules, and still explain the crystal packing very convincingly. The color code has been chosen in a way to clearly distinguish positive (blue) from negative (red) regions of the ESP, separated by white zero-potential lines. The major interactions and contacts of the molecules in the crystal are due to a match of positive and negative regions of the molecular surfaces. This determines the molecules’ orientations with respect to each other. Remarkably, a continuity of the zero-potential line between different molecules persists to a large extent showing how well the surface regions of opposite potential match.
 | ||
| Fig. 7 Electrostatic complementarity upon crystal packing in 1: electrostatic potential mapped onto the Hirshfeld surfaces of three symmetry-related neighboring molecules, two different orientations shown. Colour scale is −0.01 (red) to 0 (white) to 0.01 a.u. (blue). | ||
The very prominent belt-like region of negative electrostatic potential caused by atoms O1, O2 and N1 with the highly positive region around H2 that is very close to the negative region (Fig. 6(d)) is common to all molecules and seems to be practically unchanged by the substitution pattern. These strong ESP features are only present on one side of the molecule, Fig. S2 (ESI†), which means that a nucleophilic attack at the double bond must occur from the other side. The lack of local sensitivity of the ESP to changes at other parts in the molecule has recently been described in ref. 30 which also summarizes electrostatic-complementarity considerations for biologically active molecules in general.
A quantification of the substituent effects on the distribution of the ESP is attempted by way of a Politzer-type statistical analysis of the ESP on the HSs.15,31 This kind of analysis has been developed for the ESP on 0.001 a.u. electron density isosurfaces, and it has been shown that it is related to biological activity,15,32 but to the best of our knowledge it has not yet been described for HSs. Electron density isosurfaces of molecules in the crystal can overlap, but HSs do not by definition, so that HSs are better suited to explore properties of interacting systems. Moreover, Hirshfeld surfaces change in interacting systems because their shapes depend on the arrangement of all other molecules around the molecule in question. However, the shapes of 0.001 a.u. electron density isosurfaces and HSs of the same compound are similar (see an example in the ESI†). Therefore, the entries for compound 1 in Table 3 show that the Politzer properties for both definitions of molecular surfaces are virtually identical.
![[V with combining overline]](https://www.rsc.org/images/entities/i_char_0056_0305.gif) S+ and S−) together with their variances (σ+2 and σ−2), and the average deviation from the mean surface potential Π. Obtained using the program CrystalExplorer22 based on Hirshfeld surfaces. Second row in 1 refers to a 0.001 a.u. electron-density isosurface
S+ and S−) together with their variances (σ+2 and σ−2), and the average deviation from the mean surface potential Π. Obtained using the program CrystalExplorer22 based on Hirshfeld surfaces. Second row in 1 refers to a 0.001 a.u. electron-density isosurface
|
S
+/e Å−1 |
σ + 2/e Å−2 |
S
−/e Å−1 |
σ − 2/e Å−2 | Π/e Å−1 | |
|---|---|---|---|---|---|
| 1 | 0.051 | 0.0008 | −0.065 | 0.0024 | 0.056 |
| 0.051 | 0.0007 | −0.065 | 0.0019 | 0.056 | |
| 2(A) | 0.043 | 0.0006 | −0.068 | 0.0028 | 0.051 |
| 2(B) | 0.051 | 0.0007 | −0.067 | 0.0026 | 0.055 |
| 3 | 0.046 | 0.0005 | −0.069 | 0.0028 | 0.053 |
| 4 | 0.043 | 0.0008 | −0.052 | 0.0020 | 0.046 |
| 5 | 0.050 | 0.0007 | −0.063 | 0.0026 | 0.052 |
