
Synthesis and use of “clickable” bay-region tetrasubstituted perylene tetracarboxylic tetraesters and a perylene monoimide diester as energy acceptors†
Edanur
Aydin
,
Bilal
Nisanci
,
Murat
Acar
,
Arif
Dastan
and
Özgür Altan
Bozdemir
*
Department of Chemistry, Ataturk University, Erzurum, 25240, Turkey. E-mail: oaltanbozdemir@atauni.edu.tr
First published on 22nd October 2014
Abstract
Two novel bay-region chlorine- or phenoxy-substituted perylene tetracarboxylic tetrapropargylesters (PTTEs) and a phenoxy-substituted perylene monoimide dipropargylester (PMIDE) were synthesized, photophysically characterized, and decorated with energy donating coumarin chromophores by means of microwave-assisted Cu(I)-catalyzed 1,3-dipolar cycloaddition reactions.
Introduction
In recent years, perylene tetracarboxylic tetraesters (PTTEs), being close relatives of perylene bisimide (PBI) dyes, have attracted great attention among researchers on account of their high absorption coefficients in the visible region and their good fluorescence properties.1,2 The much better solubility of bay-region-unsubstituted PTTEs in common organic solvents, compared to that of similar PBIs, allows for facile purification and easy functionalization at the bay-region by means of bromination and nitration.1 Subsequent nucleophilic aromatic substitution or transition metal-catalyzed coupling reactions carried out on these derivatives have opened up novel pathways for the synthesis of more extended aromatic structures.2 In addition to these types of applications, their high propensity for self-aggregation into one-dimensional supramolecular polymers and columnar liquid crystals has also been demonstrated.3 Currently used PTTE derivatives bear one or two substituents at the bay-region2 and more recently, a couple of derivatives with four chlorine atoms were employed as intermediates en route to other perylene structures.4 It is obvious that increasing the number of substituents placed at the bay-region would be an important contribution to the chemistry of this class of fluorescent dyes as it may lead to improved photophysical properties. Another interesting structure possessing a perylene skeleton is the perylene monoimide diester (PMIDE).5 As the electron-withdrawing ability of these compounds lies in between that of PBIs and PTTEs, they have the potential to be implemented for the fine tuning of device performances in novel nanoelectronic devices.The development of artificial systems capable of collecting and transferring light energy into suitable energy absorbing sites continues to be the subject of a great number of research activities pursuing the ultimate goals of mimicking nature's photosynthetic reaction centers and producing efficient solar energy concentrators.6 The efficient transfer of solar energy in the form of electronic energy transfer (EET) can be accomplished with systems consisting of rationally designed energy donor and energy acceptor chromophores. In this manner, dendrimers offer one of the most efficient synthetic structures for the arrangement of energy donor chromophores around energy acceptors, so as to channel the light energy after its collection from a light source. To date, several light-harvesting dendrimers have been synthesized by exploiting well-known dye molecules,7 among which several members of the rylene dyes played important roles both as energy donors and acceptors.8 However, to the best of our knowledge, although there is an example for the use of perylene tetraesters in light-harvesting dendrimers,8g bay-region tetrasubstituted PTTE and PMIDE derivatives have never been considered as acceptor cores in dendritic energy transfer systems. In this study, for the first time, two novel tetrasubstituted PTTE derivatives and a PMIDE derivative with propargyloxy ester functionalities were constructed to explore their behaviors as energy acceptors in light-harvesting systems.
Results and discussion
In the construction of the target light harvesters, we employed a convergent approach to attach well-known 4-methyl-7-alkoxycoumarin energy donor chromophores via microwave-assisted Cu(I)-catalyzed 1,3-dipolar cycloaddition, relying on the fact that “click chemistry” has been one of the most reliable means for the synthesis of dendrimers.9 As shown in Scheme 1, our synthetic studies commenced with the synthesis of the first core energy acceptor molecule 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylicpropargyl ester 4. 1,6,7,12-tetrachloroperylenetetracarboxylic acid dianhydride 3 was reacted with a mixture of propargyl bromide and propargyl alcohol in DMF at 60 °C in the presence of a strong base DBU, to synthesise 4. In the last step, this alkyne-functionalized PTTE 4 and compound 2 were reacted in a microwave reactor using a solvent mixture (1/1/2, H2O/EtOH/CHCl3) at 65 °C in the presence of sodium ascorbate and CuSO4 for 1 h, affording LH-1 in 90% yield after chromatographic purification. To obtain our phenoxy substituted longer wavelength light-harvesting structures LH-2 and LH-3, we first synthesized the tetraphenoxy-perylene bisimide 5 by following a literature procedure.3d A partial basic hydrolysis reaction was then carried out in the presence of KOH in t-butanol at reflux temperature for 5 h.10 This reaction, after acidic work-up, apart from the starting material resulted in the formation of dianhydride 6 and monoimide-monoanhydride 7 as a mixture, which was directly used in the next step without further purification. Following the same protocol as above, this mixture was reacted with a mixture of propargyl bromide and propargyl alcohol in DMF at 60 °C for 4 h in the presence of DBU. After chromatographic purifications, perylene tetraphenoxy tetrapropargyl ester 8 and perylene tetraphenoxy monoimide bispropargyl ester 9 were obtained in 40% and 35% yield from 5, respectively. Employing the same microwave-assisted alkyne-azide cycloaddition reaction, between compounds 2 and 8 or 2 and 9, in the final step resulted in the formation of LH-2 and LH-3. After chromatographic purification, each target molecule was obtained in 90% and 95% yield, respectively.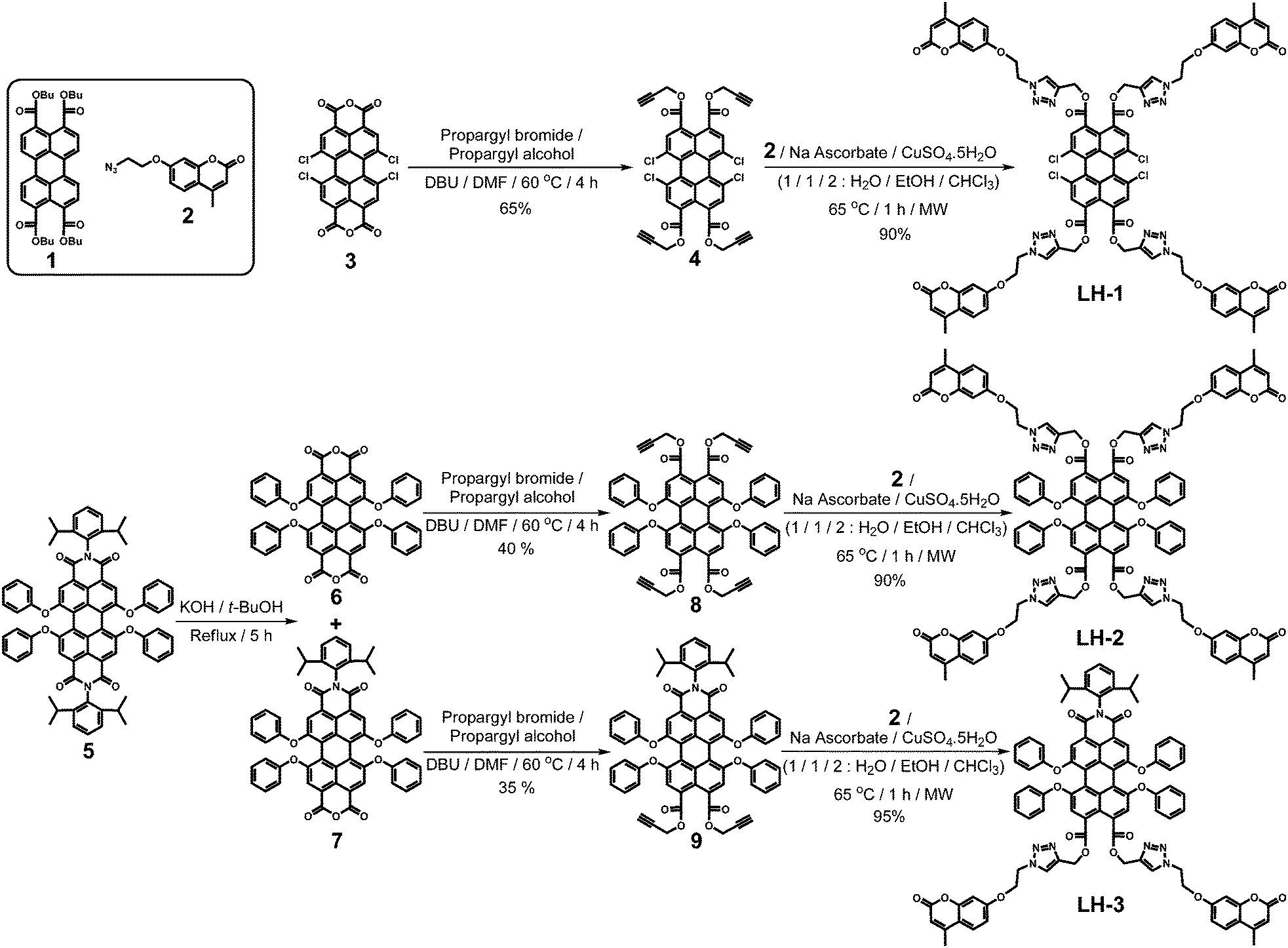 | ||
| Scheme 1 Synthesis of the propargylated perylene derivatives and their conversion into light-harvesting systems LH-1, LH-2, and LH-3. | ||
It should be noted that among the perylenetetracarboxylic acid derivatives, PTTEs are the weakest electron acceptors as a result of the poor orbital interactions between the ester carbonyls and the perylene ring, compared to the imide carbonyls of PBIs. PMIDEs, on the other hand, are stronger electron acceptors than PTTEs, while they are weaker than PBIs. Accordingly, absorption and emission wavelength maxima of these compounds follow the same trend; PTTEs being the shortest wavelength absorbers and emitters. One of the strategies frequently employed in order to shift the absorption and emission maxima of fluorescent dyes to the red end of the visible spectrum is to attach electron-donating functionalities at suitable positions in order to achieve maximum conjugation. If we take the perylene-3,4:9,10-tetra(n-butoxy)tetracarboxylic acid tetraester 1 as the parent PTTE molecule (λabs = 471 nm, λems = 516 nm) for our comparisons, it is obvious that adding substituents at the bay region would increase the solubility of the resulting compounds and, depending on the electron-withdrawing or -releasing abilities of the substituents, would alter the absorption and emission maxima of the PTTEs (Table 1). As expected, a hypsochromic shifting in the absorption and emission maxima for compound 4 (λabs = 459 nm, λems = 499 nm) was observed due to the presence of electron-withdrawing chlorine substituents, while a bathochromic shifting was detected for 8 (λabs = 492 nm, λems = 534 nm) due to the attachment of electron-releasing phenoxy substituents. For PMIDE 9, it is not surprising that the presence of only one imide group is sufficient to endow longer wavelength absorption and emission maxima (λabs = 540 nm, λems = 572 nm) in comparison to PTTEs.
| Compound | λ abs (nm)/εmax (M−1 cm−1) | λ ems (nm) | τ (ns)/λexc (nm) | χ 2 | Φ F/λexc (nm) | J/1014 M−1 cm−1 nm4 | R o (nm) | E (%) | k T(r)/ns−1 |
|---|---|---|---|---|---|---|---|---|---|
| a All spectra were recorded as dilute solutions in CHCl3. λabs/λems: maximum absorption/emission wavelengths. λexc: excitation wavelength. τ: fluorescence lifetime. ΦF: fluorescence quantum yield. Ro: Förster distance. J: spectral overlap integral. E: energy transfer efficiency. kT(r): energy transfer rate constant. b 0.1 M aqueous fluorescein solution (ΦF = 0.95) was used as a reference. c 0.5 M aqueous quinine sulfate solution (ΦF = 0.55) was used as a reference. | |||||||||
| 1 | 471 (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 410) 410) |
516 | 3.6 (457) | 1.08 | 1.00b (450) | ||||
| 2 | 320 (10968) |
378 | 0.6 (337) | 0.98 | 0.09c (315) | ||||
| 4 | 459 (18587) |
499 | 1.8 (457) | 0.91 | 0.16b (450) | ||||
| 0.04c (315) | |||||||||
| LH-1 | 459 (18744) |
499 | 1.5 (337) | 0.95 | 0.10c (315) | 1.86 | 2.46 | 90 | 16.4 |
| 8 | 492 (29180) |
534 | 5.7 (457) | 1.05 | 0.57b (496) | ||||
| 0.20c (315) | |||||||||
| LH-2 | 492 (29265) |
534 | 5.4 (337) | 0.95 | 0.25c (315) | 2.54 | 2.54 | 93 | 22.0 |
| 9 | 540 (20853) |
572 | 5.9 (500) | 0.93 | 0.30b (496) | ||||
| LH-3 | 540 (20679) |
572 | 5.4 (337) | 1.01 | 0.10c (315) | 1.73 | 2.43 | 87 | 11.4 |
In order to assess the efficiency of electronic energy transfer, several steady-state and time-resolved spectroscopic measurements were carried out on each light-harvesting system (Table 1, Fig. 1 and 2, and the ESI† Fig. S1–S6). The absorption spectrum of LH-1 appears as a summation of transitions emanating from coumarin 2 and PTTE 4 (ESI,† Fig. S1).
 | ||
| Fig. 1 (a) Fluorescence emission spectra of compounds 2, 4 and LH-1 upon excitation at 315 nm in CHCl3. The concentrations of compounds 2 and 4 were adjusted so that they have equal absorbances at 320 nm and 459 nm to LH-1. (b) Fluorescence emission spectra of compounds 2, 8 and LH-2 upon excitation at 315 nm in CHCl3. The concentrations of compounds 2 and 8 were adjusted so that they have equal absorbances at 320 nm and 492 nm to LH-2. (c) Fluorescence emission spectra of compounds 2, 9 and LH-3 upon excitation at 315 nm in CHCl3. The concentrations of compounds 2 and 9 were adjusted so that they have equal absorbances at 320 nm and 540 nm to LH-3. | ||
 | ||
| Fig. 2 Absorption (black) and excitation (red) spectra of (a) LH-1, (b) LH-2, and (c) LH-3, normalized at the absorption maxima of the core acceptor units (459 nm for LH-1, 492 nm for LH-2, and 540 nm for LH-3). The blue lines represent the efficiency of the percentage energy transfer as a function of the excitation wavelength. | ||
That an efficient electronic energy transfer takes place between the coumarin donors and the PTTE acceptor unit upon excitation of the peripheral chromophores at 315 nm is evident from the near complete quenching of the coumarin centered fluorescence at 378 nm, compared to the emission from the model compound 2 with an equal absorption intensity (Fig. 1a). Also, a comparison of the emission intensity of LH-1 at 499 nm with the PTTE 4 reveals a 10-fold increase, demonstrating a powerful antenna effect (Fig. 1a). It should be noted here that this donor unit, with a quantum yield of less than 1%, can be referred to as a “dark donor”; however, when it is excited, the excitation energy is transferred to the acceptor faster than the other quenching pathways.11
Similar measurements were carried out for LH-2. In this system, absorption features characteristic of 2 and PTTE 8 are observed in the absorption spectrum (ESI,† Fig. S2). The excitation of the coumarin part at 315 nm resulted in a strong fluorescence, with nearly a 3.1-fold increase in the emission intensity at 534 nm compared to the emission of the reference dye PTTE 8, which was accompanied by a complete quenching of the coumarin part (Fig. 1b). This somewhat smaller increase in the acceptor emission is due to the presence of vibronic transitions observed at higher energies in PTTE 8 and LH-2 (300–350 nm region), leading to a relatively intense fluorescence emanating from the possibility of direct excitation of the PTTE part in LH-2 at 315 nm.
In LH-3, where only two donor coumarin units are present, the absorption spectrum is again a combination of transitions from the coumarin 2 and PMIDE 9 units, covering a broad wavelength range stretching from nearly 600 nm to 200 nm (ESI,† Fig. S3). It is apparent that the excitation of LH-3 at 315 nm leads to an increased fluorescence at 572 nm as a result of an energy transfer, revealing itself as a 2.4-fold enhanced fluorescence compared to the emission intensity of the reference dye PMIDE 9 at equal concentrations (Fig. 1c).
In order to investigate the likelihood of intermolecular energy transfer between the components of light-harvesters and to confirm that the observed energy transfer is intramolecular in nature, suitable donor–acceptor mixtures – representing each light-harvester as a combination of separate entities – were prepared and subjected to the same spectroscopic measurements (ESI,† Fig. S4–S6). Hence, the excitation of mixtures of donor and acceptor molecules (2 and 4, 2 and 8, and 2 and 9) at the donor absorption maximum (315 nm) revealed no intermolecular energy transfer between individual chromophores, as evidenced by the low intensity of the acceptor emissions in each case and the absence of coumarin quenching.
Table 1 gives valuable photophysical parameters which can be used for estimation of the efficiency of energy transfer in all light-harvesting systems obtained in this study: acceptor emission lifetimes (τ) and fluorescence quantum yields (ΦF) increase compared to each acceptor model compound when excited at the donor site (315 nm), whereas donor lifetimes and quantum yields decrease to nearly zero, indicating an electronic energy transfer. The fluorescence decays of LH-1, LH-2, and LH-3 (ESI,† Fig. S19–S26) were found to be single-exponential with χ2 values smaller than 1.2, as a sign of good fit. The Förster radii (Ro) determined for all compounds are very close to each other as predicted from their structural similarity. Due to the close proximity of donor and acceptor units, fast energy transfer rates (k(r)) were calculated based on the fluorescence intensity data for LH-1, LH-2, and LH-3, and the values are 1.64 × 1010 s−1, 2.20 × 1010 s−1, and 1.14 × 1010 s−1, respectively. The energy transfer efficiencies were then calculated as 90% for LH-1, 93% for LH-2, and 87% for LH-3.
One of the possible pitfalls of using donor quantum yields and lifetimes in calculating energy transfer efficiencies is the preassumption that the changes in these values are merely related to the intramolecular energy transfer taking place between the donor and acceptor chromophores, ignoring the other common quenching pathways. In order to correctly determine the energy transfer efficiency, one has to compare the absorption and excitation spectra of the light-harvesting system – normalized at the acceptor absorption wavelength maximum – over the entire spectral range, in order to be sure that the harvested photons by the donor are quantitatively transferred to the acceptor unit. Indeed, this way of determining energy transfer efficiency has been proven more reliable by many researchers as it gives directly the ratio of the number of photons absorbed by the donor molecules to the number of excitations generated in the acceptor.12
When we applied this method to our light-harvesting systems, in each case, we observed a poor match between the excitation – acquired for the respective acceptor emission maximum – and donor absorption regions of the spectrum (Fig. 2). In contrast to the values calculated by means of donor quantum yields and lifetimes, we determined 25%, 24%, and 13% efficiency of energy transfer for LH-1, LH-2, and LH-3, respectively. Similar to the many other dendritic light-harvesters constructed by means of flexible bonding units,12b these low values must be due to the presence of various nonradiative quenching pathways in these conformationally flexible supramolecules, as we did not observe any aggregation or photodecomposition in these systems after several fluorescence measurements. Nevertheless, up to 10-fold fluorescence intensity increments in the presence of coumarin donor groups can safely be regarded as a sign of an efficient transfer of energy within these light-harvesters.
Conclusions
In summary, we have demonstrated here that bay-region tetrasubstituted perylene tetraester and perylene monoimide diester derivatives can be prepared with reactive ester functionalities which are ready to be exploited in the construction of various functional supramolecular systems, such as light-harvesting dendrimers or biolabels, by employing fast, microwave-assisted click chemistry. As evaluated by various spectroscopic techniques, excitation energy can be channeled into these novel dyes when they take part in light-harvesting systems and they have the potential to be used as donor sites if suitable donor–acceptor combinations are designed. Additionally, self aggregation abilities and liquid crystallinity properties are other important features of these derivatives, on which work by ourselves is in progress.Experimental
General
Compounds 213 and 314 were synthesized following procedures reported in the literature. All chemicals and solvents were purchased from Sigma-Aldrich and used without further purification. All solvents were dried and distilled before use. Reactions were monitored by thin layer chromatography using Merck TLC Silica gel 60 F254 and the plates were inspected using 254 nm UV-light and/or by acquiring 1H-NMR spectra. Column chromatography was performed over Merck Silica gel 60F (70–230 mesh ASTM). The 1H- and 13C-NMR spectra were recorded on a Varian-400 or a Bruker-400 spectrometer in CDCl3 using tetramethylsilane as the internal reference. All spectra were recorded at 25 °C and coupling constants (J values) are given in Hz. Chemical shifts are given in parts per million (ppm). Abbreviations used to define the multiplicities are as follows: s = singlet; d = doublet; t = triplet; q = quartet; hept = heptet; m = multiplet; br = broad. Mass spectra were recorded on an Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS. Microwave reactions were performed in a CEM Discover LabMate 200W microwave reactor. Absorption spectrometry was performed using a Shimadzu spectrophotometer. Steady-state fluorescence measurements were conducted using a Shimadzu RF-5301PC spectrofluorometer. Fluorescence decays for the lifetime measurements were carried out by means of a LaserStrobe Model TM-3 lifetime fluorophotometer from Photon Technology International (PTI).where Φ is the fluorescence quantum yield, I is the integrated fluorescence intensity, n is the refractive index of the solvent, and OD is the optical density (absorption). The subscript R refers to the reference fluorophore of known quantum yield.

The Förster distances (Ro), energy transfer efficiencies (E), and energy transfer rate constants (kT(r)) were calculated using the formulae below:15
| Ro = 0.211(K2n−4ΦDJ(λ))1/6 (in Å) |
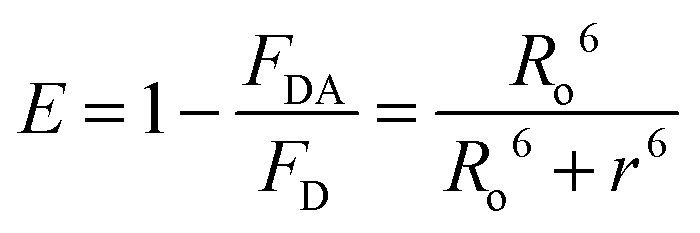
where Ro stands for the Förster distance, ΦD is the quantum yield of the donor in the absence of the acceptor, n is the refractive index of the medium, r is the distance between the donor and acceptor, and τD is the lifetime of the donor in the absence of the acceptor. K2 is usually assumed to be equal to 2/3. The overlap integral (J(λ)) expresses the degree of spectral overlap between the donor emission and the acceptor absorption, kT(r) is the rate of energy transfer from donor to acceptor.
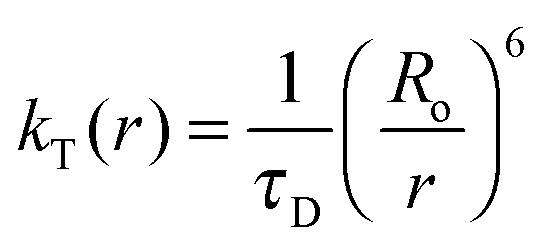
Syntheses
:Hexanes: 60:40). The first fraction was compound 9 (158 mg, 0.154 mmol, 35%). 1H NMR (400 MHz, CDCl3, 298 K): δ = 8.15 (s, 2H), 7.66 (s, 2H), 7.35 (t, J = 7.8 Hz, 1H), 7.23–7.15 (m, 10H), 7.06–6.98 (m, 4H), 6.88–6.85 (m, 8H), 4.79–4.78 (m, 4H), 2.62 (hept, J = 6.8 Hz, 2H), 2.41 (t, J = 2.5 Hz, 2H), 1.03 (d, J = 6.7 Hz, 12H). 13C NMR (101 MHz, CDCl3 298 K): δ = 166.9, 163.3, 155.4, 155.3, 155.1, 154.3, 145.6, 135.4, 133.1, 130.9, 130.7, 129.9, 129.8, 129.6, 129.4, 128.8, 124.33, 124.31, 123.8, 122.2, 121.1, 121.0, 120.6, 119.8, 119.7, 118.4, 77.1, 75.5, 53.0, 29.0, 24.0. HR-ESI-MS: m/z calcd: 1013.3200; found: 1013.3237. The second fraction was compound 8 (180 mg, 0.19 mmol, 40%). 1H NMR (400 MHz, CDCl3, 298 K): δ = 7.69 (s, 4H), 7.27–7.23 (m, 8H), 7.08 (t, J = 7.4 Hz, 4H), 6.91–6.88 (m, 8H), 4.85–4.83 (m, 8H), 2.46 (t, J = 2.4 Hz, 4H). 13C NMR (101 MHz, CDCl3 298 K): δ = 167.2, 155.7, 153.7, 135.7, 131.1, 130.0, 129.0, 124.3, 121.5, 119.9, 118.9, 77.4, 75.6, 53.1. HR-ESI-MS: m/z calcd: 948.2207; found: 948.2255.
Acknowledgements
The authors would like to thank the Ataturk University Research Fund for its support of this project (2011/375).References
- (a) Z. Yuan, Y. Xiao, Z. Li and X. Qian, Org. Lett., 2009, 11, 2808–2811 CrossRef CAS PubMed; (b) Y. Xie, X. Zhang, Y. Xiao, Y. Zhang, F. Zhou, J. Qi and J. Qu, Chem. Commun., 2012, 48, 4338–4340 RSC; (c) Y. Zhang, Z. Zhao, X. Huang, Y. Xie, C. Liu, J. Li, X. Guan, K. Zhang, C. Cheng and Y. Xiao, RSC Adv., 2012, 2, 12644–12647 RSC.
- (a) Z. Yuan, Y. Xiao and X. Qian, Chem. Commun., 2010, 46, 2772–2774 RSC; (b) Y. Li, C. Wang, C. Li, S. Di Motta, F. Negri and Z. Wang, Org. Lett., 2014, 14, 5278–5281 CrossRef PubMed.
- (a) A. Arnaud, J. Belleney, F. Boue, L. Bouteiller, G. Carrot and V. Wintgens, Angew. Chem., Int. Ed., 2004, 43, 1718–1721 CrossRef CAS PubMed; (b) X. Mo, H.-Z. Chen, M.-M. Shi and M. Wang, Chem. Phys. Lett., 2006, 417, 457–460 CrossRef CAS PubMed; (c) T. Martinsky, R. Hertmanowski, R. Stolarski and D. Bauman, Thin Solid Films, 2008, 516, 8834–8838 CrossRef PubMed; (d) X. Mo, M.-M. Shi, J.-C. Huang, M. Wang and H.-Z. Chen, Dyes Pigm., 2008, 76, 236–242 CrossRef PubMed.
- Y. Zagranyarski, L. Chen, D. Jansch, T. Gessner, C. Li and K. Mullen, Org. Lett., 2014, 16, 2814–2817 CrossRef CAS PubMed.
- (a) X. Zhang, Y. Wu, J. Li, F. Li and M. Li, Dyes Pigm., 2008, 76, 810–816 CrossRef CAS PubMed; (b) C. Xue, R. Sun, R. Annab, D. Abadi and S. Jin, Tetrahedron Lett., 2009, 50, 853–856 CrossRef CAS PubMed; (c) J. Keiber, H. Bock, O. Thiebaut, E. Grelet and H. Langhals, Eur. J. Org. Chem., 2011, 707–712 Search PubMed; (d) R. Wang, Z. Shi, C. Zhang, A. Zhang, J. Chen, W. Guo and Z. Sun, Dyes Pigm., 2013, 98, 450–458 CrossRef CAS PubMed.
- (a) N. S. Lewis, Nature, 2001, 414, 589–590 CrossRef CAS PubMed; (b) Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831–845 RSC; (c) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed; (d) M. J. Currie, J. K. Mapel, T. D. Heidel, S. Goffri and M. A. Baldo, Science, 2008, 321, 226–228 CrossRef CAS PubMed; (e) R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611–631 RSC; (f) D. Hablot, A. Harriman and R. Ziessel, Angew. Chem., Int. Ed., 2011, 50, 7833–7836 CrossRef CAS PubMed; (g) D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed; (h) S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Liobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- (a) H. Imahori, J. Phys. Chem. B, 2004, 108, 6130–6143 CrossRef CAS PubMed; (b) W.-S. Li and T. Aida, Chem. Rev., 2009, 109, 6047–6076 CrossRef CAS PubMed; (c) Y. Zeng, Y.-L. Li, J. Chen, G. Yang and Y. Li, Chem. – Asian J., 2010, 5, 992–1005 CrossRef CAS PubMed; (d) M. Kozaki, S. Morita, S. Suzuki and K. J. Okada, J. Org. Chem., 2012, 77, 9447–9457 CrossRef CAS PubMed; (e) R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330–11344 CrossRef CAS PubMed; (f) M. Kozaki, S. Suzuki and K. Okada, Chem. Lett., 2013, 42, 1112–1118 CrossRef CAS.
- (a) J. M. Serin, D. W. Brousmiche and J. M. J. Frechet, Chem. Commun., 2002, 2605–2607 RSC; (b) M. Cotlet, T. Vosch, S. Habuchi, T. Weil, K. Mullen, J. Hofkens and F. De Schryver, J. Am. Chem. Soc., 2005, 127, 9760–9768 CrossRef CAS PubMed; (c) J. H. Hurenkamp, W. R. Browne, R. Augilis, A. Pugzlys, P. H. M. van Lousdrecht, J. H. van Esch and B. L. Feringa, Org. Biomol. Chem., 2007, 5, 3354–3362 RSC; (d) O. A. Bozdemir, M. D. Yilmaz, O. Buyukcakir, A. Siemiarczuk, M. Tutas and E. U. Akkaya, New J. Chem., 2010, 34, 151–155 RSC; (e) T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed; (f) H. Langhals, A. J. Esterbauer, A. Walter, E. Riedle and I. Pugliesi, J. Am. Chem. Soc., 2010, 132, 16777–16782 CrossRef CAS PubMed; (g) M. Takahashi, H. Morimoto, K. Miyake, H. Kawaki, Y. Sei, K. Yamaguchi, T. Sengoku and H. Yoda, New J. Chem., 2008, 32, 547–553 RSC.
- (a) P. Apukuttan, W. Dehaen, V. V. Fokin and E. Van der Eycken, Org. Lett., 2004, 6, 4223–4225 CrossRef PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- Y. Nagao, T. Naito, Y. Abe and T. Misono, Dyes Pigm., 1996, 32, 71–83 CrossRef CAS.
- D. Su, J. Oh, S.-C. Lee, J. M. Lim, S. Sahu, X. Yu, D. Kim and Y.-T. Chang, Chem. Sci., 2014, 5, 4812–4818 RSC.
- (a) D. V. Roberts, B. P. Wittmershaus, Y.-Z. Zhang, S. Swan and M. P. J. Klinosky, J. Lumin., 1998, 79, 225–231 CrossRef CAS; (b) Z. Kostereli, T. Ozdemir, O. Buyukcakir and E. U. Akkaya, Org. Lett., 2012, 14, 3636–3639 CrossRef CAS PubMed; (c) S. Guo, L. Ma, J. Zhao, B. Kucukoz, A. Karatay, M. Hayvali, H. G. Yaglioglu and A. Elmali, Chem. Sci., 2014, 5, 489–500 RSC.
- Y.-C. Duan, Y.-C. Ma, E. Zhang, X.-J. Shi, M.-M. Wang, X.-W. Ye and H.-M. Liu, Eur. J. Med. Chem., 2013, 62, 11–19 CrossRef CAS PubMed.
- R. S. Sanchez, R. Gras-Charles, J. L. Bourdelande, G. Guirado and J. Hernando, J. Phys. Chem. C, 2012, 116, 7164–7172 CAS.
- Principals of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, Singapore, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of new compounds, and spectra. See DOI: 10.1039/c4nj01565g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |