
Organogels for low-power light upconversion†
Roberto
Vadrucci
,
Christoph
Weder
and
Yoan C.
Simon
*
Adolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, CH-1700 Fribourg, Switzerland. E-mail: yoan.simon@unifr.ch; christoph.weder@unifr.ch
First published on 6th November 2014
Abstract
We herein report new organogels that permit efficient optical upconversion (UC) by triplet–triplet annihilation. The materials studied consist of a liquid organic phase, composed of a mixture of N,N-dimethylformamide and dimethyl sulfoxide in which the UC chromophore pair Pd(II) mesoporphyrin IX and 9,10-diphenylanthracene was dissolved, and a three-dimensional polymer network formed by covalently cross-linking poly(vinyl alcohol) with hexamethylene diisocyanate. The new gels are highly transparent, shape-persistent, and display efficient green-to-blue upconversion with UC quantum yields of >0.6 and 14% under ambient and oxygen-free conditions, respectively. The design approach presented here permits the fabrication of a hitherto unexplored class of materials with a unique combination of properties. The framework can easily be extended to other materials based on other solvents, polymer networks, and/or chromophore pairs.
Conceptual insightsOptical upconversion by triplet–triplet annihilation is a process that is potentially useful for a plethora of applications that range from solar harvesting to in vivo bioimaging. While this effect had been demonstrated in organic solutions already over 50 years ago, its adaptation to the solid state is more recent. However, the quantum efficiency of solid-state upconverting materials is usually low. We herein report upconverting organogels, which combine the best features of upconverting solutions and solids, i.e., high quantum efficiency, transparency, low dye-content, shape persistence, and considerable oxygen tolerance. The design approach appears to be general and applicable to other materials. |
Triplet–triplet annihilation upconversion (TTA-UC) is an intriguing sequence of photophysical processes that collectively transform low-power density incident light into blue-shifted radiation by combining the energy of two photons.1,2 The mechanism is useful for various applications that range from soft actuators,3 to solar harvesting,4 to in vivo bioimaging.5,6 Materials displaying TTA-UC usually comprise two organic dyes, a sensitizer, which harvests incident light and upon intersystem crossing forms triplet excited states, and an emitter, to which these triplets are transferred and which, after an annihilation process, is responsible for delayed fluorescence from its singlet excited state (Fig. 1). TTA-UC involves Dexter-type triplet energy transfer steps that require close proximity between the involved molecules.7 Accordingly, significant UC emission is observed in liquid solutions8–11 as well as rubbery polymers,12–15 even at low dye concentrations, on account of appreciable translational mobility of the chromophores in such systems. By contrast, high dye contents are typically required to achieve upconversion in glassy materials, where exciton diffusion between densely packed chromophores is believed to compensate the lack of molecular mobility.16–21 However, a review of the pertinent literature suggests that the TTA-UC quantum yield in the most efficient solid state materials is generally much lower than in dilute liquid solutions of the same chromophores.22–24 Various hard-shell soft/liquid-core particles have recently been reported with the objective to combine the upconverting characteristics provided by a high mobility core with the mechanical stability of a rigid polymeric shell.5,25,26 The latter also can serve as a barrier for O2, which is an efficient triplet quencher and substantially reduces the UC quantum efficiency.27
 | ||
| Fig. 1 Energy-level diagram displaying energy states and electronic processes occurring during sensitized triplet–triplet annihilation upconversion (TTA-UC). ISC: intersystem crossing, TTET: triplet–triplet energy transfer, TTA: triplet–triplet annihilation, GS: ground state, 1S*, 1E*: first singlet excited states of the sensitizer S and the emitter E, 3S*, 3E*: first triplet excited states of these species. Green light with a 543 nm wavelength is used for excitation, red sensitizer phosphorescence (667 nm) is observed in case of incomplete energy transfer, and blue fluorescence (433 nm) is observed in case of successful TTA-UC. | ||
Herein we present a novel design concept for upconverting organogels, which combine several attractive features of upconverting solutions and solids, i.e., high quantum efficiency and shape persistence. The materials consist of a continuous polymer network and a liquid organic phase in which the upconverting chromophores are dissolved (Fig. 2). This architecture proves to be ideal for efficient UC by providing simultaneously good mechanical integrity, high molecular mobility, low dye content and efficient light harvesting. Furthermore, we speculated that the use of DMF and/or DMSO, which both exhibit low oxygen solubility (equilibrium concentration = 0.65 mM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and 0.33 mM,29 respectively), as the liquid phase may impart substantial tolerance against triplet–quenching by oxygen.
28 and 0.33 mM,29 respectively), as the liquid phase may impart substantial tolerance against triplet–quenching by oxygen.
 | ||
| Fig. 2 Graphic representation of the structure of the upconverting organogels studied and chemical structures of the constituents used. | ||
High optical quality gels were created by cross-linking poly(vinyl alcohol) (PVOH) in a 2:1 w/w DMF–DMSO mixture by reaction with hexamethylene diisocyanate (HMDI).30 This was readily achieved, either under ambient or under air-free conditions, by vigorous mixing of a DMSO solution containing PVOH with a DMF solution containing HMDI and optionally the upconverting chromophore pair (Fig. 2). Initially, we investigated organogels with a PVOH content of 5 wt% and cross-linking 10 mol% of the PVOH hydroxyl-groups. The mixture was then filled into 10 × 10 mm fluorescence cuvettes or poly(tetrafluoroethylene) moulds, and left to cure under ambient conditions for at least 1.5 h. The progress of the gelation reaction was monitored by FT-IR spectra (ESI Fig. S1†), which revealed that the characteristic isocyanate band of HMDI (2270 cm−1) had completely disappeared after 20 min of reaction time. The resulting PVOH–HMDI gels were highly transparent and exhibited a transmittance of >95% in the visible range (ESI Fig. S4†).
The selection of the sensitizer/emitter pair was inspired by a plethora of studies that employed palladium(II) octaethylporphyrin (PdOEP) and 9,10-diphenylanthracene (DPA) to achieve green-to-blue upconversion.12,13,31,32 To promote the solubility of the porphyrin in the DMF–DMSO mixture and to limit aggregation of this sensitizer, the dicarboxylate derivative palladium(II) mesoporphyrin IX (PdMesoIX) (Fig. 2) was employed instead of PdOEP. PdMesoIX exhibits UV-vis absorption and emission spectra that are identical to those of PdOEP (ESI Fig. S5,† and 3) and the two porphyrins have comparable phosphorescence lifetimes (PdMesoIX in THF: 1.33 ms at 298 K,33 PdOEP in toluene: 0.65 ms at 295 K (ref. 34)). Thus, we surmised that PdMesoIX can be used to sensitize the blue-light emitting DPA (Fig. 3) and that the upconversion characteristics of this hitherto unexplored sensitizer/emitter pair are similar to those of PdOEP–DPA.
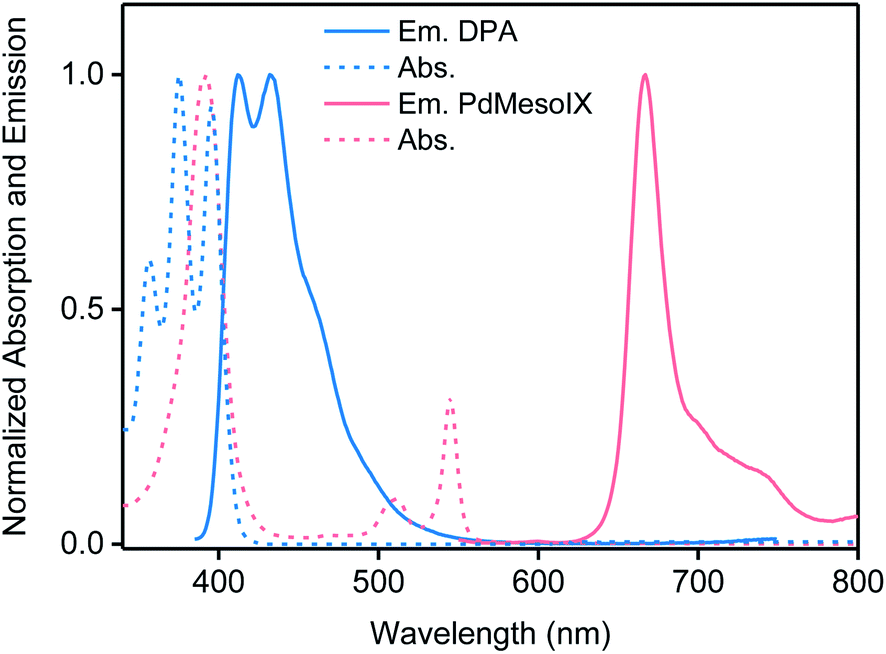 | ||
| Fig. 3 Normalized absorption (dotted lines) and emission (solid lines) spectra of PVOH organogels prepared under ambient conditions containing only DPA (blue) or PdMesoIX (red), both of a concentration 2 × 10−5 M. Emission spectra were recorded by exciting the samples at 375 (DPA-containing gel) and 543 nm (PdMesoIX-containing gel). | ||
To validate this hypothesis, a degassed solution containing PdMesoIX (2 × 10−5 M) and DPA (10−2 M) in a 2:1 w/w DMF–DMSO mixture was prepared and the upconverted emission upon excitation with a green laser emitting at 543 nm (incident power density 180 mW cm−2) was monitored (ESI Fig. S6†). The PdMesoIX concentration was chosen to efficiently harvest the irradiated light (absorbance A ≈ 0.9) and to obtain maximum upconverted light output. The PdMesoIX:DPA ratio (1:500) was selected to maximize sensitizer-to-emitter energy transfer and minimize energy back-transfer.24 The comparison with the upconverted emission spectrum of a corresponding solution of PdOEP (2 × 10−5 M) and DPA (10−2 M) in toluene reveals identical emission spectra, which exclusively display the characteristic blue DPA emission (ESI Fig. S7†), although the high DPA concentration caused some self-absorption on the high-energy end of the spectrum. Using the reported upconversion quantum yield of 36% for a vacuum-degassed PdOEP–DPA solution in toluene11 and the fluorescence quantum yield of 91.5% for a rhodamine 101 solution in ethanol as references,35 a quantum yield of 22% was determined for the argon-degassed PdMesoIX/DPA solution in DMF–DMSO by comparison of the respective absorbances, integrated emission intensities, and refractive indices (ESI Fig. S2, Section 3.2. eqn (E1) and (E2)†). The PdMesoIX/DPA solution was subsequently used as quantum yield standard for DMF–DMSO-based gels and solutions (ESI Fig. S3 and eqn (E3) and (E4)†).
We further investigated the upconversion characteristics of the PdMesoIX/DPA pair in DMF–DMSO solution by probing the upconverted emission intensity as a function of excitation power density (ESI Fig. S8†). A log–log plot of the respective data for air-free solutions (Fig. 4b) shows two different regimes, in which the data can be fitted with two separate linear regressions. The intersection of the corresponding lines marks the transition from a quadratic (low excitation intensity, slope: 1.86 ± 0.03) to a linear regime (high excitation intensity, slope: 1.20 ± 0.01) and denotes the power density where TTA becomes the dominant decay pathway for the DPA triplets, ca. 30 mW cm−2 (Fig. 4b).3,36 The experiment was repeated with solutions prepared under ambient conditions (ESI Fig. S9†). In this case, the upconverted emission intensity was, at all excitation power densities probed, lower than that of the corresponding air-free solutions and all data fit well to a single quadratic function (Fig. 4b, slope: 1.90 ± 0.03). These results indicate that triplet quenching, likely from dissolved O2, is at play for the samples prepared under ambient conditions. However, we note that also in the case of samples prepared and handled under ambient conditions TTA-UC is observed at an excitation power density as low as 10 mW cm−2 and that the quantum yield of 10% at 180 mW cm−2 is very appreciable. This behaviour is in stark contrast to that of a corresponding solution of PdOEP (2 × 10−5 M) and DPA (10−2 M) in toluene, which, if prepared under ambient conditions displays no detectable upconversion at excitation power densities up to 350 mW cm−2 (ESI Fig. S6b†), and supports the conclusion that the DMF–DMSO mixture chosen for the present indeed contributes to a reduction of the triplet-quenching by oxygen.1
 | ||
| Fig. 4 (a) Upconverted emission spectra recorded upon excitation of a PVOH–HMDI/DMF/DMSO organogel containing PdMesoIX (2 × 10−5 M) and DPA (10−2 M). The gel was prepared under air-free conditions. The sample was excited at 543 nm with a 2 mW HeNe laser at incident power densities of between 6 and 275 mW cm−2. (b) Double logarithmic plot of the integrated upconverted emission spectra of DMF–DMSO solutions (black) and PVOH–HMDI/DMF/DMSO organogels (blue) containing PdMesoIX (2 × 10−5 M) and DPA (10−2 M). The samples were prepared under air-free (triangles) or ambient (squares) conditions. Solid lines represent least-square fits to the data. | ||
We next prepared cross-linked PVOH–HMDI/DMF/DMSO gels containing PdMesoIX (2 × 10−5 M) and DPA (10−2 M) in the same concentration as the DMF–DMSO solutions discussed above and studied the gels' upconverting capabilities in an analogous manner. Gratifyingly, the UC organogels prepared under air-free conditions exclusively displayed the characteristic blue upconverted DPA emission (Fig. 4a, full spectrum ESI Fig. S10†), even if excited at power densities as low as 6 mW cm−2. Similarly to the air-free solutions, the log upconversion intensity vs. log excitation power density plot displays quadratic (slope: 1.81 ± 0.02) and linear (slope: 1.09 ± 0.01) regimes, with a transition at a power density of ca. 100 mW cm−2 (Fig. 4b). Particularly at high-excitation densities, the process is very efficient; using degassed PdMesoIX/DPA in a 2:1 w/w DMF–DMSO solution as a reference (vide supra), we estimated a quantum yield of 14% at an excitation power density of 180 mW cm−2 (ESI Fig. S3, eqn (E3) and (E4)†). We suspect that molecular mobility is somewhat restricted by the presence of the cross-linked polymer network, which results in a lower efficiency and in a higher transition power density compared to the air-free solutions. Interestingly, no major changes of transition power densities or UC intensities in the linear regime were observed when the cross-link density of the gel was reduced to 5 or 2.5 mol% or when the PVOH content was reduced to 2.5 wt% (ESI Fig. S12 and S13†).
Most intriguingly, also the chromophore-containing gels prepared and operated under ambient conditions displayed appreciable upconversion (ESI Fig. S11,† and 5). The pictures shown in Fig. 5a and b show strikingly that the materials are shape-persistent when removed from the moulds and display, in contrast to reference gels that contain only the sensitizer (Fig. 5c) or only the emitter (Fig. 5d), bright upconverted blue emission when excited at 543 nm with a power density of 350 mW cm−2. Interestingly, a strong decay of the upconverted emission intensity over time was observed upon continuous irradiation (ca. 5 min) at 180 mW cm−2 (ESI Fig. S15†). Such decay might be attributed to local photo-bleaching (e.g. reaction of PdOEP-sensitized singlet oxygen with DPA)37,38 and/or the formation of polymer degradation products since it was not observed in the air-free gels (ESI Fig. S14†), which displayed only minor intensity reduction under the same conditions. However, the effect was fully reversible; if the sample was left to rest for 4 days and excited anew, the original upconversion properties were restored, likely on account of diffusion of degradation products out of the irradiated volume as well as diffusion of “fresh” chromophores into that zone. The initial UC-intensity peak-values of ca. 106 counts (ESI Fig. S15†) are slightly lower than the peak-values found for the corresponding solutions prepared under ambient conditions (ESI Fig. S3b and S6b†), suggesting a maximum quantum efficiency of ca. 10%. However, in view of the above-discussed transient behavior, TTA-UC intensities were measured after >4 min of continuous irradiation, estimating the quantum efficiency to 10 > Φ > 0.6% at an excitation power density of 180 mW cm−2 (ESI Fig. S3, eqn (E3) and (E4)†). Very much like the solutions prepared under ambient conditions, the TTA-UC intensity, detectable at power densities ≥30 mW cm−2, increased in a quadratic fashion (slope: 1.81 ± 0.04) with the incident power density, although a deviation towards linear behaviour appears to be at play at power densities above 100 mW cm2 (Fig. 4b). We speculate that this deviation originates from mild photobleaching taking place during the timeframe of the measurement.
 | ||
| Fig. 5 Pictures of free-standing PVOH–HMDI/DMF/DMSO organogels irradiated with a 2 mW green HeNe laser at 543 nm (power density = 350 mW cm−2). The sample shown in (a) and (b) contains PdMesoIX (2 × 10−5 M) and DPA (10−2 M) and emits blue upconverted light that is clearly visible, even under ambient illumination. The samples shown in (c) and (d) contain only PdMesoIX ((c), 2 × 10−5 M) or DPA ((d), 10−2 M) and display diffracted porphyrin phosphorescence (c) or excitation light (d). All gels were produced in a mould under ambient conditions and were probed under ambient conditions after release from the mould. | ||
Liquid nitrogen freezing of the UC gel into glasses led to the immediate suppression of upconversion and the appearance of phosphorescence (ESI Fig. S16b and c†). This disappearance thus demonstrates the importance of molecular mobility for upconversion in organogels doped with low dye concentrations. By contrast, the red phosphorescence of the sample containing only the porphyrin was not affected by freezing the gels (Fig. 5c and ESI Fig. S16a†).
Conclusions
In summary, we have presented the first examples of very efficient upconverting gels. These systems consist of a small weight fraction of a cross-linked network, which bestows the material with mechanical integrity, and a solvent phase, which provides high chromophore diffusivity and protects the process to a certain extent from oxygen quenching. Such organogels are easily accessible and can be readily be transformed in any desirable shape. The materials should lend themselves to (two-photon) 2D or even 3D photopatterning,39,40 and appear to be particularly useful for soft actuators,3 gels for guided 3D cell-growth,40 or as a matrix for upconversion enhancing photonic structures, which are impossible or difficult to realize with liquid or solvent-free solid UC material.41–43 It appears that the general concept can readily be applied to gels based on other solvents, polymer networks, and chromophore systems. In particular the hygroscopic and volatile nature of the solvents employed call for further improvements. Upconverting organogels broaden the range of upconverting materials.Acknowledgements
The authors are grateful for the financial support of the Swiss National Foundation SNF (project numbers 135405 and 152968) and the Adolphe Merkle Foundation.Notes and references
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS PubMed.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed.
- Q. Liu, W. Feng, T. Yang, T. Yi and F. Li, Nat. Protoc., 2013, 8, 2033–2044 CrossRef CAS PubMed.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- C. A. Parker and C. G. Hatchard, Proc. Chem. Soc., 1962, 386–387 CAS.
- C. A. Parker, C. G. Hatchard and T. A. Joyce, Nature, 1965, 205, 1282–1284 CrossRef CAS.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
- T. N. Singh-Rachford, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2009, 131, 12007–12014 CrossRef CAS PubMed.
- Y. C. Simon, S. Bai, M. K. Sing, H. Dietsch, M. Achermann and C. Weder, Macromol. Rapid Commun., 2012, 33, 498–502 CrossRef CAS PubMed.
- A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139–143 CrossRef CAS.
- R. Vadrucci, C. Weder and Y. C. Simon, J. Mater. Chem. C, 2014, 2, 2837–2841 RSC.
- S. H. Lee, J. R. Lott, Y. C. Simon and C. Weder, J. Mater. Chem. C, 2013, 1, 5142–5148 RSC.
- A. Monguzzi, R. Tubino and F. Meinardi, J. Phys. Chem. A, 2009, 113, 1171–1174 CrossRef CAS PubMed.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303–306 CrossRef CAS PubMed.
- P. B. Merkel and J. P. Dinnocenzo, J. Phys. Chem. A, 2008, 112, 10790–10800 CrossRef CAS PubMed.
- S. H. Lee, M. A. Ayer, R. Vadrucci, C. Weder and Y. C. Simon, Polym. Chem., 2014, 5, 6898–6904 RSC.
- J.-H. Kim, F. Deng, F. N. Castellano and J.-H. Kim, Chem. Mater., 2012, 24, 2250–2252 CrossRef CAS.
- A. Monguzzi, F. Bianchi, A. Bianchi, M. Mauri, R. Simonutti, R. Ruffo, R. Tubino and F. Meinardi, Adv. Energy Mater., 2013, 3, 680–686 CrossRef CAS.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- J.-H. Kang and E. Reichmanis, Angew. Chem., Int. Ed., 2012, 51, 11841–11844 CrossRef CAS PubMed.
- C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446–450 CrossRef CAS.
- S. M. Borisov, C. Larndorfer and I. Klimant, Adv. Funct. Mater., 2012, 22, 4360–4368 CrossRef CAS.
- H. J. James and R. F. Broman, Anal. Chim. Acta, 1969, 48, 411–417 CrossRef CAS.
- C. Franco and J. Olmsted III, Talanta, 1990, 37, 905–909 CrossRef CAS.
- M. Krumova, D. Lopez, R. Benavente, C. Mijangos and J. M. Perena, Polymer, 2000, 41, 9265–9272 CrossRef CAS.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS.
- S. Baluschev, V. Yakutkin, T. Miteva, G. Wegner, T. Roberts, G. Nelles, A. Yasuda, S. Chernov, S. Aleshchenkov and A. Cheprakov, New J. Phys., 2008, 10, 013007 CrossRef.
- J. B. Callis, J. M. Knowles and M. Gouterman, J. Phys. Chem., 1973, 77, 154–157 CrossRef CAS.
- V. N. Knyukshto, A. M. Shul'ga, E. I. Sagun and É. I. Zen'kevich, Opt. Spectrosc., 2006, 100, 590–601 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- W. Fudickar, A. Fery and T. Linker, J. Am. Chem. Soc., 2005, 127, 9386–9387 CrossRef CAS PubMed.
- J. W. Chung, S.-J. Yoon, S.-J. Lim, B.-K. An and S. Y. Park, Angew. Chem., 2009, 121, 7164–7168 CrossRef.
- Y. Luo and M. S. Shoichet, Nat. Mater., 2004, 3, 249–253 CrossRef CAS PubMed.
- C. Chen, Y. Zhu, H. Bao, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2010, 2, 1499–1504 CAS.
- C. Hofmann, B. Herter, J. Gutmann, J. Löffler, S. Fischer, S. Wolf, R. Piper, N. Ekins-Daukes, N. Treat and J. C. Goldschmidt, in Photonics for Solar Energy Systems V, 2014, p. 91400H Search PubMed.
- M. Peters, J. C. Goldschmidt, P. Löper, B. Groß, J. Üpping, F. Dimroth, R. Wehrspohn and B. Bläsi, Energies, 2010, 3, 171–193 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Production of gels and solutions, characterization and supplementary experimental data. See DOI: 10.1039/c4mh00168k |
| This journal is © The Royal Society of Chemistry 2015 |