| 6 | 0.059 | 0.0010 | −0.061 | 0.0019 | 0.057 |
| 7 | 0.048 | 0.0007 | −0.066 | 0.0028 | 0.053 |
| 8 | 0.046 | 0.0008 | −0.050 | 0.0024 | 0.047 |
| 9(A) | 0.047 | 0.0006 | −0.068 | 0.0026 | 0.053 |
| 9(B) | 0.043 | 0.0005 | −0.063 | 0.0024 | 0.050 |
The descriptors listed in Table 3 are the mean positive and negative surface potentials (S+ and S−) together with their variances (σ+2 and σ−2). Additionally, the magnitude Π (the average deviation from the mean surface potential) is listed, which is a measure of the internal charge separation or local polarity of a molecule.15 The mean potentials as well as Π are remarkably similar, showing that substituent effects play a minor role with respect to the ESP distribution and that the ESP effects caused by atoms O1, O2, N1 and H2 in the active region of the molecules indeed dictate. In a similar comparative study on protease inhibitor model compounds, the differences in Π were found to be as large as 0.054 e Å−1; here, the maximum difference is 0.011 e Å−1.32
Studies on reaction with 2-phenylethanethiol (PET)
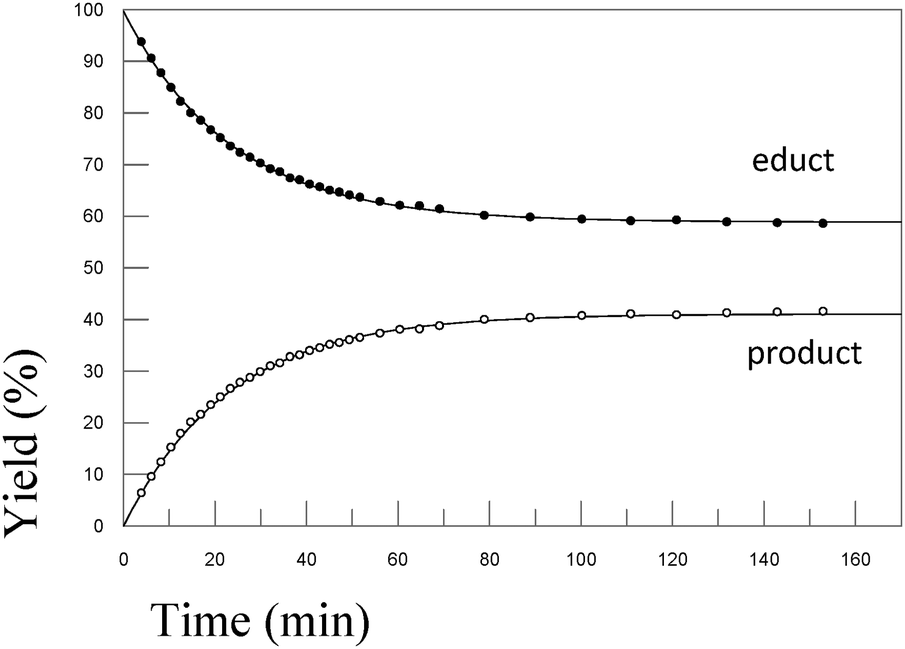
In order to test the results of the quantum-chemical computations, compound 1 was reacted with PET. The reaction was monitored via NMR (see Experimental part) and IR spectroscopy (see Experimental part), and the product was confirmed by mass spectrometry (see Experimental part). Upon the dissolution of 1 (10 mM) and PET (78 mM), exchange of the mercaptomethyl residue by PET was observed (see Fig. 8). No other side reaction occurred as also proven by LC/MS analysis (liquid chromatography/mass spectrometry, see Experimental part). In order to quantify educts and products the resonance signals of the NCH3 (δ = 3.05 ppm and 3.19 ppm) and SCH3 (δ = 2.50 ppm) substituents of the vinyl sulfone were integrated as a function of time. Within 60 minutes, the reaction equilibrated at a limit of approximately 88% (see Fig. 9) yielding an equilibrium constant K = 0.93 and a free reaction energy of 0.041 kcal mol−1.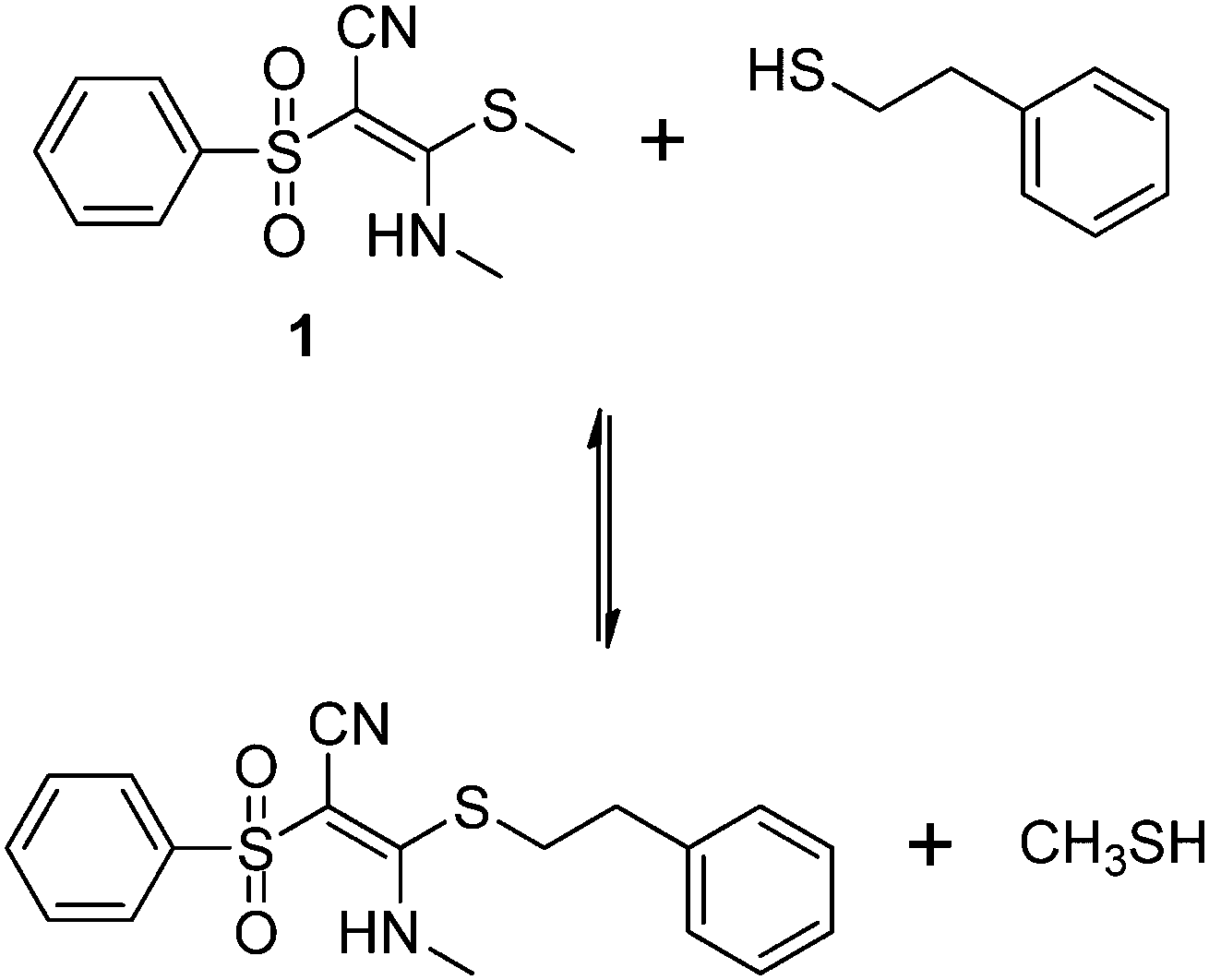 | ||
| Fig. 8 Reversible vinylic substitution reaction of 1 and 2-phenylethanethiol. | ||
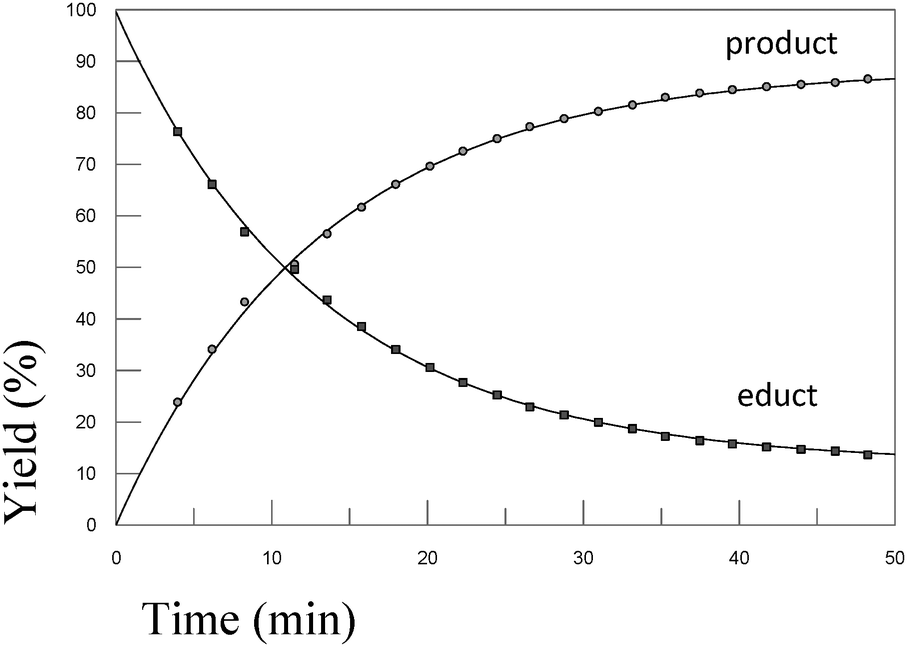 | ||
| Fig. 9 Kinetics of the reaction of 1 (10 mM) and 2-phenylethanethiol (78 mM) upon dissolution in d4-MeOD. The experimental data were extracted from 1H-NMR spectra (27 °C, 400.13 MHz) in which the NCH3 resonance signals of 1 served as the probe. | ||
The 1H-NMR spectra measured 7 days after the dissolution of 1 and PET showed no remarkable change of the ratio of the integrals. To obtain information about the influence of the concentration of PET on the kinetics of the vinylic substitution, another experiment under second-order conditions, i.e. with an equal amount of PET and 1 was performed (see Fig. 10). The reaction equilibrated within 80 minutes at a limit of 44% product. These results are consistent with our presumed thermoneutral reaction that should result in a 50:50 steady state and a K value of ca. 1.
 | ||
| Fig. 10 Kinetics of the reaction of 1 (10 mM) and 2-phenylethanethiol (10 mM) upon dissolution in d4-MeOD. The experimental data were extracted from 1H-NMR spectra (27 °C, 400.13 MHz) in which the NCH3 resonance signals of 1 served as the probe. | ||
The same experiments were also performed with vinyl sulfones 2 and 7. For these compounds the equilibrium constant was determined to K(2) = 0.58 (free reaction energy 0.32 kcal mol−1), and K(7) = 0.69 (free reaction energy 0.22 kcal mol−1) confirming the quantum-chemical computations which predict the thermoneutral vinylic substitution as the only pathway.
Additional LC/MS studies with compounds 4, 5 and 6 and PET in methanol also proved that besides the products of the vinylic substitution no other side products were formed. For this purpose, samples of the reactions with an excess of PET were analyzed after the reaction was finished. In all cases, the LC/MS spectra (see Experimental part) show only two peaks, educt and product. The mass spectra confirm the identity of the educt and the expected product, namely the vinyl sulfone resulting from the substitution of the methylthiolate by PET. The ratios of educts and products were determined by integration of the peaks. Again, the equilibrium adjusts at close to 50:50 in all cases. However, the individual values (4 → 51:49; 5 → 40:60; 6 → 41:59) might suggest a small trend with respect to the electron-withdrawing properties of the substituent at the nitrogen atom.
Furthermore, we performed an analysis of the reaction of compound 1 and PET in methanol by IR spectroscopy (see Experimental part). We used the wavenumber 1540 cm−1 to measure the amount of the starting material and the wavenumber 1642 cm−1 to measure the formation of the product. After 80 minutes the reaction was completed and equilibration was adjusted confirming the NMR studies with an orthogonal spectroscopic method.
Conclusion
Based on computed reaction energies of several reactions of vinyl sulfones by quantum-chemical methods we identified substituted vinyl sulfones which react nearly thermoneutrally with a thiol along the wanted covalent reversible mechanism while all other possible reaction paths are thermodynamically too unfavorable to take place. With the syntheses of compounds 1–9, a series of corresponding substituted vinyl sulfones was prepared. The mechanism as well as the thermodynamics of their reaction with a model thiol were investigated by monitoring the reactions of the compounds with the low-molecular weight thiol PET via NMR- and IR-spectroscopy, as well as mass spectrometry. It was verified that the reactions are thermoneutral – as predicted by the quantum-chemical calculations. As such, our investigations reveal a route from irreversibly to reversibly interacting covalent warheads.Crystal-structure analyses were performed to check the outcome of quantum-chemical calculations about the relative stability of E- and Z-configuration and to investigate possible reasons experimentally. The structures of compounds 1–9 showed that the existence of a strong resonance assisted hydrogen bond leads to a rigid six-membered ring that is the structure- and electrostatic-potential-determining kernel fragment common to all investigated molecules. The variation of substituents has little influence on the geometry of the ring or the electrostatic potential in the reactive region. Hirshfeld-surface analysis showed how electrostatic complementarity manifests itself in the crystal packing of the investigated array of compounds with matching patches of negative and positive electrostatic potential facing each other and exhibiting continuous zero-potential lines across different molecular surfaces.
The present paper represents the first step within approaches which design covalent reversibly acting inhibitors out of known covalent irreversible ones. The presented protocol is able to predict those variations in the substitution pattern which are necessary to shift the reaction energy in the desired magnitude. Additionally, it suggests experiments which are able to verify these predictions. The method was applied to sulfur containing residues, but a generalization only requires some variations in the experiments. The performed X-ray diffraction experiments provide information about the interaction of a given molecule with its crystal environment. They are important because they can be transferred to the interactions between the given ligand and the enzyme environment, i.e. are needed to improve the affinity of a given ligand.
Future investigations will concentrate on the covalent and non-covalent interactions between the new warheads and the targeted enzymes, i.e. contain the step from solution to enzyme. This will be facilitated by combining the presented vinyl sulfones with appropriate recognition units and investigate their applicability as covalent but still reversible warheads targeting catalytic or non-catalytic Cys residues.
Experimental section
General procedures
All syntheses were carried out under dry argon. The organic solvents were dried and purified according to standard procedures and stored under nitrogen. The melting points were determined with a Stuart Melting Point SMP3 apparatus using samples in sealed capillaries. The solution 1H and 13C NMR spectra were recorded at 23 °C on a Bruker Avance 400 (1H: 400.1 MHz; 13C: 100.6 MHz) using [D6]DMSO, CDCl3 or d4-MeOD as the solvent. Chemical shifts (ppm) were determined relative to internal [D5]DMSO (1H: d = 2.49 ppm, [D6]DMSO), d3-MeOD (1H: d = 4.70 ppm, MeOD), [D6]DMSO (13C: d = 39.5 ppm, [D6]DMSO), or external tetramethylsilane (TMS) (13C: d = 0 ppm, [D4]MeOD) standards. Assignment of the 13C NMR data was supported by DEPT 135, 13C, 1H HMQC, and 13C, 1H HMBC experiments.Crystals were mounted within a nitrogen-gas stream on a four-circle kappa-geometry Oxford Diffraction Xcalibur diffractometer equipped with a CCD area detector. During the measurements with a conventional Mo-Kα radiation source, a temperature of 100 K was maintained. High maximum resolutions in the range of 0.58 to 0.45 Å as well as high redundancy and completeness were reached. Crystallographic details can be found below, in the ESI† and from the deposited cif files (CCDC numbers 852449, 897056–897063).
General method of the syntheses
Isothiocyanate and (phenylsulfonyl)acetonitrile (1.0–1.2 eq.) were solved in 40 ml of dry dioxane at 0 °C. Potassium hydroxide (2.0–2.6 eq.) was added and the mixture was stirred at ambient temperature for 2 h. Methyl iodide (1.0–1.2 eq.) was added to the resulting suspension and the mixture was stirred for additional 10 h. The solution was poured into cold water and the precipitate was removed. The clear solution was neutralized by adding acetic acid. The product was isolated by filtration and recrystallized from ethanol.19
1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 2.50 (s, 3H, SCH3), 3.14 (s, 3H, NCH3), 7.46–7.52 (m, 2H, 3/5-Ph–CH), 7.54–7.60 (m, 1H, 4-Ph–CH), 7.86 (dd, J = 8.41, 1.38 Hz, 2H, 2/6-Ph–CH); 13C-NMR: (100.61 MHz, CDCl3): δ (ppm) = 18.07 (SCH3), 32.92 (NCH3), 83.47 (CC–N), 116.30 (CN), 126.62 (2C, 2/6-Ph–CH), 129.33 (2C, 3/5-Ph–CH), 133.47 (4-Ph–CH), 141.94 (1-Ph–C), 170.00 (CC–N); M.p. 137 °C; LOOP-ESI-MS calcd for C11H12N2O2S2: 268.03; found: 269.1 [M + H]+.
Space group = monoclinic P21/n, a = 7.930(2) Å, b = 11.306(2) Å, c = 14.113(3) Å, β = 99.94(3)°, V = 1246.3(4) Å3, Z = 4, max. resolution d = 0.45 Å, redundancy = 28.5, no. unique reflections = 14320, no. observed reflections (I2 > 2σ(I)) = 12324, Rint = 4.98%, completeness = 100.0%, R(1) = 2.65%, CCDC 852449.
1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 1.22 (t, J = 7.22 Hz, 3H, CH3), 2.47 (s, 3H, SCH3), 3.53 (qd, J = 7.24, 5.65 Hz, 2H, NCH2), 7.43–7.52 (m, 2H, 3/5-Ph–CH), 7.52–7.60 (m, 1H, 4-Ph–CH), 7.81–7.88 (m, 2H, 2/6-Ph–CH); 13C-NMR: (100.61 MHz, CDCl3): δ (ppm) = 15.43 (CH3), 18.09 (SCH3), 41.39 (CH2), 83.13 (CCCN), 116.29 (Cq, CCN), 126.42 (2C, 2/6-Ph–CH), 129.23, (2C, 3/5-Ph–CH) 133.35 (4-Ph–CH), 141.98 (1-Ph–C), 169.07 (CCSCH3); M.p. 85 °C; LOOP-ESI-MS calcd for C12H14N2O2S2: 282.05; found: 283.1 [M + H]+.
Space group = triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.0971(1) Å, b = 12.4447(2) Å, c = 13.3768(3) Å, α = 87.181(2)°, β = 80.232(1)°, γ = 88.150(1)°, V = 1326.39(4) Å3, Z = 4, max. resolution d = 0.49 Å, redundancy = 4.1, no. unique reflections = 22625, no. observed reflections (I2 > 2σ(I)) = 15703, Rint = 3.18%, completeness = 99.5%, R(1) = 3.20%, CCDC 897056.
, a = 8.0971(1) Å, b = 12.4447(2) Å, c = 13.3768(3) Å, α = 87.181(2)°, β = 80.232(1)°, γ = 88.150(1)°, V = 1326.39(4) Å3, Z = 4, max. resolution d = 0.49 Å, redundancy = 4.1, no. unique reflections = 22625, no. observed reflections (I2 > 2σ(I)) = 15703, Rint = 3.18%, completeness = 99.5%, R(1) = 3.20%, CCDC 897056.
1H-NMR: (400.13 MHz, CDCl3): δ (ppm) = 2.06 (s, 3H, CH3), 2.33 (s, 3H, SCH3), 7.01–7.07 (m, 2H, 3/5-Ph–CH), 7.12–7.18 (m, 2H, 2/6-Ph–CH), 7.50 (t, J = 7.65 Hz, 2H, 3/5-PhS–CH), 7.56–7.64 (m, 1H, 4-PhS–CH), 7.80–7.98 (m, 2H, 2/6-PhS–CH); 13C-NMR: (100.61 MHz, CDCl3): δ (ppm) = 17.08 (SCH3), 21.06 (CH3), 87.55 (CCCN), 115.37 (CN), 124.52 (2/6-Ph–CH), 126.62 (2/6-PhS–CH), 129.47 (2C, 3/5-PhS–CH), 130.29 (2C, 3/5-Ph–CH), 133.74 (4-PhS–CH), 134.91 (1-Ph–C), 137.68 (4-Ph–C), 141.59 (1-PhS–C), 167.05 (CCSCH3); M.p. 167 °C; LOOP-ESI-MS calcd for C17H16N2O2S2: 344.07; found: 345.1 [M + H]+.
Space group = monoclinic P21/c, a = 8.4275(2) Å, b = 16.8324(4) Å, c = 11.6886(2) Å, β = 97.869(2)°, V = 1642.47(6) Å3, Z = 4, max. resolution d = 0.54 Å, redundancy = 7.3, no. unique reflections = 10664, no. observed reflections (I2 > 2σ(I)) = 5529, Rint = 7.07%, completeness = 98.3%, R(1) = 3.94%, CCDC 897057.
1H-NMR: (400.13 MHz, CDCl3): δ (ppm) = 2.49 (s, 3H, SCH3), 4.70 (d, J = 5.90 Hz, 2H, NCH2), 7.19 (dd, J = 7.84, 1.57 Hz, 2H, Ph), 7.27–7.39 (m, 3H, Ph), 7.41–7.50 (m, 2H, 3/5-PhS-CH), 7.53–7.61 (m, 1H, 4-PhS–CH), 7.77 (dd, J = 8.47, 1.19 Hz, 2H, 2/6-PhS–CH); 13C-NMR: (100.61 MHz, CDCl3): δ (ppm) = 18.34 (SCH3), 50.08 (CH2), 85.03 (CCCN), 116.05 (CN), 126.72 (2/6-PhS–CH), 127.33 (2/6-Ph–CH), 128.46 (4-Ph–CH), 129.22, 129.31 (4C, 3/5-PhS–CH, 3/5-Ph–CH), 133.49 (4-Ph–CH), 136.07 (1-Ph–C), 141.76 (1-PhS–C), 169.04 (CCSCH3); M.p. 84 °C; LOOP-ESI-MS calcd for C17H16N2O2S2: 344.07; found: 345.1 [M + H]+.
Space group = monoclinic C2/c, a = 29.3586(16) Å, b = 5.6119(2) Å, c = 24.1160(15) Å, β = 122.985(8)°, V = 3332.8(3) Å3, Z = 8, max. resolution d = 0.49 Å, redundancy = 3.8, no. unique reflections = 14231, no. observed reflections (I2 > 2σ(I)) = 7127, Rint = 6.46%, completeness = 99.5%, R(1) = 4.44%, CCDC 897058.
1H-NMR (400.13 MHz, CDCl3): δ (ppm) = 2.10 (s, 3 H, SCH3), 7.01–7.09 (m, 2H, 3/5-PhF–CH), 7.11–7.18 (m, 2H, 2/6-PhF–CH), 7.48–7.55 (m, 2H, 3/5-Ph–CH), 7.57–7.66 (m, 1H, 4-PhS–CH), 7.89 (dd, J = 8.41, 1.13 Hz, 2H, 2/6-Ph–CH); 13C-NMR: (100.61 MHz, CDCl3): δ (ppm) = 17.14 (SCH3), 88.27 (CC–CN), 115.14 (CN), 116.58, 116.81 (2C, 3/5-PhN–CH), 126.68, 126.76 (2C, 2/6-PhN–CH), 126.88 (2C, 2/6-PhS–CH), 129.54 (2C, 3/5-PhS–CH), 133.47, 133.50 (1-PhN–C), 133.93 (4-PhS–CH), 141.32 (1-PhS–C), 160.19, 162.66 (4-PhN–CF), 167.12 (CC–SCH3); M.p. 137–138 °C; LOOP-ESI-MS calcd for C16H13FN2O2S2: 348.04; found: 349.0 [M + H]+.
Space group = monoclinic P21/c, a = 8.3288(2) Å, b = 11.2945(2) Å, c = 17.1609(4) Å, β = 100.643(2)°, V = 1586.55(6) Å3, Z = 4, max. resolution d = 0.50 Å, redundancy = 4.2, no. unique reflections = 13383, no. observed reflections (I2 > 2σ(I)) = 8950, Rint = 5.76%, completeness = 99.8%, R(1) = 6.12%, CCDC 897059.
1H-NMR (400.13 MHz, DMSO-d6): δ (ppm) = 2.24 (br. s, 3H, SCH3), 7.30–7.35 (m, 2H, 2/6-PhN–CH), 7.60–7.69 (m, 2H, 3/5-PhS–CH), 7.74 (t, J = 7.28 Hz, 1H, 4-PhS–CH), 7.84–7.88 (m, 2H, 2/6-PhS–CH), 8.19 (d, J = 8.78 Hz, 2H, 3/5-PhN–CH); 13C-NMR: (100.61 MHz, DMSO-d6): δ (ppm) = 16.10 (SCH3), 66.34 (CC–N), 115.20 (CN), 121.50 (2C, 2/6-PhN–CH), 125.06 (2C, 3/5-PhN–CH), 126.79 (2C, 2/6-PhS–CH), 129.51 (2C, 3/5-PhS–CH), 133.80 (4-PhS–CH), 141.13 (1-PhS–C), 143.39 (1-PhN–C), 145.79 (4-PhN–C), 154.69 (CC–N); M.p. 161–162 °C; LOOP-ESI-MS calcd for C16H13N3O4S2: 375.03; found: 374.1 [M − H]−.
Space group = monoclinic P21/n, a = 11.226(2) Å, b = 10.807(2) Å, c = 14.909(4) Å, β = 110.24(3)°, V = 1697.2(6) Å3, Z = 4, max. resolution d = 0.47 Å, redundancy = 7.5, no. unique reflections = 17254, no. observed reflections (I2 > 2σ(I)) = 14297, Rint = 3.57%, completeness = 97.7%, R(1) = 4.60%, CCDC 897060.
1H-NMR: (400.13 MHz, CDCl3): δ (ppm) = 2.08 (s, 3H, SCH3), 3.78 (s, 3H, OCH3), 6.83–6.89 (m, 2H, 2/6-Ph–CH), 7.02–7.11 (m, 2H, 3/5-Ph–CH), 7.47–7.55 (m, 2H, 3/5-PhS–CH), 7.56–7.64 (m, 1H, 4-PhS–CH), 7.87–7.92 (m, 2H, 2/6-PhS–CH); 13C-NMR: (100.61 MHz, DMSO-d6): δ (ppm) = 16.20 (SCH3), 55.33 (OCH3), 75.78 (CC–N), 114.35 (2C, 2/6-PhN–CH), 115.65 (CN), 125.64 (2C, 3/5-Ph–CH), 126.42 (2C, 2/6-PhS–CH), 129.48 (2C, 3/5-Ph–CH), 131.38 (4-PhN–C), 133.46 (4-PhS–CH), 142.04 (1-PhS–C), 157.71 (1-PhN–C), 167.71 (CC–N); M.p. 131 °C; LOOP-ESI-MS calcd for C17H16N2O3S2: 360.06; found: 361.1 [M + H]+.
Space group = monoclinic P21/c, a = 8.6156(2) Å, b = 20.5840(4) Å, c = 9.6518(2) Å, β = 101.108(2)°, V = 1679.62(6) Å3, Z = 4, max. resolution d = 0.58Å, redundancy = 3.9, no. unique reflections = 8593, no. observed reflections (I2 > 2σ(I)) = 5829, Rint = 3.81%, completeness = 95.9%, R(1) = 3.42%, CCDC 897061.
1H-NMR: (400.13 MHz, DMSO-d6): δ (ppm) = 2.10 (s, 3H, SCH3), 6.63 (d, J = 8.41 Hz, 2H, 2/6-PhI–CH), 7.49–7.64 (m, 5H, Ph), 7.73–7.90 (m, 2H, 2/6-PhS–CH); 13C-NMR: (100.61 MHz, DMSO-d6): δ (ppm) = 15.90 (SCH3), 66.37 (CC–NH), 86.93 (Cq, 4-Ph–C), 119.09 (Cq, CN), 124.39 (2C, 2/6-Ph–CH), 126.48 (2/6-PhS–CH), 128.66 (2C, 3/5-PhS–CH), 131.99 (4-PhS–CH), 137.33 (2C, 3/5-Ph–CH), 144.15 (1-Ph–C), 145.48 (1-PhS–C), 168.89 (CC–NH); M.p. 169 °C; LOOP-ESI-MS calcd for C16H13IN2O2S2: 455.95; found: 455.0 [M − H]−.
Space group = monoclinic C2/c, a = 28.3814(8) Å, b = 6.2777(1) Å, c = 21.5020(7) Å, β = 117.043(2)°, V = 3412.15(16) Å3, Z = 8, max. resolution d = 0.50 Å, redundancy = 7.5, no. unique reflections = 14388, no. observed reflections (I2 > 2σ(I)) = 11464, Rint = 4.23%, completeness = 99.7%, R(1) = 3.16%, CCDC 897062.
1H-NMR: (400.13 MHz, DMSO-d6): δ (ppm) = 2.13 (s, 3H, CH3) 7.18 (d, J = 7.53 Hz, 2H, 2/6-Ph–CH), 7.25 (t, J = 7.40 Hz, 1H, 4-Ph–CH), 7.38 (t, J = 7.78 Hz, 2H, 3/5-Ph–CH), 7.61–7.70 (m, 2H, 3/5-PhS–CH), 7.71–7.79 (m, 1H, 4-PhS–CH), 7.84–7.94 (m, 2H, 2/6-PhS–CH); 13C-NMR: (100.61 MHz, DMSO-d6): δ (ppm) = 16.15 (CH3), 87.16 (CC–NH), 115.46 (CN), 123.49 (4-Ph–CH), 126.30 (2C, 2/6-Ph–CH), 126.56 (2C, 2/6-PhS–CH), 129.30 (2C, 3/5-PhS–CH), 129.54 (2C, 3/5-Ph–CH), 133.59 (4-Ph–CH), 138.81 (1-Ph–C), 141.85 (1-PhS–C); M.p. 116 °C; LOOP-ESI-MS calcd for C16H14N2O2S2: 330.05; found: 331.1 [M + H]+.
Space group = triclinic P, a = 11.0502(3) Å, b = 11.5540(4) Å, c = 13.7697(4) Å, α = 66.109(3)°, β = 84.184(2)°, γ = 77.048(2)°, V = 1566.40(8) Å3, Z = 4, max. resolution d = 0.58 Å, redundancy = 3.4, no. unique reflections = 15966, no. observed reflections (I2 > 2σ(I)) = 9736, Rint = 5.67%, completeness = 96.4%, R(1) = 3.97%, CCDC 897063.
Reaction with PET
Upon the dissolution of 1 (10 mM) and PET (78 mM) in d4-MeOD at 27 °C, exchange of the mercaptomethyl residue by PET was observed. No other side reaction occurred as also proven by LC/MS analysis (liquid chromatography/mass spectrometry). In order to quantify educts and products the resonance signals of the NCH3 (δ = 3.05 ppm and 3.19 ppm) and SCH3 (δ = 2.50 ppm) substituents of the vinyl sulfone were integrated as a function of time. Within 60 minutes the reaction equilibrated at a limit of approximately 88% yielding an equilibrium constant K = 0.93 and a free reaction energy of 0.041 kcal mol−1. The 1H-NMR spectra measured 7 days after the dissolution of 1 and PET in methanol (d4-MeOD; sample was kept at 23 °C) showed no remarkable change of the ratio of the integrals. The resonance signal at δ = 3.05 ppm can be assigned to the substituted vinyl sulfone, and the one at δ = 2.50 ppm to the redundant methyl mercaptane (see Fig. 11). To obtain information about the influence of the concentration of 2-phenylethanethiol on the kinetics of the vinylic substitution, another experiment under second-order conditions, i.e. with an equal amount of PET (10 mM) and 1 (10 mM) was performed. The reaction equilibrated within 80 minutes at a limit of 44% product. These results are consistent with our presumed thermoneutral reaction that should result in a 50:50 steady state and a K value of ca. 1.
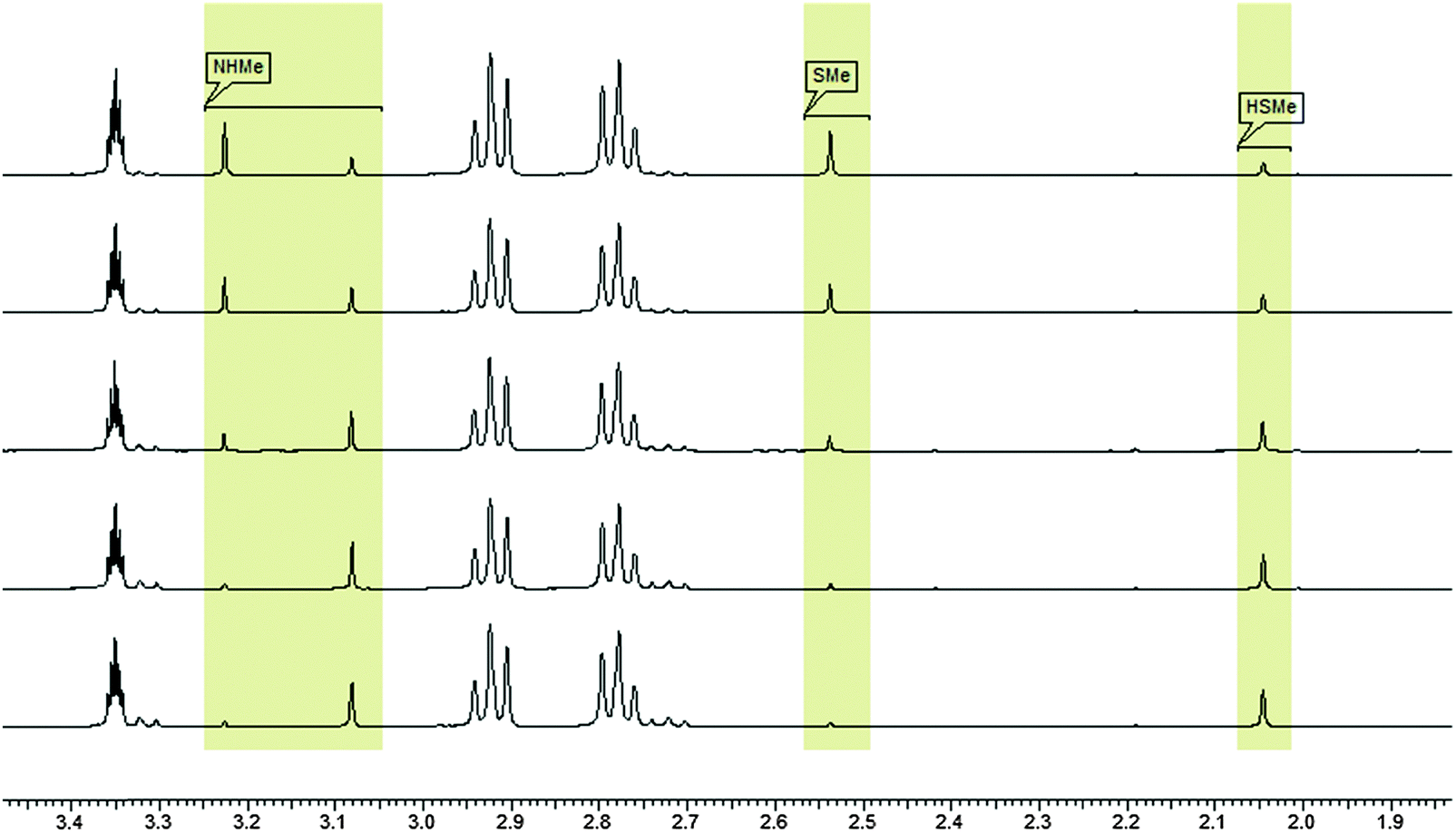 | ||
| Fig. 11 1H-NMR-spectra (27 °C, 400.13 MHz) of a 78 mM solution of 2-phenylethanethiol and a 10 mM solution of 1 in methanol (d4-MeOD), showing the resonance signals of the CH3-groups of 1 and CH3SH as a function of time. | ||
To confirm the stability of 1 after dissolution in methanol (d4-MeOD) a probe of 1 (10 mM) was prepared and investigated by 1H-NMR spectroscopy immediately after the dissolution and after 7 days (sample was kept at 23 °C). No differences were observed.
Additional LC/MS studies with compounds 4, 5 and 6 and PET in methanol (d4-MeOD) also proved that besides the products of the vinylic substitution no other side products were formed. For this purpose, samples of the reactions with an excess of PET were analyzed at 20 °C after the reaction was finished. In all cases the LC/MS spectra (Fig. 12 for cpd. 6) show two peaks, educt and product. The mass spectra (Fig. 13 for cpd. 6) confirm the identity of the educt (m/z = [6 + H]+) and the expected product (m/z = [X + H]+), namely the vinyl sulfone resulting from the substitution of the methylthiolate by PET. The ratios of educts and products were determined by integration of the peaks again proving that the equilibrium adjusted at nearly 50:50 in all cases (4 → 51:49; 5 → 40:60; 6 → 41:59).
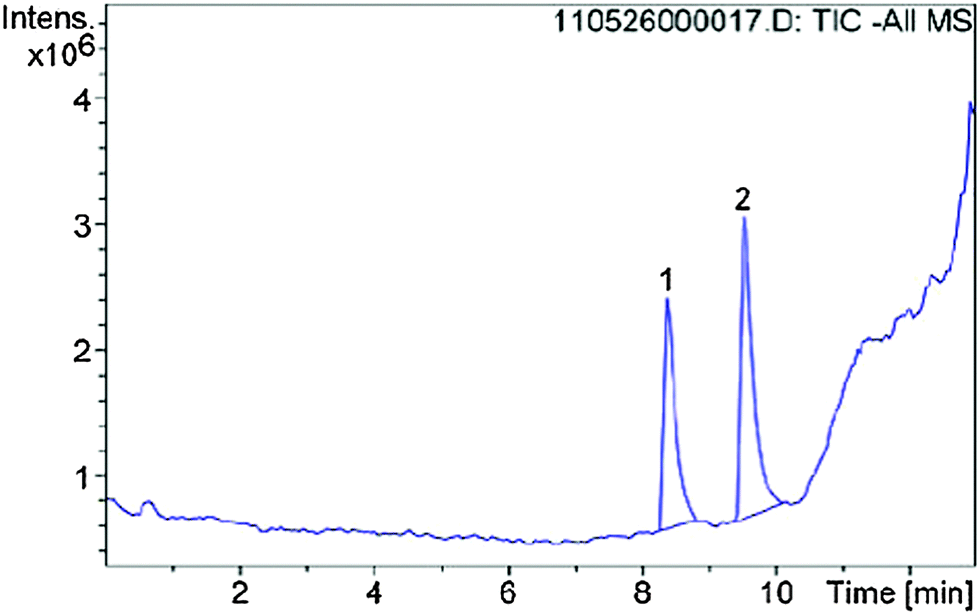 | ||
| Fig. 12 LC/MS spectrum of the reaction of inhibitor 6 with 2-phenylethanethiol, peak 1 refers to compound 6, peak 2 to the product of the vinylic substitution with PET. | ||
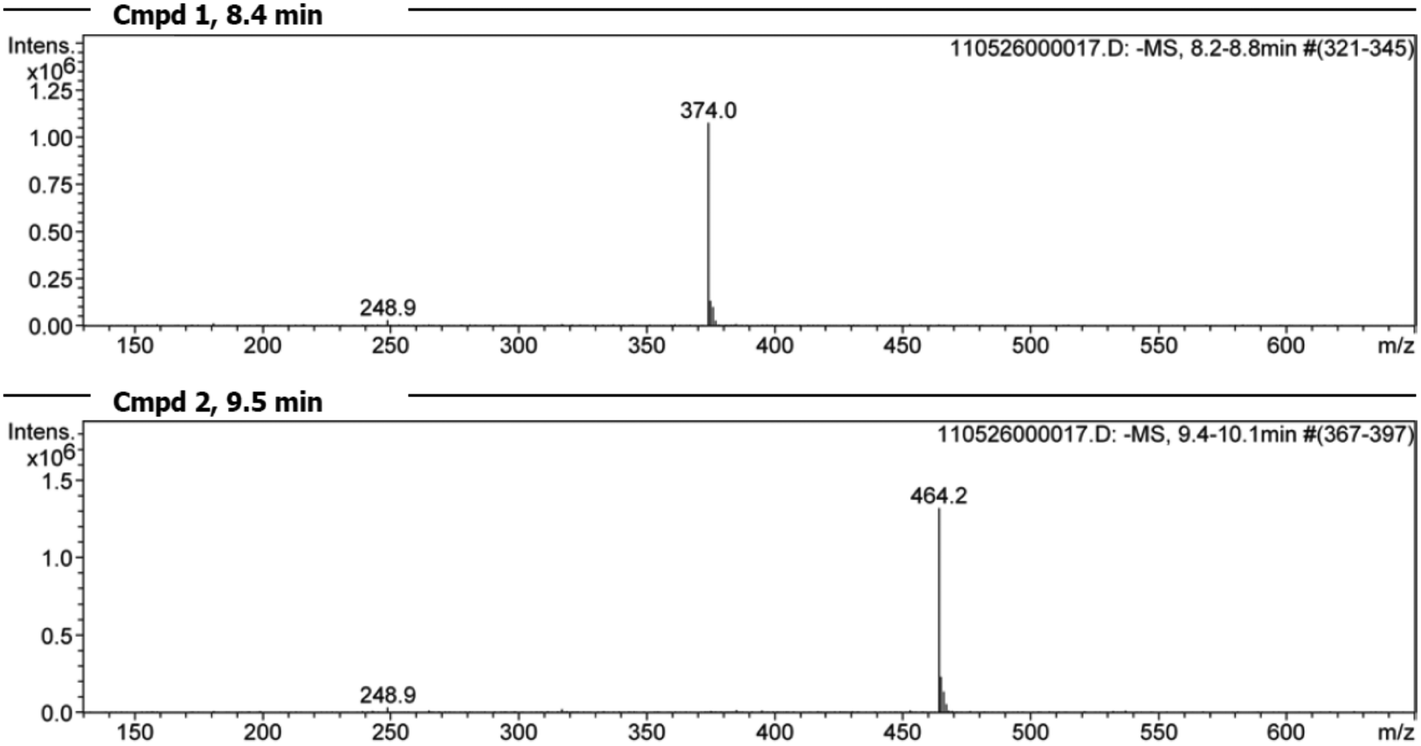 | ||
| Fig. 13 Mass spectra of the two peaks found by LC/MS analysis (Fig. 12). Spectrum for peak 1 at 8.4 min: vinyl sulfone 6 [(m/z) = 374.0] and the reaction product peak 2 at 9.5 min [(m/z) = 464.2]. | ||
Furthermore we performed an analysis of the reaction of compound 1 (20 mM) and PET (160 mM) in methanol by IR spectroscopy using the ABB MB-Rx (Fig. 14). We used the wavenumber 1540 cm−1 to measure the amount of the starting material and the wavenumber 1642 cm−1 to measure the formation of the product. After 80 minutes the reaction was completed and equilibration was adjusted confirming the NMR studies with an orthogonal spectroscopic method.
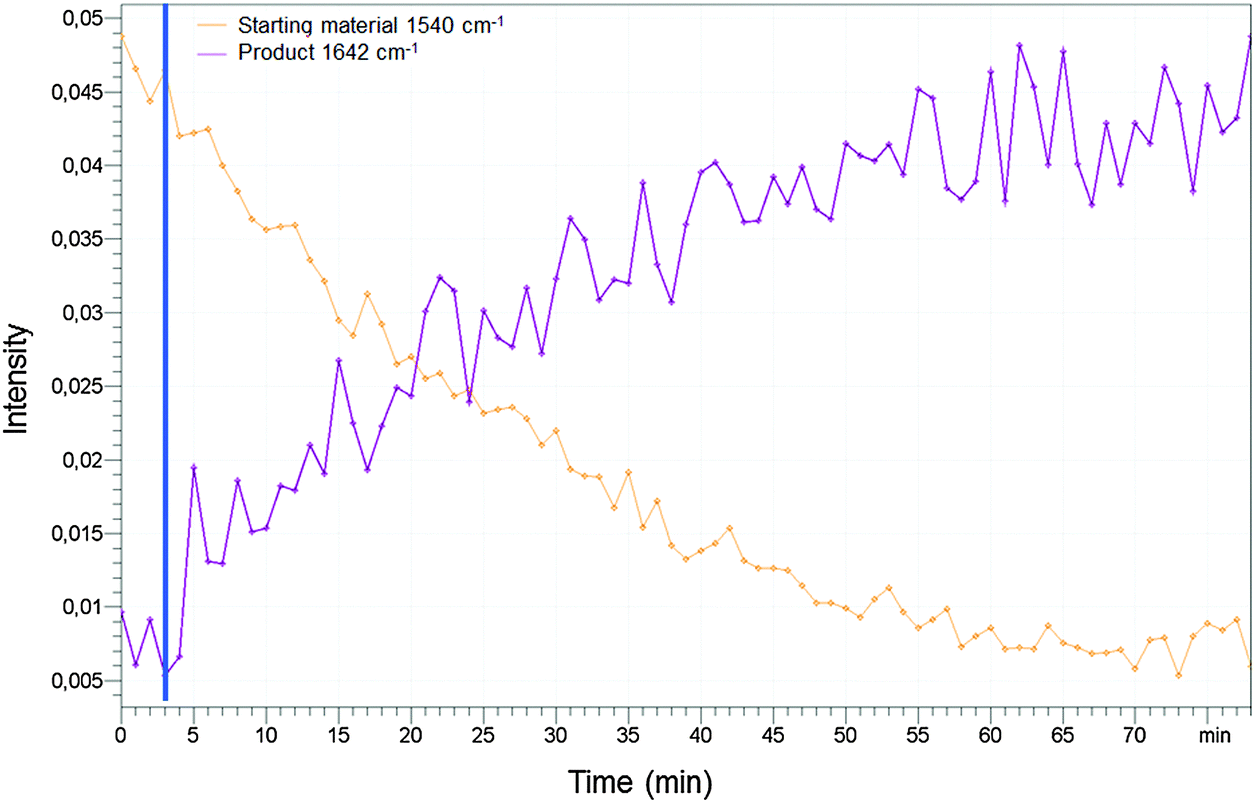 | ||
| Fig. 14 Kinetics of the reaction of 1 (10 mM) and 2-phenylethanethiol (78 mM) upon dissolution in MeOD. The experimental data were extracted from IR spectra (20 °C, 1540 and 1642 cm−1) measured by the ABB MB-Rx. | ||
Theoretical calculations
All quantum chemical computations were performed using the Turbomole program package.33 The geometrical parameters of the relevant stationary points of the potential energy surface were computed with the RI-DFT34 utilizing the functional BLYP35 in combination with a TZVP basis set.36 The reaction energies were obtained from B3LYP37 single point calculations in combination with the same basis set. In all computations, the COSMO approach with ε = 78.39 was employed to account for bulk effects of the polar physiological environment.38 Comparable computations had been successfully employed for the prediction of the reactivity of enzyme inhibitors in the past. They showed that computations which combine the B3LYP functional with TZVP basis sets are sufficiently accurate for trends in reaction energies.39–41 Hirshfeld surfaces were calculated from a superposition of tabulated spherical atomic electron densities, whereas the electrostatic potential is based on BLYP/6-31G(d) quantum-chemical calculations for isolated molecules at the experimental geometries.Acknowledgements
Financial support by the DFG (Deutsche Forschungsgemeinschaft) is gratefully acknowledged within special priority program SPP1178. S. Grabowsky thanks the Australian Research Council for funding within the Discovery Early Career Researcher Award (DECRA) DE140101330, the Discovery Project DP130103488, and the DFG within Emmy Noether project GR4451/1-1. S. Grabowsky also thanks Prof. Mark A. Spackman for his help and support.References
-
(a) R. Ettari, C. Bonaccorso, N. Micale, C. Heindl, T. Schirmeister, M. L. Calabrò, S. Grasso and M. Zappalà, ChemMedChem, 2011, 6, 1228–1237 CrossRef CAS PubMed
; (b) R. Ettari, E. Nizi, M. E. Di Francesco, A.-A. Dude, G. Pradel, R. Vicik, T. Schirmeister, N. Micale, S. Grasso and M. Zappala, J. Med. Chem., 2008, 51, 988–996 CrossRef CAS PubMed
- P. J. Rosenthal, J. E. Olson, G. K. Lee, J. T. Palmer, J. L. Klaus and D. Rasnick, Antimicrob. Agents Chemother., 1996, 40, 1600–1603 CAS
- J. T. Palmer, D. Rasnick, J. L. Klaus and D. Broemme, J. Med. Chem., 1995, 38, 3193–3196 CrossRef CAS
- P. S. Doyle, Y. M. Zhou, I. Hsieh, D. C. Greenbaum, J. H. McKerrow and J. C. Engel, PLoS Pathog., 2011, 7, e1002139, DOI:10.1371/journal.ppat.1002139
- L. U. Buxbaum, H. Denise, G. H. Coombs, J. Alexander, J. C. Mottram and P. Scott, J. Immunol., 2003, 171, 3711–3717 CrossRef CAS
- J. C. Engel, P. S. Doyle, I. Hsieh and J. H. McKerrow, J. Exp. Med., 1998, 188, 725–734 CrossRef CAS
- S. C. Barr, K. L. Warner, B. G. Kornreich, J. Piscitelli, A. Wolfe, L. Benet and J. H. McKerrow, Antimicrob. Agents Chemother., 2005, 49, 5160–5161 CrossRef CAS PubMed
- I. D. Kerr, J. H. Lee, C. J. Farady, R. Marion, M. Rickert, M. Sajid, K. C. Pandey, C. R. Caffrey, J. Legac, E. Hansell, J. H. McKerrow, C. S. Craik, P. J. Rosenthal and L. S. Brinen, J. Biol. Chem., 2009, 284, 25697–25703 CrossRef CAS PubMed
- A. J. T. Smith, X. Zhang, A. G. Leach and K. N. Houk, J. Med. Chem., 2009, 52, 225–233 CrossRef CAS PubMed
- J. F. Morrison and C. T. Walsh, Adv. Enzymol. Relat. Areas Mol. Biol., 1988, 61, 201–301 CAS
-
R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery, John Wiley & Sons, Inc., Hoboken, New Jersey, 1st edn, 2005 Search PubMed
- J. V. Schloss, Acc. Chem. Res., 1988, 21, 348–353 CrossRef CAS
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 CrossRef CAS PubMed
- M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef CAS
- P. Politzer, J. S. Murray and Z. Peralta-Inga, Int. J. Quantum Chem., 2001, 85, 676–684 CrossRef CAS PubMed
- C. F. Bernasconi, R. J. Ketner, M. L. Ragains, X. Chen and Z. Rappoport, J. Am. Chem. Soc., 2001, 123, 2155–2164 CrossRef CAS PubMed
- C. F. Bernasconi and Z. Rappoport, Acc. Chem. Res., 2009, 42, 993–1003 CrossRef CAS PubMed
- B. A. Shainyan, J. Phys. Org. Chem., 1993, 6, 59–63 CrossRef CAS PubMed
- A. V. Ivachtchenko, D. E. Dmitriev, E. S. Golovina, A. G. Koryakova, V. M. Kysil, O. D. Mitkin, I. M. Okun, S. E. Tkachenko and A. A. Vorobiev, J. Med. Chem., 2010, 53, 5186–5196 CrossRef CAS PubMed
- C. J. Shishoo, M. B. Devani, K. S. Jain, V. S. Bhadti, S. M. Shishoo, U. S. Pathak, S. Ananthan and I. S. Rathod, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1989, 28, 42–47 Search PubMed
- S. Grabowsky, P. Luger, J. Buschmann, T. Schneider, T. Schirmeister, A. N. Sobolev and D. Jayatilaka, Angew. Chem., Int. Ed., 2012, 51, 6776–6779 CrossRef CAS PubMed
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer (Version 3.0), University of Western Australia, 2012 Search PubMed
-
(a) G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023–1028 CrossRef CAS
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC
- S. K. Panigrahi and G. R. Desiraju, Proteins: Struct., Funct., Bioinf., 2007, 67, 128–141 CrossRef CAS PubMed
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed
-
(a) M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS
- G. Naray-Szabo and G. G. Ferenczy, Chem. Rev., 1995, 95, 829–847 CrossRef CAS
- C. F. Matta, J. Comput. Chem., 2014, 35, 1165–1198 CrossRef CAS PubMed
- C. B. Hübschle, B. Dittrich, S. Grabowsky, M. Messerschmidt and P. Luger, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 363–374 Search PubMed
- S. Grabowsky, T. Pfeuffer, W. Morgenroth, C. Paulmann, T. Schirmeister and P. Luger, Org. Biomol. Chem., 2008, 6, 2295–2307 CAS
-
R. Ahlrichs, TURBOMOLE V 6.1, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 2009 Search PubMed
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS PubMed
-
(a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed
- A. Klamt and G. Scheuermann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC
-
(a) H. Helten, T. Schirmeister and B. Engels, J. Org. Chem., 2005, 70, 233–237 CrossRef CAS PubMed
- R. Vicik, H. Helten, T. Schirmeister and B. Engels, ChemMedChem, 2006, 1, 1021–1028 CrossRef CAS PubMed
- V. Buback, M. Mladenovic, B. Engels and T. Schirmeister, J. Phys. Chem. B, 2009, 113, 5282–5289 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, summary of hydrogen bonds in the crystals, electrostatic potentials mapped onto the Hirshfeld surfaces of all compounds 1–9. CCDC 852449, 897056–897063. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00368g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |