
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extremely tough composites from fabric reinforced polyampholyte hydrogels†
Daniel R.
King
a,
Tao Lin
Sun
b,
Yiwan
Huang
c,
Takayuki
Kurokawa
b,
Takayuki
Nonoyama
b,
Alfred J.
Crosby
*a and
Jian Ping
Gong
*b
aPolymer Science and Engineering Department, University of Massachusetts, Amherst, MA 01003, USA. E-mail: crosby@mail.pse.umass.edu
bFaculty of Advanced Life Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: gong@mail.sci.hokudai.ac.jp
cGraduate School of Life Science, Hokkaido University, Sapporo 060-0810, Japan
First published on 10th August 2015
Abstract
Ligaments are unique wet biological tissues with high tensile modulus and fracture stress, combined with high bending flexibility. Developing synthetic materials with these properties is a significant challenge. Hydrogel composites made from high stiffness fabrics is a strategy to develop such unique materials; however, the ability to produce these materials has proven difficult, since common hydrogels swell in water and interact poorly with solid components, limiting the transfer of force from the fabric to the hydrogel matrix. In this work, for the first time, we successfully produce extraordinarily tough hydrogel composites by strategically selecting a recently developed tough hydrogel that de-swells in water. The new composites, consisting of polyampholyte hydrogels and glass fiber woven fabrics, exhibit extremely high effective toughness (250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 J m−2), high tear strength (∼65 N mm−1), high tensile modulus (606 MPa), and low bending modulus (4.7 MPa). Even though these composites are composed of water-containing, biocompatible materials, their mechanical properties are comparable to high toughness Kevlar/polyurethane blends and fiber-reinforced polymers. Importantly, the mechanical properties of these composites greatly outperform the properties of either individual component. A mechanism is proposed based on established fabric tearing theory, which will enable the development of a new generation of mechanically robust composites based on fabrics. These results will be important towards developing soft biological prosthetics, and more generally for commercial applications such as tear-resistant gloves and bulletproof vests.
000 J m−2), high tear strength (∼65 N mm−1), high tensile modulus (606 MPa), and low bending modulus (4.7 MPa). Even though these composites are composed of water-containing, biocompatible materials, their mechanical properties are comparable to high toughness Kevlar/polyurethane blends and fiber-reinforced polymers. Importantly, the mechanical properties of these composites greatly outperform the properties of either individual component. A mechanism is proposed based on established fabric tearing theory, which will enable the development of a new generation of mechanically robust composites based on fabrics. These results will be important towards developing soft biological prosthetics, and more generally for commercial applications such as tear-resistant gloves and bulletproof vests.
Conceptual insightsThis work demonstrates that robust composite materials can be fabricated by systematically combining isotropic, tough hydrogels, and woven fabrics. Fabrics have been used extensively for millennia, yet their impact in modern material research has been minimal. By combining specially designed hydrogels with simple fabrics, we have produced composites which vastly outperform the mechanical properties achievable by neat hydrogel materials. The anisotropy introduced by the fabric allows for high tensile and low bending stiffness, and the combination of the tough hydrogel and fabric results in extremely high toughness and tear resistance. For many biological applications, creating isotropic materials is not desirable; indeed, many biological tissues themselves exhibit anisotropic properties. We therefore see this method of combining an isotropic, tough hydrogel with an anisotropic foundation material as a general framework for producing water-containing materials with superior, tunable, mechanical properties. |
For over a hundred years, scientists have attempted to develop materials and methods which result in functional, soft biological components. However, many of the required characteristics are contradictory; for example, they must be soft and slippery, yet capable of supporting large loads, while containing water to increase biocompatibility.1 A range of rigid materials, including metals,2 stiff fabrics,3–6 and synthetic polymers,7–10 have been proposed for ligament and tendon prosthetics. These materials have not found widespread acceptance due to their poor biocompatibility and fatigue resistance.5,11–13 In contrast hydrogel materials possess high water content, offering similar characteristics to biological materials.14–19 However, hydrogels are often brittle and lack the high modulus and ultimate tensile strength required for load bearing applications.1 To overcome these limitations, double network hydrogels have been developed that retain the benefits of traditional hydrogels (i.e. high water content and low friction), while simultaneously providing improved toughness and tensile strength.20–25 It was recently discovered that by introducing a secondary polymer system with reversible cross links, a gel’s toughness is further increased.26 One approach to take advantage of this mechanism is with polyampholyte gels, synthesized from equal amounts of oppositely charged monomers. Owing to the random distribution of monomers, the opposing charges on the polyampholyte forms multiple ionic bonds, both intra- and inter-chain, of widely varying strength. The strong bonds work as permanent cross links, imparting elasticity to the hydrogel, and the weak bonds break by loading and reform by unloading, serving as reversible sacrificial bonds. This leads to soft and wet materials with high toughness, with the added ability to be produced in one step. In addition, they are also non-toxic and biocompatible.27,28 While these new materials provide improved mechanical properties over previous hydrogel designs, they are much softer than some load-bearing biological tissues, such as ligaments. Furthermore they exhibit isotropic mechanical properties, contrasting with ligaments which are anisotropic and have extremely high tensile moduli and bending flexibility.
A new material concept is required to develop materials that are extremely stiff, yet flexible and biocompatible, with high water content. In this communication we aim to mimic the design of natural ligaments, which are made up primarily of collagen fibers (with modulus of the order of GPa29,30) in a matrix consisting of elastin, proteoglycans, glycolipids, cells, and water (65–70% by weight).31,32 This is achieved by strategically combining stiff, glass fiber fabrics with soft, tough hydrogels. Common hydrogels swell in water and the fully swollen hydrogels usually have poor adhesion and show low sliding friction on solid surfaces.17,33 Therefore, one would expect poor mechanical properties from composites of rigid fabrics and soft matrices due to weak interfacial interactions. One method that has been used to create robust hydrogel-fiber composites involves melt electrospinning writing of high-porosity 3D microfiber networks as a scaffolding material within hydrogels.34 Rather than using intricate fabrication techniques, here we utilize commercially available glass fiber fabrics, along with tough polyampholyte hydrogels as the matrix phase. Polyampholyte hydrogels are specifically chosen due to their unique de-swelling behavior. De-swelling in water provides intimate contact and adhesion with the glass fabric. In addition, polyampholyte hydrogels are easily processable as a result of one-step polymerization (Fig. 1).27 Surprisingly, we find that this combination improves the effective tearing energy up to 250000 J m−2, much greater than either individual component. Furthermore, these new materials are capable of supporting nearly three times as much load as the neat fabric, with moduli of several hundred MPa, reaching the same order of magnitude as natural ligaments.31 Even with these improved mechanical properties, the composite materials remain transversely flexible. We consider that the de-swelling aided adhesion of the polyampholyte gel on the glass fabric results in two energy dissipation mechanisms for the toughening of the composite: one is a high pull-out friction of the fabric, and the other is the energy dissipation of the polyampholyte hydrogel matrix. The mechanism employed to create these hydrogel fabric composites provides an important new framework for developing high strength and extremely tough water-containing materials, which could be extremely useful for biological prosthetics.
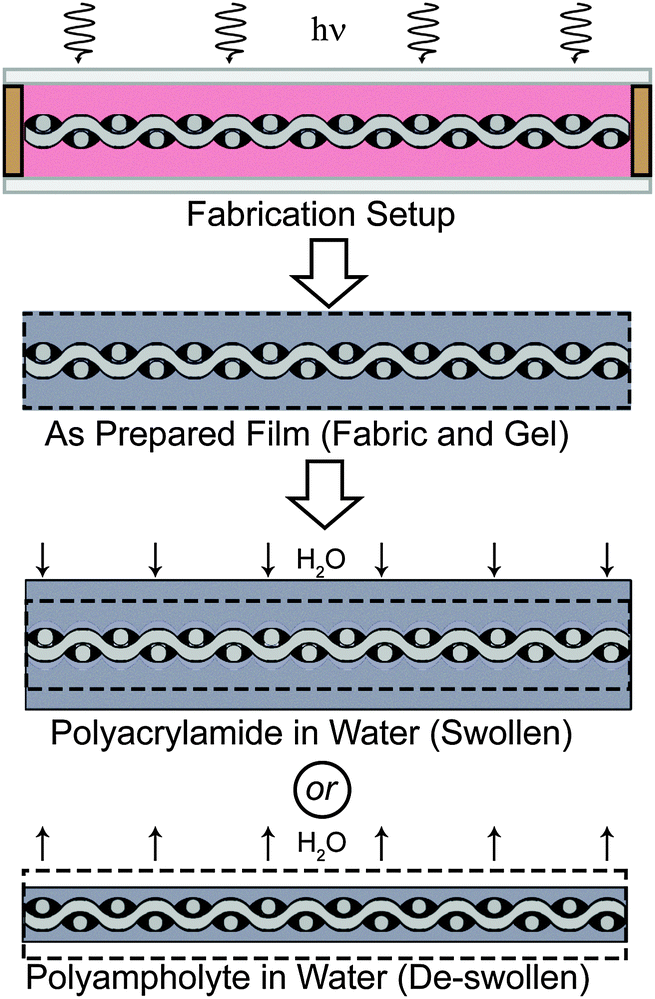 | ||
| Fig. 1 Preparation of composite samples. A fabric with spacers attached is placed in a glass mold with a monomer solution (pink), and exposed to UV light. The as-prepared film is then placed in water, and swelling or de-swelling occurs, depending on hydrogel chemistry. De-swelling, which occurs with the polyampholyte composite sample, is considered an important mechanism for fabricating mechanically superior hydrogel composites. | ||
Results
Fabrication of water-containing composites with superior mechanical properties
Glass fiber woven fabrics (effective modulus 1.3 ± 0.25 GPa) are chosen as the foundation because they are very stiff yet flexible, and have previously been used in biological prosthetics.35,36 Samples are prepared by placing 300 μm spacers on the fabric, which is inserted between two glass plates (total sample thickness, ∼1 mm, Fig. 1) to form a reaction mold. Dimethylaminoethylacrylate quaternized ammonium (DMAEA-Q) and sodium p-styrenesulfonate (NaSS) are chosen as the monomers of the polyampholyte. A formulation to obtain a tough polyampholyte hydrogel with an equal stoichiometric charge composition is chosen for the composite (see Methods section).28 After polymerization the gel composites are immersed in deionized water to reach equilibrium. During this process, the counter-ions and co-ions of the polyampholyte are dialyzed from the composite, and the sample de-swells as a result of inter- and intra- polyampholyte chain ionic bond formation.27,28 The amount of de-swelling that occurs in this system is presented in Fig. S1 (ESI†) for the neat polyampholyte hydrogel, and the glass fabric reinforced polyampholyte composite. The neat polyampholyte undergoes isotropic de-swelling, but when the fabric is introduced to reinforce the polyampholyte, de-swelling is restricted in the fabric plain direction, and therefore de-swelling only occurs in the thickness direction. For the polyampholyte hydrogels utilized here, a 30% decrease in the thickness of the composite is observed, which will play an important role in the mechanical properties of the composites. The neat polyampholyte hydrogel after de-swelling contains 50 wt% water, and shows a modulus of ∼0.1 MPa, fracture stress of ∼0.4 MPa, fracture strain of ∼30, and work of extension of ∼4 MJ m−3, which is comparable to super-tough double network gels.28 Two control groups are also prepared: a composite of traditional single network polyacrylamide hydrogels and glass fabrics (synthesis and preparation details are available in the ESI†) and neat glass fabrics (used as received).Extreme tear strength and tearing energy
First, trouser tear tests are performed to measure the toughness of the materials.23,27,37,38 Samples are prepared approximately 50 mm in length, with widths of 10 mm, 20 mm, and 40 mm. A crack is placed nominally at the center of the samples using a rotary cutter. One “leg” of the sample is attached to the base of an Instron tensile tester, and the other leg is attached to the crosshead (Fig. 2A). The crosshead is displaced at 50 mm min−1, and the load is measured, until the sample fails (Fig. 2B). A 500% increase in maximum tear strength is exhibited by the polyampholyte composite compared to the neat fabric. Interestingly, the tearing strength of the polyacrylamide composite is much less than the neat fabric. The nature of the hydrogel in the composite greatly influences the tear strength of the materials.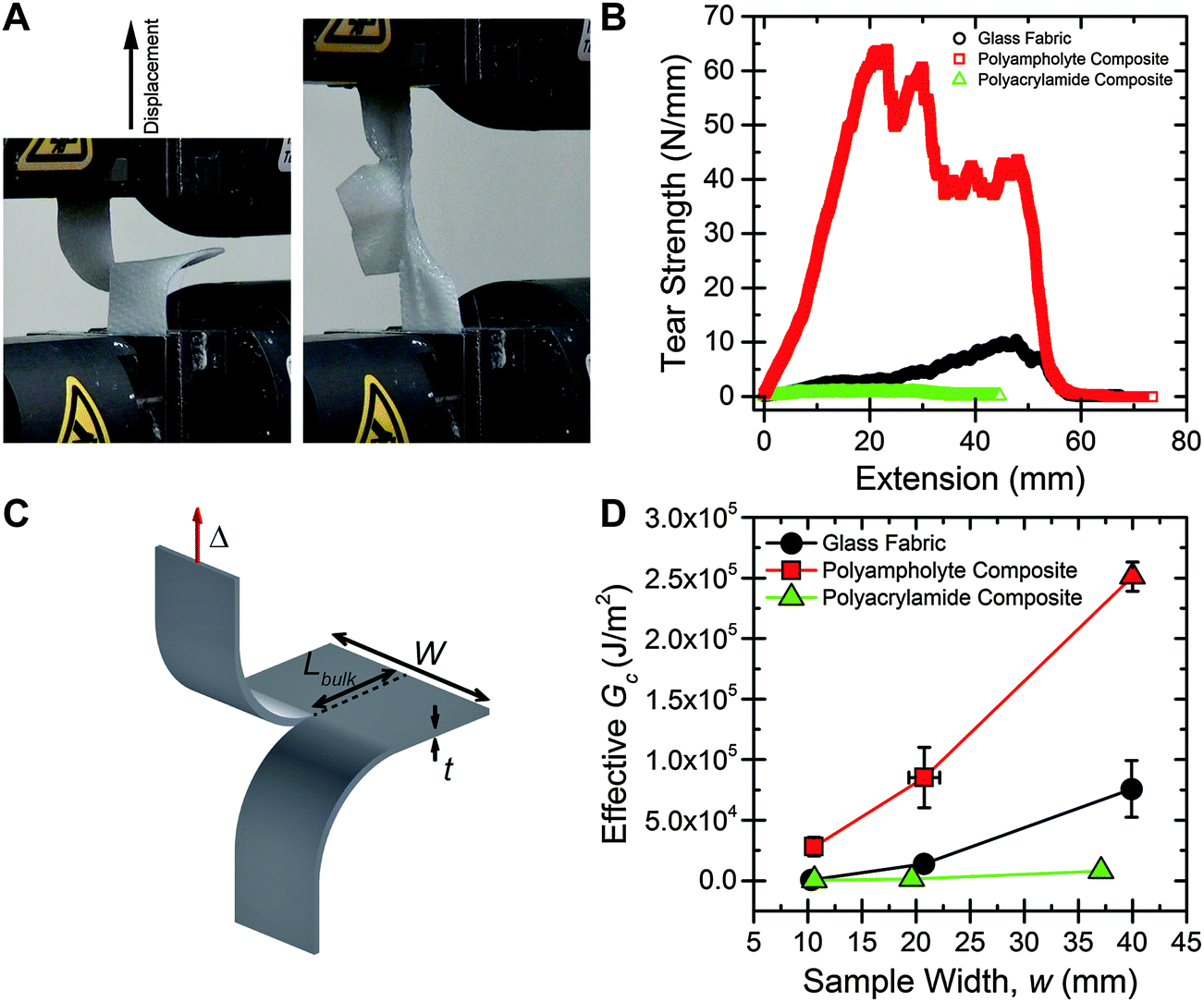 | ||
| Fig. 2 (A) A polyampholyte hydrogel composite in the grips of a tensile tester before a tearing test, and while undergoing large strain deformation in a tearing test. (B) Representative tear strength vs. displacement curves for the neat fabric, polyacrylamide hydrogel composite, and polyampholyte hydrogel composite samples, with w = 20 mm. The polyampholyte composite exhibits much higher tear strength than the two control groups. (C) Schematic of the tear test specimen, with important dimensions labelled. (D) Effective Gcvs. sample width for the three test groups. As width increases tear strength increases for all samples, due to the fiber pull-out failure mechanism. Error bars represent standard deviation, with N > 4. | ||
The tearing mechanism can be understood by observation of the sample during testing. ESI† Video S1, S2, and S3 show representative tearing tests for the neat fabric, polyacrylamide composite, and polyampholyte composite, respectively. For the neat fabric, the fibers in the transverse direction to the applied load quickly pull out of the weave as displacement increases, causing the sample to fail. In the polyacrylamide composite the gel fractures first, and then the fibers pull out of the weave, similar to the neat fabric sample. However, the polyampholyte composite sample appears to fail by a different mechanism. Here, the sample begins to stretch in both the legs and the bulk, and no tearing is observed until about 2 cm of displacement occurs. After tearing begins, the sample continues to stretch as the transverse fibers slowly pull out of the weave. The maximum tear strength values for the 20 mm wide polyampholyte composite samples (∼65 N mm−1) are very high; much higher than common elastomers (∼0.1 N mm−1 for polyisoprene and ∼0.08 N mm−1 for polydimethylsiloxane39), and approaching high toughness Kevlar/polyurethane blends (∼100 N mm−1), while consisting of approximately 50% water by mass.40
The energy required to tear, or an effective Gc, is calculated from trouser tear tests by integrating under the load, F, versus displacement, Δ, curve to determine the energy, and dividing by the projected area of new surface created, the thickness, t, multiplied by the length of the tear, Lbulk (geometry and dimensions shown in Fig. 2C):
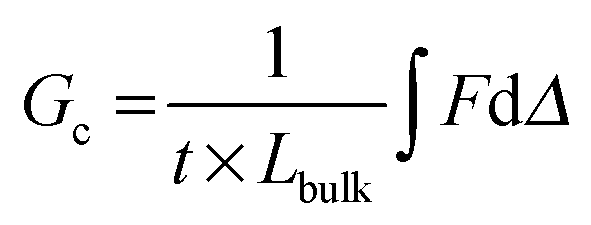 | (1) |
The Gc values exhibited by the polyampholyte composite are extremely high. For a 40 mm wide sample, a Gc of 250000 J m−2 is measured. In comparison, Gc of the neat fabric is 75000 J m−2, and Gc of the neat polyampholyte gel is about 3000 J m−2.27 From general composite theory, an averaging of mechanical properties would be expected, but in this case the composite greatly exceeds the fracture energy of either neat component. Previously, it was shown that introducing interpenetrating-network hydrogels into a woven matrix could provide a boost to mechanical properties.42 However, a dramatic increase in toughness had not been observed. Our polyampholyte composites are much tougher than other existing fiber reinforced hydrogel materials, which have Gc values ranging from 4000 to 30000 J m−2.41–43 Utilizing de-swelling, a tough polymer matrix in combination with a stiff, anisotropic foundation material is considered to be critical for creating extremely high toughness composite materials.
High load-bearing capacity
The load-bearing properties of the polyampholyte composites are also improved over the neat fabric and polyacrylamide composites. Representative load versus extension curves for the three sample materials are shown in Fig. 3A. Due to the stiffness of the glass fibers, almost all of the load is supported by the fabric. Loads can be directly compared because the maximum load occurs at a strain equal to or less than 0.08 for samples tested here, and the load supported by the gel at these strains is extremely low (about 0.075 N, Fig. S3, ESI†).28 Thus, despite differences in bulk sample thickness, because fabric geometry is similar, comparing loads represents a fair comparison between samples. The load supported by the polyampholyte composite prior to failure is nearly three times greater than that of the neat fabric (Fig. 3B). No significant change in maximum load is observed between the polyacrylamide composite and the neat fabric. Normalizing for bulk geometric properties results in a stress versus strain curve, which is presented in Fig. 3C. While the neat glass fabric exhibits high tensile strength and is very stiff, it is also very brittle, with fracture occurring at a strain of 0.02. For the hydrogel fabric composites this value is increased, with fracture strains of 0.08 and 0.04 for the polyampholyte and polyacrylamide composites, respectively. The slope of the linear portion of the stress versus strain curve is measured to determine the tensile modulus of the composites. The polyampholyte and polyacrylamide composites have tensile moduli of 606 MPa and 210 MPa, respectively, for the sample dimensions fabricated here (Fig. 3D, left columns). The resulting composites are much stiffer than the hydrogel components. For example, the modulus of the polyampholyte hydrogel is 0.1 MPa. The addition of the glass fabric increases the effective modulus by more than three orders of magnitude. It is important to note that the effective modulus of the composites can be easily tuned by changing the volume fraction of the fabric in the composite, or the percent hydration of the hydrogel. This provides a mechanism for creating robust, wet composite materials and will be explored in depth in future work.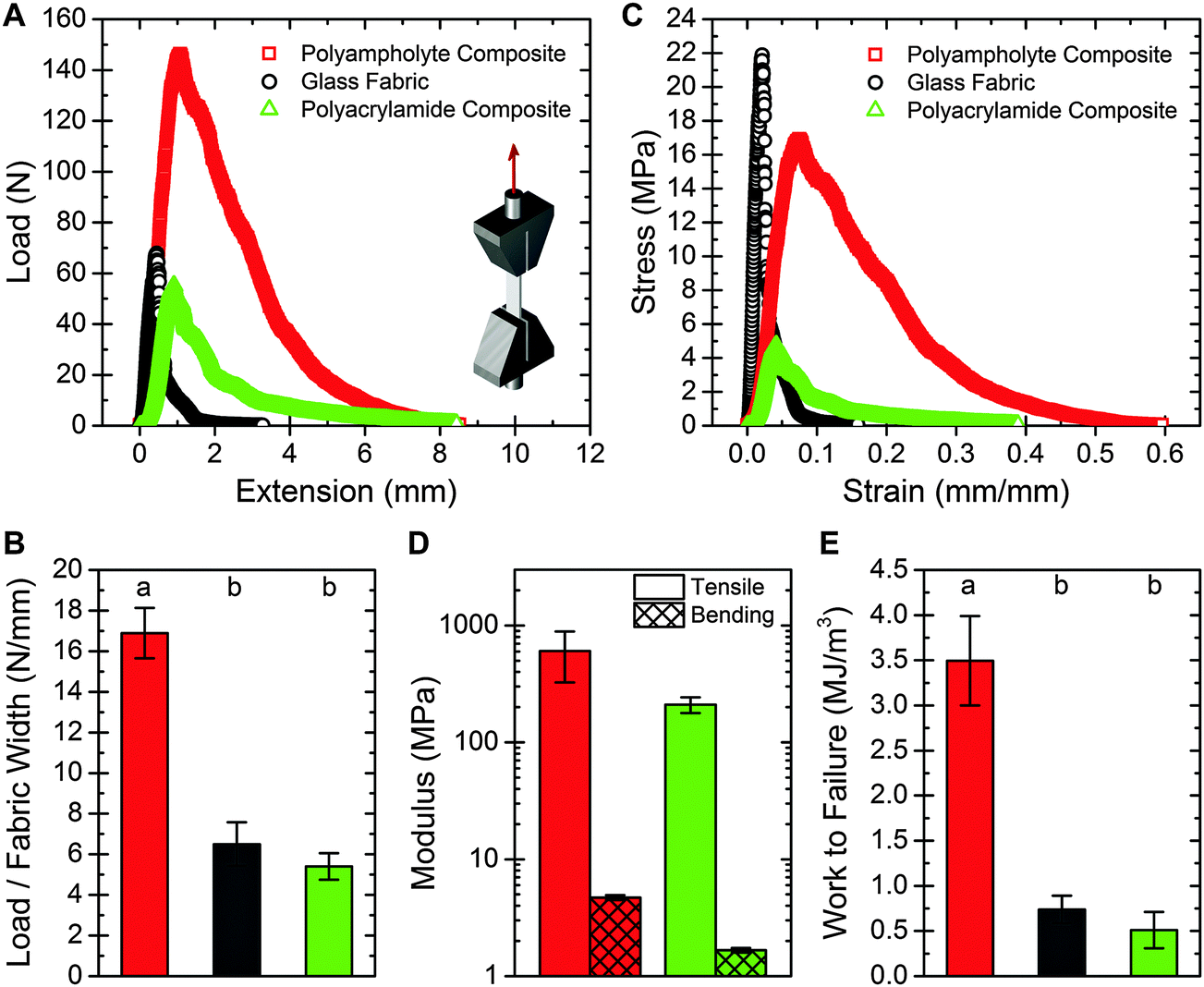 | ||
| Fig. 3 (A) Representative load vs. extension curve for the three samples tested, with t = 0.31 mm w = 10.0 mm and L = 20.5 mm for the neat fabric, t = 0.94 mm, w = 9.2 mm, and L = 14.3 mm for the polyampholyte composite, and t = 1.16 mm, w = 10.0 mm, and L = 21.7 mm for the polyacrylamide composite. Testing geometry is shown as an inset. (B) Maximum load normalized by the fabric width, for the three samples tested. The polyampholyte composite supports more than 2.5× the load per sample width when compared to the neat fabric or the polyacrylamide composite. (C) Stress versus strain curve for the samples presented in (A). (D) A comparison of the average tensile and bending modulus for the two composite samples. The fabricated composites exhibit highly anisotropic properties. (E) Work to failure for the three samples tested. The results agree with the tear strength results. For (B) and (E), ANOVA testing was used with a post-hoc Tukey test to determine significance, with P < 0.001. Error bars represent standard deviation, with N > 5. | ||
Material toughness can also be probed through tensile tests. By integrating the stress versus strain curves, the work to failure per unit volume is calculated. This failure energy for the polyampholyte composite is about seven times greater than the neat fabric or polyacrylamide composite (Fig. 3E). These findings agree with the tearing energy results, demonstrating the dramatically increased toughness of the prepared polyampholyte composites.
Due to the slippery nature of the hydrogel fabric composites, a large amount of force is required to grip the samples during tensile testing. Failure of the composites often occurred at the interface between the grip and the sample, and therefore the mechanical properties measured via tensile testing represent a lower bound; a more preferential mounting method could result in higher ultimate tensile strength and work to failure.
Highly anisotropic mechanical properties
Three-point bend tests are performed on the hydrogel fabric composites to characterize the flexibility. Bending test results for the composite materials are presented in Fig. 3D (right columns), with the polyampholyte composite and polyacrylamide composite exhibiting bending moduli of 4.7 MPa and 1.7 MPa respectively. For both composites, the bending modulus is about two orders of magnitude less than the tensile modulus, demonstrating strong anisotropy. The composite structures developed here are capable of supporting high loads and are extremely tear resistant, yet are still able to bend easily like a common elastomer, such as Sylgard 184 PDMS (Fig. S4, ESI†).35A mechanism for mechanically robust hydrogel composites
The polyampholyte composites developed here exhibit mechanical properties that have not previously been demonstrated with water-containing materials, including exceedingly high tear energy and work to failure, high load bearing capacity, and low flexural modulus. Through development of composites with glass fiber fabrics, their mechanical properties greatly exceed the properties of either neat fabric or neat hydrogel. To understand this synergistic combination, we propose a deformation model that builds upon previously known deformation mechanisms of both neat components, while introducing a new concept that leads to a general design strategy for high strength gel composites.The inherent flexible yet high strength properties of fabrics are a result of its woven structure, and are critical to designing high strength hydrogel fabric composites. Tear strength of fabrics has been researched for many years and toughening mechanisms are well understood.44,45 In a neat fabric tearing test, as the legs displace, the transverse fibers at the crack tip change orientation to align with the loading direction. As these transverse fibers undergo loading, they form a region called the del zone (Fig. 4A).44,45 In the del zone, the first fiber experiences maximum tension, while the degree of tension decreases rapidly on neighboring fibers, leaving those in the bulk relatively unperturbed. The transverse fibers in the del zone attempt to pull out of the legs, with friction between the leg fibers and transverse fibers resisting the slippage, leading to significant stretching in the del zone. When the load of the first fiber in the del zone exceeds the maximum tensile strength, the fiber breaks, and the del zone propagates into the bulk of the sample, leaving the second fiber in the del zone as the new first fiber undergoing maximum tension. This process repeats until the entire sample fails.
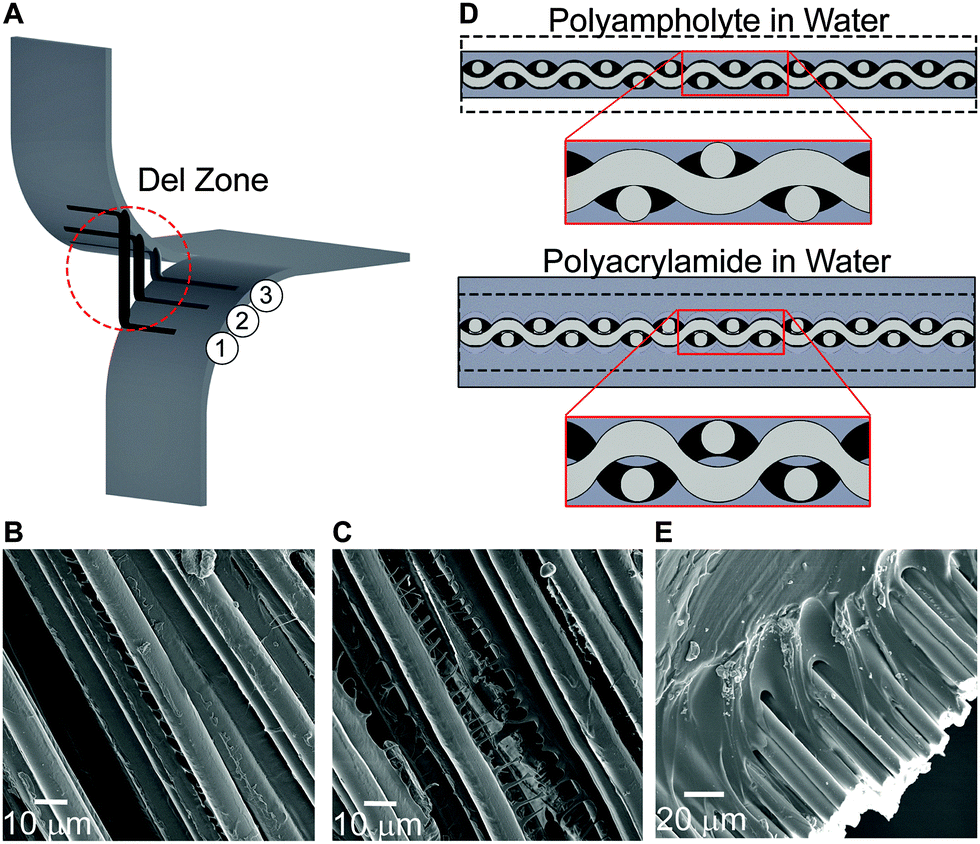 | ||
| Fig. 4 (A) A schematic of the failure mechanism of a fabric undergoing tear, emphasizing the del zone. Three fibers are shown, with fiber 1 supporting maximum tension, fiber 2 at the center of the del zone and fiber 3 just entering the del zone. The tensile strength is greater than the frictional force, resulting in fiber pullout of fibers 1 and 2. Del zone exaggerated for clarity. (B) SEM micrographs of fibers covered in the polyampholyte gel, with polyampholyte fibrils between fibers, and (C) cohesive failure of the polyampholyte gel through a fibrillation process. The results of (B) and (C) show good adhesion of the polyampholyte gel. (D) A schematic representation of the impact of de-swelling and swelling on the fabric. (E) SEM micrograph of the interface between the polyacrylamide and the glass fibers. The polyacrylamide has separated from the glass fibers. | ||
The tearing mechanism of fabrics is based on a tradeoff between fiber strength and friction from the fabric weave. Traditional fabrics are thin fibers bound in a very tight weave, where frictional force is much greater than the tensile strength of the fibers. The glass fiber fabrics used here are very stiff, and due to the sample widths tested (less than 40 mm), the frictional force generated on transverse fibers is low. Therefore, failure of the neat fabric occurs due to transverse fiber pull out, rather than fracture of the del zone fibers (Fig. 4A). To increase the tearing strength, another mechanism must account for the increased frictional forces of the del zone fibers in our composite system.
During the synthesis of the hydrogels, the fabric becomes encapsulated in a swollen gel. After polymerization and dialysis of the composite containing the polyampholyte, the hydrogel de-swells. This non-intuitive phenomenon only occurs when the true stoichiometric charge ratio is 1:1, and is the result of ionic bond formation between the oppositely charged moieties of the polymer after the dialysis of the small, mobile counter-ions and co-ions.27,28 This de-swelling mechanism also enhances the adhesion between the gel and the fibers (Fig. 4B and C). In addition, the result of de-swelling is considered to play an important role on the macroscopic length-scale: the gel applies pressure to the entire fabric, which is especially important at the weave junctions, increasing friction and resisting fiber motion (Fig. 4D top). This effectively anchors the fibers, making the pull out failure mechanism more difficult. This also allows more fibers to participate in the del zone. The combination of these events results in extremely high energy to fracture. Comparatively for polyacrylamide composites, after polymerization additional water is taken in by the hydrogel, resulting in a more greatly swollen sample. Despite containing many functional groups, due to the large amount of hydration, the polyacrylamide gel has relatively weak adhesion to solid glass surfaces (Fig. 4E).17 Additionally, the swelling process decreases the inter-fiber friction at the fabric weave junctions (Fig. 4D bottom). The importance of an appropriate swelling ratio and material toughness of the equilibrium hydrogels is prominently visible from the tearing energy plots: compared to the neat fabric, the polyampholyte composite is dramatically tougher, while the polyacrylamide is much weaker.
The equilibrium hydrogel state of the composite also plays a role in tensile tests. By compressing the fabric, the polyampholyte composite becomes stiffer. Meanwhile, the swollen polyacrylamide composite is much more compliant. Furthermore, the polyampholyte composite is capable of supporting higher loads than either the neat fabric or the polyacrylamide composites. When a fabric undergoes tension, the transverse fibers minimally influence the load bearing strength of the fabric. The de-swelling mechanism of the polyampholyte hydrogel increases the fabric weave strength, and may allow transverse load sharing, resulting in simultaneous rather than serial failure of the axially loaded fibers. Additionally, the polyampholyte hydrogels have been shown to be highly dissipative with fibrillation occurring between fibers (Fig. 4B and C), which could additionally increase tensile strength.
Based on these findings, we propose a general model for creating high stiffness, high toughness composite materials. The system must be composed of two parts, a load bearing primary foundation, in this case fabric, and a dissipative secondary matrix. The matrix should be able to completely encompass the fabric, while also volumetrically contracting to increase pressure on the fabric, thus enhancing the friction at the weave junctions. The pressure applied by the matrix in this mechanism anchors the transverse fibers in the legs, resisting fiber pull-out and tear. Utilizing a high stiffness fabric allows for high loading without foundation fracture. Additionally, the ability to dissipate energy by the matrix increases the strength of the del zone, which increases toughness. In the system presented here, volume change between the as-polymerized hydrogel and the final swollen state is implemented by preferential de-swelling of the hydrogel; however this system could be envisioned to work as well by removing volatile solvents from an elastomer polymerized in solution. Furthermore, other fabrics besides the glass fabric could be used, tuning the fiber strength to influence the tear strength and stiffness of the resulting composite. These results demonstrate the strong impact of composites developed from specially selected fabrics and hydrogels. In future work we will quantitatively discuss the influence of both macroscopic and microscopic networks to better understand the toughening mechanism of fabric-reinforced hydrogels.
Conclusion
The composites developed here are inspired by the design of natural ligaments. A large percentage of the dry weight of ligament consists of aligned fibers of collagen. Collagen is very stiff, with a modulus on the order of GPa, forming the foundation of the ligament, while other components of the ligament provide a supporting role, including hydration of the tissue. In the composites created here, the glass fabric performs the role as a stiff, load bearing, anisotropic foundation. The tough polyampholyte hydrogel acts as the remainder of the extracellular matrix, providing elasticity, toughness, and strong interfacial interaction between components. The use of a polyampholyte hydrogel which de-swells in water is probably crucial to achieving strong mechanical properties, and these properties are not seen when traditional hydrogels such as polyacrylamide is used. By controlling the modulus of the fabric and the volume fraction of the hydrogel, a high degree of tunability with respect to modulus can be achieved with this system, opening up opportunities to mimic a wide range of biological tissues. Through the framework introduced here, true ligament prosthetics is possible by intelligent composite component selection.The results shown here demonstrate a simple method to take biocompatible, soft and wet gels, and make them extremely tough, with Gc values as high as 250000 J m−2, while also being capable of supporting high loads. These results will be important in the field of soft biological prosthetics, and more generally for applications such as tear-resistant gloves, bulletproof vests, or puncture-resistant tires. Importantly, the model presented in this communication is expected to work as well for elastomer-based composites, which opens up a simple, one-step method to make high tear strength materials.
Methods
Polyampholyte composite fabrication
Samples are prepared by placing 300 μm spacers on the fabric (satin weave 8.9 oz., purchased from US Composites), which are inserted between two glass plates (total sample thickness, ∼1 mm, Fig. 1) to form a reaction mold. A 2 M solution of dimethylaminoethylacrylate quaternized ammonium and sulfonated polystyrene (0.48:0.52 charge ratio in feed to give 1:1 true stoichiometric charge ratio28) is prepared using deionized water with 0.1 mol% ketoglutaric acid as an initiator, 0.1 mol% methylene bisacrylamide as a chemical crosslinker, and an excess of sodium chloride to shield the charged monomers. The solution is mixed and heated until completely dissolved. The solution is then placed in a glove box with the sample mold. The solution is injected into the mold and cured under UV light for 12 hours. After polymerization the gel composites are placed in deionized water for at least four days to allow the gel to reach equilibrium. During this process, the small counter-ions and co-ions are dialyzed from the material and the polyampholytes form tough gel as a result of inter- and intra-chain ionic bond formation.
Polyacrylamide composite (control group) fabrication
Samples are prepared by placing 300 μm spacers on the fabric (Satin Weave 8.9 oz., purchased from US Composites), which are inserted between two glass plates (total sample thickness, ∼1 mm, Fig. 1). A 2 M solution of acrylamide is prepared in deionized water with 0.1 mol% VA-086 initiator (Wako Pure Chemical Industries, Ltd) and 1 mol% methylene bisacrylamide chemical as a crosslinker. The solution is degassed and injected into the sample mold, then placed in a glove box filled with nitrogen gas. The sample is cured under UV light (hand lamp) for 20 minutes on each side. After polymerization the gel composite is placed in deionized water for at least four days to allow the gel to reach equilibrium.Tearing tests
Samples are prepared approximately 50 mm in length, with widths of 10 mm, 20 mm, and 40 mm. A crack is placed nominally in the center of the sample with a rotary cutter. Tests are performed in the open atmosphere. One leg is attached to the base, and the other leg is attached to the crosshead, which is displaced at 50 mm min−1.Tensile tests
10 mm wide samples are prepared, and the length of the sample is recorded as the distance between grips. Tests are performed in the open atmosphere. Due to the aligned fibers in the composite samples, rectangular samples are used rather than dog bone shaped samples. Mechanical grips are used, and the crosshead is displaced at 10 mm min−1.Three point bend tests
Three point bend tests are performed on a custom built testing apparatus. The sample width is recorded (approximately 7.5 mm), and the length between bottom points is 20 mm. The testing rate is 50 μm s−1. Tests are performed in the open atmosphere.Scanning electron microscopy
Micro-scale observation was carried out by Scanning Electron Microscopy (SEM) (JSM-6010LA, JEOL, Tokyo, Japan). Samples are prepared for scanning electron microscopy analysis by solvent removal during a tearing test with a 20 mm wide sample. After the crosshead has displaced approximately 1 cm, the sample is submerged in acetone, which results in dehydration of the hydrogel composites. The samples are then dried in an oven. The samples are immobilized on brass hammer mounts using double-stick carbon tape. The samples are treated in an ion sputtering machine (E-1010, Hitachi, Tokyo Japan) with an aurum–palladium target to increase conductivity. Acceleration voltage varied from 3 to 10 kV.Acknowledgements
D.R.K. would like to acknowledge H. Yin and T. Ouchi for help with sample preparation, and A. Ihsan and T. Sullivan for experimental support. This research was financially supported by a Grant-in-Aid for Scientific Research (S) (No. 124225006) from Japan Society for the Promotion of Science (JSPS), and the Human Frontiers Science Program.References
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- E. M. Corner, Proc. R. Soc. Med., 1914, 7, 120 CAS.
- D. H. R. Jenkins, J. Bone Jt. Surg., Br. Vol., 1978, 60, 520 Search PubMed.
- D. Mendes, M. Iusim, D. Angel, A. Rotem, D. Mordehovich, M. Roffman, S. Lieberson and J. Boss, J. Biomed. Mater. Res., 1986, 20, 699 CrossRef CAS PubMed.
- A. A. Amis, S. A. Kempson, J. R. Campbell and J. H. Miller, J. Bone Jt. Surg., Br. Vol., 1988, 70, 628 CAS.
- G. H. Altman, R. L. Horan, H. H. Lu, J. Moreau, I. Martin, J. C. Richmond and D. L. Kaplan, Biomaterials, 2002, 23, 4131 CrossRef CAS.
- M. Scharling, Acta Orthop. Scand., 1981, 52, 575 CrossRef CAS.
- J. Roth, J. Kennedy, H. Lockstadt, C. McCallum and L. Cunning, Am. J. Sports Med., 1985, 13, 321 CrossRef CAS PubMed.
- L. Dürselen, L. Claes, A. Ignatius and S. Rübenacker, Biomaterials, 1996, 17, 977 CrossRef.
- R. Mascarenhas and P. MacDonald, McGill J. Med., 2008, 11, 29 Search PubMed.
- D. H. R. Jenkins and B. McKibbin, J. Bone Jt. Surg., Br. Vol., 1980, 62, 497 Search PubMed.
- B. Moyen and J.-L. Lerat, J. Bone Jt. Surg., Br. Vol., 1994, 76, 173 CAS.
- J. W. Freeman, M. D. Woods, D. A. Cromer, E. C. Ekwueme, T. Andric, E. A. Atiemo, C. H. Bijoux and C. T. Laurencin, J. Biomech., 2011, 44, 694 CrossRef PubMed.
- K. Miller, J. E. Hsu and L. J. Soslowsky, Comprehensive Biomaterials, 2011, pp. 257–279 Search PubMed.
- Y. Osada and J. P. Gong, Adv. Mater., 1998, 10, 827 CrossRef CAS.
- T. Tominaga, N. Takedomi, H. Biederman, H. Furukawa, Y. Osada and J. P. Gong, Soft Matter, 2008, 4, 1033 RSC.
- J. P. Gong, Soft Matter, 2006, 2, 544 RSC.
- J. P. Gong, T. Kurokawa, T. Narita, G. Kagata, Y. Osada, G. Nishimura and M. Kinjo, J. Am. Chem. Soc., 2001, 123, 5582 CrossRef CAS.
- J. Cui, M. A. Lackey, A. E. Madkour, E. M. Saffer, D. M. Griffin, S. R. Bhatia, A. J. Crosby and G. N. Tew, Biomacromolecules, 2012, 13, 584 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS PubMed.
- K. Yasuda, J. P. Gong, Y. Katsuyama, A. Nakayama, Y. Tanabe, E. Kondo, M. Ueno and Y. Osada, Biomaterials, 2005, 26, 4468 CrossRef CAS PubMed.
- H. R. Brown, Macromolecules, 2007, 40, 3815 CrossRef CAS.
- Y. Tanaka, R. Kuwabara, Y.-H. Na, T. Kurokawa, J. P. Gong and Y. Osada, J. Phys. Chem. B, 2005, 109, 11559 CrossRef CAS PubMed.
- B. J. DeKosky, N. H. Dormer, G. C. Ingavle, C. H. Roatch, J. Lomakin, M. S. Detamore and S. H. Gehrke, Tissue Eng., Part C, 2010, 16, 1533 CrossRef CAS PubMed.
- D. Myung, D. Waters, M. Wiseman, P.-E. Duhamel, J. Noolandi, C. N. Ta and C. W. Frank, Polym. Adv. Technol., 2008, 19, 647 CrossRef CAS PubMed.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 1 CrossRef PubMed.
- A. Bin Ihsan, T. L. Sun, S. Kuroda, M. A. Haque, T. Kurokawa, T. Nakajima and J. P. Gong, J. Mater. Chem. B, 2013, 1, 4555 RSC.
- M. P. E. Wenger, L. Bozec, M. A. Horton and P. Mesquida, Biophys. J., 2007, 93, 1255 CrossRef CAS PubMed.
- S. Strasser, A. Zink, M. Janko, W. M. Heckl and S. Thalhammer, Biochem. Biophys. Res. Commun., 2007, 354, 27 CrossRef CAS PubMed.
- S. L.-Y. Woo, S. D. Abramowitch, J. C. Loh, V. Musahl and J. H.-C. Wang, Functional Tissue Engineering, 2003, pp. 17–34 Search PubMed.
- S. L.-Y. Woo, T. Q. Lee, S. D. Abramowitch and T. W. Gilber, Basic Orthopaedic Biomechanics & Mechano-Biology, 2005, pp. 301–342 Search PubMed.
- S. Rose, A. Prevoteau, P. Elzière, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382 CrossRef CAS PubMed.
- J. Visser, F. P. W. Melchels, J. E. Jeon, E. M. van Bussel, L. S. Kimpton, H. M. Byrne, W. J. A. Dhert, P. D. Dalton, D. W. Hutmacher and J. Malda, Nat. Commun., 2015, 6, 6933 CrossRef CAS PubMed.
- S. A. Pendergraph, M. D. Bartlett, K. R. Carter and A. J. Crosby, ACS Appl. Mater. Interfaces, 2012, 4, 6640 CAS.
- S. Ramakrishna and J. Mayer, Compos. Sci. Technol., 2001, 61, 1189 CrossRef CAS.
- R. S. Rivlin and A. G. Thomas, J. Polym. Sci., 1953, 10, 291 CrossRef CAS PubMed.
- M. W. Keller, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2007, 17, 2399 CrossRef CAS PubMed.
- A. N. Gent and R. H. Tobias, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 2051 CrossRef CAS PubMed.
- S. K. N. Kutty and G. B. Nando, J. Appl. Polym. Sci., 1991, 43, 1913 CrossRef CAS PubMed.
- S. Lin, C. Cao, Q. Wang, M. Gonzalez, J. E. Dolbow and X. Zhao, Soft Matter, 2014, 10, 7519 RSC.
- I.-C. Liao, F. T. Moutos, B. T. Estes, X. Zhao and F. Guilak, Adv. Funct. Mater., 2013, 23, 5833 CrossRef CAS PubMed.
- A. Agrawal, N. Rahbar and P. D. Calvert, Acta Biomater., 2013, 9, 5313 CrossRef CAS PubMed.
- W. A. Scelzo, S. Backer and M. C. Boyce, Text. Res. J., 1994, 64, 291 CrossRef PubMed.
- W. A. Scelzo, S. Backer and M. C. Boyce, Text. Res. J., 1994, 64, 321 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mh00127g |
| This journal is © The Royal Society of Chemistry 2015 |