
A review of patents (2011–2015) towards combating resistance to and toxicity of aminoglycosides
Nishad Thamban
Chandrika
and
Sylvie
Garneau-Tsodikova
*
University of Kentucky, Department of Pharmaceutical Sciences, 789 South Limestone Street, Lexington, KY, USA. E-mail: sylviegtsodikova@uky.edu; Fax: 859 257 7585; Tel: 859 218 1686
First published on 19th November 2015
Abstract
Since the discovery of the first aminoglycoside (AG), streptomycin, in 1943, these broad-spectrum antibiotics have been extensively used for the treatment of Gram-negative and Gram-positive bacterial infections. The inherent toxicity (ototoxicity and nephrotoxicity) associated with their long-term use as well as the emergence of resistant bacterial strains have limited their usage. Structural modifications of AGs by AG-modifying enzymes, reduced target affinity caused by ribosomal modification, and decrease in their cellular concentration by efflux pumps have resulted in resistance towards AGs. However, the last decade has seen a renewed interest among the scientific community for AGs as exemplified by the recent influx of scientific articles and patents on their therapeutic use. In this review, we use a non-conventional approach to put forth this renaissance on AG development/application by summarizing all patents filed on AGs from 2011–2015 and highlighting some related publications on the most recent work done on AGs to overcome resistance and improving their therapeutic use while reducing ototoxicity and nephrotoxicity. We also present work towards developing amphiphilic AGs for use as fungicides as well as that towards repurposing existing AGs for potential newer applications.
Introduction
Aminoglycosides (AGs) are broad-spectrum antibiotics commonly used for the treatment of serious bacterial infections.1 AGs, which are naturally occurring and semi-synthetic amino-modified sugars, can be classified based on their structure and clinical efficacy. Structurally, AGs can be classified based on the substitution pattern of their 2-deoxystreptamine (2-DOS) ring as, 4,5-disubstituted 2-DOS AGs (e.g., neamine (NEA), neomycin B (NEO), paromomycin (PAR), and ribostamycin (RIB)), 4,6-disubstituted 2-DOS AGs (e.g., amikacin (AMK), arbekacin (ABK), dibekacin (DBK), gentamicin (GEN), geneticin (G418), kanamycin A and B (KANA and KANB), sisomicin (SIS), tobramycin (TOB), and plazomicin (PLZ)), and other monosubstituted 2-DOS AG scaffolds (e.g., apramycin (APR) and streptomycin (STR)) (Fig. 1). Clinically, AGs are used systemically or topically for treating infections caused by Gram-positive and Gram-negative bacterial strains. The AGs AMK, GEN, KANA, NEO, PAR, STR, and TOB are currently approved by the US Food and Drug Administration (FDA) and available for clinical use in the United States. The most common clinical applications of AGs are either alone or as part of combination therapy in the treatment of serious infections caused by aerobic Gram-negative bacilli, while less common is their application in combination with other agents against select Gram-positive pathogens.2–7 In addition, certain AGs have been found to display clinically relevant activity against protozoa (PAR), Neisseria gonorrhoeae (STR), and mycobacterial infections (AMK, ABK, KANA, STR, and TOB). The recent emergence of infections due to Gram-negative bacteria and enhanced bacterial resistance have prompted a revived interest in the use of AGs while addressing the bacterial resistance, nephrotoxicity, and ototoxicity problems associated with their clinical use.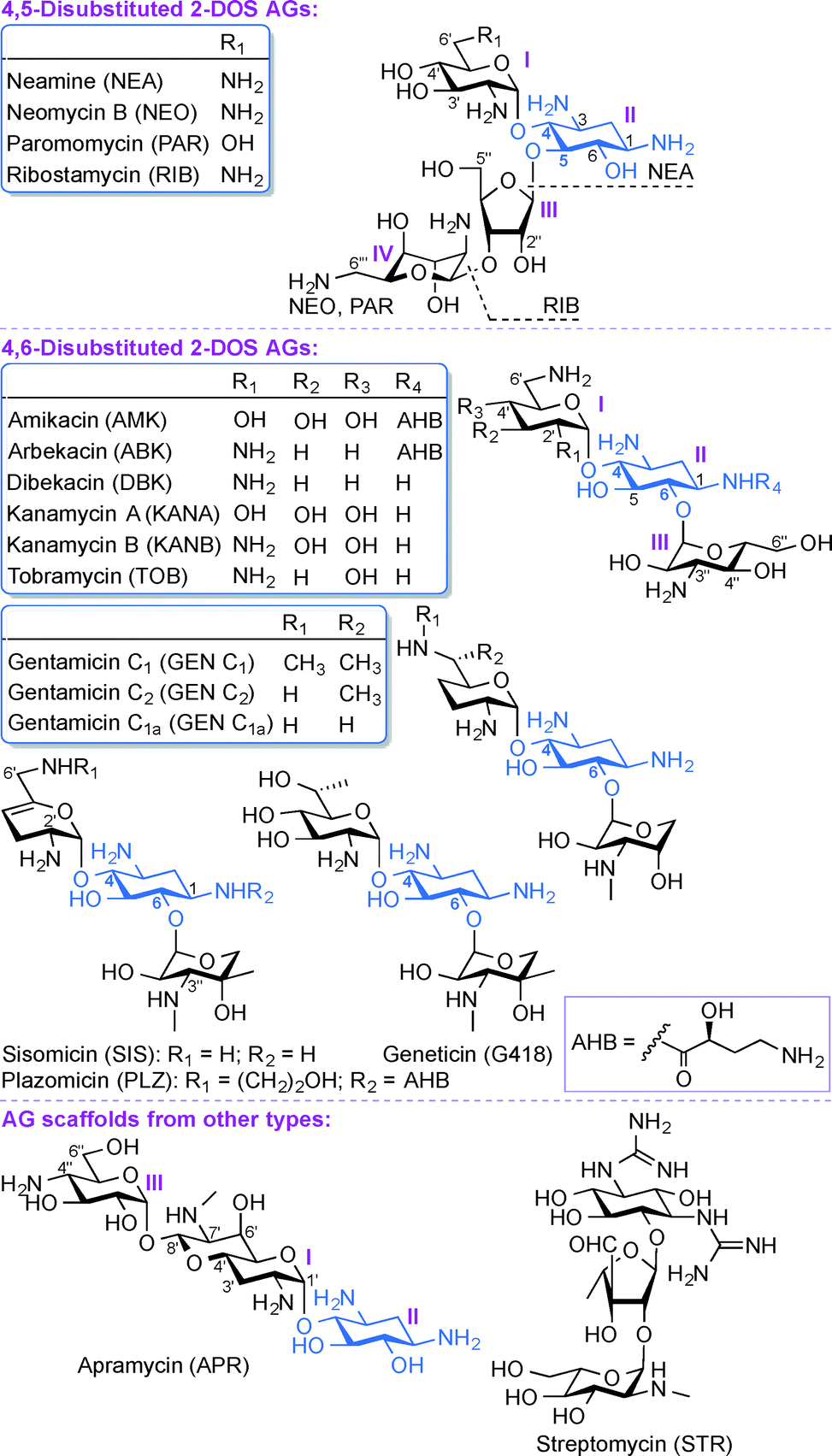 | ||
| Fig. 1 Structures of AGs presented in this review. | ||
As with any antibiotics, bacterial resistance has also contributed to the problems associated with AGs.8 The most prevalent modes of action leading to AG bacterial resistance are (i) reduction in the intracellular concentration of antibiotics by efflux pumps or through reduced membrane permeability, (ii) structural modification of the 16S ribosomal RNA (rRNA) that lead to reduced target affinity, and (iii) structural modification of AGs by AG-modifying enzymes (AMEs). The AMEs, which include AG N-acetyltransferases (AACs), AG O-phosphotransferases (APHs), and AG O-nucleotidyltransferases (ANTs) act by chemically modifying the AGs, which adversely affects their clinical efficacy.9 AACs catalyze the acetyl coenzyme A-dependent acetylation of amine groups, while APHs and ANTs catalyze the ATP and/or GTP-dependent phosphorylation and nucleotidylation of hydroxyl groups of AGs, respectively.10–12 The above classes of AMEs are typically monofunctional enzymes, but the emergence of bifunctional enzymes is another aspect relevant to the clinical use of AGs. Four genes encoding bifunctional AMEs have been identified so far: AAC(6′)-Ie/APH(2′′)-Ia from Staphylococcus aureus,13–16 ANT(3′′)-Ii/AAC(6′)-IId from Serratia marcescens,17–19 AAC(3)-Ib/AAC(6′)-Ib′ from Pseudomonas aeruginosa,20,21 and AAC(6′)-30/AAC(6′)-Ib from P. aeruginosa.22,23 Unlike other regiospecific AMEs, the newly discovered enhanced intracellular survival (Eis) protein found in Mycobacterium tuberculosis as well as other mycobacterial and non-mycobacterial strains is a versatile enzyme that can multiacetylate different amine positions of AGs.24–35
To overcome antibiotic resistance, various strategies are currently employed: (i) development of newer AG scaffolds; (ii) derivatization of existing FDA approved AGs, and (iii) repurposing existing AGs for new applications.36 In order to create more potent and less toxic AGs capable of evading resistance mechanisms, several approaches can be used, including chemical and enzymatic modification of hydroxyl and amine groups at different positions of AG scaffolds, designing structurally constrained AGs, coupling of AGs to form AG dimers, AG-lipid conjugates, and AG-drug conjugates by synthetic or semi-synthetic methods.37
To highlight the great importance of this old class of antibiotics and the regain in interest for their usage and development, herein, we decided to write a review in a non-traditional fashion by focusing on the multiple patents (19 of them!) on AGs that appeared in the last four years alone. In this review, all recent patents (filed from 2011–2015) covering synthetic modifications of AGs as well as current and potential new applications of AGs as antibacterial (15 patents), antifungal (2 patents), and antiviral (1 patent) agents, as well as their use as therapeutics for genetic disorders (1 patent) will be discussed along with scientific literature related to these patents. The main focus of these patents is on the work done to overcome resistance and increasing the potency and spectrum of activity of these AGs, thus improving their therapeutic index by decreasing their nephrotoxicity and ototoxicity.
Aminoglycosides as antibacterial agents
AGs exert their antibacterial activity by binding to bacterial ribosomes (helix 44 (h44) of the 16S rRNA or to the major groove of helix 69 (H69) of the 23S rRNA of the 50S subunit), thereby preventing the initiation of the complex process of protein synthesis.38–42 Examples of usage of AGs include: STR, which has been used extensively as a primary drug in the treatment of tuberculosis; GEN, which is active against multiple strains of Gram-positive and Gram-negative bacteria, including P. aeruginosa; KAN, which is active at low concentrations against many Gram-positive bacteria including penicillin-resistant Staphylococci; as well as GEN and TOB, which are mainstays for the treatment of Pseudomonas infections. With the emergence of multidrug-resistant (MDR) bacterial strains (and extensively-drug resistant (XDR), in the case of M. tuberculosis), many of the currently marketed AGs are not effective against numerous of these bacterial infections. There is therefore an urgent need for new and improved AG antibiotics. In order to circumvent the problems associated with antibacterial susceptibility and toxicity, extensive research was performed and is still underway to improve the bactericidal activity of AGs. This section on AGs as antibacterial agents will focus on the challenges associated with their chemical synthesis, with emphasis on (i) developing single-step derivatization as well as stereo- and regioselective modifications of AGs, (ii) reducing the number of synthetic steps involved in formation of AG scaffolds, and (iii) designing AG scaffolds based on their mechanism(s) of action in bacteria and/or aimed at improving their antibacterial activity, overcoming the action of resistance enzymes, and reducing the toxicity associated with their usage.Patent WO 2013/191550 A1 (ref. 43)
Most regioselective modifications of AGs require multistep chemical syntheses involving tedious and cost ineffective functional group protection/deprotection manipulations due to presence of numerous hydroxyl and amine groups with similar reactivity on AG scaffolds.44–48 In contrast, a more facile introduction of functionalities in structurally complex molecules generally depends on non-covalent protecting group strategies49,50 and enzymatic approaches.51In patent WO 2013/191550 A1,43 a novel one-step regioselective chemical diazotation of the C3-amine functionality of the 2-DOS ring is presented (Fig. 2A). This selective diazotation of AGs employs imidazole-1-sulfonyl azide, a shelf-stable, non-explosive, and water-soluble reagent.52 This method is cost efficient, scalable, and can be performed at neutral conditions without requiring extensive protection and deprotection chemistry. This methodology was proven successful by modifying the AGs AMK, APR, PAR, RIB, NEA, and NEO. To demonstrate the applicability of this new synthetic transformation in generating useful AG molecules, the C3-azido-NEO derivative 1 produced was further modified in 2–3 steps, and the resulting compounds were tested against a NEO-sensitive Escherichia coli strain and its resistant counterpart containing an AAC(3) enzyme (Fig. 2A). The C3-NEO derivatives were found to be active against the resistant strain, demonstrating that modification at the C3-position can overcome the action of the AAC(3) resistance enzyme. A similar observation was previously reported for AGs modified by AAC(3)-IV that were found to resist the action of AAC(3) and AAC(6′) enzymes.53 Selective transformation of the amino group at the 3-position of ring I of AGs into an azide, followed by azide modification into a moiety that cannot be N-acetylated by AAC is therefore a promising tool to protect AGs against AMEs.
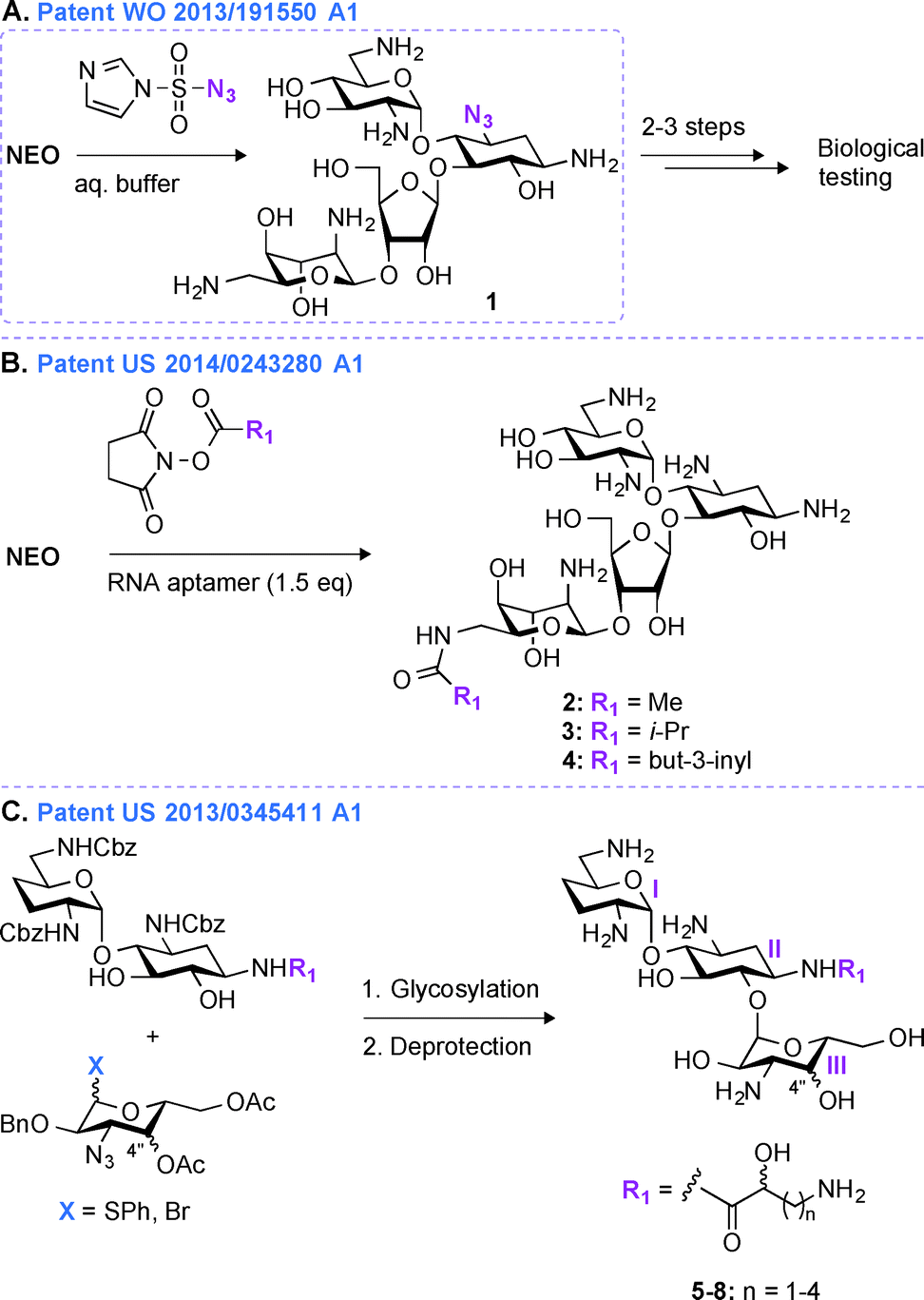 | ||
| Fig. 2 Representative scheme for the A. single-step C3-azide derivatization of AGs, B. single-step 6′′′-amide derivativation of AGs using RNA aptamers, and C. multistep synthesis of ABK derivatives from carbohydrate building blocks. | ||
Patent US 2014/0243280 A1 (ref. 54)
An alternative method for derivatizing AGs in one regioselective chemical reaction is highlighted in patent US 2014/0243280 A1 (Fig. 2B).54 The approach presented relies on the use of non-covalent supramolecular protecting groups (SPGs) based on host-guest interaction and offers a new and appealing perspective for single-step modifications of AGs. Prior to this patent, calixarenes had been reported as non-covalent protecting groups.55 However, due to the small size of the calixarenes' cavity, their use was limited to modifications of small molecules containing a maximum of two chemically equivalent functional groups. In order to overcome this drawback and to generalize this concept to AGs, in patent US 2014/0243280 A1, a novel class of SPGs based on oligonucleotides56–58 like Apt1 (23mer) and Apt2 (21mer) was developed. These RNA aptamers (1.5 equivalent) in conjunction with N-hydroxysuccinimide esters (methyl, iso-propyl, or but-3-inyl) were efficiently used for the highly chemoselective (6′′′-position) and regioselective (89 to >99%) transformation of AGs in very good yields (59–83%) (Fig. 2B). The efficacy of the compounds generated (2–4) was determined against one E. coli strain, and both NEO and PAR derivatives were found to display MIC values (6.3 μM and 12.5 μM for NEO and PAR derivatives, respectively) similar to that of their corresponding parent AGs. As the oligonucleotide-based SPGs could potentially modify a variety of AG scaffolds in one chemical step, this method could present an effective alternative for their rapid derivatization towards combating bacterial resistance. However, although single-step modifications show promise in derivatizing the 3- and 6′′′-positions of some AGs, modifications at other positions still dependent on multistep synthetic routes.Patent US 2013/0345411 A1 (ref. 59)
In patent US 2013/03455411 A1,59 a mutlistep strategy involving the use of basic carbohydrate building blocks and a glycosylation reaction between the 1′′-position of ring III and the O6 of ring II to develop novel ABK analogues is presented (Fig. 2C). Building AG analogues by using glycosylation has obviously been previously reported.60–65 In this patent, by using this method, twelve ABK analogues (5–8) were synthesized and eleven of them were tested for their antibacterial activity against nine strains of MRSA. These ABK derivatives contained a hydroxyl moiety at the 4′′-position oriented either axially or equatorially (Note: this orientation was introduced prior to glycosylation) as well as various side-chains ((S) or (R)-3-amino-2-propionate, (S) or (R)-AHB, (S) or (R)-5-amino-2-valerate, and (S) or (R)-6-amino-2-hexanoate) at the N1-position. The MIC values for these compounds were found to be 0.5–2 μg mL−1 (4–8 μg mL−1 for ABK) and 2–32 μg mL−1 (128 μg mL−1 for ABK) against ABK-sensitive and ABK-resistant MRSA, respectively. By measuring the concentration of N-acetylglucosamine in mouse urine, these compounds were found to display reduced nephrotoxicity (74.7 mIU) when compared to ABK (135.0 mIU).In the past 20 years, derivatives of ABK, originally synthesized from DBK in 1973, have also been generated via other chemical approaches. Synthetic 2′′-amino-2′′-deoxy-ABK and its 5-epiamino analogue were prepared and found to display excellent antibacterial activity against MRSA.66 The enzymatic modification of ABK by an N-acetyltransferase from Streptomyces griseus resulted in 3′′-N-acetyl-ABK, but unfortunately this compound displayed 2 to 10-fold lower activity than ABK against 59 Gram-positive and Gram-negative bacterial strains.67 The substitution of the 5-hydroxyl group of ABK by an amine moiety resulted in an analogue, which was superior to ABK against MRSA.68 Synthetic 4′′-deoxy-4′′-episubstituted ABK and 4′′-epi-5-deoxy-5-episubstituted ABK derivatives were also prepared, and among them 5,4′′-di-epi-ABK was found to be a better antibacterial agent than ABK or 5-episubstituted ABK.69 Finally, the 4′′- and 6′′-hydroxyl groups of ABK were regioselectively modified and 6′′-amino-6′′-N-[(S)-4-amino-2-hydroxybutyryl]-6′′-deoxy-ABK was found to display excellent antibacterial activity, even better than that of ABK.70
Patent WO 2011/143497 A1 (ref. 71)
A common multistep approach for derivatizing AGs consists, as previously stated, of decorating parent AG scaffolds. In patent WO 2011/143497 A1,71 140 mono- and difunctionalized derivatives of GEN were synthesized by modifying its N1- and/or N6′-positions (Fig. 3A). These compounds were tested against one representative bacterial strain of each E. coli, S. aureus, Klebsiella pneumoniae, P. aeruginosa, and Acinetobacter baumannii, which altogether contained AMEs that modify the N3- and O2′′-positions of GEN. It was observed that 1,6′-disubstitution was preferable to overcome the action of AAC(6′) enzymes in E. coli, S. aureus, P. aeruginosa, and A. baumannii. Overall, the two best GEN analogues identified were compounds 9 and 10 both containing a (S)-3-amino-2-hydroxypropynyl moiety at the 1-position and a (R)- or (S)-propane-1,2-diol at the 6′-position, respectively. In general, compound 9 was frequently found to be 2 to 4-fold more active than 10. These compounds displayed no nephrotoxicity after 14 days at doses of 60 and 30 mg kg−1 when tested in rats. When tested for acute toxicity and cytotoxicity, compound 9 was found to present less acute toxicity than 10, and cytotoxicity studies in HK-2 cells showed compound 10 to be at least 2-fold more cytotoxic than 9. | ||
| Fig. 3 Representative scheme for the preparation of A. 1,6′-dimodified GEN derivatives, B. 1,5,6′′-trisubstituted TOB derivatives (Note: 5,6′′-disubstituted AMK derivatives were also similarly produced), and C. amphiphilic TOB derivatives with various linker types at the 6′′-position. | ||
Patent US 2013/0203693 A1 (ref. 72)
Following up on the work done on derivatizing GEN in patent WO 2011/143497 A1,71 further investigations on decorating different positions of other parent AGs were performed. In patent US 2013/0203693 A1,72 structural modifications were made at the N1-, O5-, and O6′′-positions of TOB and AMK (Fig. 3B). In all cases, the final molecules contained a (S)-4-amino-2-hydroxybutyrate (AHB) group at the N1-position, whereas F, N3, and NH2 were found in place of the hydroxyl at the 5-position. Various substituents were introduced at the 6′′-position by displacing a tosyl group with structurally diverse aliphatic amines. For evaluation of their clinical efficacy, these molecules were tested against a panel of Gram-negative bacterial strains including P. aeruginosa, E. coli, K. pneumoniae, and A. baumannii. As P. aeruginosa is a prominent nosocomial pathogen and has acquired multiple mechanisms of resistance, these compounds were tested against four strains of P. aeruginosa and MIC90 values were found to be in single digit compared to ≥64 μg mL−1 for the parent AGs. When tested against four strains of K. pneumoniae and A. baumannii, the structural modifications on these compounds were shown to result in an increased potency. Compound 11 was found to be most effective against AMK-resistant P. aeruginosa in an animal model. Overall, from this patent, it can be concluded that structural modifications at the N1-, O5-, and O6′′-positions of TOB and AMK are beneficial.Patent WO 2014/013495 A1 (ref. 73)
To expand the scope of structural modifications of parent AGs, in patent WO 2014/013495 A1,73 work was done to synthesize various cationic amphiphilic derivatives of TOB, KANA, and PAR without the use of an extensive number of synthetic steps to target the negatively-charged lipopolysaccharides present in bacterial cell wall through ionic interactions (Fig. 3C).74,75 Membrane-targeting AGs offer certain advantages over antibiotics targeting intracellular components as they are not dependent on bacterial cell cycle state, can evade intracellular resistance mechanism resulting in prolonged clinical efficacy, and do not display cell permeability issue.76,77 The sugar core, number of positive charges, length of aliphatic chains, and the type of chemical bond (thioether, sulfone (12–14), sulfonyl (15–17), amides, and triazolyl (18–20)) between the hydrophobic and hydrophilic parts of the molecules were taken into account while designing these compounds. These molecules were tested against eleven strains of Gram-positive and Gram-negative bacteria and MIC values for TOB against S. pyogenes M12 was found to be 64 μg mL−1, whereas thioether, triazole, and amide derivatives were found to be 16 to 32-times more potent. The triazole derivative with a C12 chain (MIC 64 μg mL−1) and its counterpart amide (MIC 32 μg mL−1) showed improved potency compared to TOB (MIC 128 μg mL−1). The minimal biofilm inhibition concentration (MBIC) for TOB and its synthetic derivatives were found to be 64–128 and 4–32 μg mL−1, respectively. An hemolytic assay using erythrocytes from rats at concentrations 32, 64, and 128 μg mL−1 showed that there is no linear correlation between the antimicrobial potency and the hemolytic activity of the amphiphilic TOB derivatives. Overall, the optimal aliphatic chain lengths were found to be 12–16 and 4–8 carbons for mono- and disubstitution, respectively. More details about the results presented in patent WO 2014/013495 A1 can be found in the literature.78–80Patent WO 2011/044501 A2 (ref. 81)
The pharmaceutical company Achaogen was early on involved in multistep synthetic efforts towards derivatization of AGs like NEO. In patent WO 2011/044501 A2,81 in order to accelerate the process of developing new antibiotics, a chemical strategy was developed for synthesizing various NEO analogues from PAR (Fig. 4A, 21–24). A library of N1-substituted,3′-deoxy-NEO, N1-substituted,4′-deoxy-NEO, and N1-substituted,3′,4′-dideoxy-NEO analogues was synthesized by using acylation with acids or activated esters, sulfonylation, reductive amination, or epoxide opening chemistry. The MIC values were determined for a select group of analogues against one strain of P. aeruginosa and E. coli and were found to be ≤1 μg mL−1 and ≤1–16 μg mL−1, respectively.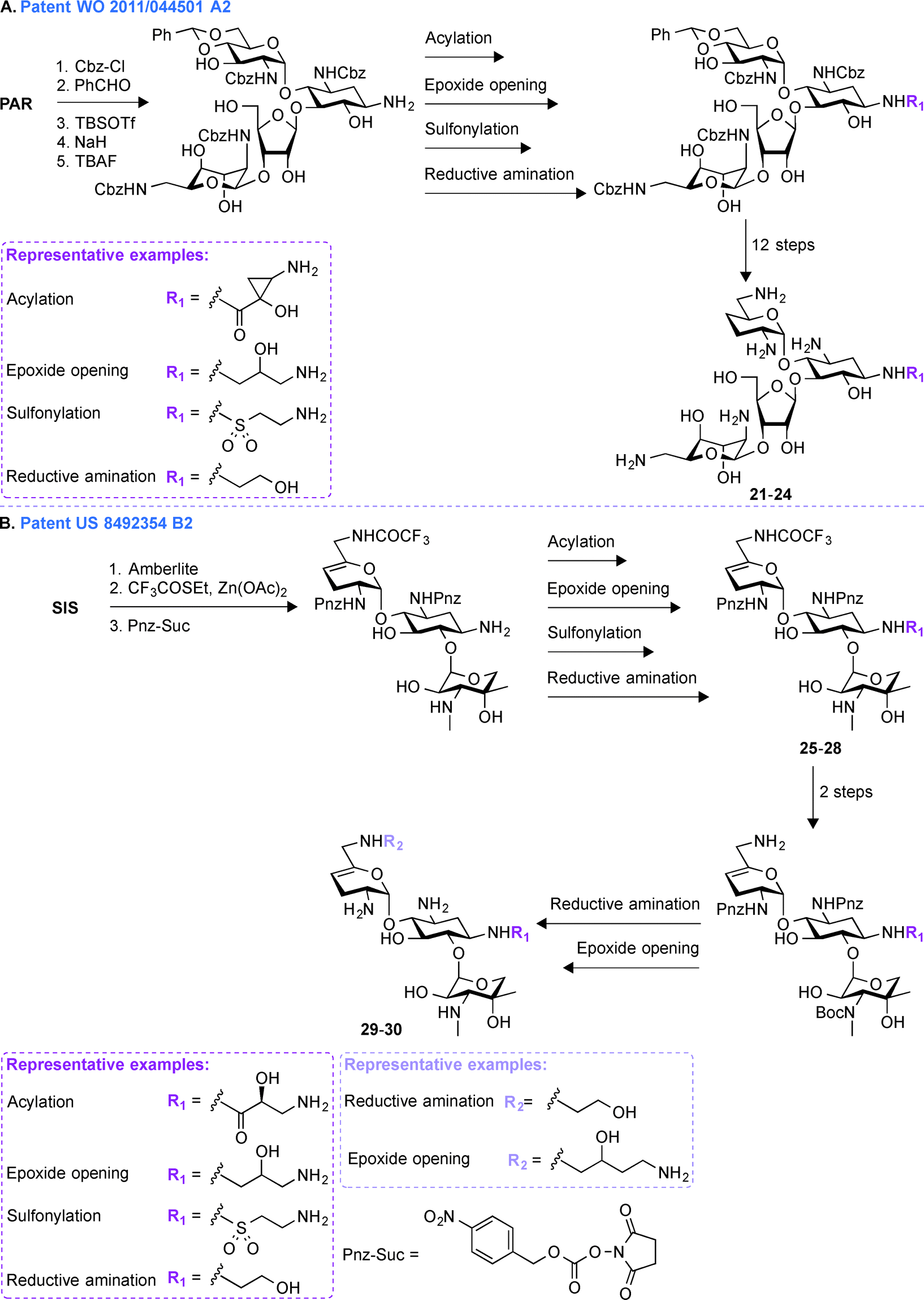 | ||
| Fig. 4 Representative scheme for the preparation of A. NEO N1-substituted,3′,4′-dideoxy-NEO analogues from PAR, and B. 1,6′-disubstituted SIS derivatives. | ||
Patent US 849354 B2 (ref. 82)
Later on, in addition to the NEO derivatives of patent WO 2011/044501 A2,81 Achaogen was successful in generating the SIS derivative PLZ (formally known as ACHN-490), which was found to evade the resistance normally associated with AGs, and more importantly to display little to no toxicity.83 In patent US 849354 B2,82 various 1,6′-disubstituted (Fig. 4B, 29–30, including PLZ) and 1,2′-disubstituted SIS derivatives were synthesized. Introduction of substituents at the 1-, 2′-, and 6′-positions was achieved by a combination of acylation (with acids or activated esters), epoxide opening, sulfonylation, and reductive amination chemistry.After the development of the very promising PLZ, which is currently in phase 3 clinical trials, Achaogen published a number of studies describing the activities of this latest addition to the AG family of drugs. In brief, PLZ was evaluated against 3050 E. coli clinical isolates and 102 MDR strains of K. pneumoniae, including a subset of 25 strains producing K. pneumoniae carbapenemase (KPC) resistance enzymes. Against K. pneumoniae, MIC50 values for GEN, TOB, and AMK were 8, 32, and 2 μg mL−1, respectively; whereas that of PLZ was 0.5 μg mL−1. Against E. coli, MIC90 values for GEN, TOB, and AMK were 32, 8, and 4 μg mL−1, respectively; whereas that of PLZ was 1 μg mL−1.84–86 PLZ was also evaluated against 493 MRSA isolates collected in 2009–2010 from 23 US hospitals and the MIC50 and MIC90 values found to be 1 and 2 μg mL−1, respectively.87 Synergy time-kill studies performed on 47 MRSA strains with different resistance phenotypes showed that combinations of sub-inhibitory concentrations of PLZ and daptomycin yielded synergy against 43/47 strains at 24 h, PLZ and ceftobiprole showed synergy in 17/47 strains, while 6/47 strains showed synergy for sub-inhibitory combinations of PLZ and linezolid.88 PLZ was also found to be active against most enterobacteriaceae species at a concentration below 4 μg mL−1 as well as carbapenem-resistant enterobacteriaceae isolates (2 μg mL−1) and was found to be effective against AG-resistant staphylococci (MIC90 = 2 μg mL−1).89,90 Finally, PLZ was found to be moderately active against the nosocomial pathogens A. baumannii and P. aeruginosa with MIC50/90 values of 8/16 μg mL−1 and 8/32 μg mL−1, respectively.91
These promising in vitro results prompted the in vivo efficacy testing of PLZ in animals.92 When tested in combination with cefepime, doripenem, imipenem, or piperacillin-tazobactam in a synergy time-kill analysis against 25 P. aeruginosa strains with different resistance phenotypes, synergy was observed in ≥70% and ≥80% of strains at 6 and 12 h, respectively, and in ≥68% at 24 h.93 The pharmacokinetic evaluation and safety monitoring of PLZ injection in healthy subjects showed no nephrotoxicity and ototoxicity in humans.94
Patent WO 2014/1454713 A2 (ref. 95)
Despite their ototoxic and nephrotoxic effects, AGs remain some of the most widely used antibiotics worldwide. Various techniques such as modulating dosage plans and the use of free radical scavengers were employed as attempts to reduce the overall toxicity associated with AGs.96 The unexpected discovery that AGs enter hair cells at high level by passing through specific ion channel prompted postulation of a similar mechanism of action in the case of kidney.97 In patent WO 2014/1454713 A2,95 a novel chemical synthetic approach was used to develop analogues of SIS for alleviating AG ototoxicity while preserving antibacterial activity (Fig. 5A, 31–39). The AGs were modified based on the notion that hindering AG entry via the hair cell MET channel would prevent ototoxicity.98 Based on the biophysical properties of the MET channel where electrochemical gradients drive cations through the channel, it was presumed that conversion of the free amines to sulfonamides would result in decrease in net charge and in turn reduced ototoxicity.99 Functional groups such as SO2Me, SO2Ph, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OPh were introduced at the N1- and N3′′-positions. MIC and minimal bactericidal concentration (MBC) values were determined for these SIS derivatives. Substitution by SO2Me was more effective than either SO2Ph or COPh, and compound 31 showed decreased toxicity against hair cells in P. aeruginosa species (22 times less ototoxic than SIS) and S. aureus species (18 times less ototoxic than SIS). Modification at the N3′′-position abolished antibacterial activity of all six SIS derivatives.
OPh were introduced at the N1- and N3′′-positions. MIC and minimal bactericidal concentration (MBC) values were determined for these SIS derivatives. Substitution by SO2Me was more effective than either SO2Ph or COPh, and compound 31 showed decreased toxicity against hair cells in P. aeruginosa species (22 times less ototoxic than SIS) and S. aureus species (18 times less ototoxic than SIS). Modification at the N3′′-position abolished antibacterial activity of all six SIS derivatives.
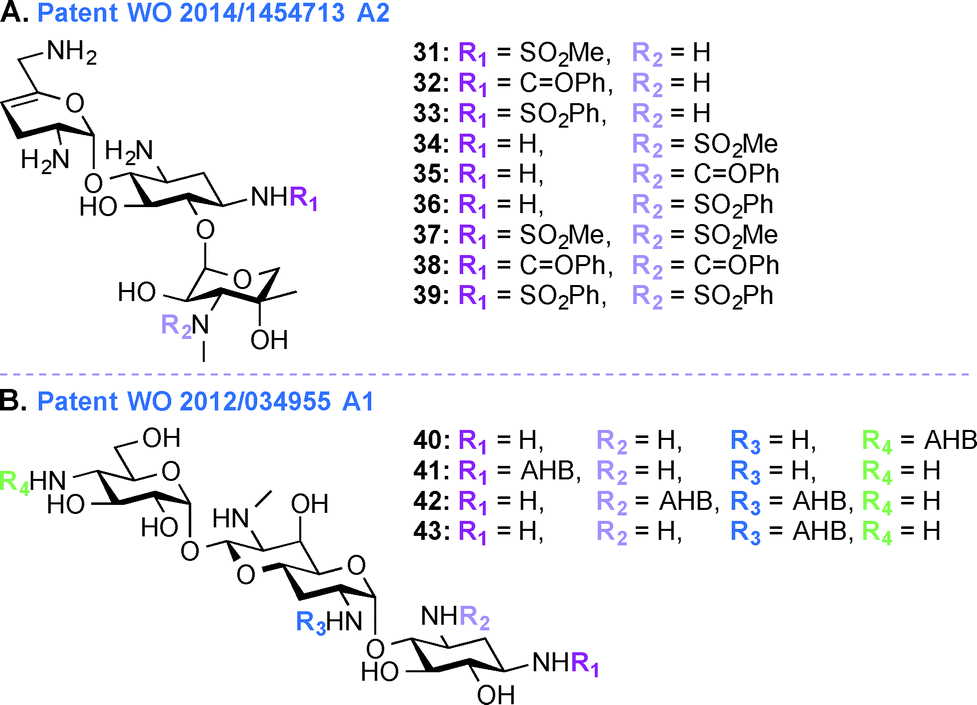 | ||
| Fig. 5 Representative structures of A. 1,3′′-disubstituted SIS derivatives, and B. APR derivatives. | ||
Patent WO 2013/170985 A1 (ref. 100)
APR, an AG that has so far only been used in veterinary medicine, has more recently been explored as it was found to display reduced ototoxicity. AGs affect the protein synthesis through binding to specific sites of the rRNA and ring I of the 2-DOS derivatives binds to the rRNA through specific and nonspecific interactions irrespective of the 4,5- or 4,6-substitution pattern. Recent evidence implies mitochondrial protein synthesis as a key element in AG ototoxicity, and the defective mitochondrial function leads to reactive oxygen species (ROS) generation resulting in ototoxicity.101,102 In patent WO 2013/170985 A1,100 the activities and ototoxicity of various parent AGs (depicted in Fig. 1), with a special focus on APR, is presented. APR, GEN, TOB, and KANA were tested for ribosomal specificity and were found to be similarly active towards bacterial ribosomes (IC50 0.02–0.08 μM) and eukaryotic cytosolic ribosomes (IC50 cytosolic hybrids 40.4–91.2 μM). In contrast, when compared to 4,6-disubstituted AGs, APR displayed a 4-fold difference in IC50 values in the case of mitochondrial ribosomes (115.6 μM for APR versus 11.1–32.6 μM for 4,6-disubstituted AGs) and 50-fold difference in the case of mitohybrid ribosomes with mutation at A-site (47.5 μM for APR versus 0.69–1.12 μM for 4,6-disusbstituted AGs). Crystallographic studies showed that the bicyclic ring II of APR stacks well to the guanine base of the C1409–G1491 base pair compared to the flexible monocyclic ring I of 4,6-disubstituted derivatives. In vitro and in vivo toxicity studies of AGs were carried out to explore the damage to the cochlea in both human and experimental animals. The dose response curve showed that 50% of all hair cells in the base of the cochlea were destroyed at 0.2 mM GEN, and complete elimination of outer hair cell was observed at 0.5 mM. In contrast, APR at a dose of 0.2 mM did not adversely affect cells even in the base, but was shown to affect basal hair cells at 2 mM concentration.Patent WO 2012/034955 A1 (ref. 103)
In addition to the work done on APR in patent WO 2013/170985 A1,103 APR derivatives were developed towards treatment of human infections (Fig. 5B, 40–43). In the past, APR has not been used in human because it was thought to have relatively high toxicity. However, work is currently underway to explore the use of APR derivatives as antibiotics (e.g., acylated and alkylated derivatives of APR have been tested for their broad spectrum activity).104 In patent WO 2012/034955 A1,103 APR and its derivatives were tested against various Gram-negative bacterial strains and their activities were compared to those of parent AGs such as TOB, GEN, NEO, and PAR. More specifically APR was tested against clinical isolates of MRSA and its MIC values were found to be similar to those of TOB, SIS, KANA, AMK, NEO, and PAR. APR was also found to display affinity for wild-type bacterial ribosomes similar to those of TOB, NEO, and GEN, but the APR affinity for hybrid ribosomes was reduced when compared to these drugs. In in vitro cochleatoxicity studies, APR was found to cause <5% hair loss in guinea pigs. After publication of this patent, Crich and co-workers synthesized various O6′- and N7′-analogues of APR, which were screened for their ability to inhibit protein synthesis in cell-free translation assays. All derivatives prepared were less active than APR, which emphasized the role of the 6′-hydroxyl group and its configuration for APR activity.105Patent WO 2012/097454 A1 (ref. 106)
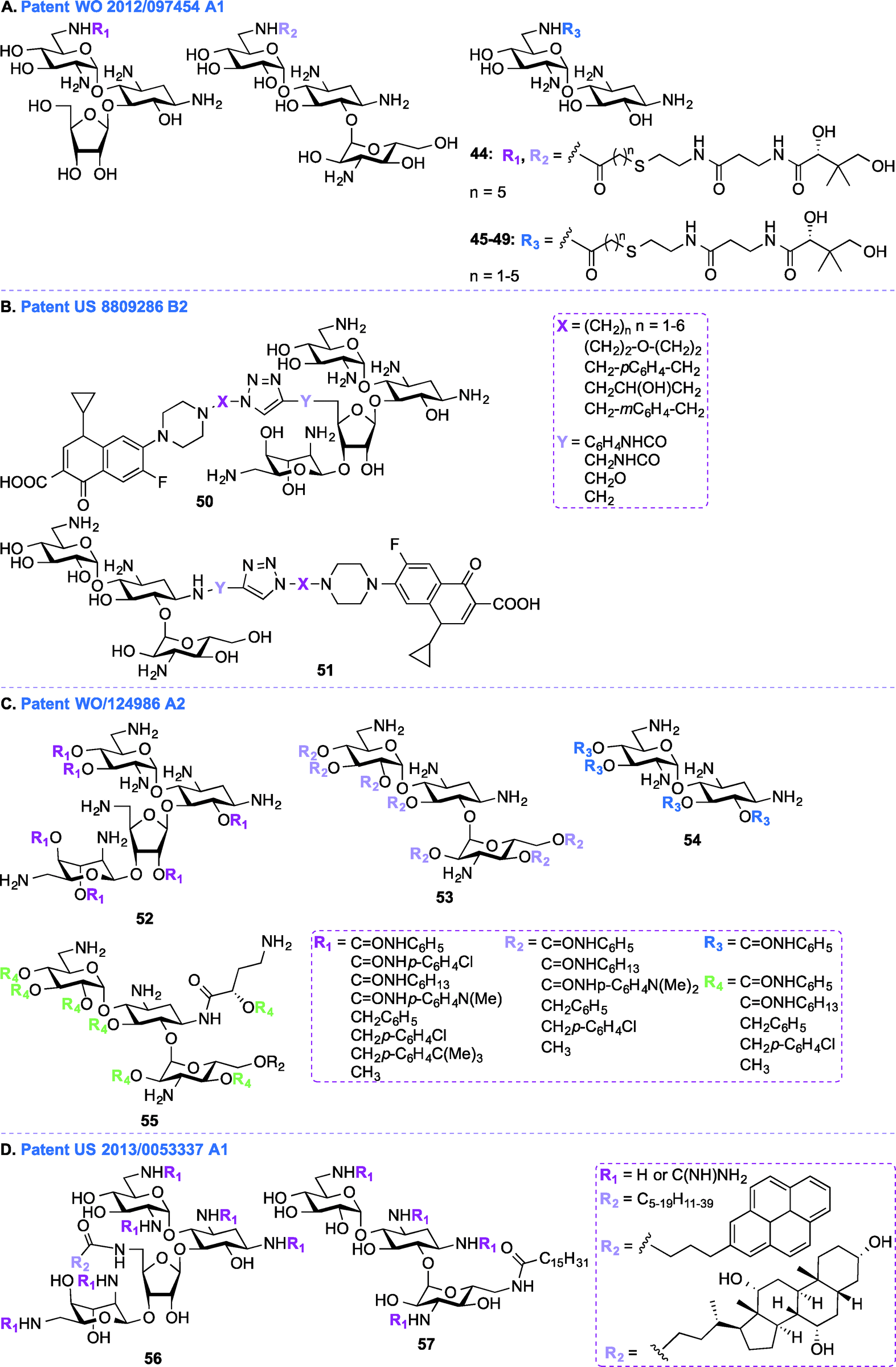
In addition to the general synthetic approaches for derivatizing AGs, conjugation of AGs with other biologically relevant molecules or drugs has been explored as an interesting alternative strategy for the development of newer AGs.107–111 Nanomolar bisubstrate inhibitors where an AG is conjugated to coenzyme A (CoA) were reported.112,113 Structure–activity relationship (SAR) studies of truncated CoA bisubstrate revealed that the pantetheinyl group is not necessary for recognition by the enzyme, but at least one phosphate group is needed.114 Following up with amide-linked analogues, various sulfonamide-, sulfoxide-, and sulfone-containing bisubstrate inhibitors were synthesized and found to be 40-fold less potent against AAC(6′)-Ii when compared to their amide-linked counterparts.115 A similar trend was observed for phosphonate-linked AGs and the decreased inhibitory potency was explained by proximity catalysis by enzymes.116 AG-CoA bisubstrates are highly potent AAC(6′) inhibitors, however their inability to penetrate cells preclude in vivo studies.In patent WO 2012/097454 A1,106 a class of AAC inhibitors comprised of pantetheine conjugated to the 6′-amine of AGs has been investigated (Fig. 6A, 44–49). Among all the AMEs, AAC(6′)s are some of the most frequently observed in clinical isolates. There is therefore an urgent need for compounds capable of inhibiting AAC(6′)s and reversing the bacterial resistance associated with these enzymes. NEA, RIB, and KANA conjugated to the pantetheinyl arm of CoA were found to undergo intracellular biotransformation by pantothenate kinase (PanK) prior to inhibiting AAC(6′) enzymes. When tested against an AG-resistant Enterococcus faecium strain, two of these conjugates (48 and 49) were found to partially resensitize the strain to KANA and TOB.117
 | ||
| Fig. 6 Representative structures of A. NEA, RIB, and KANA conjugated to the pantetheinyl arm of CoA at their 6′-position, B. NEO- and KANA-CIP hybrids, C. NEO-, KANA-, AMK-, and NEA-lipid conjugates, and D. NEO- and KANA-polycationic lipid conjugates. | ||
Patent US 8809286 B2 (ref. 118)
Besides conjugating AGs to CoA, as described above, in patent US 8809286 B2,118 work was done to conjugate NEO and KANA to the FDA approved fluoroquinolone ciprofloxacin (CIP) by using click chemistry (Fig. 6B, 50–51). The efficacy of these conjugates was tested against various strains of E. coli, Bacillus subtilis, and MRSA. The NEO-CIP hybrids were found to be 2 to 253-fold and 8 to 128-fold better than NEO against E. coli and MRSA, respectively. Similarly, notable antibacterial activity was observed in the case of KANA derivatives in comparison to the parent AG. MIC values were determined against E. coli strains expressing APH(3′) and the bifunctional AAC(6′)-Ie/APH(2′′)-Ia enzymes, and the conjugates (6–48 μg mL−1 in the case of AAC(6′)-Ie/APH(2′′)-Ia and 3–96 μg mL−1 in the case of APH(3′)) were found to be less effective than CIP (0.38 and 0.10 μg mL−1), but significantly better than NEO (>384 and 96 μg mL−1). The emergence of bacterial resistance was tested by using CIP, NEO, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/CIP :NEO mixture, and the AG-CIP conjugates against E. coli and B. subtilis strains. The relative MIC values of CIP, NEO, and the 1:1/CIP:NEO mixture were increased by 75, 4, and 20-fold against E. coli and by 37.5, 8, and 7.6-fold against B. subtilis, whereas the conjugates remained unchanged in both cases.
:1/CIP :NEO mixture, and the AG-CIP conjugates against E. coli and B. subtilis strains. The relative MIC values of CIP, NEO, and the 1:1/CIP:NEO mixture were increased by 75, 4, and 20-fold against E. coli and by 37.5, 8, and 7.6-fold against B. subtilis, whereas the conjugates remained unchanged in both cases.
Patent WO 2011/124986 A2 (ref. 119)
In patent WO 2011/124986 A2,119 a new class of AGs conjugated to lipids through their hydroxyl groups was synthesized (Fig. 6C, 52–55). Oligocationic antibacterials such as AGs, which contain multiple positively-charged amine functional moieties, as well as peptides, lipopeptides, and their synthetic mimics are known to display a broad spectrum of activity.120–124 In order to bind intracellular targets such as RNA, DNA, and proteins, the non-amphiphilic AGs have to cross the bacterial membrane. Therefore, co-administration of AGs and membrane-permeabilizing agents (e.g., ionic lipids) can result in synergistic enhancement of antibacterial activity.125,126 In patent WO 2011/124986 A2, twenty different moieties of four different oligocationic AG-based polyol (NEO, KANA, AMK, NEA) were synthesized by converting the parent AGs to their polycarbamates and polyethers derivatives. The nature of the polyol scaffold, number of positive charges, and hydrophobic groups had a significant impact on their antibacterial activity. The NEO-based hexacationic heptaphenylcarbamate (52 with R1 = CONHC6H5) was found to exhibit antibacterial activity against Gram-positive MRSE, S. aureus, MRSA, and S. epidermidis (MIC ≤ 1 μg mL−1). A remarkable 256-fold enhancement in antibacterial activity was observed for NEO-based hexacationic heptaphenylcarbamate against MRSA when compared to NEO while maintaining potent activity against NEO-susceptible strains, such as MRSE, S. aureus, S. epidermidis, and K. pneumoniae. In contrast, the tetracationic KANA, AMK, and NEA-based polyphenylcarbamates exhibited up to 64-fold reduced Gram-positive activity. Replacement of the polyphenylcarbamate by polybenzylether also resulted in potent antibacterial AGs. The most potent hexacationic NEO-based heptabenzylether (52 with R1 = CH2C6H5) displayed slightly decreased Gram-positive activity (MIC = 2–4 μg mL−1) against S. aureus, MRSA, S. epidermidis, and MRSE in comparison to its polycarbamate counterpart. In the case of both polycarbamates and polyethers, para-substitution on the aromatic ring of NEO, KANA, and AMK resulted in a 4 to 64-fold decrease in antibacterial activity. The effects of substitution pattern on the aromatic ring on antibacterial activity suggest contribution of factors other than polycationic charges and amphiphilicity on their mechanism(s) of action.127
Patent US 2013/0053337 A1 (ref. 128)
In addition to the AG-lipid conjugates presented in patent WO 2011/124986 A2,119 KANA- and NEO-lipid conjugates, with free or guanidinylated amines are presented in patent US 2013/0053337 A1 (ref. 128) (Fig. 6D, 56–57).129,130 Fourteen NEO- and two KANA-based polycationic lipid hybrids were synthesized. A variety of lipophilic moieties (e.g., C6, C12, C16, C20, doubly unsaturated C18, pyrene, and cholic acid) were selected to investigate the effects of the hydrophobic tails on the antibacterial activity of these AGs. The antibacterial activity of these hybrids was tested against both Gram-positive and Gram-negative bacterial strains, including S. aureus, MRSA, S. epidermidis, E. faecalis, E. faecium, E. coli, and P. aeruginosa. Overall, it was found that, in order to be active, the lipid chains of these hybrids needed to comprise at least 12 carbons, and 16 carbons was shown to be optimal. It was also observed that, when derivatized with the same lipid chain, the NEO scaffold was always more active than its KANA counterpart. Finally, the guanidinylated hybrids were found to be more active than their non-modified counterparts. Other examples of amphiphilc NEO-peptide derivatives have also been reported in the literature.131–133Aminoglycosides as antifungal agents
As described in the section above, traditionally AGs are used for treating bacterial infections in humans and animals, but continuous setbacks associated with their antibacterial activity have prompted scientists to explore new targets for AGs. The lack of efficient antifungal agents, the increase in the number of immunocompromised patients, as well as the widespread resistance to existing antifungal agents have culminated in the emergence of fungal disease as a major public concern.134–136 Common fungal infections like athlete's foot, ringworm, candidiasis, and serious systemic infections such as cryptococcal meningitis are treated by using FDA approved antifungal agents (e.g., amphotericin B), but the toxicity levels of these antifungals are a common concern for medical practitioners.137 Fungal and oomycete infections are responsible for plant diseases, and direct application of biocides to plant fungi is a common and efficient strategy used for their treatment. Unfortunately, inconsistent and declining efficacy, environmental impact, and toxicity have resulted in lower impact of these agents on plant fungal infections. An early report by Lee and co-workers on the activity of AGs against six crop pathogenic oomycetes (Phytophthora and Pythium species) and ten common fungi provided insight into the use of AGs as an alternative for treating fungal infections.138The widely used antifungal agents are either sterol-binding polyene compounds (e.g., amphotericin B) or sterol biosynthesis disrupting azoles (e.g., fluconazole (FLC), itraconazole (ITC), posaconazole (POS), and voriconazole (VOR)), but toxicity and resistance associated with these molecules have created a shortage of effective antifungal agents.139,140 Thus, the development of newer antifungals based on AGs possessing novel mechanism(s) of action is currently underway. Generating novel amphiphilic AGs by attaching alkyl or other hydrophobic groups to parent AGs was found to alter the microbial properties of these compounds. Extensive efforts towards generating amphiphilic AGs by attachment of hydrophobic groups to AGs have recently been reported.77,78,127,141–148
The length of the alkyl chain, the chemical nature of the hydrophobic moiety, and the site of attachment on the AG scaffold significantly influence the behavior of these amphiphilic AGs towards microbial infections. The work from the Chang and Takemoto groups has highlighted the discovery of amphiphilic KANA and NEO analogues that are antifungal, but not antibacterial and they inhibit the fungal growth by membrane perturbation.149,150
The recent development of amphiphilic AGs with mechanisms of action, which differ from traditional AGs, can represent a significant advancement towards more effective AGs.151 There are two common strategies for the synthetic production of amphiphilic AGs (i) direct modification using AGs as starting materials and (ii) diversification of ring III using glycosylation.152–158 The direct modification approach was employed for generating KANA and TOB analogues by using Boc chemistry to protect amines followed by nucleophilic substitution reaction at the only primary 6′′-OH using various functional groups such as thioether, alkoxyl, alkylamino, alkylamido, and esters. Modifications on ring III of KANB at the 3′′-, 4′′-, and 6′′-positions yielded little to no antibacterial activity, but resulted in inhibition of various yeasts, oomycetes, and fungi with MIC values ranging from 3.9 μg mL−1 to 31.3 μg mL−1, highlighting the significance of the alkyl chain in antifungal switch.149,159 Recent work from Garneau-Tsodikova and co-workers has highlighted the in vitro antifungal activity, cytotoxicity, and membrane-disruptive action of amphiphilic KANB and TOB analogues bearing a thioether linkage at their 6′′-position.143,160,161 TOB analogues with C12 and C14 chains showed promising activities against fungal strains with MIC values ranging from 1.95 to 62.5 mg L−1 and 1.95 to 7.8 mg L−1, respectively.161 In the case of KANB, analogues with C12, C14, and C16 chains were 4, 16, and 32-times more effective than KANB against several of the yeast strains with MIC values ranging from 1.95 to 31.2 μg mL−1.160 TOB C12 and C14 analogues were tested for synergistic combination effect against C. albicans strains with four azoles.162 The combination effects of C12 or C14 and azoles appears non toxic to mammalian cells at higher or equal to synergistic antifungal MIC values of these drugs against fungi. This section of the review summarizes the recent application of AGs as antifungal agents exploring their synthesis using direct modification and glycodiversification strategies.
Patent US 2011/0130357 A1 (ref. 163)
In patent US 2011/0130357 A1, a glycodiversification strategy was used to synthesize various analogues of KANA and KANB as antifungal agents (Fig. 7A, 58–61). The present invention relates to novel analogues of AGs with substituents at the 6′′-position of ring III which exhibit enhanced antifungal activity, but with minimum antibacterial property. Modifications at the 3′′-, 4′′-, and 6′′-positions of the antibacterial KANB resulted in the antifungal agent 59. Compound 59 and its analogues were synthesized via successive regio- and stereoselective glycosylation of an azido-neamine core, deacetylation, Staudinger reduction of the azido groups, and hydrogenolysis of the benzyl groups. Different alkyl chains ranging from C4 to C12 in length were introduced at the O4-position of ring III and C8 was found to be the optimum chain length for efficient antifungal activity. 59 and its derivatives were tested against various fungal strains and for 59, the MIC value was found to be 31.3 μg mL−1 against Fusarium graminearum. To understand the specificity of compound 59 for fungi, its mechanism of action was studied using yeast Saccharomyces cerevisiae and the result showed the influence of plasma membrane permeability for growth inhibition.150 To further elucidate SARs for this phenomenon, KANB derivatives were synthesized using glycodiversification and the 3′′-OH was found to be better than the 3′′-NH2, and deoxygenation at the 6′′-OH was found to be unnecessary for their activity.159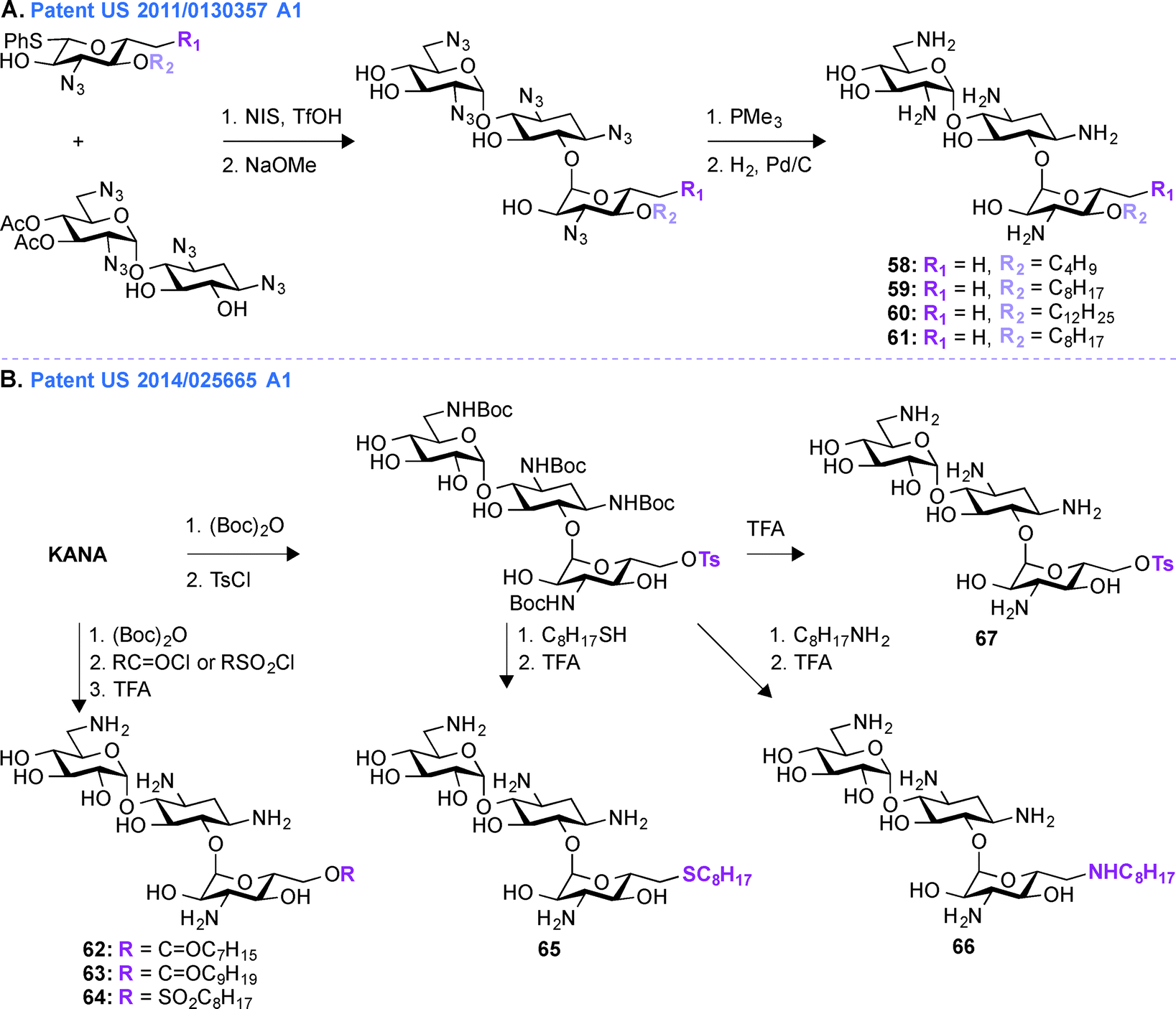 | ||
| Fig. 7 A. Representative scheme for the preparation of A. 6′′-alkylated KANB amphiphiles, and B. 6′′-alkylated KANA amphiphiles with ether, thioether, and amine linkages. | ||
Patent US 2014/0256665 A1 (ref. 164)
In patent US 2011/0130357 A1, the synthesis of compound 59 and its derivatives were carried out using glycodiversification reactions and, even though these molecules showed considerable antifungal activity, the complex synthetic protocol previously patented was found to be not suitable for scale-up. Thus, based on the SAR study of 59, a simplified method of direct modification of AG was developed in patent US 2014/0256665 A1 (ref. 164) to produce another set of amphiphilic molecules from tetra Boc-protected KANA (Fig. 7B, 62–67). The synthesis followed direct introduction of esters and sulfones to the Boc-protected KANA as well as introduction of amines and thioethers using substitution reactions. These AGs were used in combination with azoles in different combinations ranging from 1:1 to 1:1000 (AG:azole). The most active KANA derivative was found to be compound 64. The relative growth inhibitory activities for 64 were tested against various bacterial and fungal strains and the MIC value for 64 against F. graminarum was found to be between 7.8 and 15.6 μg mL−1. Compound 64 was tested against E. coli and Micrococcus luteus and its MIC values were found to be 250 and 62.5 μg mL−1, respectively (Note: KANA MIC values = 0.98 and 1.95 μg mL−1, respectively). These data demonstrated the inherent lower antibacterial activity and greater antifungal property of 64. Synergistic inhibition assays were performed with 64 and a variety of azoles (ITC, FLC, POS, VOR, clotrimazole, tioconazole, ketoconazole, metconazole, and pyraclostrobin) against an azole-resistant Candida albicans strain. These combinations showed synergistic effect with FICI values ranging from 0.13–0.31 μg mL−1. Finally the synergistic effect was confirmed by time-kill curve study against that C. albicans strain. To further understand the mechanism of action of compound 64, fungal models were used and it was found that the fungicide action results from membrane perturbation, which is influenced by lipid modulation.165
Newer applications of aminoglycosides
Apart from their use in treatment of microbial infections, research has recently focused on exploring AGs for potential treatment of genetic disorders. The genetic disorders for which AGs are currently investigated result from nonsense mutations where a premature termination codon (PTC) replaces an amino acid coding-codon to interfere with protein translation resulting in truncated and inactive proteins.166–169 Base pair insertion, deletion, or substitution result in the formation of PTCs, which ultimately result in a number of genetic disorders such as cystic fibrosis (CF),170 Lafora disease, Duchenne muscular dystrophy (DMD),171 Rett syndrome,172 spinal muscular dystrophy,173 congenital muscular dystrophy,174 Type I Usher disease,175 ataxia-telangiectasia,176 Hurler syndrome,177 hemophelia A, hemophelia B,178 and Tay-sachs. At the present, there are no effective treatments available for these disorders. Even though gene therapy has been considered a potential solution for these disorders, there are still many critical problems to be solved before using this technique in humans. AGs act against microbial infections by binding to A-site of the 16S rRNA and this mechanism of action has been exploited for their ability to readthrough stop codon mutations, generating full-length proteins from part of the mRNA molecules.179–182 Studies have shown several parent AGs, such as AMK and GEN, to promote readthrough of the PTC in mouse models and patient samples.183–185 Additionally, derivatives of PAR such as NB30, NB54, NB74, NB84, and NB124 were reported to suppress PTCs and display lower toxicity when compared to their parent AGs (Fig. 8).186–191 Thus, research on AGs and their derivatives' ability to suppress PTCs while improving their potency and decreasing their toxicity is a promising direction to pursue for the discovery of treatment of the resulting disorders.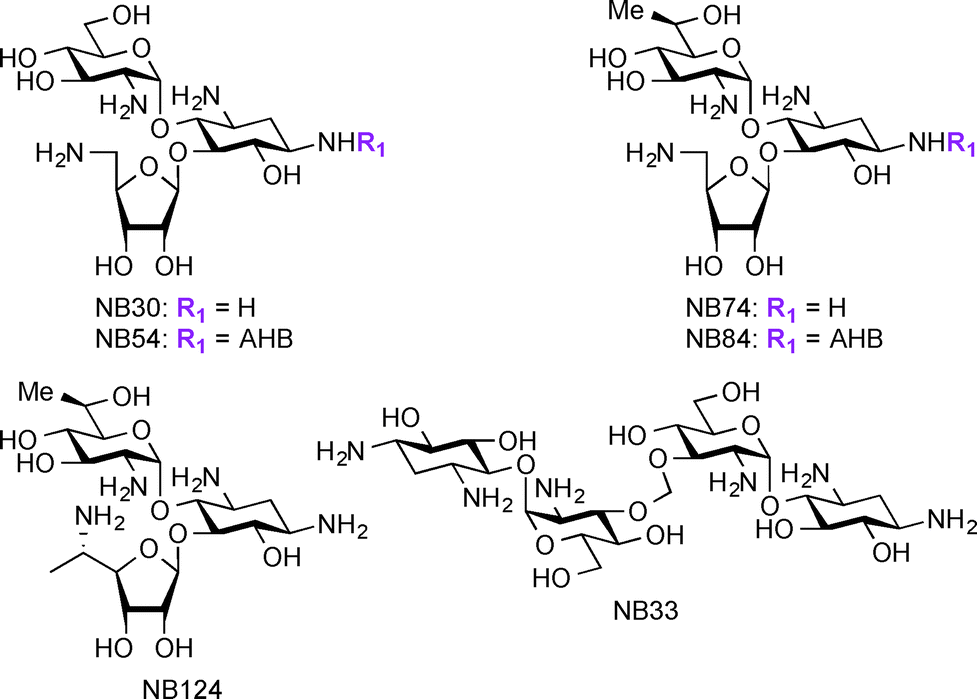 | ||
| Fig. 8 Structures of NB derivatives discussed in the section “newer applications of AGs”. | ||
In addition to treatment of genetic disorders resulting from PTCs, AGs have been explored as potential anti-HIV (Human Immunodeficiency Virus) agents. Research has shown that AGs and AG conjugates are capable of interfering with many steps of the HIV life cycle, thus providing a new application for AG-based compounds.192,193 The AGs conjugates NEO-Hoechst 33258 have been found to have higher affinity and specificity to HIV-1 trans-activation response element (TAR) RNA.194 AG-arginine conjugates (AArCs) and AG-polyarginine conjugates (APArCs) have also been extensively investigated as potential treatment of HIV.195,196 NEO and APR have also been found to bind HIV-1 dimerization initiation (DIS), which is responsible for genome initiation.197,198 Homo- and hetero-dimers of KANA, NEO, and TOB, as well as naphthalene-based diimide-conjugated AGs were reported to bind to HIV-1 Rev response element (RRE).199,200 NEA dimers as well as nucleobase-AG conjugates were found to inhibit TAR-Tat binding201 and guanidine derivatives of NEO were shown to bind the RNA helix of HIV-1 frameshift.202 Overall, these studies demonstrate the additional novel uses of AGs, which will be the focus of this section.
Patent US 2014/0357590 A1 (ref. 203)
In the treatment of genetic disorders associated with PTCs, the mechanism of action by which AGs bind to the 16S rRNA is used for enhancement of termination suppression by stop codons. AGs have been shown to suppress various stop codons with notably different efficiencies (UGA > UAG > UAA).204 The first genetic disease examined was CF, and the experiments demonstrated AG-mediated suppression of CF transmembrane conductance regulator (CFTR) mutations especially by the members of the GEN family and G418.170,205 Although G418 displayed the best termination suppression activity in in-vitro experiments, its toxicity, even at low concentrations, was a major drawback. In patent US 2014/0357590 A1,203 PAR-derived pseudotrisaccharide AGs were designed to replace GEN, which exhibited high premature stop-codon mutation readthrough activity while exerting low cytotoxicity and antimicrobial activity (Fig. 9A, 68–75). Based on the results from patent WO 2007/113841,206 where the derivative NB30 exhibited reduced cytotoxicity compared to PAR and GEN while promoting dose-dependent suppression of nonsense mutation of the PCDH15 gene in the case of Usher's disease but its suppression potency was lower than PAR and GEN,175,207 NB54 was developed as a second-generation concept structure, which exhibited substantially higher readthrough efficiency than PAR and GEN. Furthermore, NB30 and NB54 were evaluated for readthrough efficiency for treatment of USH1C nonsense mutation, and in vivo administration of NB54 induced the recovery of full-length harmonin 1a with enhanced efficiency.208 NB54 was tested for its ototoxicity and enhanced suppression of CF PTCs. NB54 was shown to partially restore halide efflux in a CF bronchial epithelial cell line, but not in a CF cell line lacking a PTC.186 In related research efforts, two novel analogues, NB30 and NB33, which are derivatives of G418 were synthesized and were found to display increased selectivity towards eukaryotic cells over prokaryotic cells. Cell toxicity tests using three kidney cell lines confirmed that NB30 is 6–15 fold less toxic than GEN and PAR. NB30 was shown to produce full-length proteins more efficiently than PAR or GEN. To address the enhanced affinity for eukaryotic cells, a comparative biochemical and structural analysis of NB30, NB33, together with a series of different 4-, 4,5-, and 4,6-substituted 2-DOS AGs in both eukaryotic and prokaryotic systems was performed. From these studies, it was revealed that NB33 is the strongest inhibitor of eukaryotic protein translation and it was found to preferentially bind the eukaryotic RNA decoding-site.191,209,210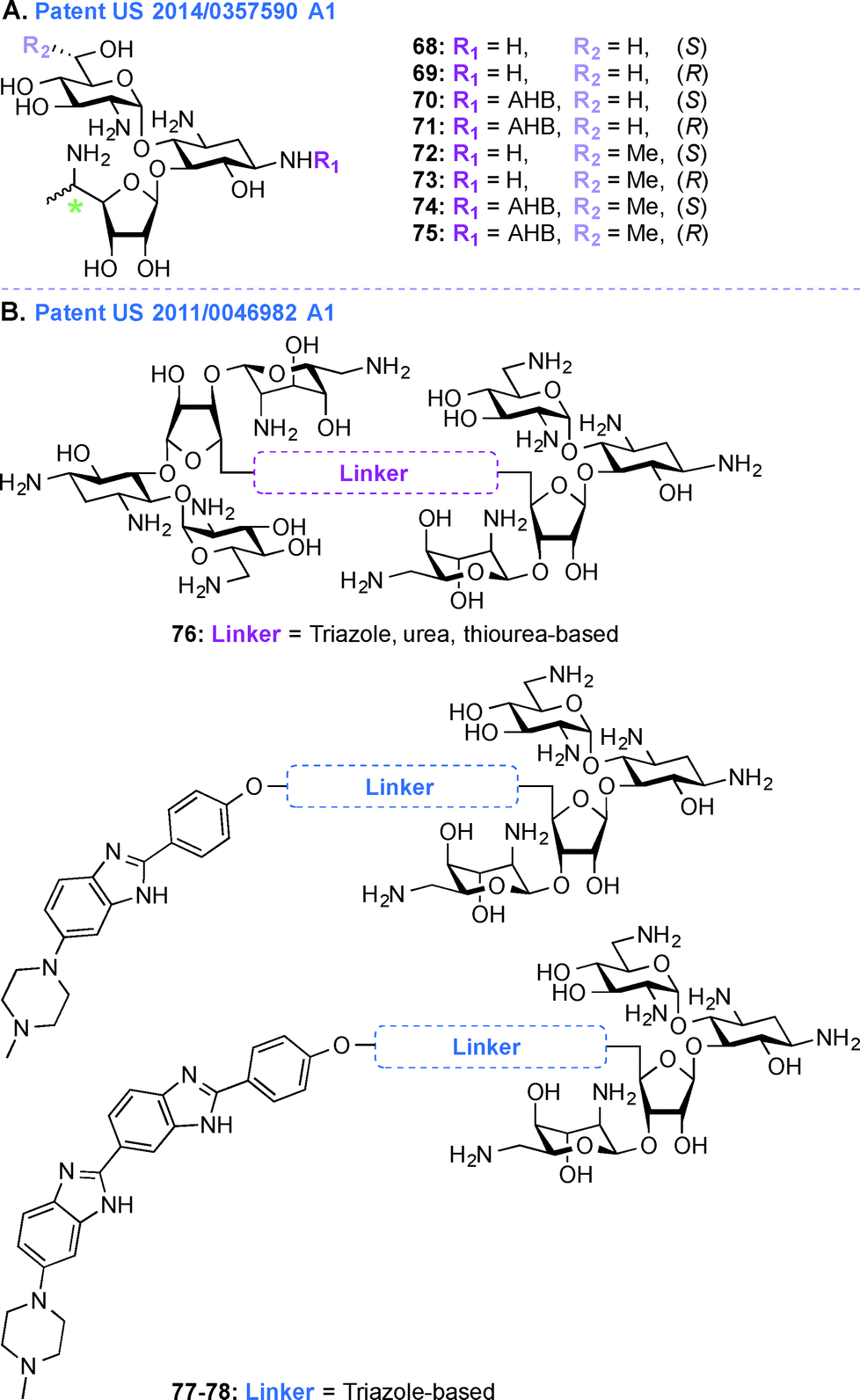 | ||
| Fig. 9 Representative structures of A. pseudotrisaccharide NB derivatives, and B. NEO dimers as well as NEO-benzimidazole and NEO-bisbenzimidazole conjuagtes with various linkers. | ||
In patent US 2014/0357590 A1, newer analogues 68–75 were synthesized by modifying ring I, ring II, and ring III of the pseudotrisaccharide (Fig. 9A). For the readthrough assays, the prevalent nonsense mutations chosen were R3X and R245X for USH1, G542X and W1282X for CF, Q70X for HS, and R3381X for DMD. For all the mutations tested, the efficacy for AG-induced readthrough was 70 > 71 > NB54 > GEN, suggesting better cell permeability for 70 and 71 over NB54 due to the presence of a 5′′-methyl group. A similar trend was observed for another NB derivative, compound 74, where two methyl groups ((R)-6′-methyl group (S)-5′′-methyl groups) were shown to operate synergistically to enhance readthrough activity. The derivatives 68–75 were investigated as antibacterial agents against both Gram-negative and Gram-positive bacterial strains (E. coli, B. subtilis) and were found to have lower specificity for prokaryotic ribosome (IC50 values ranging from 160 ± 20 to 2266 ± 196 nM) compared to GEN (28 ± 4 nM) and PAR (51 ± 5 nM), explaining their lower antibacterial activity. Cell toxicity data demonstrated that 68 and 70, which were formed by installation of a (S)-5′′-methyl group on NB30 and NB54, respectively, did not significantly affect their cytotoxicity values (LC50 values 21.4 and 23.5 μM for NB30 and 68, respectively, and 6.1 and 10.1 μM for NB54 and 70, respectively) whereas 1.9-fold and 4.6-fold increase in their stop codon suppression activity was observed. The ratios for prokaryotic versus eukaryotic selectivity were found to be 2214 for GEN, 1118 for PAR, and 225 for G418, whereas compounds 68–75 displayed values between 0.4–151, which explained their lower cytotoxicity values.
Patent US 2011/0046982 Al (ref. 211)
Acquired Immunodeficiency Syndrome (AIDS), which is caused by the HIV, is a global health issue and there is a great necessity for new therapies to treat this disease. In patent US 2011/0046982 A1,211 AG-small molecule combinations were evaluated for their binding affinity and specificity to viral RNA, such as HIV TAR RNA. As NEO is able to inhibit Tat peptide binding to the TAR RNA and studies on the interactions of NEO with TAR helped identify three binding sites,212,213 in this patent, NEO dimers were investigated to enhance TAR specificity (Fig. 9B, 76). Various aspects of different AG-small molecule conjugates in terms of their method of synthesis, efficiency for binding DNA, RNA, and correlation of efficacy of these molecules with respect to the linkers used were explored in details.194,214–222 Additionally, as the benzimidazole Hoechst 33258 is capable of inhibiting TAR region of HIV RNA, NEO-benzimidazole (analogues of Hoechst 33258) conjugates were also investigated (Fig. 9B, 77–78). Conjugation of the AG NEO and benzimidazoles tethered by different alkyl linkers displayed high affinity binding to TAR with nanomolar IC50 values. UV melting studies were performed in the presence and absence of various ligands, NEO-benzimidazole derivatives 77–78 (4.4 × 107 M−1) were found to have a higher binding constant than NEO (1.7 × 105 M−1), Hoechst 33258 (4.2 × 105 M−1), and benzimidazoles (5.2 × 105 M−1) alone. The length of the linker affected the change in melting temperature and NEO-benzimidazole conjugate with 24 backbone carbon atoms showed the biggest change in melting temperature. Similarly, in the case of NEO dimers, the length of the linkers appeared to display a direct correlation with their change in melting temperature. These NEO conjugates provided maximum protection from HIV cytopathic effects (2–44%) at a varied range of concentrations (2–206 μM). These compounds were also tested for their efficiency to control the release of reverse transcriptase (RT), even though the cells were positive for HIV after 6 days of experiment they were shown to inhibit the RT release up to a limit. Finally, all these NEO derivatives were used to study competition-binding affinity against HIV-1 TAR RNA. The IC50 values for these compounds ranged from micro to nanomolar concentrations and their binding free energy were calculated. The dimers with less flexible linkers were found to have better binding values.Perspective and conclusions
In the last decade, the field of AG antibiotics has experienced a renewed interest among the scientific community and efforts were made to counter the resistance and toxicity associated with these drugs. In that ten-year period, more traditional reviews on AGs have been published in the literature.1,6,8,36,37,151,223–232 In this review, by summarizing all recent patents on AGs, we highlighted their use/development as antibacterials and antifungals, as well as those for potential newer applications (genetic disorders and HIV).Even though extensive work has been done in the field of AGs, there are still many avenues left to explore. In recent years, a thorough study of the mechanism of action of AGs on bacterial ribosomes led to the discovery of the H69 binding site, a new target now available for further exploration. Several approaches have also been explored to counter the resistance caused by AMEs, including designing AME inhibitors, synthesizing AG scaffolds capable of evading the action of AMEs, and regulating AME expression.37,233–235 Even though metal cations have lately been shown to inhibit AAC activity and increase AG efficacy in AG-resistant bacterial strains,236,237 more detailed mechanistic investigations remain to be performed. The discovery of the lack of ototoxicity of APR also offers multiple prospects for the development of this relatively unexplored scaffold. The fields of AGs as antifungal agents and as treatments for PTC disorders are still in their infancy and surely provide a lot of opportunities for investigation. These areas of study will probably be some of the most exciting for AG developers to pursue in the future. In conclusion, the area of research on AGs has recently experienced a renaissance and promise to remain for a long time a valuable source for drug discovery and development.
Acknowledgements
Our work on aminoglycosides and bacterial resistance is supported by the National Institutes of Health (NIH) Grant AI090048 (S. G.-T.) and by startup funds from the College of Pharmacy at the University of Kentucky (S. G.-T.). We appreciate the numerous efforts of those working in the field of AGs and apologize for the work not cited because of the scope of this review.Notes and references
- J. L. Houghton, K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 880–902 CrossRef CAS PubMed.
- I. A. Bliziotis, G. Samonis, K. Z. Vardakas, S. Chrysanthopoulou and M. E. Falagas, Clin. Infect. Dis., 2005, 41, 149–158 CrossRef CAS PubMed.
- P. D. Tamma, S. E. Cosgrove and L. L. Maragakis, Clin. Microbiol. Rev., 2012, 25, 450–470 CrossRef CAS PubMed.
- J. C. Graham and F. K. Gould, J. Antimicrob. Chemother., 2002, 49, 437–444 CrossRef CAS PubMed.
- G. P. Bodey, R. Feld and M. A. Burgess, Am. J. Med. Sci., 1976, 271, 179–186 CrossRef CAS PubMed.
- L. Leibovici, L. Vidal and M. Paul, J. Antimicrob. Chemother., 2009, 63, 246–251 CrossRef CAS PubMed.
- J. A. Martinez, N. Cobos-Trigueros, A. Soriano, M. Almela, M. Ortega, F. Marco, C. Pitart, H. Sterzik, J. Lopez and J. Mensa, Antimicrob. Agents Chemother., 2010, 54, 3590–3596 CrossRef CAS PubMed.
- S. Garneau-Tsodikova and K. J. Labby, Med. Chem. Commun., 2016 10.1039/C5MD00344J.
- M. S. Ramirez and M. E. Tolmasky, Drug Resist. Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- E. H. Serpersu, C. Ozen and E. Wright, Methods Mol. Med., 2008, 142, 261–271 CAS.
- G. D. Wright, Curr. Opin. Microbiol., 1999, 2, 499–503 CrossRef CAS PubMed.
- K. J. Shaw, P. N. Rather, R. S. Hare and G. H. Miller, Microbiol. Rev., 1993, 57, 138–163 CAS.
- D. D. Boehr, D. M. Daigle and G. D. Wright, Biochemistry, 2004, 43, 9846–9855 CrossRef CAS PubMed.
- D. M. Daigle, D. W. Hughes and G. D. Wright, Chem. Biol., 1999, 6, 99–110 CrossRef CAS PubMed.
- S. J. Caldwell and A. M. Berghuis, Antimicrob. Agents Chemother., 2012, 56, 1899–1906 CrossRef CAS PubMed.
- H. Frase, M. Toth and S. B. Vakulenko, J. Biol. Chem., 2012, 287, 43262–43269 CrossRef CAS PubMed.
- D. Centron and P. H. Roy, Antimicrob. Agents Chemother., 2002, 46, 1402–1409 CrossRef CAS PubMed.
- C. Kim, D. Hesek, J. Zajicek, S. B. Vakulenko and S. Mobashery, Biochemistry, 2006, 45, 8368–8377 CrossRef CAS PubMed.
- K. D. Green and S. Garneau-Tsodikova, Biochimie, 2013, 95, 1319–1325 CrossRef CAS PubMed.
- C. Kim, A. Villegas-Estrada, D. Hesek and S. Mobashery, Biochemistry, 2007, 46, 5270–5282 CrossRef CAS PubMed.
- V. Dubois, L. Poirel, C. Marie, C. Arpin, P. Nordmann and C. Quentin, Antimicrob. Agents Chemother., 2002, 46, 638–645 CrossRef CAS PubMed.
- R. E. Mendes, M. A. Toleman, J. Ribeiro, H. S. Sader, R. N. Jones and T. R. Walsh, Antimicrob. Agents Chemother., 2004, 48, 4693–4702 CrossRef CAS PubMed.
- K. D. Green, W. Chen and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2011, 55, 3207–3213 CrossRef CAS PubMed.
- W. Chen, T. Biswas, V. R. Porter, O. V. Tsodikov and S. Garneau-Tsodikova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9804–9808 CrossRef CAS PubMed.
- J. L. Houghton, K. D. Green, R. E. Pricer, A. S. Mayhoub and S. Garneau-Tsodikova, J. Antimicrob. Chemother., 2013, 68, 800–805 CrossRef CAS PubMed.
- K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemMedChem, 2012, 7, 73–77 CrossRef CAS PubMed.
- W. Chen, K. D. Green, O. V. Tsodikov and S. Garneau-Tsodikova, Biochemistry, 2012, 51, 4959–4967 CrossRef CAS PubMed.
- B. C. Jennings, K. J. Labby, K. D. Green and S. Garneau-Tsodikova, Biochemistry, 2013, 52, 5125–5132 CrossRef CAS PubMed.
- W. Chen, K. D. Green and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2012, 56, 5831–5838 CrossRef CAS PubMed.
- K. H. Kim, D. R. An, J. Song, J. Y. Yoon, H. S. Kim, H. J. Yoon, H. N. Im, J. Kim, J. Kim do, S. J. Lee, K. H. Kim, H. M. Lee, H. J. Kim, E. K. Jo, J. Y. Lee and S. W. Suh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7729–7734 CrossRef CAS PubMed.
- R. E. Pricer, J. L. Houghton, K. D. Green, A. S. Mayhoub and S. Garneau-Tsodikova, Mol. BioSyst., 2012, 8, 3305–3313 RSC.
- O. V. Tsodikov, K. D. Green and S. Garneau-Tsodikova, PLoS One, 2014, 9, e92370 Search PubMed.
- J. L. Houghton, T. Biswas, W. Chen, O. V. Tsodikov and S. Garneau-Tsodikova, ChemBioChem, 2013, 14, 2127–2135 CrossRef CAS PubMed.
- K. D. Green, R. E. Pricer, M. N. Stewart and S. Garneau-Tsodikova, ACS Infect. Dis., 2015, 1, 272–283 CrossRef CAS.
- K. D. Green, T. Biswas, C. Chang, R. Wu, W. Chen, B. K. Janes, D. Chalupska, P. Gornicki, P. C. Hanna, O. V. Tsodikov, A. Joachimiak and S. Garneau-Tsodikova, Biochemistry, 2015, 54, 3197–3206 CrossRef CAS PubMed.
- M. Y. Fosso, Y. Li and S. Garneau-Tsodikova, Med. Chem. Commun., 2014, 5, 1075–1091 RSC.
- K. J. Labby and S. Garneau-Tsodikova, Future Med. Chem., 2013, 5, 1285–1309 CrossRef CAS PubMed.
- D. Moazed and H. F. Noller, Nature, 1987, 327, 389–394 CrossRef CAS PubMed.
- J. Kondo, K. Pachamuthu, B. Francois, J. Szychowski, S. Hanessian and E. Westhof, ChemMedChem, 2007, 2, 1631–1638 CrossRef CAS PubMed.
- M. A. Borovinskaya, R. D. Pai, W. Zhang, B. S. Schuwirth, J. M. Holton, G. Hirokawa, H. Kaji, A. Kaji and J. H. Cate, Nat. Struct. Mol. Biol., 2007, 14, 727–732 CAS.
- L. Wang, A. Pulk, M. R. Wasserman, M. B. Feldman, R. B. Altman, J. H. Cate and S. C. Blanchard, Nat. Struct. Mol. Biol., 2012, 19, 957–963 CAS.
- M. R. Wasserman, A. Pulk, Z. Zhou, R. B. Altman, J. C. Zinder, K. D. Green, S. Garneau-Tsodikova, J. H. Doudna Cate and S. C. Blanchard, Nat. Commun., 2015, 6, 7896 CrossRef CAS PubMed.
- A. A. Bastian and A. Hermann, W.O. Pat. 2013/191550 A1, 2013 Search PubMed.
- P. B. Alper, M. Hendrix, P. Sears and C.-H. Wong, J. Am. Chem. Soc., 1998, 120, 1965–1978 CrossRef CAS.
- J. Roestamadji, I. Grapsas and S. Mobashery, J. Am. Chem. Soc., 1995, 117, 11060–11069 CrossRef CAS.
- J. Haddad, S. B. Vakulenko and S. Mobashery, J. Am. Chem. Soc., 1999, 121, 11922–11923 CrossRef CAS.
- R. Pathak, D. Perez-Fernandez, R. Nandurdikar, S. K. Kalapala, E. C. Böttger and A. Vasella, Helv. Chim. Acta, 2008, 91, 1533–1552 CrossRef CAS.
- S. Hanessian, J. Szychowski, N. B. Campos-Reales Pineda, A. Furtos and J. W. Keillor, Bioorg. Med. Chem. Lett., 2007, 17, 3221–3225 CrossRef CAS PubMed.
- J. Li, F. I. Chiang, H. N. Chen and C. W. Chang, J. Org. Chem., 2007, 72, 4055–4066 CrossRef CAS PubMed.
- S. H. Lee and C. S. Cheong, Tetrahedron, 2001, 57, 4801–4815 CrossRef CAS.
- S. Samantaray, U. Marathe, S. Dasgupta, V. K. Nandicoori and R. P. Roy, J. Am. Chem. Soc., 2008, 130, 2132–2133 CrossRef CAS PubMed.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- K. D. Green, W. Chen, J. L. Houghton, M. Fridman and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 119–126 CrossRef CAS PubMed.
- A. Hermann, A. A. Bastian and A. Marcozzi, U.S. Pat. 2014/0243280 A1, 2014 Search PubMed.
- D. Coquiere, A. de la Lande, O. Parisel, T. Prange and O. Reinaud, Chemistry, 2009, 15, 11912–11917 CrossRef CAS PubMed.
- K. Y. Wang, S. McCurdy, R. G. Shea, S. Swaminathan and P. H. Bolton, Biochemistry, 1993, 32, 1899–1904 CrossRef CAS PubMed.
- W. Sun, L. Du and M. Li, Curr. Pharm. Des., 2010, 16, 2269–2278 CrossRef CAS PubMed.
- L. Jiang, A. K. Suri, R. Fiala and D. J. Patel, Chem. Biol., 1997, 4, 35–50 CrossRef CAS PubMed.
- Y. Kobayashi, Y. Akiyama, T. Murakami, N. Minowa, M. Tsushima, Y. Hiraiwa, S. Murakami, M. Abe, K. Sasaki, S. Hoshiko, T. Miyake, K. Takahashi and D. Ikeda, U.S. Pat. 2013/0345411 A1, 2013 Search PubMed.
- F. S. Liang, S. K. Wang, T. Nakatani and C. H. Wong, Angew. Chem., Int. Ed., 2004, 43, 6496–6500 CrossRef CAS PubMed.
- B. Clique, A. Ironmonger, B. Whittaker, J. Colley, J. Titchmarsh, P. Stockley and A. Nelson, Org. Biomol. Chem., 2005, 3, 2776–2785 CAS.
- R. B. Yan, M. Yuan, Y. Wu, X. You and X. S. Ye, Bioorg. Med. Chem., 2011, 19, 30–40 CrossRef CAS PubMed.
- K. Tatsuta, J. Antibiot., 2013, 66, 107–129 CrossRef CAS PubMed.
- L. Chen, M. Hainrichson, D. Bourdetsky, A. Mor, S. Yaron and T. Baasov, Bioorg. Med. Chem., 2008, 16, 8940–8951 CrossRef CAS PubMed.
- W. A. Greenberg, E. S. Priestley, P. S. Sears, P. B. Alper, C. Rosenbohm, M. Hendrix, S. C. Hung and C. H. Wong, J. Am. Chem. Soc., 1999, 121, 6527–6541 CrossRef CAS.
- S. Kondo, Y. Ikeda, D. Ikeda, T. Takeuchi, T. Usui, M. Ishii, T. Kudo, S. Gomi and S. Shibahara, J. Antibiot., 1994, 47, 821–832 CrossRef CAS PubMed.
- K. Hotta, A. Sunada, J. Ishikawa, S. Mizuno, Y. Ikeda and S. Kondo, J. Antibiot., 1998, 51, 735–742 CrossRef CAS PubMed.
- Y. Hiraiwa, T. Usui, Y. Akiyama, K. Maebashi, N. Minowa and D. Ikeda, Bioorg. Med. Chem. Lett., 2007, 17, 3540–3543 CrossRef CAS PubMed.
- Y. Hiraiwa, N. Minowa, T. Usui, Y. Akiyama, K. Maebashi and D. Ikeda, Bioorg. Med. Chem. Lett., 2007, 17, 6369–6372 CrossRef CAS PubMed.
- K. Sasaki, Y. Kobayashi, T. Kurihara, Y. Yamashita, Y. Takahashi, T. Miyake and Y. Akamatsu, J. Antibiot., 2015 DOI:10.1038/ja.2015.61.
- T. Glinka and O. Rodny, W.O. Pat. 2011/143497 A1, 2011 Search PubMed.
- M. G. Chaparian, M. Brady, B. R. Renikuntla, S. Gadthula, S. Meneni and V. S. P. Chaturvedula, U.S. Pat. 2013/0203693 A1, 2013 Search PubMed.
- M. Fridman, I. M. Herzog, M. Feldman and Y. Berkov-Zrihen, W.O. Pat. 2014/013495 A1, 2014 Search PubMed.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, J. Med. Chem., 2000, 43, 3085–3092 CrossRef CAS PubMed.
- H. Tsubery, I. Ofek, S. Cohen, M. Eisenstein and M. Fridkin, Mol. Pharmacol., 2002, 62, 1036–1042 CrossRef CAS PubMed.
- J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef CAS PubMed.
- M. Ouberai, F. El Garch, A. Bussiere, M. Riou, D. Alsteens, L. Lins, I. Baussanne, Y. F. Dufrene, R. Brasseur, J. L. Decout and M. P. Mingeot-Leclercq, Biochim. Biophys. Acta, 2011, 1808, 1716–1727 CrossRef CAS PubMed.
- I. M. Herzog, K. D. Green, Y. Berkov-Zrihen, M. Feldman, R. R. Vidavski, A. Eldar-Boock, R. Satchi-Fainaro, A. Eldar, S. Garneau-Tsodikova and M. Fridman, Angew. Chem., Int. Ed., 2012, 51, 5652–5656 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, I. M. Herzog, M. Feldman and M. Fridman, Org. Lett., 2013, 15, 6144–6147 CrossRef CAS PubMed.
- Y. Berkov-Zrihen, I. M. Herzog, R. I. Benhamou, M. Feldman, K. B. Steinbuch, P. Shaul, S. Lerer, A. Eldar and M. Fridman, Chemistry, 2015, 21, 4340–4349 CrossRef CAS PubMed.
- J. B. Aggen, P. Dozzo, A. A. Goldblum, D. J. Hildebrandt, T. R. Kane, M. H. Gliedt and M. S. Linsell, W.O. Pat. 2011/044501 A1, 2011 Search PubMed.
- P. Dozzo, A. A. Goldblum, J. B. Aggen and M. S. Linsell, U.S. Pat. 849354 B2, 2013 Search PubMed.
- P. Dozzo and H. E. Moser, Expert Opin. Ther. Pat., 2010, 20, 1321–1341 CrossRef CAS PubMed.
- A. Endimiani, K. M. Hujer, A. M. Hujer, E. S. Armstrong, Y. Choudhary, J. B. Aggen and R. A. Bonomo, Antimicrob. Agents Chemother., 2009, 53, 4504–4507 CrossRef CAS PubMed.
- D. Landman, E. Babu, N. Shah, P. Kelly, M. Backer, S. Bratu and J. Quale, J. Antimicrob. Chemother., 2010, 65, 2123–2127 CrossRef CAS PubMed.
- I. Galani, M. Souli, G. L. Daikos, Z. Chrysouli, G. Poulakou, M. Psichogiou, T. Panagea, A. Argyropoulou, I. Stefanou, G. Plakias, H. Giamarellou and G. Petrikkos, J. Chemother., 2012, 24, 191–194 CrossRef CAS PubMed.
- F. C. Tenover, I. Tickler, E. S. Armstrong, A. Kubo, S. Lopez, D. H. Persing and G. H. Miller, Int. J. Antimicrob. Agents, 2011, 38, 352–354 CrossRef CAS PubMed.
- G. Lin, L. M. Ednie and P. C. Appelbaum, Antimicrob. Agents Chemother., 2010, 54, 2258–2261 CrossRef CAS PubMed.
- J. B. Aggen, E. S. Armstrong, A. A. Goldblum, P. Dozzo, M. S. Linsell, M. J. Gliedt, D. J. Hildebrandt, L. A. Feeney, A. Kubo, R. D. Matias, S. Lopez, M. Gomez, K. B. Wlasichuk, R. Diokno, G. H. Miller and H. E. Moser, Antimicrob. Agents Chemother., 2010, 54, 4636–4642 CrossRef CAS PubMed.
- D. M. Livermore, S. Mushtaq, M. Warner, J. C. Zhang, S. Maharjan, M. Doumith and N. Woodford, J. Antimicrob. Chemother., 2011, 66, 48–53 CrossRef CAS PubMed.
- D. Landman, P. Kelly, M. Backer, E. Babu, N. Shah, S. Bratu and J. Quale, J. Antimicrob. Chemother., 2011, 66, 332–334 CrossRef CAS PubMed.
- N. Reyes, J. B. Aggen and C. F. Kostrub, Antimicrob. Agents Chemother., 2011, 55, 1728–1733 CrossRef CAS PubMed.
- G. A. Pankuch, G. Lin, A. Kubo, E. S. Armstrong, P. C. Appelbaum and K. Kosowska-Shick, Antimicrob. Agents Chemother., 2011, 55, 2463–2465 CrossRef CAS PubMed.
- R. T. Cass, C. D. Brooks, N. A. Havrilla, K. J. Tack, M. T. Borin, D. Young and J. B. Bruss, Antimicrob. Agents Chemother., 2011, 55, 5874–5880 CrossRef CAS PubMed.
- A. J. Ricci, R. J. Greenhouse and A. G. Cheng, W.O. Pat. 2014/1454713 A1, 2014 Search PubMed.
- L. P. Rybak and V. Ramkumar, Kidney Int., 2007, 72, 931–935 CrossRef CAS PubMed.
- A. Alharazneh, L. Luk, M. Huth, A. Monfared, P. S. Steyger, A. G. Cheng and A. J. Ricci, PLoS One, 2011, 6, e22347 CAS.
- H. C. Ou, S. Keating, P. Wu, J. A. Simon, D. W. Raible and E. W. Rubel, J. Assoc. Res. Otolaryngol., 2012, 13, 759–770 CrossRef PubMed.
- B. Pan, J. Waguespack, M. E. Schnee, C. LeBlanc and A. J. Ricci, J. Neurophysiol., 2012, 107, 2408–2420 CrossRef CAS PubMed.
- E. C. Böttger and A. Vasella, W.O. Pat. 2013/170985 A1, 2013 Search PubMed.
- H. Jiang, S. H. Sha and J. Schacht, J. Neurosci. Res., 2006, 83, 1544–1551 CrossRef CAS PubMed.
- S. H. Sha and J. Schacht, Hear. Res., 1999, 128, 112–118 CrossRef CAS PubMed.
- E. C. Böttger and A. Vasella, W.O. Pat. 2012/034955 A1, 2012 Search PubMed.
- H. A. Kirst, U.S. Pat. 4370665, 1982 Search PubMed.
- A. R. Mandhapati, D. Shcherbakov, S. Duscha, A. Vasella, E. C. Böttger and D. Crich, ChemMedChem, 2014, 9, 2074–2083 CrossRef CAS PubMed.
- K. Auclair and K. Vong, W.O. Pat. 2012/097454 A1, 2012 Search PubMed.
- D. M. Johnson and R. N. Jones, Clin. Microbiol. Infect., 1997, 3, 335–344 CrossRef CAS PubMed.
- C. Zhi, Z. Y. Long, A. Manikowski, J. Comstock, W. C. Xu, N. C. Brown, P. M. Tarantino Jr., K. A. Holm, E. J. Dix, G. E. Wright, M. H. Barnes, M. M. Butler, K. A. Foster, W. A. LaMarr, B. Bachand, R. Bethell, C. Cadilhac, S. Charron, S. Lamothe, I. Motorina and R. Storer, J. Med. Chem., 2006, 49, 1455–1465 CrossRef CAS PubMed.
- D. Sriram, P. Yogeeswari, J. S. Basha, D. R. Radha and V. Nagaraja, Bioorg. Med. Chem., 2005, 13, 5774–5778 CrossRef CAS PubMed.
- C. Hubschwerlen, J. L. Specklin, C. Sigwalt, S. Schroeder and H. H. Locher, Bioorg. Med. Chem., 2003, 11, 2313–2319 CrossRef CAS PubMed.
- R. J. Kerns, M. J. Rybak, G. W. Kaatz, F. Vaka, R. Cha, R. G. Grucz and V. U. Diwadkar, Bioorg. Med. Chem. Lett., 2003, 13, 2109–2112 CrossRef CAS PubMed.
- F. Gao, X. Yan, O. M. Baettig, A. M. Berghuis and K. Auclair, Angew. Chem., Int. Ed., 2005, 44, 6859–6862 CrossRef CAS PubMed.
- X. Yan, F. Gao, S. Yotphan, P. Bakirtzian and K. Auclair, Bioorg. Med. Chem., 2007, 15, 2944–2951 CrossRef CAS PubMed.
- F. Gao, X. Yan, T. Shakya, O. M. Baettig, S. Ait-Mohand-Brunet, A. M. Berghuis, G. D. Wright and K. Auclair, J. Med. Chem., 2006, 49, 5273–5281 CrossRef CAS PubMed.
- F. Gao, X. Yan, O. Zahr, A. Larsen, K. Vong and K. Auclair, Bioorg. Med. Chem. Lett., 2008, 18, 5518–5522 CrossRef CAS PubMed.
- F. Gao, X. Yan and K. Auclair, Chemistry, 2009, 15, 2064–2070 CrossRef CAS PubMed.
- K. Vong, I. S. Tam, X. Yan and K. Auclair, ACS Chem. Biol., 2012, 7, 470–475 CrossRef CAS PubMed.
- T. Baasov, V. Pokrovskaya, V. Belakhov and M. Hainrichson, U.S. Pat. 8809286 B2, 2014 Search PubMed.
- F. Schweizer, S. Bera and G. Zhanel, W.O. Pat. 2011/124986 A2, 2011 Search PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 417–428 CrossRef CAS PubMed.
- P. B. Savage, C. Li, U. Taotafa, B. Ding and Q. Guan, FEMS Microbiol. Lett., 2002, 217, 1–7 CrossRef CAS PubMed.
- D. Liu, S. Choi, B. Chen, R. J. Doerksen, D. J. Clements, J. D. Winkler, M. L. Klein and W. F. DeGrado, Angew. Chem., Int. Ed., 2004, 43, 1158–1162 CrossRef CAS PubMed.
- S. A. David, J. Mol. Recognit., 2001, 14, 370–387 CrossRef CAS PubMed.
- S. A. Shelburne, D. M. Musher, K. Hulten, H. Ceasar, M. Y. Lu, I. Bhaila and R. J. Hamill, Antimicrob. Agents Chemother., 2004, 48, 4016–4019 CrossRef CAS PubMed.
- K. R. P. Drew, L. K. Sanders, Z. W. Culumber, O. Zribi and G. C. L. Wong, J. Am. Chem. Soc., 2009, 131, 486–493 CrossRef PubMed.
- S. Bera, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2010, 53, 3626–3631 CrossRef CAS PubMed.
- F. Schweizer, S. Bera and G. Zhanel, U.S. Pat. 2013/0053337 A1, 2013 Search PubMed.
- S. Bera, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2008, 51, 6160–6164 CrossRef CAS PubMed.
- S. Bera, G. G. Zhanel and F. Schweizer, J. Antimicrob. Chemother., 2010, 65, 1224–1227 CrossRef CAS PubMed.
- S. Bera, G. G. Zhanel and F. Schweizer, Bioorg. Med. Chem. Lett., 2010, 20, 3031–3035 CrossRef CAS PubMed.
- S. Bera, G. G. Zhanel and F. Schweizer, Carbohydr. Res., 2011, 346, 560–568 CrossRef CAS PubMed.
- S. Bera, R. Dhondikubeer, B. Findlay, G. G. Zhanel and F. Schweizer, Molecules, 2012, 17, 9129–9141 CrossRef CAS PubMed.
- M. C. Fisher, D. A. Henk, C. J. Briggs, J. S. Brownstein, L. C. Madoff, S. L. McCraw and S. J. Gurr, Nature, 2012, 484, 186–194 CrossRef CAS PubMed.
- G. W. Procop and G. D. Roberts, Clin. Lab. Med., 2004, 24, 691–719 CrossRef PubMed , vi-vii.
- R. V. Fleming, T. J. Walsh and E. J. Anaissie, Infect. Dis. Clin. North. Am., 2002, 16, 915–933 CrossRef PubMed , vi-vii.
- G. J. Alangaden, Infect. Dis. Clin. North. Am., 2011, 25, 201–225 CrossRef PubMed.
- H. B. Lee, Y. Kim, J. C. Kim, G. J. Choi, S. H. Park, C. J. Kim and H. S. Jung, J. Appl. Microbiol., 2005, 99, 836–843 CrossRef CAS PubMed.
- F. C. Odds, A. J. Brown and N. A. Gow, Trends Microbiol., 2003, 11, 272–279 CrossRef CAS PubMed.
- M. A. Ghannoum and L. B. Rice, Clin. Microbiol. Rev., 1999, 12, 501–517 CAS.
- S. Hanessian, K. Pachamuthu, J. Szychowski, A. Giguere, E. E. Swayze, M. T. Migawa, B. Francois, J. Kondo and E. Westhof, Bioorg. Med. Chem. Lett., 2010, 20, 7097–7101 CrossRef CAS PubMed.
- J. Zhang, F. I. Chiang, L. Wu, P. G. Czyryca, D. Li and C. W. Chang, J. Med. Chem., 2008, 51, 7563–7573 CrossRef CAS PubMed.
- M. Y. Fosso, H. Zhu, K. D. Green, S. Garneau-Tsodikova and K. Fredrick, ChemBioChem, 2015, 16, 1565–1570 CrossRef CAS PubMed.
- L. Zimmermann, A. Bussiere, M. Ouberai, I. Baussanne, C. Jolivalt, M. P. Mingeot-Leclercq and J. L. Decout, J. Med. Chem., 2013, 56, 7691–7705 CrossRef CAS PubMed.
- S. Hanessian, J. Szychowski, S. S. Adhikari, G. Vasquez, P. Kandasamy, E. E. Swayze, M. T. Migawa, R. Ranken, B. Francois, J. Wirmer-Bartoschek, J. Kondo and E. Westhof, J. Med. Chem., 2007, 50, 2352–2369 CrossRef CAS PubMed.
- J. Szychowski, J. Kondo, O. Zahr, K. Auclair, E. Westhof, S. Hanessian and J. W. Keillor, ChemMedChem, 2011, 6, 1961–1966 CrossRef CAS PubMed.
- J. Zhang, K. Keller, J. Y. Takemoto, M. Bensaci, A. Litke, P. G. Czyryca and C. W. Chang, J. Antibiot., 2009, 62, 539–544 CrossRef CAS PubMed.
- R. Dhondikubeer, S. Bera, G. G. Zhanel and F. Schweizer, J. Antibiot., 2012, 65, 495–498 CrossRef CAS PubMed.
- C. W. Chang, M. Fosso, Y. Kawasaki, S. Shrestha, M. F. Bensaci, J. Wang, C. K. Evans and J. Y. Takemoto, J. Antibiot., 2010, 63, 667–672 CrossRef CAS PubMed.
- S. Shrestha, M. Grilley, M. Y. Fosso, C. W. Chang and J. Y. Takemoto, PLoS One, 2013, 8, e73843 CAS.
- C. W. Chang and J. Y. Takemoto, Med. Chem. Commun., 2014, 5, 1048–1057 RSC.
- J. Li, J. Wang, P. G. Czyryca, H. Chang, T. W. Orsak, R. Evanson and C. W. Chang, Org. Lett., 2004, 6, 1381–1384 CrossRef CAS PubMed.
- J. Wang, J. Li, H. N. Chen, H. Chang, C. T. Tanifum, H. H. Liu, P. G. Czyryca and C. W. Chang, J. Med. Chem., 2005, 48, 6271–6285 CrossRef CAS PubMed.
- C. H. Chou, C. S. Wu, C. H. Chen, L. D. Lu, S. S. Kulkarni, C. H. Wong and S. C. Hung, Org. Lett., 2004, 6, 585–588 CrossRef CAS PubMed.
- J. Li, F. I. Chiang, H. N. Chen and C. W. Chang, Bioorg. Med. Chem., 2007, 15, 7711–7719 CrossRef CAS PubMed.
- B. Elchert, J. Li, J. Wang, Y. Hui, R. Rai, R. Ptak, P. Ward, J. Y. Takemoto, M. Bensaci and C. W. Chang, J. Org. Chem., 2004, 69, 1513–1523 CrossRef CAS PubMed.
- J. Wang, J. Li, D. Tuttle, J. Y. Takemoto and C. W. Chang, Org. Lett., 2002, 4, 3997–4000 CrossRef CAS PubMed.
- C. W. Chang, Y. Hui, B. Elchert, J. Wang, J. Li and R. Rai, Org. Lett., 2002, 4, 4603–4606 CrossRef CAS PubMed.
- M. Y. Fosso, M. N. Alfindee, Q. Zhang, V. N. Nziko, Y. Kawasaki, S. K. Shrestha, J. Bearss, R. Gregory, J. Y. Takemoto and C.-W. T. Chang, J. Org. Chem., 2015, 80, 4398–4411 CrossRef CAS PubMed.
- M. Y. Fosso, S. K. Shrestha, K. D. Green and S. Garneau-Tsodikova, J. Med. Chem., 2015 DOI:10.1021/acs.jmedchem.5b01375.
- S. K. Shrestha, M. Y. Fosso, K. D. Green and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4861–4869 CrossRef CAS PubMed.
- S. K. Shrestha, M. Y. Fosso and S. Garneau-Tsodikova, Sci. Rep., 2015, 5, 17070 CrossRef PubMed.
- C.-W. T. Chang, C. K. Evans and J. Y. Takemoto, U.S. Pat. 2011/0130357 A1, 2011 Search PubMed.
- J. Y. Takemoto, C.-W. T. Chang, S. K. Shrestha and M. Grilley, U.S. Pat. 2014/0256665 A1, 2014 Search PubMed.
- S. K. Shrestha, C. W. Chang, N. Meissner, J. Oblad, J. P. Shrestha, K. N. Sorensen, M. M. Grilley and J. Y. Takemoto, Front. Microbiol., 2014, 5, 671 Search PubMed.
- L. Linde and B. Kerem, Trends Genet., 2008, 24, 552–563 CrossRef CAS PubMed.
- L. V. Zingman, S. Park, T. M. Olson, A. E. Alekseev and A. Terzic, Clin. Pharmacol. Ther., 2007, 81, 99–103 CrossRef CAS PubMed.
- N. Venkataraman, A. L. Cole, P. Ruchala, A. J. Waring, R. I. Lehrer, O. Stuchlik, J. Pohl and A. M. Cole, PLoS Biol., 2009, 7, e95 Search PubMed.
- R. Kellermayer, Eur. J. Med. Genet., 2006, 49, 445–450 CrossRef PubMed.
- D. M. Bedwell, A. Kaenjak, D. J. Benos, Z. Bebok, J. K. Bubien, J. Hong, A. Tousson, J. P. Clancy and E. J. Sorscher, Nat. Med., 1997, 3, 1280–1284 CrossRef CAS PubMed.
- V. Malik, L. R. Rodino-Klapac, L. Viollet and J. R. Mendell, Ther. Adv. Neurol. Disord., 2010, 3, 379–389 CrossRef CAS PubMed.
- A. C. Popescu, E. Sidorova, G. Zhang and J. H. Eubanks, J. Neurosci. Res., 2010, 88, 2316–2324 CAS.
- E. C. Wolstencroft, V. Mattis, A. A. Bajer, P. J. Young and C. L. Lorson, Hum. Mol. Genet., 2005, 14, 1199–1210 CrossRef CAS PubMed.
- V. Allamand, L. Bidou, M. Arakawa, C. Floquet, M. Shiozuka, M. Paturneau-Jouas, C. Gartioux, G. S. Butler-Browne, V. Mouly, J. P. Rousset, R. Matsuda, D. Ikeda and P. Guicheney, J. Gene Med., 2008, 10, 217–224 CrossRef PubMed.
- A. Rebibo-Sabbah, I. Nudelman, Z. M. Ahmed, T. Baasov and T. Ben-Yosef, Hum. Genet., 2007, 122, 373–381 CrossRef CAS PubMed.
- C. H. Lai, H. H. Chun, S. A. Nahas, M. Mitui, K. M. Gamo, L. Du and R. A. Gatti, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15676–15681 CrossRef CAS PubMed.
- G. Gunn, Y. Dai, M. Du, V. Belakhov, J. Kandasamy, T. R. Schoeb, T. Baasov, D. M. Bedwell and K. M. Keeling, Mol. Genet. Metab., 2014, 111, 374–381 CrossRef CAS PubMed.
- P. D. James, S. Raut, G. E. Rivard, M. C. Poon, M. Warner, S. McKenna, J. Leggo and D. Lillicrap, Blood, 2005, 106, 3043–3048 CrossRef CAS PubMed.
- S. M. Rowe and J. P. Clancy, BioDrugs, 2009, 23, 165–174 CrossRef CAS PubMed.
- V. B. Mattis, R. Rai, J. Wang, C. W. Chang, T. Coady and C. L. Lorson, Hum. Genet., 2006, 120, 589–601 CrossRef CAS PubMed.
- B. Chawla, A. Jhingran, A. Panigrahi, K. D. Stuart and R. Madhubala, PLoS One, 2011, 6, e26660 CAS.
- M. Manuvakhova, K. Keeling and D. M. Bedwell, RNA, 2000, 6, 1044–1055 CrossRef CAS PubMed.
- I. Sermet-Gaudelus, M. Renouil, A. Fajac, L. Bidou, B. Parbaille, S. Pierrot, N. Davy, E. Bismuth, P. Reinert, G. Lenoir, J. F. Lesure, J. P. Rousset and A. Edelman, BMC Med., 2007, 5, 5 CrossRef PubMed.
- L. Linde, S. Boelz, M. Nissim-Rafinia, Y. S. Oren, M. Wilschanski, Y. Yaacov, D. Virgilis, G. Neu-Yilik, A. E. Kulozik, E. Kerem and B. Kerem, J. Clin. Invest., 2007, 117, 683–692 CAS.
- M. Du, K. M. Keeling, L. Fan, X. Liu, T. Kovacs, E. Sorscher and D. M. Bedwell, J. Mol. Med., 2006, 84, 573–582 CrossRef CAS PubMed.
- S. M. Rowe, P. Sloane, L. P. Tang, K. Backer, M. Mazur, J. Buckley-Lanier, I. Nudelman, V. Belakhov, Z. Bebok, E. Schwiebert, T. Baasov and D. M. Bedwell, J. Mol. Med., 2011, 89, 1149–1161 CrossRef CAS PubMed.
- I. Nudelman, A. Rebibo-Sabbah, D. Shallom-Shezifi, M. Hainrichson, I. Stahl, T. Ben-Yosef and T. Baasov, Bioorg. Med. Chem. Lett., 2006, 16, 6310–6315 CrossRef CAS PubMed.
- I. Nudelman, A. Rebibo-Sabbah, M. Cherniavsky, V. Belakhov, M. Hainrichson, F. Chen, J. Schacht, D. S. Pilch, T. Ben-Yosef and T. Baasov, J. Med. Chem., 2009, 52, 2836–2845 CrossRef CAS PubMed.
- C. Brendel, V. Belakhov, H. Werner, E. Wegener, J. Gartner, I. Nudelman, T. Baasov and P. Huppke, J. Mol. Med., 2011, 89, 389–398 CrossRef CAS PubMed.
- M. Vecsler, B. Ben Zeev, I. Nudelman, Y. Anikster, A. J. Simon, N. Amariglio, G. Rechavi, T. Baasov and E. Gak, PLoS One, 2011, 6, e20733 CAS.
- I. Nudelman, D. Glikin, B. Smolkin, M. Hainrichson, V. Belakhov and T. Baasov, Bioorg. Med. Chem., 2010, 18, 3735–3746 CrossRef CAS PubMed.
- K. F. Blount and Y. Tor, ChemBioChem, 2006, 7, 1612–1621 CrossRef CAS PubMed.
- K. F. Blount, F. Zhao, T. Hermann and Y. Tor, J. Am. Chem. Soc., 2005, 127, 9818–9829 CrossRef CAS PubMed.
- B. Willis and D. P. Arya, Biochemistry, 2006, 45, 10217–10232 CrossRef CAS PubMed.
- A. Lapidot, A. Berchanski and G. Borkow, FEBS J., 2008, 275, 5236–5257 CrossRef CAS PubMed.
- R. Hegde, G. Borkow, A. Berchanski and A. Lapidot, FEBS J., 2007, 274, 6523–6536 CrossRef CAS PubMed.
- E. Ennifar, J. C. Paillart, S. Bernacchi, P. Walter, P. Pale, J. L. Decout, R. Marquet and P. Dumas, Biochimie, 2007, 89, 1195–1203 CrossRef CAS PubMed.
- S. Bernacchi, S. Freisz, C. Maechling, B. Spiess, R. Marquet, P. Dumas and E. Ennifar, Nucleic Acids Res., 2007, 35, 7128–7139 CrossRef CAS PubMed.
- J. B. Tok, L. J. Dunn and R. C. Des Jean, Bioorg. Med. Chem. Lett., 2001, 11, 1127–1131 CrossRef CAS PubMed.
- J. B. Tok and J. Fenker, Bioorg. Med. Chem. Lett., 2001, 11, 2987–2991 CrossRef CAS PubMed.
- E. Riguet, J. Desire, O. Boden, V. Ludwig, M. Gobel, C. Bailly and J. L. Decout, Bioorg. Med. Chem. Lett., 2005, 15, 4651–4655 CrossRef CAS PubMed.
- D. W. Staple, V. Venditti, N. Niccolai, L. Elson-Schwab, Y. Tor and S. E. Butcher, ChemBioChem, 2008, 9, 93–102 CrossRef CAS PubMed.
- T. Baasov, D. Atia-Glikin, J. Kandasamy and V. Belakhov, U.S. Pat. 2014/0357590 A1, 2014 Search PubMed.
- M. Hainrichson, I. Nudelman and T. Baasov, Org. Biomol. Chem., 2008, 6, 227–239 CAS.
- M. Howard, R. A. Frizzell and D. M. Bedwell, Nat. Med., 1996, 2, 467–469 CrossRef CAS PubMed.
- T. Baasov, T. Ben-Yosef, I. Nudelman, A. Rebibo-Sabbah, D. Shallom-Shezifi and M. Hainrichson, W.O. Pat. 2007/113841, 2007 Search PubMed.
- T. Baasov, T. Ben-Yosef, I. Nudelman, A. Rebibo-Sabbah, D. Shallom-Shezifi and M. Hainrichson, W.O. Pat. 2007/11384183 A3, 2007 Search PubMed.
- T. Goldmann, N. Overlack, F. Moller, V. Belakhov, M. van Wyk, T. Baasov, U. Wolfrum and K. Nagel-Wolfrum, EMBO Mol. Med., 2012, 4, 1186–1199 CrossRef CAS PubMed.
- J. Kondo, M. Hainrichson, I. Nudelman, D. Shallom-Shezifi, C. M. Barbieri, D. S. Pilch, E. Westhof and T. Baasov, ChemBioChem, 2007, 8, 1700–1709 CrossRef CAS PubMed.
- T. Goldmann, A. Rebibo-Sabbah, N. Overlack, I. Nudelman, V. Belakhov, T. Baasov, T. Ben-Yosef, U. Wolfrum and K. Nagel-Wolfrum, Invest. Ophthalmol. Visual Sci., 2010, 51, 6671–6680 Search PubMed.
- D. P. Arya, N. Ranjan and S. Kumar, U.S. Pat. 2011/0046982 A1, 2011 Search PubMed.
- N. Tassew and M. Thompson, Org. Biomol. Chem., 2003, 1, 3268–3270 CAS.
- S. Wang, P. W. Huber, M. Cui, A. W. Czarnik and H. Y. Mei, Biochemistry, 1998, 37, 5549–5557 CrossRef CAS PubMed.
- N. Ranjan and D. P. Arya, Molecules, 2013, 18, 14228–14240 CrossRef CAS PubMed.
- S. B. Rajur, J. Robles, K. Wiederholt, R. G. Kuimelis and L. W. McLaughlin, J. Org. Chem., 1997, 62, 523–529 CrossRef CAS PubMed.
- N. Ranjan, E. Davis, L. Xue and D. P. Arya, Chem. Commun., 2013, 49, 5796–5798 RSC.
- B. Willis and D. P. Arya, Biochemistry, 2010, 49, 452–469 CrossRef CAS PubMed.
- S. Kumar, P. Kellish, W. E. Robinson Jr., D. Wang, D. H. Appella and D. P. Arya, Biochemistry, 2012, 51, 2331–2347 CrossRef CAS PubMed.
- S. Kumar, M. N. Spano and D. P. Arya, Bioorg. Med. Chem., 2015, 23, 3105–3109 CrossRef CAS PubMed.
- B. Willis and D. P. Arya, Bioorg. Med. Chem., 2014, 22, 2327–2332 CrossRef CAS PubMed.
- L. Jiang, D. Watkins, Y. Jin, C. Gong, A. King, A. Z. Washington, K. D. Green, S. Garneau-Tsodikova, A. K. Oyelere and D. P. Arya, ACS Chem. Biol., 2015, 10, 1278–1289 CrossRef CAS PubMed.
- D. Watkins, S. Kumar, K. D. Green, D. P. Arya and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 3899–3905 CrossRef CAS PubMed.
- O. W. Guthrie, Toxicology, 2008, 249, 91–96 CrossRef PubMed.
- J. Xie, A. E. Talaska and J. Schacht, Hear. Res., 2011, 281, 28–37 CrossRef CAS PubMed.
- M. X. Guan, Mitochondrion, 2011, 11, 237–245 CrossRef CAS PubMed.
- E. S. Armstrong and G. H. Miller, Curr. Opin. Microbiol., 2010, 13, 565–573 CrossRef CAS PubMed.
- P. Poulikakos and M. E. Falagas, Expert Opin. Pharmacother., 2013, 14, 1585–1597 CrossRef CAS PubMed.
- S. Jana and J. K. Deb, Appl. Microbiol. Biotechnol., 2006, 70, 140–150 CrossRef CAS PubMed.
- B. Becker and M. A. Cooper, ACS Chem. Biol., 2013, 8, 105–115 CrossRef CAS PubMed.
- S. Magnet and J. S. Blanchard, Chem. Rev., 2005, 105, 477–498 CrossRef CAS PubMed.
- G. F. Busscher, F. P. Rutjes and F. L. van Delft, Chem. Rev., 2005, 105, 775–791 CrossRef CAS PubMed.
- G. G. Zhanel, C. D. Lawson, S. Zelenitsky, B. Findlay, F. Schweizer, H. Adam, A. Walkty, E. Rubinstein, A. S. Gin, D. J. Hoban, J. P. Lynch and J. A. Karlowsky, Expert Rev. Anti-Infect. Ther., 2012, 10, 459–473 CrossRef CAS PubMed.
- M. S. Ramirez, N. Nikolaidis and M. E. Tolmasky, Front. Microbiol., 2013, 4, 121 Search PubMed.
- K. Shi, S. J. Caldwell, D. H. Fong and A. M. Berghuis, Front. Cell. Infect. Microbiol., 2013, 3, 22 Search PubMed.
- K. Vong and K. Auclair, Med. Chem. Commun., 2012, 3, 397–407 RSC.
- D. L. Lin, T. Tran, J. Y. Alam, S. R. Herron, M. S. Ramirez and M. E. Tolmasky, Antimicrob. Agents Chemother., 2014, 58, 4238–4241 CrossRef CAS PubMed.
- Y. Li, K. D. Green, B. R. Johnson and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4148–4156 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |