
A strategic approach to the synthesis of functionalized spirooxindole pyrrolidine derivatives: in vitro antibacterial, antifungal, antimalarial and antitubercular studies†
Saoussen
Haddad
ab,
Sarra
Boudriga
a,
Tarunkumar Nanjibhai
Akhaja
c,
Jignesh Priyakant
Raval
c,
François
Porzio
b,
Armand
Soldera
b,
Moheddine
Askri
*a,
Michael
Knorr
d,
Yoann
Rousselin
e,
Marek M.
Kubicki
e and
Dhanji
Rajani
f
aLaboratory of Heterocyclic Chemistry Natural Products and Reactivity/LCHPNR, Department of Chemistry, Faculty of Science of Monastir, 5000 Monastir, Tunisia. E-mail: moheddine.askri@fsm.rnu.tn; Tel: +216 98676187
bDepartment of Chemistry, Quebec Center for Functional Materials, University of Sherbrooke, Sherbrooke, Québec, Canada J1K 2R1
cDepartment of Chemistry, UkaTarsadia University, Maliba Campus, Bardoli – Mahuva Road, Dist: Surat 394350, Gujarat, India
dInstitute UTINAM - UMR CNRS 6213, University of Franche-Comté, 16 Route de Gray, F-25030 Besançon, France
eInstitute of Molecular Chemistry - UMR CNRS 6302, University of Bourgogne, 9 Avenue A. Savary, F-21078 Dijon, France
fMicrocare Laboratories, Surat – 395001, Gujarat, India
First published on 27th October 2014
Abstract
A series of spiro[pyrrolidin-2,3′-oxindoles] has been synthesized by exo-selective 1,3-dipolar cycloaddition reaction of a stabilized azomethine ylide, generated in situ by thermal [1,5]-prototropy, across various (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones. The stereochemistry of these N-heterocycles has been confirmed using an X-ray diffraction study. To rationalize the observed regio- and stereoselectivity, DFT calculations at the B3LYP/6-31G(d,p) level were employed. It was found that this reaction preferentially affords the kinetic product. The compounds have been screened for their in vitro antibacterial, antifungal, antimalarial and antitubercular activities. Several compounds exhibited good activities comparable to those of established standard drugs.
Introduction
During the past decades, multicomponent reactions (MCRs) have emerged as a powerful tool in organic, combinatorial and medicinal chemistry. They actually offer rapid and easy access to complex N-heterocyclic systems in a single eco-friendly synthetic operation with high atom economy, wide structural diversity and multiple-bond-forming efficiency.1 Among the various N-heterocycles, spirooxindole derivatives are widespread in numerous natural products2 and are pharmacologically important compounds.1h,3 In particular, the spiro[pyrrolidin-2,3′-oxindole] core is a privileged heterocyclic ring system, which is featured in a large number of bioactive naturally occurring alkaloids,4 and also in some compounds synthesized for medicinal purposes (Fig. 1). These spiro-molecules exhibit a wide spectrum of important bioactivities such as antitumoral,5 anti-inflammatory,6 antimycobacterial,7 antimicrobial,8 antitubercular,7a,d and acetylcholinesterase-inhibitory9 activities. | ||
| Fig. 1 Some examples of biologically active compounds containing the spiro[pyrrolidin-2,3′-oxindole] motif. | ||
Recently, the multicomponent 1,3-dipolar cycloaddition of azomethine ylides with alkenes has been employed as a strategy for the synthesis of a variety of spiro[pyrrolidin-2,3′-oxindoles] with high regio- and stereoselectivity.10 These reactions are limited to the use of non-stabilized azomethine ylides, generated in situ by decarboxylative condensation of isatin with α-amino acids or cyclic amino acids (Scheme 1). However, we are aware of just one report on the use of stabilized azomethine ylides as reagents for multicomponent 1,3-dipolar cycloaddition (Scheme 1).11 These stabilized azomethine ylides have been generated in situ by thermal [1,5]-prototropy of the corresponding iminoesters derived from isatin. Grigg12 and Tsuge13 outlined in their extensive studies on the generation of stabilized ylides via prototropic shift of the proton to the imine nitrogen that the latter undergo preferentially endo-cycloaddition. Liu and co-workers11 reported very recently on the first example of an exo-selective dipolar cycloaddition reaction using a stabilized azomethine ylide.
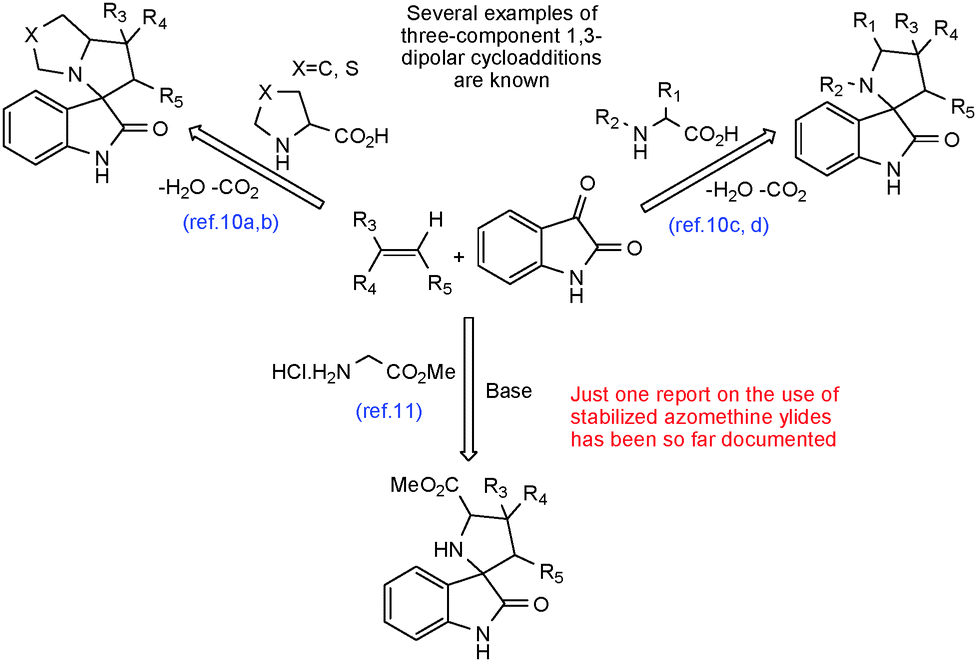 | ||
| Scheme 1 Multicomponent 1,3-dipolar cycloaddition for the synthesis of spiro[pyrrolidin-2,3′-oxindole] derivatives. | ||
In the continuity of our studies on the development of cycloaddition reactions,14 we herein present the synthesis of an extended series of novel spirooxindole pyrrolidine derivatives via one-pot three-component 1,3-dipolar cycloaddition. Since no theoretical and biological studies on that type of spirooxindoles have been reported yet, we explicitly investigated from a theoretical point of view the regio- and stereochemistry by means of Density Functional Theory (DFT) calculations. Furthermore, the newly synthesized heterocyclic compounds were screened in vitro to evaluate their antibacterial, antifungal, antitubercular and antimalarial activities.
Results and discussion
Synthetic chemistry
The targeted compounds were obtained by one-pot three-component 1,3-dipolar cycloaddition of (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 4, with a stabilized azomethine ylide 3. The latter was generated in situ by thermal [1,5]-prototropy of the corresponding iminoesters derived from isatin 1 and glycine methyl ester 2a or sarcosine methyl ester 2b. The (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 4a–e chosen for our study as dipolarophiles have been prepared by the Wittig reaction between N-phenyl maleimide and aromatic aldehydes. The alkenes 4 were assigned to have the E configuration on the basis of their NMR spectra, in accordance with the literature data.15 To address a possible influence of the electronic propensity of the substituent at the para-position of the aryl group of dipolarophile 4 on the yield and stereochemical outcome of the reaction, both electron-donating and electron-withdrawing substituents (H, CH3, OCH3, Cl and Br) have been employed.As a sample model reaction, the three-component azomethine ylide cycloaddition reaction between isatin 1a, glycine methyl ester 2a and dipolarophile 4c was investigated in detail to optimize the reaction conditions (Scheme 2, Table 1). Various solvents with different polarities, such as methanol, ethanol, acetonitrile, tetrahydrofuran and toluene were explored. After optimization of the reaction conditions, we observed that the best results were obtained by refluxing the reaction mixture in methanol for 2 hours, providing the spirooxindole pyrrolidine derivative 5c in excellent yield (95%) (Table 1, entry 2).
 | ||
| Scheme 2 1,3-Dipolar cycloaddition leading to spiropyrrolidineoxindole 5c. | ||
Having established suitable reaction conditions, we explored the scope of this reaction with different dipolarophiles 4, as well as with various substituted isatins 1 and amino acid methyl ester 2 (Scheme 3). As shown in Table 2, the reaction proceeded with high regio- and stereoselectivity to afford the expected novel spiropyrrolidine derivatives, the spiro[2,3′]-oxindole-spiro[3,3′′]-5-carbomethoxypyrrolidine-4-N-phenylsuccinimides 5, with moderate to high yields. We found that the desired spiropyrrolidine derivatives 5a–o, generated from glycine methyl ester 2a reactions, were obtained in satisfactory to very high yields (80–95%), regardless of the electronic properties of the p-substituent at the aryl group (H, CH3, OCH3, Cl and Br) of dipolarophile 4 (Table 2, entries 1–15). When using the sarcosine methyl ester 2b as reagent, the yields were somewhat lower (Table 2, entries 16–18). All compounds were isolated as colorless solids.
 | ||
| Scheme 3 1,3-Dipolar cycloaddition reactions for the synthesis of spiropyrrolidineoxindoles with various R1 and R2 moieties. | ||
| Entry | Compound | R1 | R2 | Ar | Yieldb (%) |
|---|---|---|---|---|---|
a The reaction was carried out in 1 mmol scale in methanol (10 mL) at 60 °C, and the ratio of 1/2/4/Et3N is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1.
b Isolated yield based on substituted isatins. :1:1:1.
b Isolated yield based on substituted isatins.
|
|||||
| 1 | 5a | H | H | C6H5 | 87 |
| 2 | 5b | H | H | p-MeC6H4 | 88 |
| 3 | 5c | H | H | p-MeOC6H4 | 95 |
| 4 | 5d | H | H | p-ClC6H4 | 80 |
| 5 | 5e | H | H | p-BrC6H4 | 82 |
| 6 | 5f | Br | H | C6H5 | 85 |
| 7 | 5g | Br | H | p-MeC6H4 | 83 |
| 8 | 5h | Br | H | p-MeOC6H4 | 87 |
| 9 | 5i | Br | H | p-ClC6H4 | 85 |
| 10 | 5j | Br | H | p-BrC6H4 | 89 |
| 11 | 5k | NO2 | H | C6H5 | 80 |
| 12 | 5l | NO2 | H | p-MeC6H4 | 88 |
| 13 | 5m | NO2 | H | p-MeOC6H4 | 85 |
| 14 | 5n | NO2 | H | p-ClC6H4 | 83 |
| 15 | 5o | NO2 | H | p-BrC6H4 | 82 |
| 16 | 5p | H | Me | C6H5 | 65 |
| 17 | 5q | H | Me | p-MeC6H4 | 67 |
| 18 | 5r | H | Me | p-ClC6H4 | 63 |
The structure of the spiroadducts was elucidated on the basis of their spectroscopic data and an X-ray structure analysis, as exemplified for cycloadduct 5c. The 1H NMR spectrum 5c shows two mutually coupled doublets at δ 2.57 and 2.76 ppm (J = 19.0 Hz) corresponding to the CH2 group, as well as two further doublets at δ 4.33 (J = 9.7 Hz) and 5.05 ppm (J = 9.7 Hz) assigned to the pyrrolidine H-4 and H-5 protons (Scheme 3), respectively. The occurrence of these two doublets clearly proves the regiochemistry of the cycloaddition reaction. If the hypothetical alternative regioisomer 6c (Scheme 3) would have been formed, the pyrrolidinyl protons should give rise to two singlets in the 1H NMR spectrum. The 13C NMR spectrum of the cycloadduct 5c exhibits peaks at δ 57.1 and 65.2 ppm corresponding to C-4 and C-5 (Scheme 3), respectively. The two spirocarbons C-3 and C-2 resonate at δ 62.1 and 75.0 ppm, respectively. In addition, two carbonyl carbons are recognized at δ 172.1 and 178.3 ppm and are assigned to the ester and oxindole carbonyl groups; two further signals resonating at δ 173.7 and 178.9 ppm are attributed to the N-phenyl succinimide carbonyls. These observed chemical shifts are in accordance with the suggested stereochemistry for 5c shown in Scheme 3 and corroborated by the elucidation of the crystal structure of derivative 5c (see below).
A plausible mechanism for the regio- and stereoselective spirooxindole formation is proposed in Scheme 4. The azomethine ylides 3 are generated by thermal [1,5]-prototropy of the iminoester obtained by condensation of the isatin derivatives 1 with amino acid methyl ester 2. The azomethine ylides 3 then undergo a 1,3-dipolar cycloaddition reaction across the dipolarophile 4 in a regioselective manner.
 | ||
| Scheme 4 Proposed mechanism for the 1,3-dipolar cycloaddition of azomethine ylides across (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 4. | ||
Crystal structure determination of cycloadduct 5c
The stereochemical outcome of the cycloaddition was also elucidated by an X-ray diffraction study performed on a single-crystal of 5c (Fig. 2). This cycloadduct is formed by an exo-approach between the (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-dione and the Z,E-dipole (Scheme 5). | ||
| Fig. 2 ORTEP of the molecular structure of cycloadduct 5c in the crystal at the 50% probability level. For clarity, only stereo-chemically significant hydrogen atoms are shown. Selected bond lengths (Å) and angles (°): C9–C21 1.531(1), C9–C23 1.534(1), C9–C8 1.572(1), N2–C11 1.468(1), N2–C10 1.463(1), C10–C14 1.557(1), C10–C20 1.513(2); C21–C9–C23 102.19(8), C8–C9–C10 103.44(8), C9–C10–N2 106.30(9), C14–C10–C20 101.52(9), C10–N2–C11 104.62(9), C14–N3–C15 111.77(9). | ||
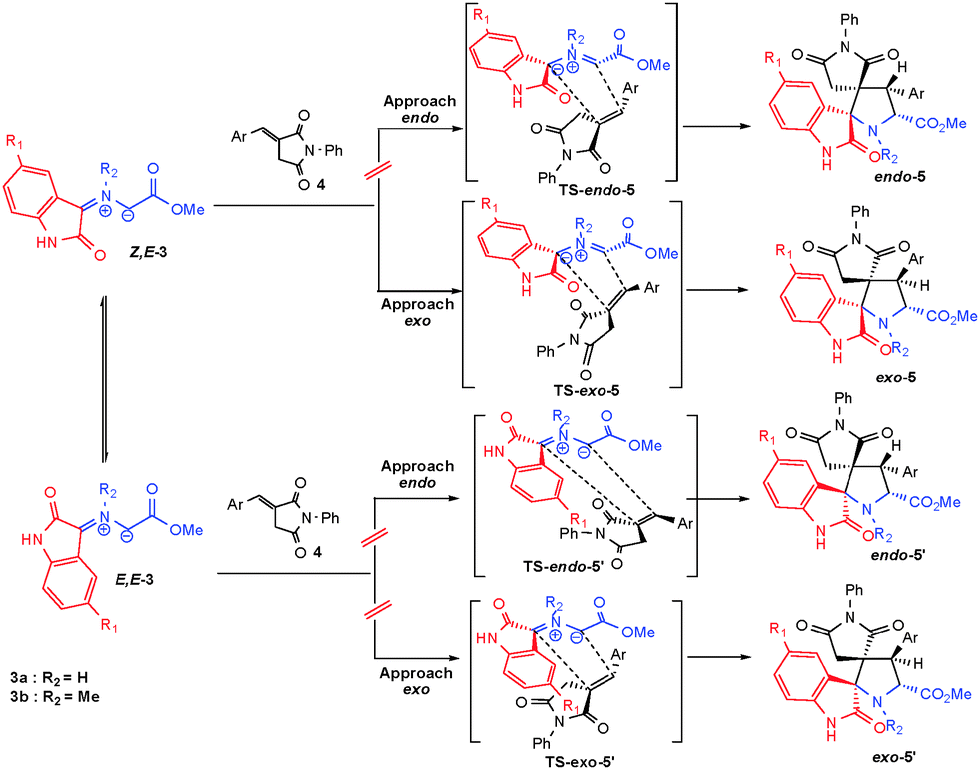 | ||
| Scheme 5 Stereoisomeric pathways for the 1,3-dipolar cycloaddition reaction between Z,E-3 and E,E-3 and dipolarophile 4. | ||
There are some interesting features in the molecular structure of 5c. The fused heterocycle formed by the 10 atoms (C10, C14, O4, N3, C15 through C20) is almost planar with the largest deviation from the best least square plane of 0.06 Å for the O4 atom. This suggests a high degree of π conjugation in this part of the molecule. The dihedral angles over the both spiranic carbon atoms C9 and C10 are very close to 90°: 88.95(7)° for the C8/C9/C10//C21/C9/C23 and 88.87(6)° the C14/C10/C20//C9/C10/N2 planes. One also notes that the N3 atom is in a perfect trigonal planar hybridization (sum of three angles around is equal to 360°), whereas the N2 atom adopts a pyramidal structure with the sum of three angles between the σ bonds reaching 323.1°. The molecule shown in Fig. 2 bears four chiral carbon atoms: C8, C9, C10 and C11, all with R configuration.
DFT simulation
In order to support the experimental results and thus to unveil the regio- and stereoselectivity of these reactions, DFT calculations were carried out using the B3LYP functional and the 6-31G(d,p) basis set in the Gaussian 0916 environment. The stereochemistry of the 1,3-dipolar cycloaddition with azomethine ylides was established by the ylide geometry and the endo/exo approach. The azomethine ylides 3 are formed from the reaction of isatin with a primary or secondary amine (Fig. 3) and exhibit four possible isomers, (E,E), (E,Z), (Z,E) and (Z,Z). Their relative stability can be estimated from steric repulsion and formation of intramolecular hydrogen bond between the carbonyl oxygen and the H–N or H–C hydrogens.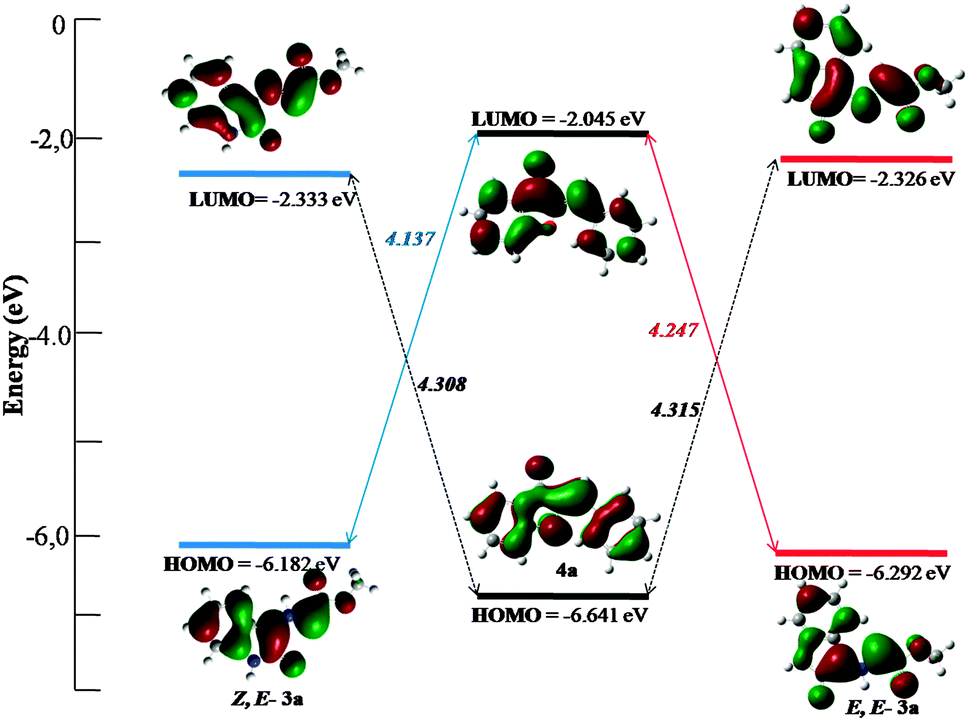 | ||
| Fig. 3 HOMO and LUMO energies calculated at the B3LYP/6-31G(d,p) level. | ||
The values of the minimized electronic energy for the four conformations are shown in Table S1 (in the ESI†). The energies of the Z,Z and E,Z conformations (16.0 and 8.0 kcal mol−13a; 7.6 and 5.1 kcal mol−1) are clearly higher than the energies of the other two isomers. These large values stem from the significant steric hindrance between the carbonyls of the ester group and the oxindole, and between the carbonyl of ester group and the isatin core, respectively (Scheme 6). Conversely, the energy difference between the Z,E and E,E isomers is in order of 0.6–0.8 kcal mol−1, close to the thermal energy, RT on the order of 0.6 kcal mol−1 at 25 °C. Thus, these two conformations cannot be energetically distinguished at this step. This increased stability of these two isomers comparatively to the two others, can be explained by the presence of two distinct hydrogen bonds, between H–N (in the case of dipole 3a) or H–C (in the case of dipole 3b) with the two ester and oxindole carbonyls (Scheme 6). The computational investigation is thus focused on the Z,E and E,E conformations (Table S1 in the ESI†). Since for both dipoles 3a and 3b it has been experimentally evidenced that the same reactive pathway toward an exo approach on a Z,E-dipole is followed, only calculations involving 3a were carried out.
 | ||
| Scheme 6 The four possible isomers for the azomethineylides 3. | ||
To clarify the regioselectivity of these reactions, analysis of the HOMO/LUMO energies was first conducted through the Frontier Molecular Orbital (FMO) theory. This widely used theory is based on the statement that a valuable estimate of the actual reactivity can be achieved by unveiling interaction between the HOMO and the LUMO of the reactants.17 As shown in Fig. 3, energies related to the transition between the HOMO of Z,E-3a or E,E-3a and the LUMO of 4a are lower (4.137 and 4.247 eV, respectively) than between the LUMO and the HOMO of the same compounds (4.308 and 4.313 eV, respectively). This suggests that the HOMOdipole–LUMOdipolarophile interaction controls the cycloaddition reaction within a normal electron demand reaction. In fact, 0.11 eV, or 2.5 kcal mol−1 that corresponds to the difference between the two HOMO–LUMO energy gaps (Fig. 3), is noticeably larger than RT (0.6 kcal mol−1 at 25 °C), revealing that cycloaddition with Z,E-3a is favoured. The occurrence of such a charge transfer from the dipole to the dipolarophile is confirmed by the calculation of the electronic chemical potential μ,18 the chemical hardness η,19 and the electrophilicity index ω20 (details of the calculations are found in the ESI†).
To disclose the most favourable attack site, the DFT-based local chemical reactivity Fukui function parameters for nucleophilic (fk+) and electrophilic (fk−) attacks, have been calculated through the NBO (Natural Bond Orbital) atomic net charges approach using eqn (1) and (2) respectively.21
| fk+ = PN+1k − PNk = qNk − qN+1k | (1) |
| fk− = PNk − PN−1k = qN−1k − qNk | (2) |
We verified that these 1,3-dipolar cycloaddition reactions take place along asynchronous concerted processes, which is confirmed by the IRC22 cycloaddition profile, using the Hessian based predictor–corrector (HPC) method.23 These results are in accordance with previous studies.24 Nevertheless, four possible transition structures exist. Their corresponding cycloadducts have been optimized and characterized under the same level of calculation by using the Berny analytical gradient method.25 The stationary points were characterized by frequency calculations in order to ensure that the minima, i.e. reactants and products, do not possess imaginary frequency. The transition states (TS) are characterized by a saddle point leading to the occurrence of one imaginary frequency. The optimized geometries of endo-transition state (TS-endo-5a and TS-endo-5′a) and related exo-transition state (TS-exo-5a and TS-endo-5′a) are displayed in Fig. 4. In this notation, 5a denotes cycloaddition with Z,E-3, while 5′a denotes cycloaddition with E,E-3. The activation energy (Ea) between reactants and transition states, as well as variations in the internal energy (ΔU), enthalpy (ΔH), entropy (ΔS) and Gibbs free energy (ΔG) between reactants and products are all compiled in Table S4 (in the ESI†). Enthalpy, entropy and Gibbs free energy values have been corrected for zero point energy.
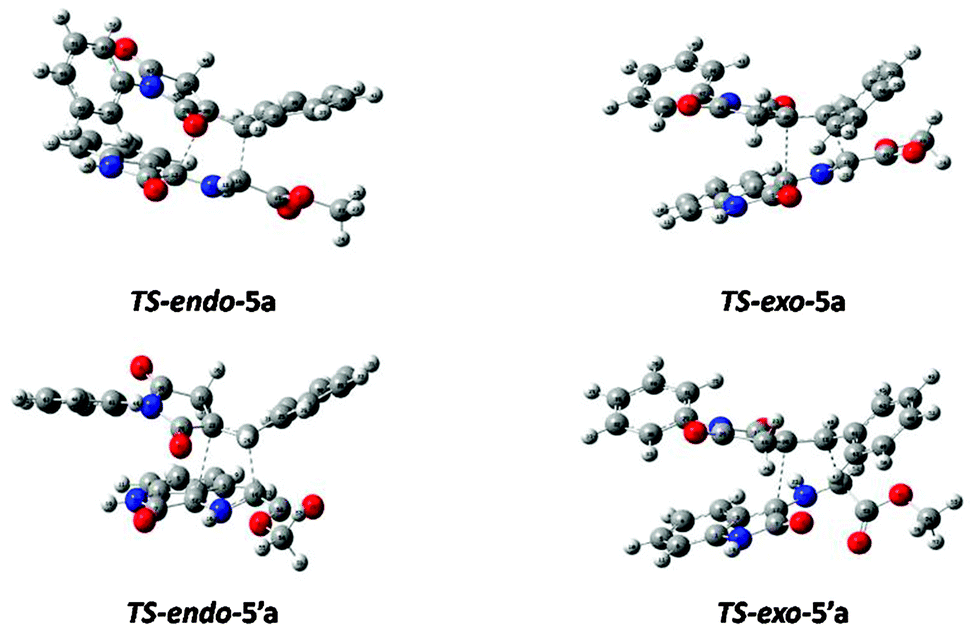 | ||
| Fig. 4 endo- and exo-transition structures corresponding to the stereoisomeric path of the 1,3-dipolar cycloaddition reaction channels of E,E-3a (5a in the figure) and Z,E-3a (5′a in the figure) with 4a. | ||
We first examined the thermodynamic aspect of these cycloadditions. An analysis of data stemming from gas-phase DFT calculations underlines that all these reactions exhibit negative relative internal energies (ΔU < 0) as well as negative reaction enthalpies (ΔH < 0). It is worth noting that in the gas phase, calculations predict that these reactions have positive free Gibbs energies (ΔG > 0). The calculated positive values of the free Gibbs enthalpies directly stem from reaction enthalpies (ΔH < 0, stabilization toward the products) that cannot make up for their respective loss of entropy (ΔS < 0).
Conversely, the experimental results in methanol show that the reaction is spontaneous toward the products. The issue of these negative values for the free Gibbs enthalpy could be settled by considering the effect of solvent, and will be carried out in forthcoming studies using the continuum approach26 for instance. Nevertheless, variations in internal energies, enthalpy as well as free Gibbs energies in the gas phase altogether (Table S4 in the ESI†) are always consistent with each other. More specifically, those thermodynamic functions correctly describe the decreasing order of stability: exo-5a > endo-5a > exo-5′a > endo-5′a. The difference in energies between the two most stable products (exo-5a and endo-5a) is 4.8, 4.7 and 5.4 kcal mol−1 for relative internal energy, reaction enthalpy and free Gibbs energy, respectively. These energy differences are much greater that RT (0.6 kcal mol−1 at 25 °C). Thus, calculations demonstrate that the cycloadduct resulting from the exo approach of Z,E-3a (exo-5a) is the most favourable thermodynamic product (Table S4 in the ESI†).
The kinetic aspect of these reactions will not be fully explored.27 The Arrhenius approach is only considered, through a comparison of the activation energies. The exo approach of Z,E-3a (exo-5a) is thus found to be favourable (Table S4 in the ESI†). The TS of exo-5a is lower in energy than the TS endo-5a, the TS endo-5′a and the TS exo-5′a which are 3.0, 4.9 and 12.9 kcal mol−1 higher in energy, respectively. Actually, the difference between the two lowest energetic barriers (3.0 kcal mol−1) is large compared to the thermal energy RT (0.6 kcal mol−1 at 25 °C). Thus, this reaction is mainly kinetic dependent and leads to exo-5a as the most probable outcome which is also the thermodynamic product since it presents the lowest value of ΔG (Table S4 in the ESI†)
To summarize, DFT calculations in the gas-phase at the B3LYP/6-31G(d,p) level of theory successfully account for both the regio- and stereochemistry experimentally observed (Scheme 4). The aforementioned calculations correctly predict the exo-5a product, i.e. the exo approach between the Z,E-3a and 4a, as the most favourable product of the 1,3-dipolar cycloaddition.
Biological activities
| Comp. | Antibacterial activity | Antifungal activity | ||||
|---|---|---|---|---|---|---|
| Gram negative bacteria | Gram positive bacteria | |||||
| E.C. | P.A. | S.A. | S.P. | C.A. | A.N. | |
| MTCC 443 | MTCC 1688 | MTCC 96 | MTCC 442 | MTCC 227 | MTCC 282 | |
| G: gentamycin, A: ampicillin, C: chloramphenicol, N: nystatin, G: greseofulvin, NT: not tested. | ||||||
| 5a | 125 | 125 | 250 | 200 | 1000 | 250 |
| 5b | 200 | 250 | 500 | 500 | 1000 | 250 |
| 5c | 125 | 125 | 500 | 500 | >1000 | 1000 |
| 5d | NT | NT | NT | NT | NT | NT |
| 5e | 200 | 250 | 250 | 250 | >1000 | 1000 |
| 5f | 100 | 200 | 62.5 | 100 | 1000 | 1000 |
| 5g | 500 | 250 | 250 | 200 | 500 | 1000 |
| 5h | 250 | 250 | 250 | 200 | 1000 | 250 |
| 5i | 62.5 | 100 | 200 | 200 | 250 | 500 |
| 5j | 100 | 125 | 100 | 125 | 250 | 1000 |
| 5k | NT | NT | NT | NT | NT | NT |
| 5l | 250 | 200 | 200 | 200 | 1000 | >1000 |
| 5m | 250 | 250 | 200 | 250 | 1000 | >1000 |
| 5n | 50 | 100 | 500 | 500 | 200 | 500 |
| 5o | 200 | 100 | 500 | 500 | 500 | 500 |
| 5p | 62.5 | 100 | 200 | 200 | 200 | 1000 |
| 5q | 100 | 125 | 200 | 200 | 500 | 1000 |
| 5r | 200 | 62.5 | 100 | 100 | 1000 | 1000 |
| G | 0.5 | 1 | 0.25 | 0.5 | — | — |
| A | 100 | 100 | 250 | 100 | — | — |
| C | 50 | 50 | 50 | 50 | — | — |
| N | — | — | — | — | 100 | 100 |
| G | 500 | 100 | ||||
In the first screening phase against MTCC 443, two compounds 5i and 5p showed excellent activity, compared to standard antibiotic ampicillin; but 5f, 5j and 5q were equally active.
In the case of Pseudomonas aeruginosa, compounds 5i, 5n, 5o and 5p were as equipotent as ampicillin. Thus, 5r was found to be the most active in vitro with a MIC of 62.5 μg mL−1 against MTCC 1688. The compounds were also evaluated against Staphylococcus aureus and Staphylococcus pyogenus. In this case, 5f was found to be the most active derivative in vitro with a MIC of 62.5 μg mL−1 against MTCC 96. All compounds were found to be equal or less active (100–500 μg mL−1), compared to standard antibiotic ampicillin.
It is confirmed from the in vitro antifungal activity data, that 5n and 5p display the highest activity against Griseofulvin, while 5i, 5j show a somewhat inferior activity, compared to compounds 5n and 5p. Compounds 5g, 5o and 5q displayed variable activity against Gram-positive strains as compared to the standard antifungal agent Griseofulvin.
| Compound | Mean IC50 values (μg mL−1) | Compound | Mean IC50 values (μg mL−1) |
|---|---|---|---|
| C: chloroquinine,Q: quinine, NT: not tested. | |||
| 5a | 0.680 | 5k | NT |
| 5b | 0.980 | 5l | 0.670 |
| 5c | 1.330 | 5m | 0.910 |
| 5d | NT | 5n | 1.120 |
| 5e | 1.470 | 5o | 0.970 |
| 5f | 1.190 | 5p | 1.230 |
| 5g | 0.570 | 5q | 0.044 |
| 5h | 0.940 | 5r | 0.860 |
| 5i | 1.270 | C | 0.020 |
| 5j | 1.560 | Q | 0.268 |
Only compound 5q showed much better IC50 values against P. falciparum than quinine as a standard drug; the other compounds exhibited only a weak antimalarial activity (MIC = 0.68–1.56 μg mL−1). This promising antimalarial activity of 5q may be due to sufficient hydrogen bonding capacity with the desired lipophilicity or with favorable steric hinderance.29
| Comp. | M. tuberculosis H37Rv | % Inhibition | Comp. | M. tuberculosis H37Rv | % Inhibition |
|---|---|---|---|---|---|
| MTCC 200 | MTCC 200 | ||||
| I: isoniazid, NT: not tested. | |||||
| 5a | 500 | 98 | 5k | NT | NT |
| 5b | 25 | 99 | 5l | 100 | 98 |
| 5c | 500 | 98 | 5m | 25 | 99 |
| 5d | NT | NT | 5n | 1000 | 98 |
| 5e | 500 | 98 | 5o | 100 | 98 |
| 5f | 1000 | 98 | 5p | 250 | 98 |
| 5g | 62.5 | 99 | 5q | 1000 | 98 |
| 5h | 50 | 99 | 5r | 1000 | 98 |
| 5i | 250 | 98 | I | 0.20 | 99 |
| 5j | 500 | 98 | |||
Structural activity relationship – SAR
The results of the biological evaluation revealed that the activity was considerably affected by introducing a methyl group at the N − 1 position in pyrrolidine scaffold and the substituents at the isatin core as well as on the aryl ring (Scheme 3). For antibacterial activity, it was observed that the introduction of electron-withdrawing groups on the isatin ring showed considerable increase in the antibacterial potency of the compounds. It was observed from the screening data that the compounds 5i and 5n containing bromo and nitro substituents showed significant potency against the bacteria E. coli.Compound 5r containing a methyl group at the N − 1 position showed maximum inhibition against P. aeruginosa at MIC 62.5 μg mL−1.
The compound 5f containing bromo substitution on the isatin ring showed excellent activity against S. aureus and S. pyogenes at MIC 62.5 μg mL−1 and 100 μg mL−1, respectively.
According to the MIC values of antifungal activity, compounds with the nitro group at the isatin ring 5n and 5p containing a methyl group at the N − 1 position showed excellent activity against C. albicans.
In contrast, the presence of a methyl group at the N − 1 position of pyrrolidene ring as well as on the aryl ring illustrated superior antimalarial activity.
On the other hand, the results of the antitubercular screening demonstrated that compounds 5b, 5g, 5h and 5m with 99% inhibition displayed excellent activity against M. Tuberculosis H37Rv, which could be credited by the presence of electron-donating groups (methyl or methoxy) on the aromatic ring.
Conclusions
We have synthesized in good to excellent yields a series of novel spirooxindole pyrrolidines using as synthetic route a one-pot, three-component 1,3-dipolar cycloaddition of azomethine ylides. These latter have been generated in situ by thermal prototropy of the corresponding iminoesters with (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones as dipolarophiles. The cycloadducts were obtained after a short reaction time with high regio- and stereoselectivity, regardless of the electronic propensities of the substituents at the para-position of the aryl groups. The regiochemistry and the stereochemistry of the cycloadducts have been rationalized using DFT calculations. An analysis based on FMO calculations, Fukui functions, electronic chemical potential, chemical hardness, global electrophilicity, as well as relative internal energy, reaction enthalpy, free Gibbs enthalpy and activation energy, predicts the same outcome as noticed in the experiment, i.e. an exo-approach between the Z,E-dipole and dipolarophile. The DFT study reveals that the spirocycloadducts 5 are obtained through a 1,3-dipolar cycloaddition reaction via a highly asynchronous mechanism with very low activation energy. The computational results thus support the experimentally observed regioselectivity and stereoselectivity. Noteworthy, some of these heterocycles exhibit promising antibacterial, antifungal, antitubercular, and even antimalarial activities. So, in a next step the use of enantiomeric pure compounds deserves to be investigated.Experimental section
Apparatus and general information
NMR spectra were recorded with a Bruker-Spectrospin AC 300 spectrometer operating at 300 MHz for 1H and 75 MHz for 13C using tetramethylsilane (TMS) as the internal standard (0.00 ppm) in CDCl3 as solvent. The following abbreviations were used to explain the multiplicities: bs = broad singlet, s = singlet, d = doublet, m = multiplet. IR spectra were recorded on a Perkin-Elmer Spectrum TwoTM FT-IR in the ATR mode. Elemental analyses were performed on a Perkin Elmer 2400 Series II Elemental CHNS analyzer. The ESI HRMS spectrum of 5i was obtained on a LTQ Orbitrap XL mass spectrometer (Thermo Scientific) in the positive mode. Materials: thin-layer chromatography (TLC): TLC plates (Merck, silica gel 60 F2540.2 mm 200 × 200 nm); substances were detected using UV light at 254 nm.General procedure for the preparation of cycloadducts 5
A mixture of isatin 1 (1.0 mmol), α-amino ester hydrochloride 2a (0.12 g, 1.0 mmol) or 2b (1.14 g, 1.0 mmol), (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 4 (1 mmol) and Et3N (0.10 g, 1.0 mmol) was heated in methanol (10 mL) at 60 °C for 2–3 hours. After completion of the reaction (TLC), the solvent was removed under vacuum. The crude product was purified by column chromatography on silica gel using cyclohexane/ethyl acetate (3:2) as the eluent to give analytical pure 5. Spectroscopic data for the all compound are presented in the ESI.†
X-ray crystallography
A colourless bright single-crystal of 5c has been mounted on a Nonius Kappa Apex II diffractometer and the intensity data have been collected at 115 K with Mo Kα radiation of λ = 0.71073 Å. These data were further treated with the suite of SAINT V8.27B (Bruker AXS Inc., 2012) programs and within the OLEX2 frame.30 The model of the structure has been solved by direct methods with SHELXS-97 and refined with SHELXL-97.31 The molecules of 5c crystallize in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) together with those of the solvent (ethanol). The solvent molecules are disordered in an unprecedented mode over the crystallographic site of −3 local symmetry with consequent occupancies equal to 1/3 for carbon atom (covering both methylene and methyl carbons) and 1/6 for OH groups. The methylene and methyl hydrogens were not located in this disordered solvent part of the unit cell. CCDC 976702 (5c).
, a = 28.3533(11) Å, b = 28.3533(11) Å, c = 15.9781(7) Å, α = 90°, β = 90°, γ = 120°, V = 11124.0(10) Å3, Z = 18, λ(Mo-Kα) = 0.71073 Å, F(000) = 4902, μ(Mo-Kα) = 0.082 mm−3, T = 115(2) K. 75304 reflections collected, 5544 unique and 4827 with I > 2σ(I). Final agreement factors: R(F) = 0.0445 (all observed) and 0.0383 with I > 2σ(I). Final residuals ρmax = 0.539, ρmin = −0.558 e− Å−3. GOF = 1.036.
together with those of the solvent (ethanol). The solvent molecules are disordered in an unprecedented mode over the crystallographic site of −3 local symmetry with consequent occupancies equal to 1/3 for carbon atom (covering both methylene and methyl carbons) and 1/6 for OH groups. The methylene and methyl hydrogens were not located in this disordered solvent part of the unit cell. CCDC 976702 (5c).
, a = 28.3533(11) Å, b = 28.3533(11) Å, c = 15.9781(7) Å, α = 90°, β = 90°, γ = 120°, V = 11124.0(10) Å3, Z = 18, λ(Mo-Kα) = 0.71073 Å, F(000) = 4902, μ(Mo-Kα) = 0.082 mm−3, T = 115(2) K. 75304 reflections collected, 5544 unique and 4827 with I > 2σ(I). Final agreement factors: R(F) = 0.0445 (all observed) and 0.0383 with I > 2σ(I). Final residuals ρmax = 0.539, ρmin = −0.558 e− Å−3. GOF = 1.036.
Acknowledgements
Computations have been made available thanks to the Calcul Quebec, and Compute Canada.Notes and references
- For recent reviews on multicomponent reactions, see, (a) P. Dunn, Chem. Soc. Rev., 2012, 41, 1452 RSC; (b) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6324 CrossRef PubMed; (c) B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef CAS PubMed; (d) J. Zhu, in Multicomponent Reactions, ed. H. Bienayme, Wiley-VCH, Weinheim, 2005, pp. 1–32, pp. 468–469 Search PubMed; (e) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (f) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234 CrossRef CAS PubMed; (g) C. Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969 RSC; (h) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef PubMed.
- B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef CAS PubMed.
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203 CAS.
- (a) M. A. Koch, A. Schuffenhauer, M. Scheck, S. Wetzel, M. Casaulta, A. Odermatt, P. Ertland and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17272 CrossRef CAS PubMed; (b) P. R. Sebahar and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 5666 CrossRef CAS; (c) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed.
- (a) A. S. Girgis, Eur. J. Med. Chem., 2009, 44, 91 CrossRef CAS PubMed; (b) R. F. George, N. S. M. Ismail, J. Stawinski and A. S. Girgis, Eur. J. Med. Chem., 2013, 68, 339 CrossRef CAS PubMed.
- E. M. Hussein and M. I. Abdel-Monem, ARKIVOC, 2011, 10, 85 CrossRef.
- (a) S. U. Maheswari, K. Balamurugan, S. Perumal, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2010, 20, 7278 CrossRef CAS PubMed; (b) R. Kumar, S. Perumal, P. Senthilkumar, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2009, 44, 3821 CrossRef PubMed; (c) R. Kumar, S. Perumal, P. Senthilkumar, P. Yogeeswari and D. Sriram, J. Med. Chem., 2008, 51, 5731 CrossRef CAS PubMed; (d) R. S. Kumar, S. M. Rajesh, S. Perumal, D. Banerjee, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2010, 45, 411 CrossRef CAS PubMed.
- (a) A. A. Raj, R. Raghunathan, M. R. S. Kumari and N. Raman, Bioorg. Med. Chem., 2003, 11, 407 CrossRef CAS; (b) G. Periyasami, R. Raghunathan, G. Surendiran and N. Mathivanan, Bioorg. Med. Chem. Lett., 2008, 18, 2342 CrossRef CAS PubMed; (c) A. Thangamani, Eur. J. Med. Chem., 2010, 45, 6120 CrossRef CAS PubMed; (d) Y. Arun, G. Bhaskar, C. Balachandran, S. Ignacimuthuand and P. T. Perumal, Bioorg. Med. Chem. Lett., 2013, 23, 1839 CrossRef CAS PubMed.
- (a) M. A. Ali, R. Ismail, T. S. Choon, Y. K. Yoon, A. C. Wei, S. Pandian, R. S. Kumar, H. Osman and E. Manogaran, Bioorg. Med. Chem. Lett., 2010, 20, 7064 CrossRef CAS PubMed; (b) Y. Kia, H. Osman, R. S. Kumar, V. Murugaiyah, A. Basiri, S. Perumal, H. A. Wahab and C. S. Bing, Bioorg. Med. Chem., 2013, 7, 1696 CrossRef PubMed.
- (a) K. Chandraprakash, M. Sankaran, C. Uvarani, R. Shankar, A. Ata, F. Dallemer and P. S. Mohan, Tetrahedron Lett., 2013, 54, 3896 CrossRef CAS PubMed; (b) K. Alimohammadi, Y. Sarrafi, M. Tajbakhsh, S. Yeganegi and M. Hamzehloueian, Tetrahedron, 2011, 67, 1589 CrossRef CAS PubMed; (c) M. Ghandi, A. Taheri and A. Abbasi, Tetrahedron, 2010, 66, 6744 CrossRef CAS PubMed; (d) F. M. Moghaddam, M. R. Khodabakhshi, Z. Ghahremannejad, B. K. Foroushani and S. W. Ng, Tetrahedron Lett., 2013, 54, 2520 CrossRef CAS PubMed; (e) A. Dandiaa, R. Singha, J. Joshi and S. Kumari, J. Fluorine Chem., 2013, 156, 283 CrossRef PubMed; (f) R. F. George, N. S. M. Ismail, J. Stawinski and A. S. Girgis, J. Med. Chem., 2013, 68, 339 CrossRef CAS PubMed; (g) J. Xiao, H. Zhang, S. Liang, J. Ren, H. Yang and X. Chen, J. Org. Chem., 2013, 78, 11577 CrossRef CAS PubMed; (h) Y. Hu, Y. Zou, H. Wu and D. Shi, Ultrason. Sonochem., 2012, 19, 264 CrossRef CAS PubMed; (i) R. Rajesh and R. Raghunathan, Tetrahedron Lett., 2010, 51, 5845 CrossRef CAS PubMed.
- J. Liu, H. Sun, X. Liu, L. Ouyang, T. Kang, Y. Xie and X. Wang, Tetrahedron Lett., 2012, 53, 2336 CrossRef CAS PubMed.
- (a) R. Grigg and J. Kemp, J. Chem. Soc., Chem. Commun., 1977, 125 RSC; (b) R. Grigg and J. Kemp, J. Chem. Soc., Chem. Commun., 1978, 109 RSC; (c) S. Yamada, C. Hongo, R. Yoshioka and I. Chibata, J. Org. Chem., 1983, 48, 843 CrossRef CAS; (d) R. Grigg and H. Q. N. Gunaratne, Tetrahedron Lett., 1983, 24, 4457 CrossRef CAS.
- (a) O. Tsuge, S. Kanemasa, A. Hatada and K. Matsuda, Chem. Lett., 1984, 801 CrossRef CAS; (b) O. Tsuge, S. Kanemasa, T. Yamada and K. Matsuda, Heterocycles, 1985, 23, 2489 CrossRef CAS; (c) O. Tsuge, S. Kanemasa, A. Hatada and K. Matsuda, Bull. Chem. Soc. Jpn., 1986, 59, 2537 CrossRef CAS; (d) O. Tsuge, S. Kanemasa, T. Yamada and K. Matsuda, J. Org. Chem., 1987, 52, 2523 CrossRef CAS.
- (a) S. Boudriga, M. Askri, R. Gharbi, M. Rammah and K. Ciamala, J. Chem. Res., 2003, 4, 204 CrossRef; (b) M. Askri, N. Jgham, M. Rammah, K. Ciamala, K. Monnier-Jobé and J. Vebrel, Heterocycles, 2007, 71, 289 CrossRef CAS; (c) N. Wannassi, M. M. Rammah, S. Boudriga, M. Rammah, K. Monnier-Jobé, K. Ciamala, M. Knorr, M. Enescu, Y. Rousselin and M. M. Kubicki, Heterocycles, 2010, 81, 2749 CrossRef CAS PubMed; (d) N. Wannassi, H. Jelizi, M. Rammah, K. Ciamala, M. Knorr, K. Monnier-Jobé, Y. Rousselin, M. M. Kubickiand and C. Strohmann, Heterocycles, 2012, 85, 835 CrossRef CAS; (e) H. Jelizi, M. Rammah, M. Askri, K. Ciamala and K. Monnier-Jobé, Lett. Org. Chem., 2011, 8, 268 CrossRef CAS.
- L. Yan, W. Yang, L. Li, Y. Shen and Z. Jiang, Chin. J. Chem., 2011, 29, 1906 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. W. Challacombe, P. M. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.6, Gaussian, Inc., Pittsburgh PA, 1998 Search PubMed.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1971, 4, 57 CrossRef CAS; (c) K. Fukui, Molecular Orbitals in Chemistry, Physics, and Biology, Academic, New York, NY, 1964, p. 525 Search PubMed.
- L. R. Domingo, M. J. Aurell, P. Perez and R. Contreras, Tetrahedron, 2002, 58, 4417 CrossRef CAS.
- P. K. Chattaraj, U. Sarkar and D. R. Roy, Chem. Rev., 2006, 106, 2065 CrossRef CAS PubMed.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr, L. V. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- (a) S. Arulmozhiraja and P. Kolandaivel, Mol. Phys., 1997, 90, 55 CrossRef CAS; (b) P. W. Ayers, W. Yang and L. J. Bartolotti, in Chemical Reactivity Theory: A Density Functional, ed. P. K. Chattaraj, CRC Press, Boca Raton, 2009, ch. 18 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61 CrossRef CAS.
- (a) C. D. Valentin, M. Freccero, R. Gandolfi and A. Rastelli, J. Org. Chem., 2000, 65, 6112–6120 CrossRef PubMed; (b) K. N. Houk, J. Gonzalez and Y. Li, Acc. Chem. Res., 1995, 28, 81 CrossRef CAS.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (b) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS PubMed; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- F. Jensen, Introduction to Computational Chemistry, John Wiley & Sons, Chichester, UK, 2006 Search PubMed.
- P. Laflamme, F. Porzio, B. Ameduri and A. Soldera, Polym. Chem., 2012, 3, 652 RSC.
- H. D. Isenberg, Clinical Microbiology Procedures Handbook, American Society for Microbiology, Washington, DC, 1992, vol. 1 Search PubMed.
- A. Mälkiä, L. Murtomäki, A. Urtti and K. Kontturi, Eur. J. Pharm. Sci., 2004, 23, 13 CrossRef PubMed.
- OLEX2: a complete structure solution, refinement and analysis program O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 976702. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01008f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |