
Polarity-tunable and wavelength-tunable bacteriochlorins bearing a single carboxylic acid or NHS ester. Use in a protein bioconjugation model system†
Jianbing
Jiang
,
Chih-Yuan
Chen
,
Nuonuo
Zhang
,
Pothiappan
Vairaprakash
and
Jonathan S.
Lindsey
*
Department of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu
First published on 31st October 2014
Abstract
To broaden the scope of near-infrared (NIR)-active chromophores available for bioconjugation with proteins, 10 new bacteriochlorins have been synthesized: six are lipophilic and bear a carboxylic acid tether; four are hydrophilic and bear four carboxylic acids and one N-hydroxysuccinimido (NHS) ester tether. Each bacteriochlorin exhibits a sharp long-wavelength absorption (Qy) band in the NIR region (727–823 nm). The lipophilic bacteriochlorins were examined in DMF (fluorescence quantum yield Φf = 0.037–0.19) whereas the hydrophilic bacteriochlorins were examined in DMF (Φf also = 0.037–0.19) or aqueous phosphate buffer (Φf = 0.0011–0.13). Two bacteriochlorins were conjugated to myoglobin (Mb), which contains ∼14 accessible amino groups. Use of 2, 10, or 50 equivalents of a hydrophilic bacteriochlorin–NHS ester (BC7) gave average loadings of 0.62, 1.6, or 7.1 bacteriochlorins/Mb as determined by absorption spectral comparison with the strongly absorbing heme ligand. MALDI-MS analysis showed a distribution of 0–9 bound bacteriochlorins for the conjugate sample with average loading of 7.1. The Mb–BC7 conjugates exhibited characteristic absorption and fluorescence spectra in aqueous buffer, yet the Φf value was markedly low (Φf ∼ 0.02) regardless of loading versus that of the BC7 monomer (Φf = 0.12), attributed in part to heme quenching. Removal of the heme revealed a loading-dependent Φf, which ranged from 0.091 (0.62 loading) to 0.023 (7.1 loading). The decrease in Φf with increased loading is attributed to self-quenching perhaps facilitated by excited-state energy transfer among the bacteriochlorins (Förster R0 = 59 Å). Taken together, the results show facile access to a collection of useful bacteriochlorins for NIR spectroscopic studies, along with a pigment–protein system that serves the dual purposes of a convenient testbed for evaluating protein bioconjugation processes as well as a nanosized architecture for use in photochemical studies.
Introduction
The conjugation of chromophores to biological molecules has a rich history both in methods and applications.1–9 Yet, a comparatively unexplored topic in this domain concerns the use of NIR-active chromophores. The challenge to filling this lacuna entails synthetic tailoring of NIR-active chromophores to achieve the molecular design requirements, which typically include adjusting the polarity of the chromophore, tuning the wavelength of absorption/emission, and incorporating a single bioconjugatable tether. Examples of synthetic NIR chromophores range from long-chain cyanine dyes to quantum dots.10–13 Nature’s NIR-active chromophores are built around the bacteriochlorin π-system, which provides the basis for bacterial photosynthesis (Bchl a, b and g)14 and unknown roles in other organisms (e.g., tolyporphins)15–21 (Chart 1). | ||
| Chart 1 Representative natural bacteriochlorins. | ||
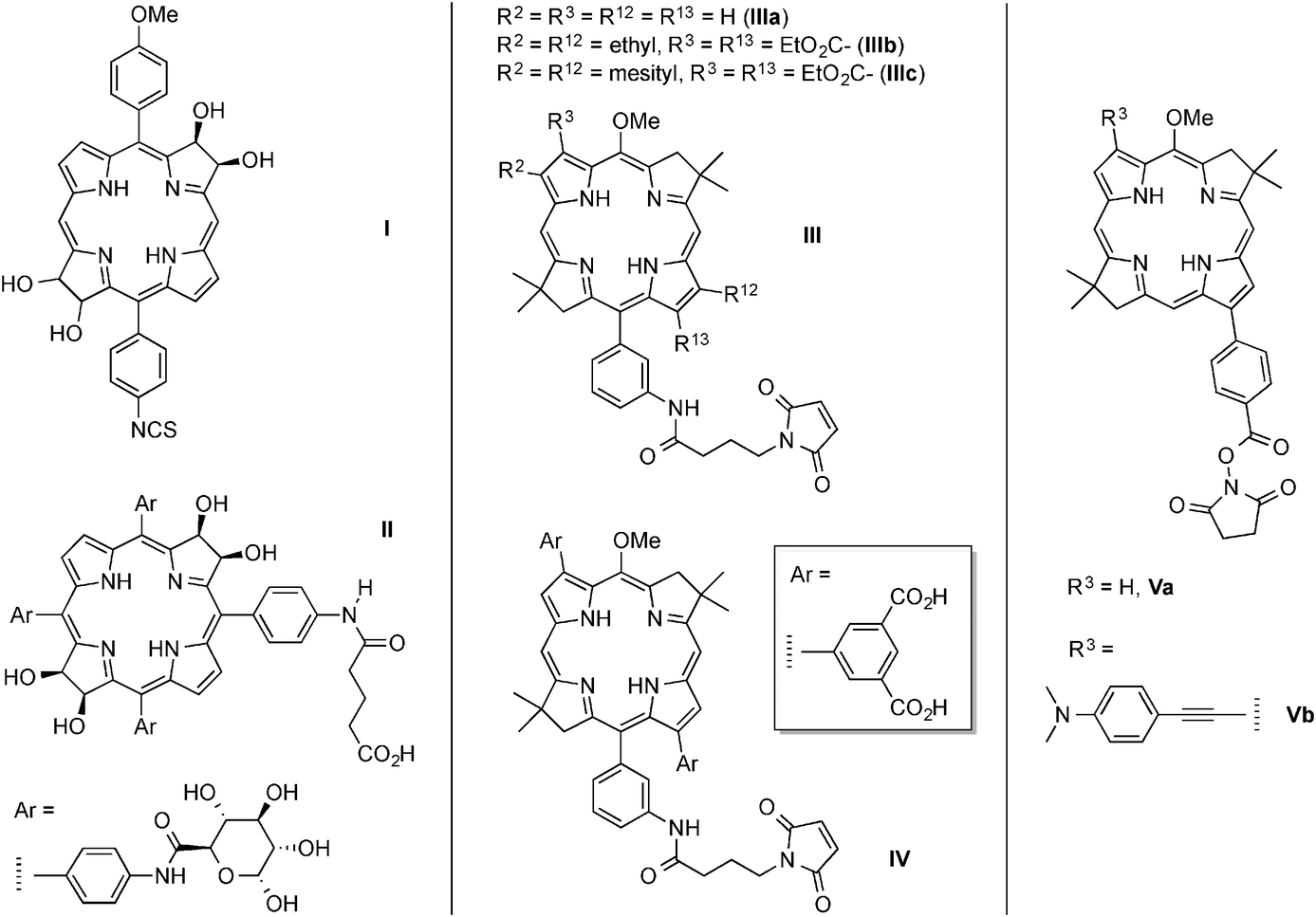
Semisynthesis beginning with naturally occurring macrocycles has been a mainstay for preparing and tailoring bacteriochlorins,22,23 but the presence of a number of substituents about the perimeter of such macrocycles limits synthetic manipulations particularly for wavelength tuning, polarity tuning, and installation of a single bioconjugatable tether. Methods to prepare synthetic bacteriochlorins are under active investigation24–44 and have been recently reviewed.45,46 Two approaches that define the range of such methods include (1) double addition to a porphyrin thereby converting two, opposite pyrrole rings to pyrroline rings, and (2) de novo synthesis wherein the pyrroline rings are incorporated as pre-made constituents upon macrocycle formation.40–44,47–50 Bioconjugatable bacteriochlorins have been prepared by both approaches, as illustrated by the examples shown in Chart 2. Bacteriochlorins I and II were prepared by OsO4 treatment of a porphyrin,25,27 whereas the set of III–V were prepared by de novo synthesis.51–55 Note the nature of the bioconjugatable groups [isothiocyanate (I), carboxylic acid (II), maleimide (III, IV), and NHS ester (V)] as well as the polarity: bacteriochlorin members of sets III and V are hydrophobic, whereas IV is hydrophilic.
 | ||
| Chart 2 Representative bioconjugatable synthetic bacteriochlorins. | ||
The ability to tune the position of the long-wavelength absorption band (and hence the position of the fluorescence emission band) relies on introduction of auxochromes at the perimeter of the macrocycle.56 The long-wavelength absorption (Qy) band stems from a transition that is polarized along the long axis of the molecule, as shown in Fig. 1. Accordingly, the introduction of substituents at the β-pyrrole positions (2, 3, 12, 13) or adjacent meso-positions (5, 15) enables the band to be shifted from ∼700 to nearly 900 nm. For the members of set III, the Qy band ranges from 713 to 756 nm.51,53 Such bacteriochlorins have been bioconjugated to analogues of the native membrane-spanning peptides of the light-harvesting complexes of photosynthetic bacteria. The resulting biohybrid light-harvesting architectures self-assemble in aqueous-detergent media. The appended synthetic bacteriochlorins – attached via a maleimide–cysteinyl linkage – absorb NIR light and funnel the resulting excited-state energy to lower-energy-absorbing chromophores as part of the light-harvesting process.51,53
 | ||
| Fig. 1 Molecular design features of synthetic bacteriochlorins. | ||
We set out to develop a more broadly viable set of wavelength-tunable and polarity-tunable bacteriochlorins. Because one objective is to be able to conjugate multiple copies of a bacteriochlorin to a given peptide, we turned to the use of carboxylic acid or NHS esters (conjugatable with amines)57 to avoid the problems anticipated if multiple cysteines were employed to accommodate bacteriochlorin–maleimides. To our knowledge, the only bacteriochlorin–NHS esters prepared by de novo synthesis are Va and Vb of Chart 2.
Two sets of target bacteriochlorins were identified (Chart 3). The members of the first set are lipophilic and bear a carboxylic acid for bioconjugation (BC1–BC6). The members of the second set are hydrophilic (each contains four carboxylic acid groups for aqueous solubilization) and bear a single NHS ester for bioconjugation (BC7–BC10). All of the bacteriochlorins are free base macrocycles, except for one zinc bacteriochlorin (BC4). BC7 and BC8 differ only in the meta- versus para-substitution of the tether. On the basis of the spectral properties of analogous bacteriochlorins (lacking a carboxylic acid tether), absorption in the NIR region (726–823 nm) is expected.
 | ||
| Chart 3 Structures of six lipophilic monocarboxy-bacteriochlorins (top) and four hydrophilic tetracarboxy-bacteriochlorins bearing an NHS ester (below). | ||
In this paper, we report the synthesis of the 10 bacteriochlorins along with their absorption and fluorescence properties in DMF and/or aqueous solution. Many applications of the bacteriochlorins can be envisaged. We have employed myoglobin (Mb) as a globular protein for bioconjugation with selected hydrophilic bacteriochlorins, thereby affording a nanoscale analogue of pheophytin-coated polystyrene particles described by Cellarius and Mauzerall nearly a half-century ago.58 The absorption and fluorescence properties of the resulting Mb–bacteriochlorin conjugates have been examined in aqueous solution in the presence or absence of the heme ligand. Taken together, the studies afford a new set of synthetic bacteriochlorins for use in cases where one seeks aqueous or membrane solubility, wavelength tunability (726 to 823 nm), and bioconjugation via one of the simplest joining reactions (amidation).
Results and discussion
Synthesis of lipophilic bacteriochlorins
The six monocarboxy-bacteriochlorins (BC1–BC6) were derivatized from three known bacteriochlorin building blocks (BC11,51BC12,42BC1353) bearing substituents at the 2, 3, 12, 13, 15 positions for wavelength tailoring and derivatization (Chart 4). Three distinct methods were employed to introduce the carboxylic acid group. (1) For bacteriochlorins with a 3-aminophenyl group, nucleophilic ring-opening of succinic anhydride gave the carboxylic acid group directly (BC1, BC4 and BC5). (2) For 15-brominated bacteriochlorins, Suzuki coupling (with compounds bearing a protected carboxylic acid group) followed by deprotection with trifluoroacetic acid (TFA) unveiled the carboxylic acid group (BC2 and BC3). (3) Pd-mediated carbonylation with a BOC-protected amine formed the bacteriochlorin–imide, which upon TFA deprotection unveiled the carboxylic acid group (BC6). All of these methods proceeded smoothly to afford the monocarboxy-bacteriochlorins in good to excellent yields. | ||
| Chart 4 Three known bacteriochlorin building blocks. | ||
Reaction of aminophenylbacteriochlorin BC11 with succinic anhydride in CHCl3 afforded BC1 in 67% yield (Scheme 1). Metalation44 of bacteriochlorin BC11 with zinc triflate in the presence of sodium hydride afforded BC14 in 52% yield. Reaction of BC14 with succinic anhydride gave the carboxy-bacteriochlorin BC4 in 62% yield. In a similar manner to that of BC1 and BC4, treatment of bacteriochlorin BC13 with succinic anhydride afforded BC5 in 71% yield.
 | ||
| Scheme 1 Synthesis of monocarboxy-bacteriochlorins BC1, BC4 and BC5. | ||
Suzuki coupling of bacteriochlorin BC12 with compound 1 failed to give the desired carboxy-bacteriochlorin BC2, presumably because of the presence of the free carboxylic acid group of 1 (Scheme 2). Alternatively, protection of the free carboxylic acid group of 1 by treatment with di-tert-butyl dicarbonate [(Boc)2O] in the presence of MgCl259 afforded the tert-butyl ester 2 in 60% yield. Suzuki coupling of bacteriochlorin BC12 with 2 gave pro-BC2 in excellent yield (93%). Cleavage of the tert-butyl protecting group in 20% TFA gave BC2 in 74% yield.52
 | ||
| Scheme 2 Synthesis of monocarboxy-bacteriochlorin BC2. | ||
BC3 was obtained in a similar manner as for BC2, using the known Suzuki coupling partner 341 (Scheme 3).
 | ||
| Scheme 3 Synthesis of monocarboxy-bacteriochlorin BC3. | ||
Bacteriochlorin-13,15-dicarboximides with a methoxy group at the 5-position have been synthesized previously.43 The imide-forming reaction entails treatment of a bacteriochlorin (bearing a 13-carboethoxy group and a 15-bromo group; e.g., BC12) to Pd-mediated carbamoylation in the presence of an amine and CO. The reaction is carried out in the presence of a base, typically Cs2CO3. Thus, BC12 was converted to BC15 in 62% yield upon use of 3 equivalents of Cs2CO3.43 Upon repeating this synthesis, BC15 was obtained in 55% yield, and we noted the presence of a trace amount (<5%) of the corresponding bacteriochlorin–imide lacking the 5-methoxy group (BC16). When the reaction was repeated with 9 equivalents of Cs2CO3, the ratio reversed: the demethoxylated BC16 was obtained in 84% yield whereas the 5-methoxybacteriochlorin BC15 was obtained in <5% yield (Scheme 4). The reaction is readily monitored by absorption spectroscopy (as well as MALDI-MS), given that the long-wavelength absorption maximum is at 726 nm (BC12), 798 nm (BC15) and 820 nm (BC16). Removal of the 5-methoxy group thus provides a convenient means to impart a bathochromic shift of the long-wavelength absorption band of the bacteriochlorin.
 | ||
| Scheme 4 Demethoxylation upon imide formation (see text for yields). | ||
Herein, 15 equivalents of Cs2CO3 were used to form the bacteriochlorin–imide as well as remove the 5-methoxy group. The synthesis was first carried out with n-butylamine, which gave the 5-demethoxylated bacteriochlorin–imide BC17 in 52% yield (Scheme 5). Similar use of tert-butyl 4-aminobutyrate gave pro-BC6 in 65% yield. Cleavage of the protecting group with TFA gave the monocarboxy-bacteriochlorin BC6 in 90% yield.
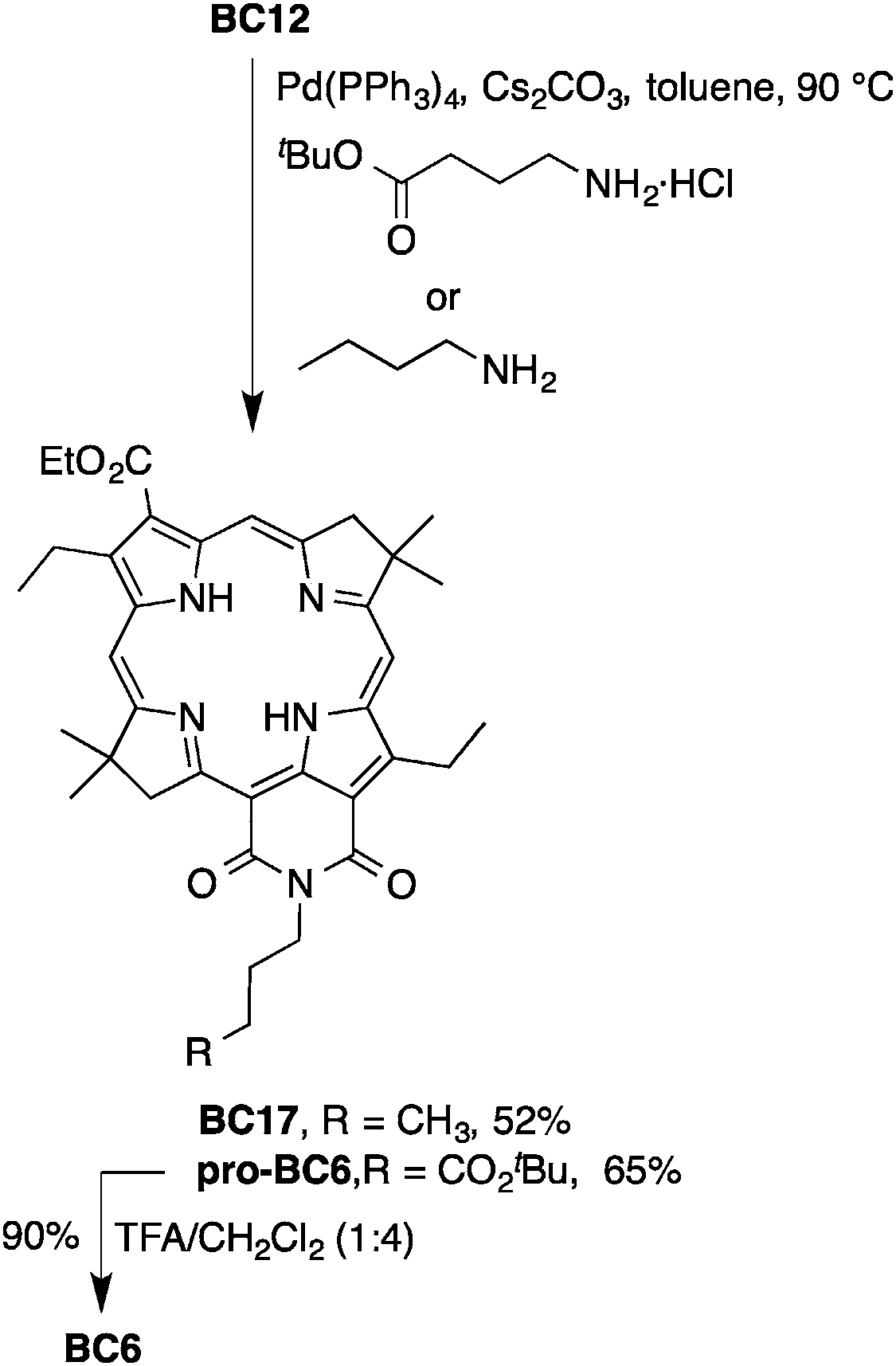 | ||
| Scheme 5 Synthesis of monocarboxy-bacteriochlorin BC6. | ||
Synthesis of hydrophilic bacteriochlorins
The generic route for the synthesis of the four tetracarboxy-bacteriochlorin–NHS esters (BC7–BC10, Chart 3) is as follows: (1) self-condensation of a bromodihydrodipyrrin–acetal to form the 3,13-dibromo-5-methoxybacteriochlorin macrocycle;40,42 (2) Pd-mediated Suzuki coupling to install the 3,13-bis(3,5-di-tert-butoxycarbonylphenyl) groups;52 (3) regioselective bromination at the 15-position;41 (4) Pd-mediated Suzuki or Sonogashira coupling to install the unprotected carboxylic acid tether at the 15-position;41 (5) reaction with N-hydroxysuccinimide (HOSu) in the presence of N,N-dicyclohexylcarbodiimide (DCC) or 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) to afford the penultimate, fully protected bacteriochlorin target bearing four tert-butyl esters and one NHS ester; and (6) selective cleavage of the tert-butyl groups to give the tetracarboxy-bacteriochlorin bearing a single NHS ester.The synthesis of the bacteriochlorin–NHS esters BC7 and BC8 is shown in Scheme 6. BC18 and BC19 were reported in our previous paper,52 and are presented here for comparison. The Suzuki coupling of bacteriochlorin BC18 with p-anilinoboronic ester 4 afforded BC20 in 71% yield. Treatment of BC19 or BC20 with succinic anhydride in CHCl3 afforded the intermediate 15-carboxybacteriochlorin, which was partially purified by column chromatography. Esterification of each crude bacteriochlorin with HOSu in the presence of DCC gave pro-BC7 or pro-BC8 in 52% or 53% yield (for two steps), respectively. Treatment of pro-BC7 or pro-BC8 with 20% TFA in CH2Cl2 unveiled the four carboxylic acid groups in 76% or 71% yield, respectively, while keeping the bacteriochlorin chromophore and NHS ester intact.
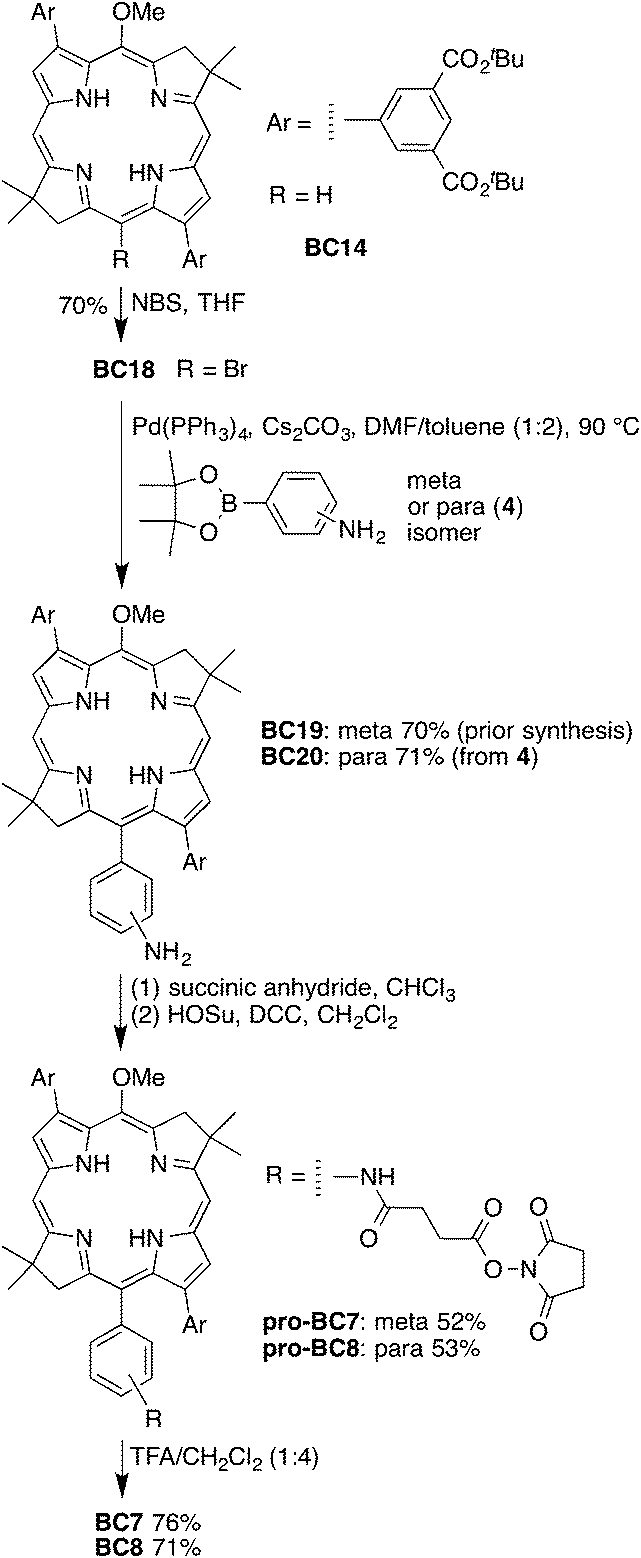 | ||
| Scheme 6 Synthesis of tetracarboxy-bacteriochlorin–NHS esters BC7 and BC8. | ||
Treatment of bacteriochlorin BC2152 with N-bromosuccinimide (NBS) in THF afforded the 15-brominated product BC22 in 42% yield. The presence of the 3,13-aryldiester substituents on the bacteriochlorin ring caused a slightly adverse effect given that the yield was lower than that of bacteriochlorin BC18 (70%).52 The copper-free Sonogashira reaction60 of BC22 and 6-heptynoic acid (5) was carried out in toluene/triethylamine (TEA) containing Pd2(dba)3 and P(o-tol)3 at 70 °C (Scheme 7). The resulting monocarboxy–bacteriochlorin was esterified with HOSu–DCC to afford the bacteriochlorin–NHS ester pro-BC9 in 19% yield for two steps. The low yield could be attributed to two factors: (1) deprotonation of the free carboxylic acid of 5 under the basic reaction conditions, which would result in low solubility; and (2) purification of bacteriochlorin–NHS ester pro-BC9 by preparative TLC (instead of column chromatography), from which recovery was poor. Finally, cleavage of the tert-butyl ester with 20% TFA in CH2Cl2 give the final bacteriochlorin BC9 in 82% yield.
 | ||
| Scheme 7 Synthesis of tetracarboxy-bacteriochlorin–NHS ester BC9. | ||
Pd-mediated carbonylation of 15-bromobacteriochlorin BC22 with 3-aminopropanoic acid in toluene afforded the bacteriochlorin–imide, which was purified by column chromatography and used directly in the next step. Treatment with HOSu–EDC and 4-dimethylaminopyridine (DMAP) gave the bacteriochlorin–NHS ester pro-BC10 in 22% yield for two steps (Scheme 8). Cleavage of the protecting group with TFA gave the free tetracarboxy-bacteriochlorin BC10 in 94% yield.
 | ||
| Scheme 8 Synthesis of the tetracarboxy-bacteriochlorin–imide bearing an NHS ester. | ||
The bacteriochlorins BC1–BC10 and precursors typically were characterized by absorption and fluorescence spectroscopy, 1H NMR spectroscopy, 13C NMR spectroscopy (where quantity and solubility allowed), MALDI mass spectrometry, and ESI mass spectrometry.
Photophysical properties
The parameters of interest include (1) the position of the Qy absorption band, (2) the position of the companion fluorescence emission band, and (3) the sharpness of each band as measured by the full-width-at-half-maximum (fwhm). The absorption and emission spectra of the lipophilic bacteriochlorins were collected in N,N-dimethylformamide (DMF) (Fig. 2). All of these parameters, including the Φf values, are listed in Table 1. Each bacteriochlorin gave characteristic absorption and fluorescence spectra,61 indicating the absence of any adverse effect due to the presence of the bioconjugatable tether. The Qy band of members of the set of six carboxy-bacteriochlorins is in the NIR region, ranging from 730–820 nm. As expected, an increase in the number of electron-withdrawing groups (e.g., ester or imide moieties) along the y-axis of the bacteriochlorin caused a bathochromic shift in the absorption and emission spectra.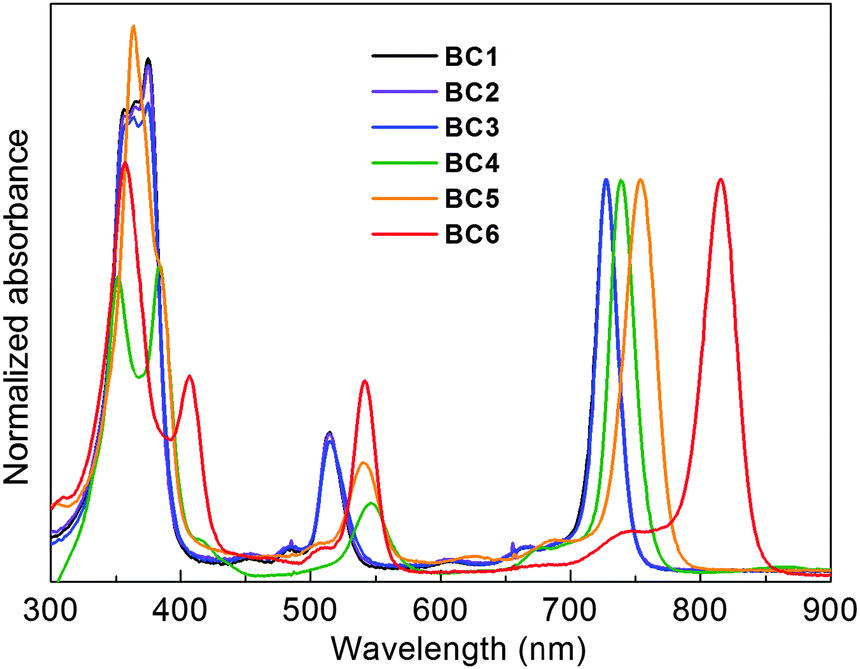 | ||
| Fig. 2 Normalized absorption spectra in DMF at room temperature. Spectral parameters are given in Table 1. | ||
| Compounds | λ abs/nm | fwhm nm (Abs) | λ em/nm | fwhm nm (Flu) | Φ f |
|---|---|---|---|---|---|
| a All spectra were recorded in DMF at room temperature. | |||||
| BC1 | 727 | 21 | 733 | 24 | 0.18 |
| BC2 | 728 | 20 | 734 | 24 | 0.18 |
| BC3 | 727 | 19 | 733 | 24 | 0.19 |
| BC4 | 737 | 24 | 745 | 39 | 0.14 |
| BC5 | 754 | 28 | 764 | 27 | 0.18 |
| BC6 | 816 | 30 | 822 | 27 | 0.037 |
The absorption and emission spectra of the hydrophilic bacteriochlorins were collected in DMF and in aqueous potassium phosphate buffer (Fig. 3). The spectroscopic parameters, along with the fluorescence quantum yield (Φf) values, are listed in Table 2. The data for the parent bacteriochlorins BC23 and BC24 (Chart 5) also are included for comparison.
 | ||
| Fig. 3 Normalized absorption and fluorescent spectra in potassium phosphate buffer (0.5 M, pH 7, for BC7–BC9) and DMF (for BC10) at room temperature. | ||
| Compounds | solvent | λ abs/nm | fwhm nm (Abs) | λ em/nm | fwhm nm (Flu) | Φ f |
|---|---|---|---|---|---|---|
| a Each sample contains 1% DMF to facilitate initial dissolution. The buffer is potassium phosphate (0.5 M, pH 7.0). b Data reported in ref. 52. c Low signal-to-noise ratio precluded the determination of the fwhm value. | ||||||
| BC23 | DMF | 729 | 22 | 735 | 23 | 0.19 |
| BC23 | Buffer | 730 | 26 | 736 | 26 | 0.078 |
| BC24 | DMF | 746 | 31 | 753 | 23 | 0.16 |
| BC24 | Buffer | 749 | 35 | 758 | 37 | 0.11 |
| BC7 | DMF | 727 | 23 | 732 | 24 | 0.17 |
| BC7 | Buffer | 729 | 24 | 735 | 26 | 0.12 |
| BC8 | DMF | 727 | 21 | 733 | 24 | 0.19 |
| BC8 | Buffer | 729 | 23 | 736 | 26 | 0.13 |
| BC9 | DMF | 754 | 26 | 760 | 24 | 0.133 |
| BC9 | Buffer | 757 | 27 | 765 | 27 | 0.13 |
| BC10 | DMF | 808 | 35 | 818 | 33 | 0.037 |
| BC10 | Buffer | 823 | 50 | 829 | N/Ac | 0.0011 |
 | ||
| Chart 5 Parent bacteriochlorins lacking bioconjugatable tethers. | ||
Each bacteriochlorin exhibited absorption and fluorescence in DMF characteristic of the bacteriochlorin chromophore: a strong B band (UV region), modest Qx band (green-yellow region), and intense Qy band (NIR region). BC7–BC9 gave similar spectra in aqueous phosphate buffer, whereas that of BC10 was significantly broadened characteristic of aggregation. Other than this lone exception, all bacteriochlorins displayed sharp absorption and emission bands with fwhm 22–35 nm. As with the lipophilic bacteriochlorins, introduction of the bioconjugatable tether in BC7 and BC8 caused little absorption or emission shift (by comparison with the parent compound BC23), while the ethynyl group in BC9 and the 13,15-imide moiety in BC10 gave the expected bathochromic shift (in comparison with BC24). The Φf values ranged from 0.037–0.19, with exception for (aggregated) BC10 in buffer, which gave 0.0011.
Bioconjugation study
Cellarius and Mauzerall employed polystyrene nanoparticles bearing surface-adsorbed pheophytins as prototypical photoreactors.58 Their ingenious strategy “combines the structural features of an interface with the simplicity of studying photochemistry in solution” and in particular enabled studies of pigment loading, pigment–pigment interactions, excited-state energy transfer, and perhaps exciton trapping.58 From the vantage of 50 years hence, the work was unavoidably limited by the polydispersity of the particles (24–260 nm diameter) and by the adsorption rather than covalent attachment of the tetrapyrrole chromophore. Accordingly, we felt that a modern analogue of the pheophytin-on-polystyrene particles could be constructed of bioconjugatable bacteriochlorins covalently attached to a globular protein, with the latter providing a ‘particle’ with known surface derivatization sites and uniform nanoscale size (∼5 nm diameter).We examined bioconjugation of selected hydrophilic bacteriochlorins with the protein Mb. The specific goals of this investigation include (1) quantitative analysis of the bacteriochlorin/Mb ratios, and (2) comparison of the spectral properties (absorption, fluorescence, Φf) of the bacteriochlorins bound to Mb with those for the bacteriochlorins free in solution. In addition to the more exacting analogue of the pheophytin-on-particles system of Cellarius and Mauzerall,58 we felt the Mb-conjugates could provide a testbed that is more simple and controlled than those in typical fluorophore–protein conjugation studies. The latter range from the widespread conjugation of fluorophores to antibodies62–66 to our own use of biomimetic light-harvesting peptides.51,53 For these experiments we chiefly examined bacteriochlorin BC7 but also looked briefly at BC8.
Mb was selected for the bioconjugation for the following reasons: (1) Mb is a water-soluble globular protein (diameter ∼50 Å) containing 19 lysine residues,67 of which six are involved in stabilizing electrostatic interactions (Lys16–Asp122, Lys47–Asp44, Lys56–Glu52, Lys77–Glu18, Lys79–Glu4 and Lys133–Glu6).68,69 The remaining 14 primary amines (13 Lys residues and 1 N-terminus amine) are considered accessible for the amine–NHS ester ligation. (2) The heme chromophore absorbs strongly at 408 nm (ε = 188![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1).70 The heme absorption is a better reference peak for calculation of intensely absorbing chromophore/protein ratios than the frequently used, weaker, broad (often non-descript) protein absorption at 280 nm (for apomyoglobin (apoMb), ε280nm = 15900 M−1 cm−1),71 a wavelength where solvent, impurities, and even the chromophore typically also absorb. A diarylbacteriochlorin40 (e.g., BC7–BC10), for example, exhibits ε280nm = 52900 M−1 cm−1, which dwarfs that of Mb even for a 1:1 loading. (3) The heme ligand can be removed from the protein binding pocket as needed by organic extraction. (4) Mb can be purchased at low price in large quantity (hundreds of mgs) and with high purity (95–100%). We chose Mb from equine skeletal muscle for bioconjugation studies, although Mbs from different organisms have similar primary, secondary (helicity, 8 helical segments) and tertiary structures (see ESI,† Fig. S1 and S2).
000 M−1 cm−1).70 The heme absorption is a better reference peak for calculation of intensely absorbing chromophore/protein ratios than the frequently used, weaker, broad (often non-descript) protein absorption at 280 nm (for apomyoglobin (apoMb), ε280nm = 15900 M−1 cm−1),71 a wavelength where solvent, impurities, and even the chromophore typically also absorb. A diarylbacteriochlorin40 (e.g., BC7–BC10), for example, exhibits ε280nm = 52900 M−1 cm−1, which dwarfs that of Mb even for a 1:1 loading. (3) The heme ligand can be removed from the protein binding pocket as needed by organic extraction. (4) Mb can be purchased at low price in large quantity (hundreds of mgs) and with high purity (95–100%). We chose Mb from equine skeletal muscle for bioconjugation studies, although Mbs from different organisms have similar primary, secondary (helicity, 8 helical segments) and tertiary structures (see ESI,† Fig. S1 and S2).
The rationale for focus on Mb versus the more prevalent use of antibodies for fluorophore conjugation warrants emphasis: Mb is more compact (∼17 kDa versus ∼150 kDa); Mb is abundantly available as a pure compound; Mb and conjugates thereof readily afford MALDI-MS data; and the presence of the heme provides a convenient (removable) absorption spectrometric internal calibrant. The attachment of fluorophores to antibodies is an essential step for use in flow cytometry or cellular staining,62,64,65 for example, yet for fundamental spectroscopic and photochemical studies, a small globular protein such as Mb (or apoMb) affords distinct advantages, as described below.
In one study, the bioconjugation of BC7 was carried out at room temperature with 2, 10, or 50 equiv. of the bacteriochlorin–NHS ester versus Mb. The conjugation was performed in aqueous solution containing 10% DMSO. Purification by gel permeation chromatography (GPC) with potassium phosphate buffer (0.5 M, pH 7.0) caused elution of the conjugate as a clear dark green band, while the free bacteriochlorin (unreacted or hydrolyzed bacteriochlorin–NHS ester) remained on top of the column. The resultant conjugate solution was subjected to centrifugal filtration, and the absence of the bacteriochlorin absorption of the filtrates indicated the thorough removal of the free bacteriochlorin.
The absorption spectrum in potassium phosphate buffer of the Mb–bacteriochlorin conjugate Mb–BC7 closely resembled the sum of the component parts in each case (2, 10 or 50 equiv.) although a small amount of tailing (to long wavelength) of the bacteriochlorin Qy band was observed. The spectrum of the conjugate prepared with 50 equiv. is shown in Fig. 4 (panel A) along with that of Mb and BC7 (the spectra for 2 and 10 equiv. are shown in the ESI,† Fig. S3 and S4).
 | ||
| Fig. 4 (A). Absorption spectra of Mb, BC7 and conjugate Mb–BC7 in potassium phosphate buffer (0.5 M, pH 7.0). The concentration of each component is chosen arbitrarily. (B). The normalized experimental (blue), reconstructed (cyan) absorption, and emission (magenta, dashed) spectra of conjugate Mb–BC7. Spectral parameters are given in Table 3. | ||
Multicomponent analysis (using the known absorption spectrum of Mb and of BC7) in each case was carried out using PhotochemCAD72 to assess the bacteriochlorin/Mb ratio. The characteristic absorption peaks of Mb (408 nm) and bacteriochlorins (362, 516, 729 nm) were selected for the calculation. Reconstruction of the absorption of the conjugate versus the experimental absorption visually shows the accuracy of the absorption deconvolution for calculation of the bacteriochlorin/Mb ratio (Fig. 4, panel B). The results are listed in Table 3. The use of 2, 10, and 50 equiv. of BC7 resulted in 0.62, 1.6, and 7.1 bacteriochlorins per Mb.
| Compound | Degree of loadingb | λ abs/nm | fwhm nm (Abs) | λ em/nm | fwhm nm (Flu) | Φ f |
|---|---|---|---|---|---|---|
| a All data determined in potassium phosphate buffer (0.5 M, pH 7.0) at room temperature. b The ratio of BC7 to Mb, determined by multicomponent absorption spectral analysis. | ||||||
| Mb–BC7 (2 equiv.) | 0.62 | 729 | 27 | 735 | 26 | 0.019 |
| apoMb–BC7 (2 equiv.) | 726 | 23 | 730 | 23 | 0.091 | |
| Mb–BC7 (10 equiv.) | 1.6 | 728 | 26 | 734 | 27 | 0.020 |
| apoMb–BC7 (10 equiv.) | 726 | 24 | 732 | 24 | 0.071 | |
| Mb–BC7 (50 equiv.) | 7.1 | 726 | 27 | 735 | 27 | 0.018 |
| apoMb–BC7 (50 equiv.) | 721 | 30 | 731 | 27 | 0.023 | |
The same three conjugates were examined by MALDI-MS using α-cyano-4-hydroxycinnamic acid (CHCA) as matrix. The data are shown in Fig. 5. The increase in loading with number of equivalents of BC7 was clearly seen, with a distribution of peaks separated by Δm = 920 Da, which corresponds to BC7 minus the NHS moiety. The distribution shifts to higher mass with increasing number of equivalents. For the conjugate prepared with 50 equiv. of BC7, which gave an average loading of 7.1 (by absorption spectroscopy), individual peaks in the progression of 0–9 were clearly observed. Since ionization efficiencies may vary with different amounts of chromophores attached, the MALDI-MS results, while insightful, are not reliable for calculations of bacteriochlorin/Mb ratios. The minimum conclusion is that the distribution is narrow for 10 equiv. (1.6 loading) yet quite broad for 50 equiv. (7.1 loading). In a separate experiment, BC7 and BC8 were conjugated at 60 equiv. relative to Mb, affording conjugates that also were quite soluble in aqueous solution. In both cases, the resulting loading was 9 and 12, respectively. The shift of the peaks in the distribution to higher mass was readily observed upon MALDI-MS analysis (see ESI,† Fig. S5).
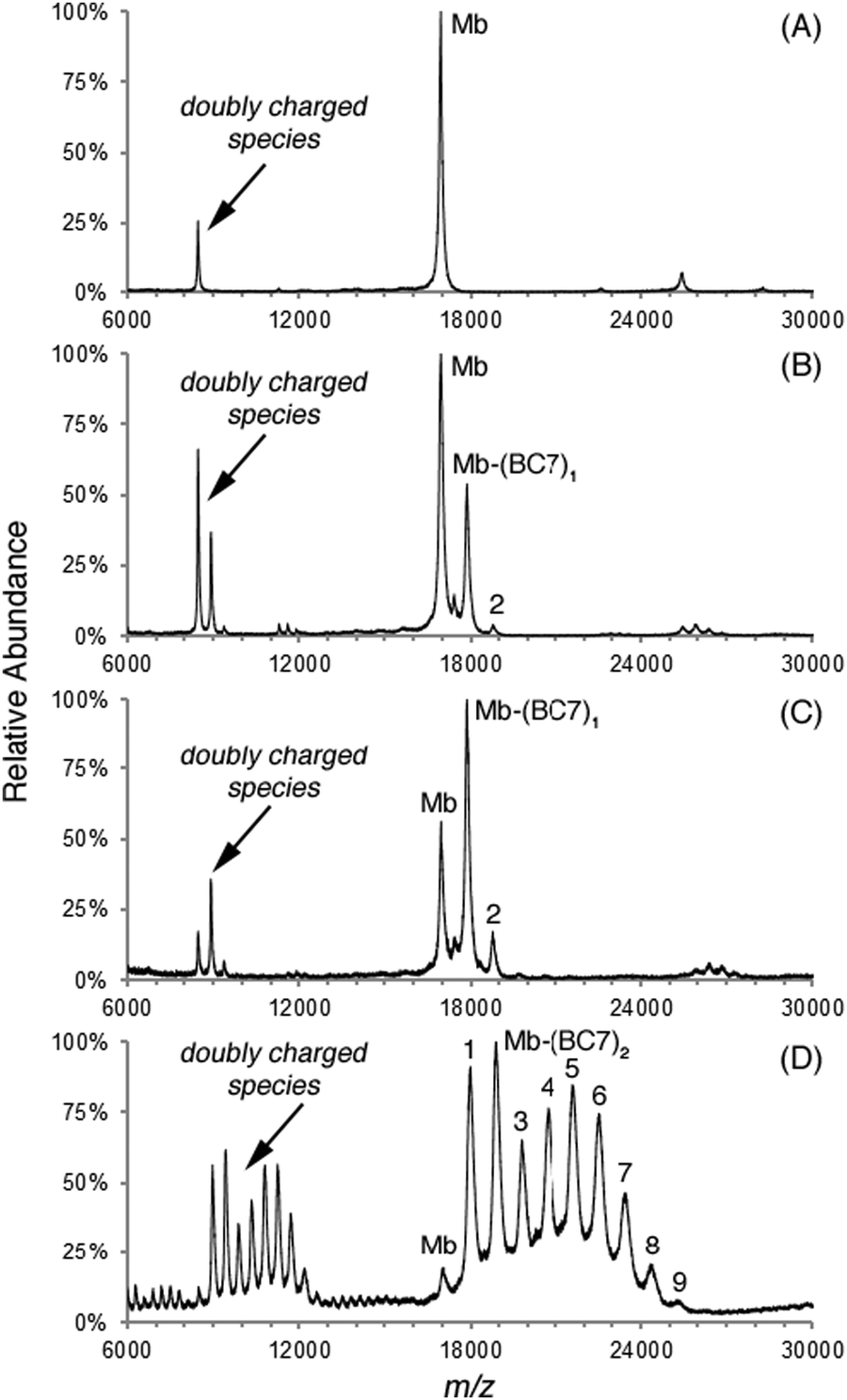 | ||
| Fig. 5 The MALDI spectra of the conjugate samples of Mb to (A) 0, (B) 2, (C) 10, and (D) 50 equiv. of BC7. Peaks that match the mass of labeled Mb were marked by Mb–(BC7)x, where x indicates the number of the bacteriochlorins attached. | ||
The fluorescence properties of the Mb–BC7 conjugates were examined. The spectrum for the conjugate derived from 50 equiv. of BC7 is shown in Fig. 4 (panel B). The Φf value upon attachment to the protein was decreased to ∼0.02, to be compared with the value of 0.12 for BC7 in aqueous solution. The Φf value was essentially indifferent to the level of loading. To distinguish possible effects of heme as a quencher, the heme was removed by extraction with 2-butanone,73 to afford the corresponding apoMb conjugates. In each case, the resulting apoMb–BC7 conjugate (derived from 2, 10 or 50 equiv. of BC7) gave a characteristic bacteriochlorin absorption spectrum (see ESI,† Fig. S6–S8). Indeed, no trace of tailing of the long-wavelength, Qy absorption band was observed. Unlike for Mb–BC7, however, the Φf values now were a function of loading (i.e., BC7/Mb ratio). The results are illustrated in Fig. 6. The Φf value for the lowest-loading conjugate (2 equiv. of BC7, average 0.62 bacteriochlorins/Mb) was 0.091, only decreased by 25% from that of the parent BC7 monomer. On the other hand, the decline with loading (to 0.023 for 50 equiv., average 7.1 bacteriochlorins/Mb) is attributed to self-quenching of the bacteriochlorins on the protein. Thus, a distinction between quenching due to the presence of heme versus quenching due to bacteriochlorin self-interactions is clearly obtained.
 | ||
| Fig. 6 Fluorescence quantum yield values as a function of loading and ±heme. | ||
The origin of self-quenching is unclear. The absorption spectra and the emission spectra of the apoMb–BC7 conjugates were essentially identical to those of the monomeric BC7. Calculations of the Förster through-space energy transfer (using PhotochemCad72) showed that the self-exchange process for bacteriochlorin–bacteriochlorin energy transfer exhibits R0 = 59 Å (the distance at which energy transfer is 50% efficient). Given that the diameter of Mb is ∼50 Å from most distant points, a considerable degree of energy transfer between bacteriochlorins attached to Mb is expected to be permissible. Hence, any excited-state trapping site(s) at/near the protein are likely to be encountered upon successive transfer steps.
Outlook
The ability to synthesize hydrophilic/hydrophobic bacteriochlorins that exhibit some degree of wavelength tunability and bear a single carboxylic acid (or NHS-ester) opens the door to a wide variety of studies, particularly upon bioconjugation to proteins. The lipophilic bacteriochlorins can be used for attachment to the hydrophobic region of membrane-spanning light-harvesting peptides to give rise to self-assembled artificial photosynthetic architectures. The attachment of a hydrophilic bacteriochlorin to Mb affords a water-soluble NIR-active protein architecture that is likely a more exacting and versatile analogue of the pheophytin-coated polystyrene particles first reported by Cellarius and Mauzerall nearly 50 years ago. The bioconjugation of bacteriochlorins to Mb described herein also affords several attractive attributes as a testbed for attachment of chromophores to proteins: (1) the presence of heme provides a convenient absorption calibration standard for determining average loading of intensely absorbing chromophores, which often is difficult when relying solely on the weakly absorbing (and typically overlapped) 280 nm band of proteins; (2) mass spectrometry of the Mb–chromophore conjugate provides a more granular view of the loading distribution; and (3) heme can be readily removed following loading determination to assess spectral properties in the resulting apoMb, including absorption and fluorescence spectra as well as Φf values.Experimental section
Protocols
The crude bacteriochlorin–Mb conjugate Mb–BC7 or Mb–BC8 was purified by passage (gravity-elution) over a PD-10 GPC column (Sephadex G-25 medium, bed dimension: 14.5 × 50 mm) with potassium phosphate buffer (0.5 M, pH 7.0) as eluent. The conjugate eluted as a clear dark green band, while free bacteriochlorin (unreacted or hydrolyzed bacteriochlorin–NHS ester) remained on top of the column. The resultant conjugate solution was subjected to centrifugal Amicon® Ultra-4 filtration (regenerated cellulose, molecular weight cutoff = 10 K) for 30 min. The resulting filtrate lacked bacteriochlorin absorption, consistent with the removal of any unconjugated bacteriochlorin. The solution that did not pass through the filter constituted the purified bacteriochlorin–Mb conjugate. The purification protocol is expected to remove all DMSO used in the bioconjugation reaction. MALDI-MS for Mb–BC7: m/z = 198812, 20695, 21509, 22484 (most intense), 23401, and 24319. MALDI-MS for Mb–BC8: m/z = 21589, 22509, 23432 (most intense), 24354 and 25330. Further data are provided in the ESI† (Fig. S9 and S10).
The following protocol describes the use of 2, 10, or 50 equiv. of bacteriochlorin–Mb. A sample of Mb (0.52 mg, 30 nmol) was dissolved in potassium phosphate buffer (0.1 M, pH 8.3, 0.15 mL). In a separate vial, bacteriochlorin BC7 (60 μg, 60 nmol, 2 equiv. or 0.30 mg, 0.30 μmol, 10 equiv., or 1.5 mg, 1.5 μmol, 50 equiv.) was initially dissolved in DMSO (30 μL) and then 120 μL of the same phosphate buffer was added with stirring to make a homogeneous bacteriochlorin solution. The resulting bacteriochlorin solution was then pipetted into the Mb solution, and incubated in the dark for 3 h at room temperature (∼23 °C). The final concentration of Mb was 0.1 mM, and DMSO accounts for 10% by volume. The remainder of the protocol is identical for that above with 60 equiv. of bacteriochlorin–Mb. The characterization data are provided in the body of the paper.
000 M−1 cm−1 for BC7 at 728 nm; Φf = 0.12 for BC7. The calculated R0 was 59 Å for BC7–BC7.
Syntheses
:1)] to afford a viscous colorless liquid (0.20 g, 60%): 1H NMR (300 MHz, CDCl3) δ 1.33 (s, 12H), 1.41 (s, 9H), 2.53 (t, J = 7.5 Hz, 2H), 2.92 (t, J = 7.5 Hz, 2H), 7.21 (d, J = 7.8 Hz, 2H), 7.73 (d, J = 7.8 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 25.1, 28.3, 31.5, 37.1, 80.6, 83.9, 100.3, 128.0, 135.2, 144.4, 172.3; ESI-MS obsd 354.2083, calcd 354.2087 [(M + Na)+, M = C19H29BO4].
:2), degassed by bubbling with argon for 30 min] was added to the Schlenk flask, and the reaction mixture was stirred at 90 °C for 18 h. The reaction mixture was cooled to room temperature, concentrated to dryness, and diluted with CH2Cl2. The resulting solution was washed with aqueous NaHCO3. The organic layer was separated, dried (Na2SO4), concentrated and chromatographed [silica, CH2Cl2/ethyl acetate (49:1)] to obtain a greenish solid (37.6 mg, 93%): 1H NMR (400 MHz, CDCl3) δ −1.81 (brs, 1H), −1.51 (brs, 1H), 1.29 (t, J = 7.5 Hz, 3H), 1.55 (s, 9H), 1.63–1.70 (m, 6H), 1.77 (t, J = 7.5 Hz, 3H), 1.83 (s, 6H), 1.95 (s, 6H), 2.77 (t, J = 7.6 Hz, 2H), 3.16 (t, J = 7.6 Hz, 2H), 3.79 (q, J = 7.5 Hz, 2H), 3.83–3.89 (m, 6H), 4.27 (s, 3H), 4.38 (s, 2H), 4.80 (q, J = 7.5 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 8.57 (s, 1H), 8.61 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 14.4, 14.9, 17.8, 20.2, 20.3, 28.5, 31.2, 31.3, 31.4, 37.4, 45.2, 46.3, 47.5, 52.3, 61.4, 62.0, 64.4, 80.8, 94.5, 94.7, 112.9, 123.2, 126.2, 127.7, 127.8, 132.4, 132.6, 133.4, 135.0, 138.6, 139.7, 140.2, 154.7, 161.0, 168.1, 168.5, 169.2, 172.7; MALDI-MS obsd 804.6556; ESI-MS obsd 805.4520, calcd 805.4535 [(M + H)+, M = C48H60N4O7]; λabs (CH2Cl2) 356, 365, 376, 515, 729 nm.
:2), degassed by bubbling with argon for 30 min] was added to the Schlenk flask, and the reaction mixture was stirred at 90 °C for 18 h. The reaction mixture was cooled to room temperature, concentrated to dryness, diluted with CH2Cl2 and washed with aqueous NaHCO3. The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2/ethyl acetate (49:1)] provided a greenish solid (29 mg, 73%): 1H NMR (400 MHz, CDCl3) δ −1.83 (brs, 1H), −1.53 (brs, 1H), 1.30 (t, J = 7.5 Hz, 3H), 1.59 (s, 9H), 1.62–1.69 (m, 6H), 1.76 (t, J = 7.5 Hz, 3H), 1.82 (s, 6H), 1.94 (s, 6H), 3.76 (q, J = 7.5 Hz, 2H), 3.82–3.89 (m, 4H), 3.95 (q, J = 7.5 Hz, 2H), 4.26 (s, 3H), 4.36 (s, 2H), 4.72 (s, 2), 4.78 (q, J = 7.2 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 8.4 Hz, 2H), 8.56 (s, 1H), 8.60 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 14.5, 14.9, 17.9, 20.2, 20.3, 28.4, 31.2, 31.4, 45.2, 46.3, 47.6, 52.3, 61.6, 62.0, 64.4, 66.1, 82.8, 94.5, 94.7, 112.4, 113.9, 123.2, 126.4, 127.7, 132.4, 132.5, 132.8, 134.4, 135.0, 135.1, 135.2, 138.5, 154.7, 157.7, 161.3, 168.2, 168.4, 168.6, 169.3; MALDI-MS obsd 806.6729; ESI-MS obsd 807.4323, calcd 807.4327 [(M + H)+, M = C47H58N4O8]; λabs (CH2Cl2) 356, 364, 376, 515, 729 nm.
:1) to CH2Cl2/methanol (4:1)] to yield a greenish solid (11.2 mg, 71%): 1H NMR (400 MHz, THF-d8, the CO2H proton was not observed) δ −0.80 (s, 1H), −0.47 (s, 1H), 0.97 (t, J = 7.2 Hz, 3H), 1.09 (t, J = 7.2 Hz, 3H), 1.77 (s, 3H), 1.81 (s, 3H), 1.83 (s, 3H), 1.87 (s, 3H), 1.93 (s, 6H), 2.07 (s, 3H), 2.09 (s, 3H), 2.24 (s, 3H), 2.47 (s, 3H), 2.59–2.67 (m, 4H), 3.63 (s, 3H), 3.75 (d, J = 2.4 Hz, 2H), 4.20–4.27 (m, 4H), 4.31 (t, J = 7.2 Hz, 2H), 6.45 (s, 1H), 6.72 (s, 1H), 6.97–7.04 (m, 2H), 7.10 (s, 2H), 7.54 (d, J = 8.0 Hz, 1H), 7.65 (s, 1H), 8.94 (s, 1H), 9.68 (s, 1H), 9.72 (s, 1H); 13C NMR (100 MHz, THF-d8) δ 16.9, 17.0, 24.2, 24.4, 24.7, 32.3, 34.0, 34.1, 34.2, 35.2, 48.9, 49.6, 50.7, 55.7, 63.59, 63.65, 65.8, 119.0, 121.1, 124.4, 124.8, 128.2, 129.1, 130.8, 131.09, 131.16, 131.20, 131.7, 136.1, 137.5, 138.1, 138.6, 138.8, 139.7, 139.9, 140.4, 140.5, 140.6, 141.2, 141.9, 144.1, 160.1, 166.3, 169.08, 169.18, 172.9, 173.8, 174.7, 177.2; MALDI-MS obsd 971.0664; ESI-MS obsd 972.4897, calcd 972.4906 [(M + H)+, M = C59H65N5O8]; λabs (CH2Cl2) 364, 543, 756 nm.
:3)]. The resulting solid was extracted with hexanes, sonicated in a benchtop sonication bath and centrifuged. The supernatant was discarded, leaving a reddish solid (13.0 mg, 65%): 1H NMR (400 MHz, CDCl3) δ −0.72 (s, 1H), −0.51 (s, 1H), 1.47 (s, 9H), 1.68–1.78 (m, 9H), 1.92 (s, 6H), 1.93 (s, 6H), 2.25–2.32 (m, 2H), 2.54 (t, J = 8.4 Hz, 2H), 4.08 (q, J = 7.2 Hz, 2H), 4.21 (q, J = 7.2 Hz, 2H), 4.33 (s, 2H), 4.50 (t, J = 7.2 Hz, 2H), 4.73 (s, 2H), 4.77 (q, J = 7.2 Hz, 2H), 8.57 (s, 1H), 8.70 (s, 1H), 9.55 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.8, 17.4, 17.6, 20.1, 20.9, 24.7, 28.4, 30.0, 31.2, 31.6, 33.8, 39.6, 45.7, 46.1, 52.1, 53.3, 61.5, 80.4, 94.7, 99.2, 99.5, 102.0, 115.0, 122.1, 133.4, 134.6, 136.2, 136.9, 140.3, 144.4, 162.8, 163.3, 165.9, 168.3, 168.5, 170.4, 172.9, 176.2; MALDI-MS obsd 709.4791; ESI-MS obsd 710.3921, calcd 710.3912 [(M + H)+, M = C41H51N5O6]; λabs (CH2Cl2) 358, 408, 544, 819 nm.
:2)] to afford a greenish solid, which was used directly in the next step. The greenish solid was dissolved in CH2Cl2 (1.23 mL) followed by addition of DCC (38.1 mg, 0.185 mmol). The mixture was stirred under argon for 3 min. Then HOSu (21.3 mg, 0.185 mmol) was added. The resulting mixture was stirred for 40 min and then filtered to remove insoluble material. The filtrate was concentrated and chromatographed [silica, CH2Cl2/ethyl acetate (9:1)] to yield a greenish solid (9.0 mg, 40%): 1H NMR (300 MHz, CDCl3) δ −1.59 (s, 1H), −1.21 (s, 1H), 1.64 (s, 18H), 1.69 (s, 18H), 1.90 (s, 6H), 2.00 (s, 6H), 2.74–2.81 (m, 6H), 3.06 (t, J = 7.5 Hz, 2H), 3.70 (s, 3H), 3.91–4.18 (m, 2H), 4.38 (s, 2H), 7.14 (t, J = 7.8 Hz, 1H), 7.38 (d, J = 5.4 Hz, 1H), 7.41–7.43 (m, 3H), 7.58 (s, 1H), 7.96 (s, 1H), 8.18 (s, 1H), 8.38 (s, 1H), 8.66 (t, J = 9.9 Hz, 2H), 8.76 (s, 1H), 8.91 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 14.4, 22.9, 25.2, 25.78, 25.81, 26.9, 28.48, 28.54, 31.4, 34.2, 45.4, 46.0, 47.8, 49.4, 52.1, 63.6, 81.9, 97.3, 97.8, 113.2, 119.6, 123.4, 125.5, 126.6, 128.16, 128.27, 128.31, 129.2, 129.6, 132.0, 132.6, 133.71, 133.75, 134.3, 134.9, 135.2, 136.0, 136.3, 137.1, 138.6, 139.0, 141.6, 155.4, 157.0, 160.8, 165.8, 168.20, 168.37, 169.1, 169.2, 169.7; MALDI-MS obsd 1242.5563; ESI-MS obsd 1241.5811, calcd 1241.5811 [(M + H)+, M = C61H71BrN4O13]; λabs (CH2Cl2) 366, 517, 730 nm.
:1)]. The resulting solid was treated with hexanes/CH2Cl2 (9:1), sonicated in a benchtop sonication bath and centrifuged. The supernatant was discarded, leaving a green solid (6.5 mg, 53%): 1H NMR (300 MHz, CDCl3, the NH proton peaks were not observed) δ −1.58 (s, 1H), −1.20 (s, 1H), 1.63 (s, 18H), 1.69 (s, 18H), 1.84 (s, 6H), 1.98 (s, 6H), 2.85–2.90 (m, 6H), 3.15 (t, J = 6.6 Hz, 2H), 3.68 (s, 3H), 3.95 (s, 2H), 4.38 (s, 2H), 7.25–7.27 (m, 1H), 7.41–7.46 (m, 3H), 8.01 (d, J = 1.8 Hz, 2H), 8.40 (s, 1H), 8.66–8.69 (m, 4H), 8.76 (t, J = 1.8 Hz, 1H), 8.91 (d, J = 1.8 Hz, 2H); MALDI-MS 1240.4706; ESI-MS obsd 1241.5795, calcd 1241.5805 [(M + H)+, M = C71H80N6O14]; λabs (CH2Cl2) 366, 518, 730 nm.
:1), deaerated by bubbling with argon for 30 min] was added to the Schlenk flask under argon and deaerated by three freeze–pump–thaw cycles. The reaction mixture was stirred at 70 °C for 18 h. The reaction mixture was cooled to room temperature, concentrated to dryness, diluted with CH2Cl2 and washed (saturated aqueous NaHCO3 solution). The organic layer was separated, dried (Na2SO4) and concentrated. Column chromatography [silica, CH2Cl2/EtOAc (19:1) to CH2Cl2/CH3OH (19:1)] provided a reddish solid (8.0 mg, total yield is given below): MALDI-MS obsd 1192.7728; ESI-MS obsd 1193.5701, calcd 1193.5693 [(M + H)+, M = C68H80N4O15]; λabs (CH2Cl2) 382, 547, 756 nm. Half of the product (4.0 mg), DCC (6.9 mg, 34 μmol) and HOSu (3.9 mg, 34 μmol) were stirred in CH2Cl2 (0.34 mL) under argon at room temperature for 40 min. The resulting mixture was filtered to remove insoluble material. The filtrate was concentrated and separated by preparative TLC [silica, CH2Cl2/methanol (99:1)] to yield a reddish solid (2.8 mg, 19%): 1H NMR (300 MHz, CDCl3) δ −1.22 (s, 1H), −0.97 (s, 1H), 1.54–1.58 (m, 2H), 1.66 (s, 36H), 1.82 (s, 6H), 1.83 (s, 6H), 2.22–2.28 (m, 2H), 2.78–2.91 (m, 8H), 4.12 (s, 3H), 4.17 (s, 3H), 4.26 (s, 3H), 4.32 (s, 2H), 4.42 (s, 2H), 8.45 (s, 1H), 8.49 (s, 1H), 8.82 (s, 1H), 8.83 (s, 1H), 8.86–8.87 (m, 4H); MALDI-MS 1289.4308; ESI-MS obsd 1290.5865, calcd 1290.5857 [(M + H)+, M = C72H83N5O17]; λabs (CH2Cl2) 381, 547, 756 nm.
:1)]. The resulting solid was mixed with EDC (9.6 mg 50 μmol), DMAP (0.20 mg, 2.0 μmol) and HOSu (5.7 mg, 50 μmol) in CH2Cl2 (0.20 mL) and stirred under argon for 3 h. The reaction residue was chromatographed [silica, CH2Cl2/ethyl acetate (19:1 to 4:1)] to afford a reddish solid (2.7 mg, 22%): 1H NMR (300 MHz, CDCl3) δ −0.43 (s, 1H), 0.09 (s, 1H), 1.66 (s, 18H), 1.67 (s, 18H), 1.79 (s, 6H), 1.82 (s, 6H), 2.83 (s, 4H), 3.31 (t, J = 6.6 Hz, 2H), 4.18 (s, 3H), 4.25 (s, 2H), 4.27 (s, 3H), 4.70 (s, 2H), 4.83 (t, J = 6.6 Hz, 2H), 8.38 (s, 1H), 8.45 (s, 1H), 8.83 (d, J = 2.1 Hz, 2H), 8.84 (d, J = 2.1 Hz, 2H), 8.89 (t, J = 1.5 Hz, 2H); MALDI-MS obsd 1248.9579; ESI-MS obsd 1249.5371, calcd 1249.5340 [(M + H)+, M = C68H76N6O17]; λabs (CH2Cl2) 377, 564, 811 nm.
:1)] to afford a reddish solid (9.1 mg, 52%): 1H NMR (300 MHz, THF-d8) δ 1.25 (t, J = 7.2 Hz, 3H), 1.51–1.69 (m, 9H), 1.81 (s, 3H), 1.83 (s, 3H), 1.94 (s, 6H), 3.61–3.76 (m, 4H), 3.89–4.02 (m, 4H), 4.13 (s, 3H), 4.37 (s, 2H), 4.55 (s, 2H), 4.61 (t, J = 7.5 Hz, 2H), 6.71–6.74 (m, 1H), 6.92–6.94 (m, 2H), 7.17 (t, J = 7.8 Hz, 1H), 8.44 (s, 1H), 8.48 (s, 1H); MALDI-MS obsd 753.39; ESI-MS obsd 754.2917, calcd 754.2947 [(M + H)+, M = C41H47N5O5Zn]; λabs (CH2Cl2) 355, 385, 553, 738 nm.
:5 to 3:7)] to afford a reddish solid (11.9 mg, 52%): 1H NMR (400 MHz, CDCl3) δ −0.74 (s, 1H), −0.54 (s, 1H), 1.10 (t, J = 7.5 Hz, 3H), 1.62–1.79 (m, 13H), 1.92 (s, 6H), 1.93 (s, 6H), 4.08 (q, J = 7.2 Hz, 2H), 4.22 (q, J = 7.2 Hz, 2H), 4.33 (s, 2H), 4.44 (t, J = 7.5 Hz, 2H), 4.74 (s, 2H), 4.78 (q, J = 7.2 Hz, 2H), 8.58 (s, 1H), 8.71 (s, 1H), 9.56 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.3, 14.8, 17.4, 17.6, 20.1, 20.9, 21.0, 30.0, 31.2, 31.3, 31.6, 40.4, 45.7, 46.1, 52.1, 53.36, 53.45, 61.5, 94.7, 99.4, 99.5, 101.9, 115.2, 122.0, 133.5, 134.6, 136.2, 136.8, 140.3, 144.3, 162.6, 163.4, 165.9, 168.2, 168.5, 170.5, 176.0; MALDI-MS obsd 622.9292; ESI-MS obsd 624.3532, calcd 624.3544 [(M + H)+, M = C37H45N5O4]; λabs (CH2Cl2) 357, 408, 543, 818 nm.
:1), deaerated by bubbling with argon for 30 min] was added to the Schlenk flask under argon and deaerated by three freeze–pump–thaw cycles. The reaction mixture was stirred at 90 °C for 18 h. The reaction mixture was cooled to room temperature, concentrated to dryness, diluted with CH2Cl2 and washed with saturated aqueous NaHCO3. The organic layer was separated, dried (Na2SO4), concentrated and chromatographed [silica, CH2Cl2/ethyl acetate (23:2)] to provide a greenish solid (38.0 mg, 71%): 1H NMR (300 MHz, CDCl3) δ −1.52 (s, 1H), −1.18 (s, 1H), 1.65 (s, 18H), 1.69 (s, 18H), 1.85 (s, 6H), 1.98 (s, 6H), 3.60 (s, 2H), 3.68 (s, 3H), 4.00 (s, 2H), 4.38 (s, 2H), 6.41 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.1 Hz, 1H), 7.26 (s, 1H), 7.62 (d, J = 8.1 Hz, 1H), 8.48 (t, J = 1.5 Hz, 1H), 8.64–8.67 (m, 4H), 8.76 (t, J = 1.5 Hz, 1H), 8.91 (d, J = 1.5 Hz, 1H) (two anilino NH protons were not observed); 13C NMR (100 MHz, CDCl3) δ 25.1, 28.5, 31.27, 31.38, 45.1, 46.0, 47.7, 52.4, 63.5, 81.4, 81.8, 83.5, 97.31, 97.37, 114.1, 114.3, 122.9, 127.0, 127.5, 128.0, 129.1, 131.07, 131.12, 131.9, 132.1, 133.9, 134.1, 134.2, 134.8, 136.0, 136.2, 136.6, 138.7, 138.9, 145.5, 154.8, 161.9, 165.6, 165.8, 169.06, 169.22; MALDI-MS 1043.6068; ESI-MS obsd 1044.5475, calcd 1044.5481 [(M + H)+, M = C63H73N5O9]; λabs (CH2Cl2) 366, 520, 729 nm.
:1)] to afford a reddish solid (20 mg, 42%): 1H NMR (300 MHz, CDCl3) δ −1.52 (s, 1H), −1.25 (s, 1H), 1.66 (s, 36H), 1.83 (s, 6H), 1.86 (s, 6H), 4.16 (s, 3H), 4.20 (s, 3H), 4.28 (s, 3H), 4.37 (s, 2H), 4.44 (s, 2H), 8.50 (s, 2H), 8.83–8.85 (m, 2H), 8.87–8.88 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 14.4, 28.5, 29.9, 31.0, 31.3, 46.0, 47.9, 53.27, 53.39, 54.7, 64.6, 82.04, 82.10, 94.6, 96.7, 97.1, 98.2, 125.2, 126.0, 129.2, 130.0, 130.4, 131.5, 132.8, 133.1, 133.4, 133.7, 133.9, 134.4, 134.5, 136.3, 136.6, 158.2, 160.8, 165.08, 165.14, 168.6, 168.8, 169.2, 173.4; ESI-MS obsd 1147.4245, calcd 1147.4274 [(M + H)+, M = C61H71BrN4O13]; λabs (CH2Cl2) 375, 531, 740 nm.
Acknowledgements
Mr Nuonuo Zhang was supported by the China Scholarship Council (CSC, 201306250076) as a visiting PhD student from Tianjin University for joint research at North Carolina State University. This research was carried out as part of the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-SC0001035. Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation.References
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef PubMed.
- R. K. V. Lim and Q. Lin, Sci. China: Chem., 2010, 53, 61–70 CrossRef CAS PubMed.
- L. I. Willems, W. A. Van der Linden, N. Li, K.-Y. Li, N. Liu, S. Hoogendoorn, G. A. Van der Marel, B. I. Florea and H. S. Overkleeft, Acc. Chem. Res., 2011, 44, 718–729 CrossRef CAS PubMed.
- Y.-X. Chen, G. Triola and H. Waldmann, Acc. Chem. Res., 2011, 44, 762–773 CrossRef CAS PubMed.
- S. S. van Berkel, M. B. van Eldijk and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806–8827 CrossRef CAS PubMed.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- F. Giuntini, C. M. A. Alonso and R. W. Boyle, Photochem. Photobiol. Sci., 2011, 10, 759–791 CAS.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- A. S. Tatikolov, J. Photochem. Photobiol., C, 2012, 13, 55–90 CrossRef CAS PubMed.
- M. Ptaszek, Prog. Mol. Biol. Transl. Sci., 2013, 113, 59–108 CAS.
- K. Umezawa, D. Citterio and K. Suzuki, Anal. Sci., 2014, 30, 327–349 CrossRef CAS.
- J. Pichaandi and F. C. J. M. van Veggel, Coord. Chem. Rev., 2014, 263–264, 138–150 CrossRef CAS PubMed.
- H. Scheer, in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 1–26 Search PubMed.
- M. R. Prinsep, F. R. Caplan, R. E. Moore, G. M. L. Patterson and C. D. Smith, J. Am. Chem. Soc., 1992, 114, 385–387 CrossRef CAS.
- C. D. Smith, M. R. Prinsep, F. R. Caplan, R. E. Moore and G. M. L. Patterson, Oncol. Res., 1994, 6, 211–218 CAS.
- M. R. Prinsep, G. M. L. Patterson, L. K. Larsen and C. D. Smith, Tetrahedron, 1995, 51, 10523–10530 CrossRef CAS.
- M. R. Prinsep, G. M. L. Patterson, L. K. Larsen and C. D. Smith, J. Nat. Prod., 1998, 61, 1133–1136 CrossRef CAS PubMed.
- P. Morlière, J.-C. Mazière, R. Santus, C. D. Smith, M. R. Prinsep, C. C. Stobbe, M. C. Fenning, J. L. Golberg and J. D. Chapman, Cancer Res., 1998, 58, 3571–3578 Search PubMed.
- T. G. Minehan, L. Cook-Blumberg, Y. Kishi, M. R. Prinsep and R. E. Moore, Angew. Chem., Int. Ed., 1999, 38, 926–928 CrossRef CAS.
- M. R. Prinsep and J. Puddick, Phytochem. Anal., 2011, 22, 285–290 CrossRef CAS PubMed.
- Y. Chen, G. Li and R. K. Pandey, Curr. Org. Chem., 2004, 8, 1105–1134 CrossRef CAS.
- M. A. Grin, A. F. Mironov and A. A. Shtil, Anti-Cancer Agents Med. Chem., 2008, 8, 683–697 CrossRef CAS.
- J. M. Sutton, N. Fernandez and R. W. Boyle, J. Porphyrins Phthalocyanines, 2000, 4, 655–658 CrossRef CAS.
- J. M. Sutton, O. J. Clarke, N. Fernandez and R. W. Boyle, Bioconjugate Chem., 2002, 13, 249–263 CrossRef CAS PubMed.
- A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, J. Org. Chem., 2005, 70, 2306–2314 CrossRef CAS PubMed.
- J. R. McCarthy, J. Bhaumik, N. Merbouh and R. Weissleder, Org. Biomol. Chem., 2009, 7, 3430–3436 CAS.
- A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2009, 13, 408–414 CrossRef.
- S. Singh, A. Aggarwal, S. Thompson, J. P. C. Tomé, X. Zhu, D. Samaroo, M. Vinodu, R. Gao and C. M. Drain, Bioconjugate Chem., 2010, 21, 2136–2146 CrossRef CAS PubMed.
- J. M. Dabrowski, L. G. Arnaut, M. M. Pereira, C. J. P. Monteiro, K. Urbanska, S. Simoes and G. Stochel, ChemMedChem, 2010, 5, 1770–1780 CrossRef CAS PubMed.
- N. A. M. Pereira, S. M. Fonseca, A. C. Serra, T. M. V. D. Pinho e Melo and H. D. Burrows, Eur. J. Org. Chem., 2011, 3970–3979 CrossRef CAS.
- J. M. Dąbrowski, K. Urbanska, L. G. Arnaut, M. M. Pereira, A. R. Abreu, S. Simões and G. Stochel, ChemMedChem, 2011, 6, 465–475 CrossRef PubMed.
- Z. Yu and M. Ptaszek, Org. Lett., 2012, 14, 3708–3711 CrossRef CAS PubMed.
- V. M. Alexander, K. Sano, Z. Yu, T. Nakajima, P. L. Choyke, M. Ptaszek and H. Kobayashi, Bioconjugate Chem., 2012, 23, 1671–1679 CrossRef CAS PubMed.
- A. Kozyrev, M. Ethirajan, P. Chen, K. Ohkubo, B. C. Robinson, K. M. Barkigia, S. Fukuzumi, K. M. Kadish and R. K. Pandey, J. Org. Chem., 2012, 77, 10260–10271 CrossRef CAS PubMed.
- L. P. Samankumara, S. Wells, M. Zeller, A. M. Acuña, B. Röder and C. Brückner, Angew. Chem., Int. Ed., 2012, 51, 5757–5760 CrossRef CAS PubMed.
- M. M. Pereira, A. R. Abreu, N. P. F. Goncalves, M. J. F. Calvete, A. V. C. Simoes, C. J. P. Monteiro, L. G. Arnaut, M. E. Eusébio and J. Canotilho, Green Chem., 2012, 14, 1666–1672 RSC.
- J. Ogikubo, E. Meehan, J. T. Engle, C. J. Ziegler and C. Brückner, J. Org. Chem., 2013, 78, 2840–2852 CrossRef CAS PubMed.
- A. Aggarwal, S. Thompson, S. Singh, B. Newton, A. Moore, R. Gao, X. Gu, S. Mukherjee and C. M. Drain, Photochem. Photobiol., 2014, 90, 419–430 CrossRef CAS PubMed.
- H.-J. Kim and J. S. Lindsey, J. Org. Chem., 2005, 70, 5475–5486 CrossRef CAS PubMed.
- D. Fan, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2007, 72, 5350–5357 CrossRef CAS PubMed.
- M. Krayer, M. Ptaszek, H.-J. Kim, K. R. Meneely, D. Fan, K. Secor and J. S. Lindsey, J. Org. Chem., 2010, 75, 1016–1039 CrossRef CAS PubMed.
- M. Krayer, E. Yang, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, New J. Chem., 2011, 35, 587–601 RSC.
- C.-Y. Chen, E. Sun, D. Fan, M. Taniguchi, B. E. McDowell, E. Yang, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, Inorg. Chem., 2012, 51, 9443–9464 CrossRef CAS PubMed.
- M. Galezowski and D. T. Gryko, Curr. Org. Chem., 2007, 11, 1310–1338 CrossRef CAS.
- C. Brückner, L. Samankumara and J. Ogikubo, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2012, vol. 17, pp. 1–112 Search PubMed.
- T. G. Minehan and Y. Kishi, Tetrahedron Lett., 1997, 38, 6811–6814 CrossRef CAS.
- T. G. Minehan and Y. Kishi, Tetrahedron Lett., 1997, 38, 6815–6818 CrossRef CAS.
- T. G. Minehan and Y. Kishi, Angew. Chem., Int. Ed., 1999, 38, 923–925 CrossRef CAS.
- W. Wang and Y. Kishi, Org. Lett., 1999, 1, 1129–1132 CrossRef CAS.
- K. R. Reddy, J. Jiang, M. Krayer, M. A. Harris, J. W. Springer, E. Yang, J. Jiao, D. M. Niedzwiedzki, D. Pandithavidana, P. S. Parkes-Loach, C. Kirmaier, P. A. Loach, D. F. Bocian, D. Holten and J. S. Lindsey, Chem. Sci., 2013, 4, 2036–2053 RSC.
- J. Jiang, P. Vairaprakash, K. R. Reddy, T. Sahin, M. P. Pavan, E. Lubian and J. S. Lindsey, Org. Biolmol. Chem., 2014, 12, 86–103 RSC.
- M. A. Harris, J. Jiang, D. M. Niedzwiedzki, J. Jiao, M. Taniguchi, C. Kirmaier, P. A. Loach, D. F. Bocian, J. S. Lindsey, D. Holten and P. S. Parkes-Loach, Photosynth. Res., 2014, 121, 35–48 CrossRef CAS PubMed.
- Z. Yu and M. Ptaszek, Org. Lett., 2012, 14, 3708–3711 CrossRef CAS PubMed.
- T. Harada, K. Sano, K. Sato, R. Watanabe, Z. Yu, H. Hanaoka, T. Nakajima, P. L. Choyke, M. Ptaszek and H. Kobayashi, Bioconjugate Chem., 2014, 25, 362–369 CrossRef CAS PubMed.
- E. Yang, C. Kirmaier, M. Krayer, M. Taniguchi, H.-J. Kim, J. R. Diers, D. F. Bocian, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2011, 115, 10801–10816 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, 1996 Search PubMed.
- R. A. Cellarius and D. Mauzerall, Biochim. Biophys. Acta, 1966, 112, 235–255 CrossRef CAS.
- G. Bartoli, M. Bosco, A. Carlone, R. Dalpozzo, E. Marcantoni, P. Melchiorre and L. Sambri, Synthesis, 2007, 3489–3496 CrossRef CAS PubMed.
- R. W. Wagner, Y. Ciringh, C. Clausen and J. S. Lindsey, Chem. Mater., 1999, 11, 2974–2983 CrossRef CAS.
- M. Kobayashi, M. Akiyama, H. Kano and H. Kise, in Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 79–94 Search PubMed.
- B. Abrams, Z. Diwu, O. Guryev, S. Aleshkov, R. Hingorani, M. Edinger, R. Lee, J. Link and T. Dubrovsky, Anal. Biochem., 2009, 386, 262–269 CrossRef CAS PubMed.
- A. S. Manjappa, K. R. Chaudhari, M. P. Venkataraju, P. Dantuluri, B. Nanda, C. Sidda, K. K. Sawant and R. S. R. Murthy, J. Controlled Release, 2011, 150, 2–22 CrossRef CAS PubMed.
- P. K. Chattopadhyay, B. Gaylord, A. Palmer, N. Jiang, M. A. Raven, G. Lewis, M. A. Reuter, A. K. M. N. Rahman, D. A. Price, M. R. Betts and M. Roederer, Cytometry, Part A, 2012, 81A, 456–466 CrossRef CAS PubMed.
- D. Majonis, O. Ornatsky, D. Weinrich and M. A. Winnik, Biomacromolecules, 2013, 14, 1503–1513 CrossRef CAS PubMed.
- F. Bryden, A. Maruani, H. Savoie, V. Chudasama, M. E. B. Smith, S. Caddick and R. W. Boyle, Bioconjugate Chem., 2014, 25, 611–617 CrossRef CAS PubMed.
- M. Dautrevaux, Y. Boulanger, K. Han and G. Biserte, Eur. J. Biochem., 1969, 11, 267–277 CrossRef CAS PubMed.
- E. B. Garcia-Moreno, L. X. Chen, K. L. March, R. S. Gurd and F. R. N. Gurd, J. Biol. Chem., 1985, 260, 14070–14082 Search PubMed.
- C. H. I. Ramos, M. S. Kay and R. L. Baldwin, Biochemistry, 1999, 38, 9783–9790 CrossRef CAS PubMed.
- A. Castro-Forero, D. Jiménez, J. López-Garriga and M. Torres-Lugo, J. Appl. Polym. Sci.: Appl. Polym. Symp., 2008, 107, 881–890 CrossRef CAS PubMed.
- S. C. Harrison and E. R. Blout, J. Biol. Chem., 1965, 240, 299–303 CAS.
- J. M. Dixon, M. Taniguchi and J. S. Lindsey, Photochem. Photobiol., 2005, 81, 212–213 CrossRef CAS.
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 3, 283–291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Sequence and structure of Mb; MALDI-MS and spectral data for bacteriochlorin–Mb conjugates. See DOI: 10.1039/c4nj01340a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |