
A sensitive photoelectrochemical immunoassay based on mesoporous carbon/core–shell quantum dots as donor–acceptor light-harvesting architectures
Rongxia
Li
,
Jian
Gao
,
Picheng
Gao
,
Sen
Zhang
,
Yixin
Liu
,
Bin
Du
and
Qin
Wei
*
Key Laboratory of Chemical Sensing & Analysis in Universities of Shandong, School of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, P. R. China. E-mail: sdjndxwq@163.com; Fax: +86-531-82765969; Tel: +86-531-82767872
First published on 4th November 2014
Abstract
Herein, we demonstrate the protocol of a label-free photoelectrochemical (PEC) immunoassay on the basis of ordered mesoporous carbon (CMK-3) and water-soluble CdSe@ZnS core–shell quantum dots (QDs) coupled with a biospecific interaction for the ultrasensitive detection of human immunoglobulin (antigen, H-IgG) as a model protein. The CMK-3 was dispersed with chitosan (CS-CMK-3), which contains a large amount of amino groups (–NH2), and the CdSe@ZnS QDs were treated with thioglycolic acid, which contains carboxylic acid groups (–COOH). The layer-by-layer assembling of CdSe@ZnS QDs and CS-CMK-3 achieved through the covalent bonding of –COOH and –NH2 was employed as a photoactive antibody (Ab) immobilization matrix. Improved sensitivity was achieved through the synergistic effect of the excellent electrical conductivity and high specific surface area of CMK-3, as well as the high photon-to-electron conversion efficiency of CdSe@ZnS QDs. The photoexcitation of a CMK-3/CdSe@ZnS QDs-modified ITO electrode potentiostated at 0 V (vs. Ag/AgCl) under white light led to a stable anodic photocurrent. To perform the immunoassay, anti-human immunoglobulin (antibody, anti-H-IgG) was conjugated onto the CdSe@ZnS QDs-modified electrode by using EDC–NHS coupling reactions between –COOH of CdSe@ZnS QDs and –NH2 of the antibody. The concentrations of H-IgG were measured through the decrease in photocurrent intensity resulting from the increase in steric hindrance due to the formation of the immunocomplex. Under the optimal conditions, a linear relationship between photocurrent decrease and H-IgG concentration was obtained in the range 10 pg mL−1–100 ng mL−1 with a detection limit of 5 pg mL−1. This strategy opens a simple perspective for the application of mesoporous conductive material and core–shell QDs as light-harvesting architecture, which might be of great significance in PEC bioanalysis in the future.
1. Introduction
Among various biosensing techniques, the newly developed yet dynamically developing photoelectrochemical (PEC) detection is of special interest for its potential application in bioanalysis.1 The PEC method evolved from electrochemistry but is different from the traditional electrochemical methods. A PEC sensor has the advantages of both optical methods and electrochemical sensors due to its coupling photoirradiation with electrochemical detection. Due to reduced background signals obtained from the separation of the excitation source and the detection signal, the PEC technique possesses potentially higher sensitivity than the conventional electrochemical methods.2 The detection principle of PEC biosensors is based on the photocurrent change caused by the biological interactions between biosensing elements and their corresponding target analytes.3 Due to the different energy forms of the excitation source and the detection signal, the PEC method is not only simple but also quite sensitive.3b,d,4 Given its desirable advantages, many efforts have been exerted to develop this method for different analytes in the past decade, such as DNA oligonucleotides,1b,5 enzyme inhibitors,4a cells and some small molecules,6 anti-cholera toxin antibody,6a,7 mouse α-fetoprotein,4c and IgG,8 which have been successfully determined accordingly. In these investigations, the label-free method has been chosen, being preferred for the purpose of simplification and low cost.1b,3b,d,6a–c Despite high simplicity, the deficiencies of these PEC label-free protocols, including low capacity for analyte loading and inability of amplification, need to be overcome promptly. Hence, employing materials with a high specific surface area and high photon-to-electron conversion efficiency might be a useful route for signal enhancement.Quantum dots (QDs), as one kind of semiconductor nanocrystals with unique size-dependent properties,9 have emerged as a significant new class of materials over the past decade. QDs exhibit a wide range of electrical and optical properties, and QDs-based bioassay has become one of the most exciting fields at the forefront in analytical chemistry. Especially, group II–VI semiconductors (e.g., CdSe, CdS, HgS, ZnS, ZnSe) have been a focus in the research field of bioelectrochemistry and bioimaging.10 The QDs are often passivated by a second semiconductor material (e.g., ZnS)11 to protect the core from oxidation and bleaching.12 The band gap energy of the shell is higher in order to confine the exciton generation and relaxation to the core, and thus increase the quantum yield.11a,b,13
Recently, it has been shown that CdSe@ZnS QDs can not only be used as fluorescence labels for biomolecules,14 but can also be attached to metal electrodes for PEC studies.15 Among these, the photocurrent can be switched by the introduction of cytochrome c15a,d and different enzymes can be combined with QDs-modified electrodes.15b,c However, not only was expensive and sophisticated instrumentation (lock-in amplifier) necessary to detect the photocurrent in these detection systems, but also the photocurrent intensity of CdSe/ZnS QDs was small (about 10−9–10−8 A). Thus, developing an indirect and sensitive PEC method with QDs based on immunoassay for the detection of general proteins is of great significance.
In recent years, carbon materials, due to their extraordinary electronic and mechanical properties, have been applied in a broad range of applications with different desirable functionalities such as energy conversion/fuel storage, catalysis, biotechnology and optoelectronic nanodevices.16 With the discovery of photoinduced charge transfer between semiconductor quantum dots (SQDs) as the donor and carbon materials as the acceptor,16a,17 carbon-based donor–acceptor architectures have been successfully fabricated as photochemical energy conversion systems with the aim of increasing the photoresponses of SQDs.18 Carbon materials can effectively increase charge generation at the interface and transport pathways for the photoinduced electrons to the electrode.16a Ordered mesoporous carbon, CMK-3, possesses the properties of excellent mechanical stability, high specific surface area, large pore volume and adjustable pore size contribution. Due to the numerous edge-plane-like defect sites which make electron transfer easier, CMK-3 shows a fast electron transfer rate and larger current response.19 Moreover, it can act as a scaffold in splitting and transferring the flow of photoinduced charge carriers,20 which could reduce the recombination probability of electron–hole pairs of QDs effectively.
In this work, a facile assembly of CdSe@ZnS QDs onto the pores and surface of CMK-3 as an efficient photocurrent conversion architecture was demonstrated. In order to obtain a uniformly modified ITO electrode as well as to facilitate further modification, CMK-3 was dispersed with chitosan (CS-CMK-3), which has a good film-forming property and contains a large amount of amino groups (–NH2). The thioglycolic acid (TGA)-capped water-soluble CdSe@ZnS QDs were assembled on the electrode simply by covalent bonding between the –COOH group of CdSe@ZnS QDs and the –NH2 group of CS-CMK-3. Then, human immunoglobulin (H-IgG) was conjugated onto the CdSe@ZnS QDs-modified electrode by using EDC–NHS coupling reactions. The H-IgG concentrations were directly (label-free) measured through the decrease in photocurrent intensity resulting from the specific immunoreaction. Results indicate that the proposed label-free PEC biosensor showed good performance in the monitoring of H-IgG with a rapid response and wide concentration range, and could successfully be applied to the detection of other proteins. The established method provides an approach for the assembly of QDs with other nanomaterials possessing a special structure for rapid electron transportation, and for further designation of simple QDs-based PEC biosensors.
2. Experimental section
2.1 Reagents and chemicals
Cadmium oxide (CdO) (99.99%), selenium (99.9%, powder), zinc dimethyldithiocarbamate (Zn(DMSC)2) (97%, powder), trioctylphosphine (TOP) (90%), oleic acid (OA) (90%) and 1-octadecene (ODE) (90%) were purchased from Sigma-Aldrich and used as received. Thioglycolic acid (TGA), NaBH4, mesoporous carbon (CMK-3) and thiourea were from Sinopharm Chemical Reagent Beijing Co., Ltd, China. Human immunoglobulin antigen (H-IgG) and anti-human immunoglobulin (anti-H-IgG) were purchased from Shanghai Guyan Technology Co. Ltd 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were obtained from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). Bovine serum albumin (BSA, 96–99%) and ascorbic acid (AA) were from Sigma-Aldrich. Phosphate buffered saline (PBS, 0.1 M, pH 7.4) was used as electrolyte for all electrochemical measurements and for the preparation of the Ag and Ab solutions. Ultrapure water was used throughout the experiments. All other chemicals were of analytical reagent grade.2.2 Apparatus
PEC measurements were performed with 30 W LED light (Tengwei light, China). Photocurrent was measured on a CHI760D electrochemical workstation (Shanghai CHI Instruments Co., China) with a three-electrode system: a modified ITO electrode with a geometrical area of 0.25 ± 0.01 cm2 used as the working electrode, a Pt wire used as the counter electrode and a saturated Ag/AgCl electrode used as the reference electrode. All the photocurrent measurements were performed at a constant potential of 0 V (versus Ag/AgCl). A 0.1 M PBS containing 0.1 M AA was used as the blank solution for photocurrent measurements. Transmission electron microscopy (TEM) images were obtained from a Hitachi H-800 microscope (Japan). The UV-vis absorption spectra were obtained on a TU-1901 UV-vis spectrophotometer (Beijing General Instrument Co. Ltd). Photoluminescence (PL) spectra were collected on an LS-45/55 PL spectrometer (PerkinElmer, America).2.3 Synthesis of water-soluble CdSe@ZnS core–shell quantum dots
CdSe@ZnS core–shell quantum dots were synthesized using a slightly modified procedure reported by Bong-Hyun Jun et al.21 Typically, CdO (1 × 10−3 M, 0.1284 g) and Zn(DMSC)2 (5 × 10−3 M, 1.520 g) were dissolved in OA (1.695 g) and ODE (20 mL), treated with N2 gas, and heated to 100 °C under vacuum for 1 h. The solution was further heated to 320 °C to form a transparent solution and injected into a precursor solution, which was prepared by dissolving Se (1 × 10−3 M, 0.078 g) in TOP (1 mL). The growth temperature was set to 300 °C for 5 min and then, the solution was cooled to room temperature. To prepare water-soluble single QDs, the purified QDs were dispersed in CHCl3 to obtain a 0.1 × 10−6 M QDs solution. Tetramethylammonium hydroxide (TMAH) (100 mg) was mixed well with TGA in CHCl3. After 15 min, a clear and colorless aqueous layer (about 10% of the total volume) formed above the CHCl3 layer. The biphasic solution was mixed by vigorous shaking and was allowed to stand for 1 h in order to equilibrate. The lower organic phase, which contained deprotonated TGA, was transferred into a vial for the ligand exchange reaction with the QDs. 100 μL TOP-capped QDs (0.1 μM in CHCl3) was added to the TGA-CHCl3 solution and mixed well. The solution was allowed to stand at room temperature for 1–5 h. After the reaction, the TGA-capped QDs that had separated from the CHCl3 solutions were collected, washed with CHCl3 (three times), and dispersed in 1.0 mL water.2.4 Fabrication of PEC Sensor
The ITO slices (type N-STN-S1-10, Zhuhai Kaivo Electronic Components Co. Ltd, Guangdong, China, sheet resistance ≤10 Ω cm−2) were sonicated in acetone, NaOH (1 M) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ethanol/water, and water for about 30 min consecutively, and dried with a nitrogen flow. To fabricate the CS-CMK-3 composite film-modified ITO, the CMK-3 powder was ultrasonically dispersed in chitosan solution (0.25%, wt) to obtain a homogeneous solution of CS-CMK-3 (1 mg mL−1). 4 μL CS-CMK-3 was coated onto the ITO electrode and dried at room temperature, then 4 μL CdSe@ZnS QDs was coated onto the CS-CMK-3-modified ITO electrode. Conjugation of antibodies onto a CdSe@ZnS QDs-modified electrode was achieved using EDC–NHS as the coupling agent. In brief, the CS-CMK-3/CdSe@ZnS QDs-modified electrode was activated by immersion in a solution containing 10 mg mL−1 EDC and 20 mg mL−1 NHS for 50 min. Then, the activated electrode was thoroughly rinsed with distilled water and dried. Following this step, 4 μL anti-H-IgG (10 μg mL−1) was dropped on the electrode surface and the electrode was incubated at 4 °C for 1 h. After incubation, the electrode was rinsed with distilled water. 4 μL BSA (1%, wt) was incubated on the modified electrode for 1 h at room temperature to block non-specific binding sites, and then washed with distilled water thoroughly. Finally, 4 μL analyte (H-IgG) solution with different concentrations was dropped on the electrode and incubated at 37 °C for 1 h followed by washing with distilled water. A detailed description of the immunosensor process is illustrated in Scheme 1A.
:1 (v/v) ethanol/water, and water for about 30 min consecutively, and dried with a nitrogen flow. To fabricate the CS-CMK-3 composite film-modified ITO, the CMK-3 powder was ultrasonically dispersed in chitosan solution (0.25%, wt) to obtain a homogeneous solution of CS-CMK-3 (1 mg mL−1). 4 μL CS-CMK-3 was coated onto the ITO electrode and dried at room temperature, then 4 μL CdSe@ZnS QDs was coated onto the CS-CMK-3-modified ITO electrode. Conjugation of antibodies onto a CdSe@ZnS QDs-modified electrode was achieved using EDC–NHS as the coupling agent. In brief, the CS-CMK-3/CdSe@ZnS QDs-modified electrode was activated by immersion in a solution containing 10 mg mL−1 EDC and 20 mg mL−1 NHS for 50 min. Then, the activated electrode was thoroughly rinsed with distilled water and dried. Following this step, 4 μL anti-H-IgG (10 μg mL−1) was dropped on the electrode surface and the electrode was incubated at 4 °C for 1 h. After incubation, the electrode was rinsed with distilled water. 4 μL BSA (1%, wt) was incubated on the modified electrode for 1 h at room temperature to block non-specific binding sites, and then washed with distilled water thoroughly. Finally, 4 μL analyte (H-IgG) solution with different concentrations was dropped on the electrode and incubated at 37 °C for 1 h followed by washing with distilled water. A detailed description of the immunosensor process is illustrated in Scheme 1A.
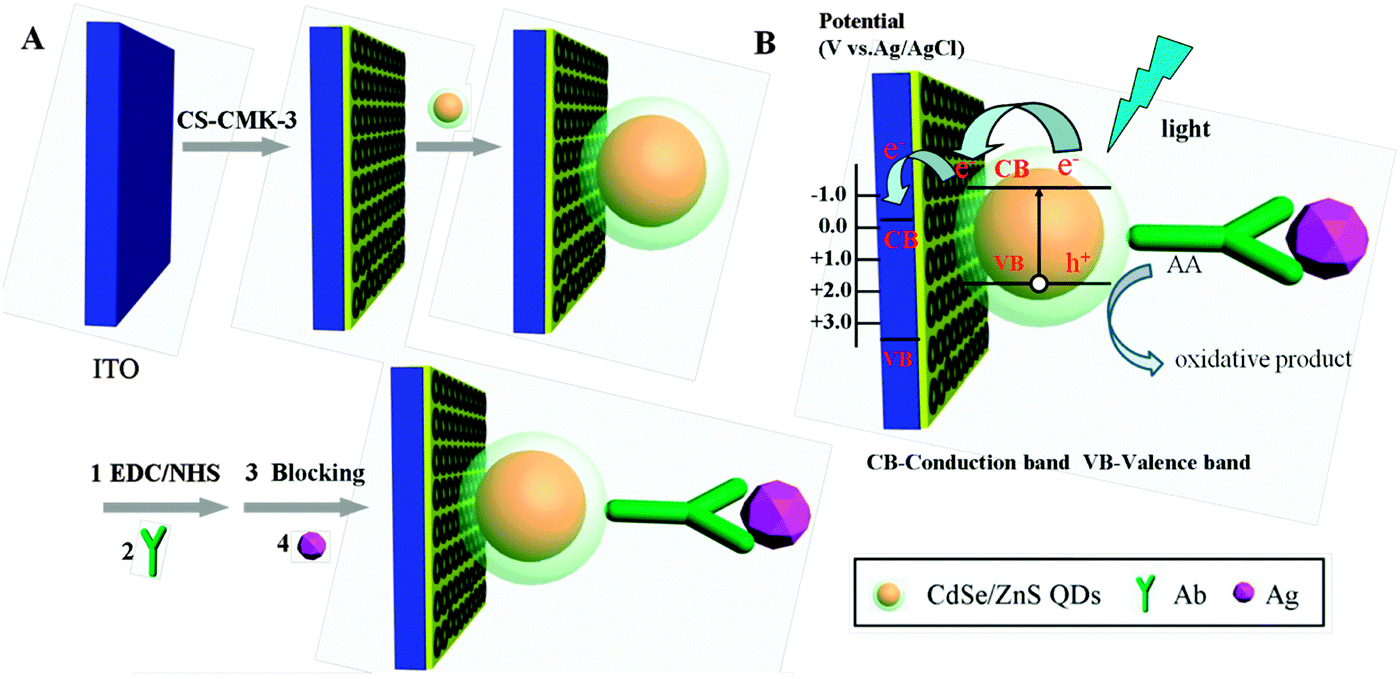 | ||
| Scheme 1 (A) Schematic diagram of the stepwise immunosensor fabrication process. (B) Photocurrent generation mechanism of CS-CMK-3/CdSe@ZnS QDs. | ||
3. Results and discussion
3.1 Characterization of water-soluble CdSe@ZnS core–shell quantum dots
After the synthesis of carboxylated CdSe@ZnS QDs according to the process mentioned above, a series of characterization technologies were used to further analyze their relevant properties. As shown from the TEM image in Fig. 1A, the size of the CdSe@ZnS QDs was found to be about 5 nm with good monodispersity. Fluorescence microscopy imaging was performed to further confirm the excellent fluorescence properties and the high quantum yield of CdSe@ZnS QDs (Fig. 1B). The optical property of QDs (the inset of Fig. 1B) exhibited a broad absorption spectrum (covering the range of wavelengths < 700 nm) with a characteristic peak at around 650 nm and a narrow PL emission band centered at 680 nm, which potentially indicated effective absorption and photo-electron transfer under visible wavelengths. The narrow emission spectrum indicated that a high degree of monodispersity of QDs was present. | ||
| Fig. 1 The TEM (A) and fluorescence microscopy images (B) of the prepared CdSe@ZnS QDs. The inset is the UV-vis spectrum (a) and room-temperature PL spectrum (b) of CdSe@ZnS QDs. | ||
3.2 Photoelectrochemical behavior of CS-CMK-3/CdSe@ZnS QDs-modified electrode
In order to confirm the effect of CS-CMK-3 on the PEC behavior of CdSe@ZnS QDs, the photocurrent responses and the corresponding fluorescence emission spectra of both substances and their assemblies were studied. As shown in Fig. 2A, compared to the CdSe@ZnS QDs-modified electrode (b), there was an obvious enhancement in the photocurrent of the CS-CMK-3/CdSe@ZnS QDs-modified electrode (c). On the basis of electron transfer in CdSe QDs–C60 nanocomposites,22 we suspect that the enhancement may be ascribed to the donor–acceptor assemblies of CMK-3 and CdSe@ZnS QDs. To further investigate the effect of CMK-3 on the PEC properties of CdSe@ZnS QDs, emission quenching of CdSe@ZnS QDs is a good measure to probe the photoinduced electron transfer process in donor–acceptor assemblies.23 For pristine CMK-3 (a), no luminescence peak is perceived, while the CdSe@ZnS QDs (b) exhibit high luminescence with a maximum emission at 680 nm. It is interesting to note that upon adding CMK-3 into the CdSe@ZnS QDs solution (c), an obviously reduced emission intensity at 680 nm was observed, which is assigned to electron–hole recombination in CdSe@ZnS QDs. The significant emission quenching seen in this experiment indicated that the dramatically increased photocurrent stems from effective charge carrier separation via electron transfer of the CMK-3. | ||
| Fig. 2 (A) Photocurrent responses of (a) CS-CMK-3, (b) CdSe@ZnS QDs and (c) CS-CMK-3/CdSe@ZnS QDs. (B) The corresponding fluorescence emission spectra. | ||
The photocurrent generation mechanism of CS-CMK-3/CdSe@ZnS QDs is shown in Scheme 1B. When CdSe@ZnS QDs absorbed photons with energy higher than that of their band gap, electrons are excited from the (occupied) valence band to the (empty) conduction band and formed electron–hole pairs. Once this process occurred, the electron–hole pairs would recombine or the charges would be transferred. In this study, the electrons transferred to the CMK-3 rapidly and further to the ITO electrode.
3.3 Photoelectrochemical immunosensor
In this study, CS-CMK-3, CdSe@ZnS QDs, Ab and BSA were modified onto the electrode successively. The interfacial behavior of each sensor fabrication step was probed by recording the electrochemical impedance spectra and photocurrent. EIS was used to characterize the fabrication process of the biosensor. As shown in Fig. 3A, compared to bare ITO (a), after the deposition of CS-CMK-3 (b), the semicircular diameter of the Nyquist plot that represents the electron transfer resistance (Ret),24 became much smaller due to better conductivity of CS-CMK-3. However, upon assembling CdSe@ZnS QDs (c), the semicircular diameter dramatically increased, suggesting successful deposition of the CdSe@ZnS QDs on the outer layer of CS-CMK-3 and in turn hindered the electron transfer. After the immobilization of Ab and the subsequent BSA blocking, the Ret increases gradually again (curves d–e), demonstrating the successful assembling of sensing elements on the electrode surface. The reason for the resistance increase is that the non-conductive properties of the proteins obstruct the mass transport and electron transfer of the electrochemical probe to the electrode surface by increasing the hindrance effect. | ||
| Fig. 3 (A) EIS of (a) CS-CMK-3-modified electrodes, (b) bare ITO electrodes, (c) CS-CMK-3/CdSe@ZnS QDs-modified electrodes, (d) after Ab immobilization, (e) after anchoring the Ag corresponding to 10 ng mL−1. The EIS measurements were carried out in 0.1 M KCl containing 5.0 mM K3Fe(CN)6/K4Fe(CN)6 (1:1). The frequency range was between 0.1 and 100000 Hz with an applied voltage of 5 mV. (B) Photocurrent response of CS-CMK-3/CdSe@ZnS QDs-modified electrodes (a) before and (b) after Ab immobilization, (c) after further blocking with BSA, (d) after anchoring Ag corresponding to 10 ng mL−1. | ||
The fabrication of the immunosensor could also be monitored by PEC experiments (Fig. 3B). After immobilization of the Ab and BSA on the CS-CMK-3/CdSe@ZnS QDs-modified electrode, the photocurrent intensity decreased (curves b and c). This could be explained by the fact that the immobilization of the proteins on the CS-CMK-3/CdSe@ZnS QDs-modified electrode hindered the diffusion of electrons and resulted in the decrease in the photocurrent intensity. After the as-obtained biosensor was incubated with the corresponding antigen (curve d), the photocurrent further decreased. On the basis of the photocurrent decrease due to the formation of the immunocomplex, a label-free PEC immunosensor was achieved.
3.4 Optimization of experimental conditions
In order to obtain high sensitivity for this PEC detection system, the experimental parameters in the fabricating and detecting processes must be investigated and optimized. The effect of pH on the assay performance was studied over a pH range from 5.4 to 10 with a universal PBS buffer. To obtain high signal-to-background ratios (Fig. 4A), pH 8 was chosen to be the optimum PEC measurement condition in this test. Besides, the concentration of CMK-3, which acted as the matrix of the ITO electrode for the subsequent combination of QDs, has a great influence on charge generation at the interface and transport pathways for the photoinduced electrons to the electrode. The photocurrent sharply increased as the concentration of CMK-3 increased from 0.5 mg mL−1 to 2 mg mL−1 and then formed a plateau at 2 mg mL−1 (Fig. 4B). Giving an overall consideration, in this study 2 mg mL−1 CMK-3 was chosen throughout the PEC measurements. | ||
| Fig. 4 (A) Effects of the pH values of 5.4, 6.1, 7.0, 8.06, 9.0, and 10.0 on the photocurrent response of the modified electrodes after incubation in 10 ng mL−1 H-IgG. (B) Effects of concentrations of CMK-3 at 0.5, 1, 2, 3, and 4 mg mL−1 on the photocurrent response of the modified electrodes after incubation in 10 ng mL−1 H-IgG. The error bars show the standard deviation of five replicate determinations. | ||
The degree of photocurrent variation in this PEC immunoassay is directly related to the concentration of target Ag. Fig. 5A presents the photocurrents after incubation with Ag of variable concentrations. The photocurrent decreases with an increase in the concentration of Ag. As shown in Fig. 5B, the photocurrent decrease was proportional to the concentration of Ag in a linear range from 10 pg mL−1 to 100 ng mL−1. Upon treatment of the sensing interface with increasing Ag concentration, more antigen–antibody immunocomplexes could be induced onto the electrode interface. The reduction in photocurrent was prone to saturation over 100 ng mL−1. The detection limit was experimentally found to be 8 pg mL−1. In addition, the sensitivity of the presented PEC immunosensor here was comparable to, and even better than, those of many reported immunoassay methods for human IgG. The analytical performances of various H-IgG immunoassays are listed in Table 1. The comparison results show that this PEC assay is promising for the determination of human IgG in clinical applications.
 | ||
| Fig. 5 (A) Effect of different concentrations of Ag on the photocurrent responses. (B) The corresponding calibration curve, ΔI is the change in photocurrent before and after Ag immobilization. All photocurrent responses were measured in 0.1 M PBS (pH = 8.06) containing 0.1 M AA with an applied potential of 0 V under white light. | ||
| Analyte | Measurement protocol | Linear range | Detection limit | Ref. |
|---|---|---|---|---|
| Human IgG | Photoelectrochemical immunoassay | 10 pg mL−1–100 ng mL−1 | 8 pg mL−1 | This work |
| Goat anti-human IgG | LRET-based immunoassay | 3–67 μg mL−1 | 0.88 μg mL−1 | 25 |
| Human IgG | Flow immunoassay | 5.0 × 10−6–9.6 × 10−4 M | 8.0 × 10−6 M | 26 |
| Human IgG | Electrochemical immunoassay | 0.01–15 nM | 5.0 pM | 27 |
| Goat anti-human IgG | Chemiluminescence resonance energy transfer immunoassay | 0.2–4.0 nM | 2.9 × 10−11 M | 28 |
| Human IgG | Fluoroimmunoassay | 0.2–12 μg mL−1 | 10 ng mL−1 | 29 |
| Human IgG | Electrochemical impedance | 10–1 μg mL−1 | 5 ng mL−1 | 30 |
Control experiments revealed that the developed PEC sensor does not exhibit any obvious changes in photocurrent when incubating the as-fabricated immunosensor in sample solutions containing a 100-fold excess of different interfering agents, such as glucose, cholesterol, human hemoglobin, and alpha-fetoprotein (Fig. 6A). The results indicated good selectivity of the developed PEC sensor. The reproducibility of this PEC H-IgG immunoassay was also assessed by an inter-assay relative standard deviation (RSD). By assaying the same concentration of H-IgG with five electrodes in identical experimental conditions, an RSD of 5.5% was obtained, indicating a good reproducibility of the fabrication protocol. Moreover, it is also worth mentioning that the photocurrent response of the PEC sensor was fairly reversible and stable under several on/off irradiation cycles for 300 seconds. As shown in Fig. 6B, the current could reproducibly increase sharply under each irradiation and recover rapidly in the dark, indicating the structural stability of the developed PEC sensors and their potential for biosensing experiments.
 | ||
| Fig. 6 (A) Selectivity of the proposed immunoassay to human IgG (H-IgG) by comparing it to interference at the 10 ng mL−1 level: glucose, cholesterol, alpha-fetoprotein, human hemoglobin, and the mixed sample, where n = 5 for each point. (B) Time-based photocurrent response of PEC immunosensor. | ||
4. Conclusions
In this work, a simple and low-potential label-free PEC immunosensor using H-IgG as a model analyte was successfully achieved based on CMK-3 and water-soluble core–shell CdSe@ZnS QDs. As compared to the conventional enzyme-labeled PEC immunoassays, this simple biosensing strategy possesses high sensitivity via CMK-3 with good ability of transmitting and accepting electrons, and core–shell CdSe@ZnS QDs with excellent absorption efficiency in visible light. In addition, this immunoassay also has the advantages of low cost (4 μL in each step and no enzyme is used) and simplicity (the label-free method is easy to operate). The fast photoelectron communication among CdSe@ZnS QDs, CMK-3 and the ITO electrode led to a novel method for the PEC detection of H-IgG with good analytical performance. This strategy shows its excellence by being simple, cost-effective and specific in immunoassays, and shows promise to open a new perspective for the combination of a mesoporous material with superior electrochemical properties and core–shell QDs for biological analysis.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21375047, 21377046 and 21245007), the Science and Technology Plan Project of Jinan (No. 201307010). The Science and Technology Development Plan of Shandong Province (No. 2014GSF120004). QW thanks the Special Foundation for Taishan Scholar Professorship of Shandong Province and UJN (No. ts20130937).References
- (a) R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602–7625 CrossRef CAS PubMed; (b) M. Liang and L.-H. Guo, Environ. Sci. Technol., 2007, 41, 658–664 CrossRef CAS; (c) M. Liang, S. Jia, S. Zhu and L.-H. Guo, Environ. Sci. Technol., 2007, 42, 635–639 CrossRef; (d) W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2014, 15, 7421–7441 CrossRef PubMed.
- (a) M. Liang, S. Liu, M. Wei and L.-H. Guo, Anal. Chem., 2006, 78, 621–623 CrossRef CAS PubMed; (b) W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2014 10.1039/C4CS00228H.
- (a) X. Zhang, S. Li, X. Jin and S. Zhang, Chem. Commun., 2011, 47, 4929–4931 RSC; (b) X. Zhang, Y. Zhao, S. Li and S. Zhang, Chem. Commun., 2010, 46, 9173–9175 RSC; (c) W. Zhu, Y.-R. An, X.-M. Luo, F. Wang, J.-H. Zheng, L.-L. Tang, Q.-J. Wang, Z.-H. Zhang, W. Zhang and L.-T. Jin, Chem. Commun., 2009, 2682–2684 RSC; (d) W.-W. Zhao, J. Wang, J.-J. Xu and H.-Y. Chen, Chem. Commun., 2011, 47, 10990–10992 RSC; (e) W.-W. Zhao, C.-Y. Tian, J.-J. Xu and H.-Y. Chen, Chem. Commun., 2011, 48, 895–897 RSC.
- (a) V. Pardo-Yissar, E. Katz, J. Wasserman and I. Willner, J. Am. Chem. Soc., 2003, 125, 622–623 CrossRef CAS PubMed; (b) G.-L. Wang, J.-J. Xu and H.-Y. Chen, Biosens. Bioelectron., 2009, 24, 2494–2498 CrossRef CAS PubMed; (c) G.-L. Wang, J.-J. Xu, H.-Y. Chen and S.-Z. Fu, Biosens. Bioelectron., 2009, 25, 791–796 CrossRef CAS PubMed.
- X. Zhang, Y. Zhao, H. Zhou and B. Qu, Biosens. Bioelectron., 2011, 26, 2737–2741 CrossRef CAS PubMed.
- (a) N. Haddour, J. Chauvin, C. Gondran and S. Cosnier, J. Am. Chem. Soc., 2006, 128, 9693–9698 CrossRef CAS PubMed; (b) G.-L. Wang, P.-P. Yu, J.-J. Xu and H.-Y. Chen, J. Phys. Chem. C, 2009, 113, 11142–11148 CrossRef CAS; (c) Q. Kang, L. Yang, Y. Chen, S. Luo, L. Wen, Q. Cai and S. Yao, Anal. Chem., 2010, 82, 9749–9754 CrossRef CAS PubMed; (d) Z. Qian, H.-J. Bai, G.-L. Wang, J.-J. Xu and H.-Y. Chen, Biosens. Bioelectron., 2010, 25, 2045–2050 CrossRef CAS PubMed; (e) D. Chen, H. Zhang, X. Li and J. Li, Anal. Chem., 2010, 82, 2253–2261 CrossRef CAS PubMed.
- N. Haddour, S. Cosnier and C. Gondran, Chem. Commun., 2004, 2472–2473 RSC.
- G.-L. Wang, J.-J. Xu, H.-Y. Chen and S.-Z. Fu, Biosens. Bioelectron., 2009, 25, 791–796 CrossRef CAS PubMed.
- (a) H. Weller, Angew. Chem., Int. Ed. Engl., 1993, 32, 41–53 CrossRef; (b) W. K. Leutwyler, S. L. Bürgi and H. Burgl, Science, 1996, 271, 933–937 Search PubMed.
- (a) A. C. C. Esteves and T. Trindade, Curr. Opin. Solid State Mater. Sci., 2002, 6, 347–353 CrossRef CAS; (b) C. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- (a) B. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. Heine, H. Mattoussi, R. Ober, K. Jensen and M. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS; (b) M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468–471 CrossRef CAS.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- (a) X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed; (b) F. Chen and D. Gerion, Nano Lett., 2004, 4, 1827–1832 CrossRef CAS.
- (a) C. Stoll, S. Kudera, W. J. Parak and F. Lisdat, Small, 2006, 2, 741–743 CrossRef CAS PubMed; (b) K. Schubert, W. Khalid, Z. Yue, W. J. Parak and F. Lisdat, Langmuir, 2009, 26, 1395–1400 CrossRef PubMed; (c) J. Tanne, D. Schafer, W. Khalid, W. Parak and F. Lisdat, Anal. Chem., 2011, 83, 7778–7785 CrossRef CAS PubMed; (d) C. Stoll, C. Gehring, K. Schubert, M. Zanella, W. Parak and F. Lisdat, Biosens. Bioelectron., 2008, 24, 260–265 CrossRef CAS PubMed.
- (a) A. Kongkanand, R. Martínez Domínguez and P. V. Kamat, Nano Lett., 2007, 7, 676–680 CrossRef CAS PubMed; (b) M. Ouyang, J.-L. Huang, C. L. Cheung and C. M. Lieber, Science, 2001, 291, 97–100 CrossRef CAS PubMed; (c) J. Kong, N. R. Franklin, C. Zhou, M. G. Chapline, S. Peng, K. Cho and H. Dai, Science, 2000, 287, 622–625 CrossRef CAS; (d) P. Chen, X. Wu, J. Lin and K. Tan, Science, 1999, 285, 91–93 CrossRef CAS.
- (a) X. Peng, J. Chen, J. A. Misewich and S. S. Wong, Chem. Soc. Rev., 2009, 38, 1076–1098 RSC; (b) S. Chaudhary, H. Lu, A. M. Müller, C. J. Bardeen and M. Ozkan, Nano Lett., 2007, 7, 1973–1979 CrossRef CAS PubMed; (c) H. Ago, K. Petritsch, M. S. Shaffer, A. H. Windle and R. H. Friend, Adv. Mater., 1999, 11, 1281–1285 CrossRef CAS; (d) D. M. Guldi, G. A. Rahman, V. Sgobba, N. A. Kotov, D. Bonifazi and M. Prato, J. Am. Chem. Soc., 2006, 128, 2315–2323 CrossRef CAS PubMed; (e) I. Robel, B. A. Bunker and P. V. Kamat, Adv. Mater., 2005, 17, 2458–2463 CrossRef CAS; (f) F. Vietmeyer, B. Seger and P. V. Kamat, Adv. Mater., 2007, 19, 2935–2940 CrossRef CAS.
- D. M. Guldi, G. A. Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471–487 RSC.
- (a) B. Zhang, X. Chen, J. Yang, D. Yu, Y. Chen, D. Wu, R. Fu and M. Zhang, Nanotechnology, 2010, 21, 045601 CrossRef PubMed; (b) J.-S. Yu, S. Kang, S. B. Yoon and G. Chai, J. Am. Chem. Soc., 2002, 124, 9382–9383 CrossRef CAS PubMed; (c) H. Kabbour, T. F. Baumann, J. H. Satcher, A. Saulnier and C. C. Ahn, Chem. Mater., 2006, 18, 6085–6087 CrossRef CAS; (d) N. Jia, Z. Wang, G. Yang, H. Shen and L. Zhu, Electrochem. Commun., 2007, 9, 233–238 CrossRef CAS PubMed; (e) C. E. Banks, R. R. Moore, T. J. Davies and R. G. Compton, Chem. Commun., 2004, 1804–1805 RSC.
- M. Zhou, L. Shang, B. Li, L. Huang and S. Dong, Electrochem. Commun., 2008, 10, 859–863 CrossRef CAS PubMed.
- B. H. Jun, D. W. Hwang, H. S. Jung, J. Jang, H. Kim, H. Kang, T. Kang, S. Kyeong, H. Lee and D. H. Jeong, Adv. Funct. Mater., 2012, 22, 1843–1849 CrossRef CAS.
- J. H. Bang and P. V. Kamat, ACS Nano, 2011, 5, 9421–9427 CrossRef CAS PubMed.
- (a) A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007–4015 CrossRef CAS PubMed; (b) B.-R. Hyun, Y.-W. Zhong, A. C. Bartnik, L. Sun, H. D. Abruna, F. W. Wise, J. D. Goodreau, J. R. Matthews, T. M. Leslie and N. F. Borrelli, ACS Nano, 2008, 2, 2206–2212 CrossRef CAS PubMed.
- G. Zhao, J.-J. Xu and H.-Y. Chen, Electrochem. Commun., 2006, 8, 148–154 CrossRef CAS PubMed.
- M. Wang, W. Hou, C.-C. Mi, W.-X. Wang, Z.-R. Xu, H.-H. Teng, C.-B. Mao and S.-K. Xu, Anal. Chem., 2009, 81, 8783–8789 CrossRef CAS PubMed.
- D. Tang, R. Niessner and D. Knopp, Biosens. Bioelectron., 2009, 24, 2125–2130 CrossRef CAS PubMed.
- Z. Zhong, M. Li, D. Xiang, N. Dai, Y. Qing, D. Wang and D. Tang, Biosens. Bioelectron., 2009, 24, 2246–2249 CrossRef CAS PubMed.
- G. Qin, S. Zhao, Y. Huang, J. Jiang and F. Ye, Anal. Chem., 2012, 84, 2708–2712 CrossRef CAS PubMed.
- H. Zhao, Y. Chang, M. Liu, S. Gao, H. Yu and X. Quan, Chem. Commun., 2013, 49, 234–236 RSC.
- H. Qi, C. Wang and N. Cheng, Microchim. Acta, 2010, 170, 33–38 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |