
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-based virtual screening for fragment-like ligands of the G protein-coupled histamine H4 receptor†
Enade P.
Istyastono‡
ab,
Albert J.
Kooistra‡
a,
Henry F.
Vischer
a,
Martien
Kuijer
a,
Luc
Roumen
a,
Saskia
Nijmeijer
a,
Rogier A.
Smits
c,
Iwan J. P.
de Esch
a,
Rob
Leurs
a and
Chris
de Graaf
*a
aDivision of Medicinal Chemistry, Amsterdam Institute for Molecules, Medicines and Systems (AIMMS), Faculty of Exact Sciences, VU University Amsterdam, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands. E-mail: C.de.Graaf@vu.nl
bCenter for Environmental Studies Sanata Dharma University (CESSDU), Division of Drug Design and Discovery, Faculty of Pharmacy, Universitas Sanata Dharma, Depok, Sleman, Yogyakarta 55282, Indonesia
cGriffin Discoveries B.V., De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands
First published on 30th March 2015
Abstract
We have explored the possibilities and challenges of structure-based virtual screening (SBVS) against the human histamine H4 receptor (H4R), a key player in inflammatory responses. Several SBVS strategies, employing different H4R ligand conformations, were validated and optimized with respect to their ability to discriminate small fragment-like H4R ligands from true inactive fragments, and compared to ligand-based virtual screening (LBVS) approaches. SBVS studies with a molecular interaction fingerprint (IFP) scoring method enabled the identification of H4R ligands that were not identified in LBVS runs, demonstrating the scaffold hopping potential of combining molecular docking and IFP scoring. Retrospective VS evaluations against H4R homology models based on the histamine H1 receptor (H1R) crystal structure did not give higher enrichments of H4R ligands than H4R models based on the beta-2 adrenergic receptor (β2R). Complementary prospective SBVS studies against β2R-based and H1R-based H4R homology models led to the discovery of different new fragment-like H4R ligand chemotypes. Of the 37 tested compounds, 9 fragments (representing 5 different scaffolds) had affinities between 0.14 and 6.3 μM at the H4R.
Introduction
The histamine H4 receptor (H4R), belonging to the family of G protein-coupled receptors (GPCRs),1 has been reported to play an important role in allergic and inflammatory processes.2–6 So far, one H4R ligand has entered Phase II clinical trials,2,4 but no marketed drug currently targets this receptor. The quest to find new ligands for H4R therefore remains attractive. Most H4R ligands (including compounds JNJ777120 (2) and VUF10497 (3); Fig. 1) have resulted from high-throughput screening (HTS) campaigns and subsequent medicinal chemistry programs.4–8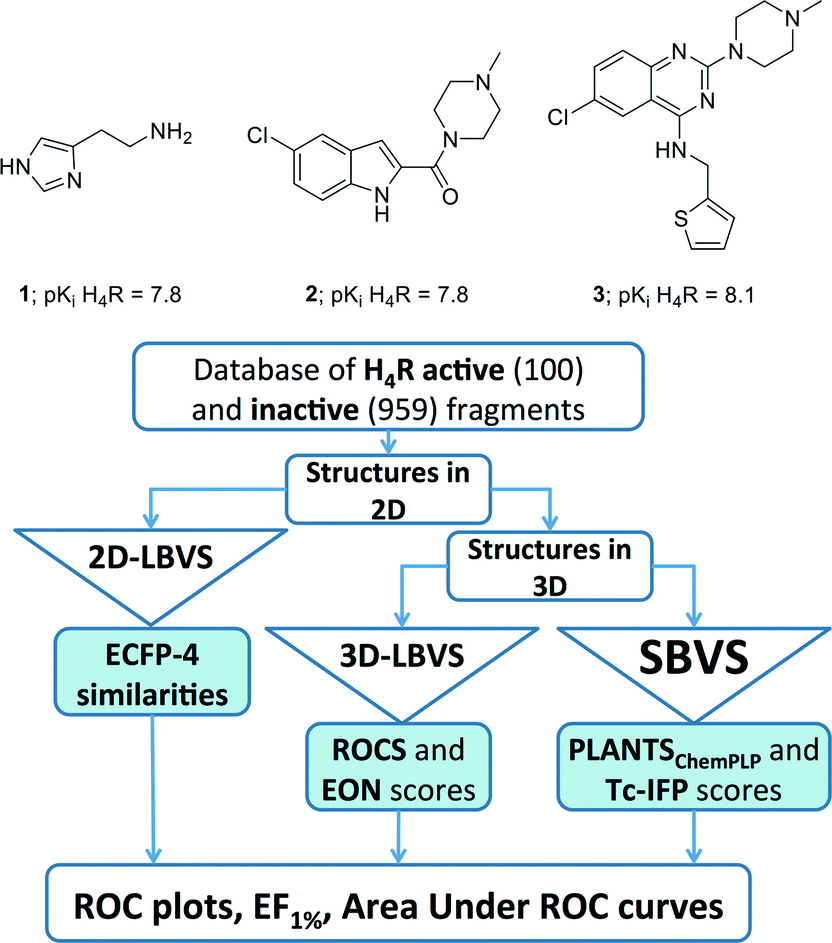 | ||
| Fig. 1 Structure and the biological activity of histamine (1), JNJ7777120 (2), and VUF10497 (3).7,8,81 and the flowchart of approaches employed in the retrospective VS protocols. See Experimental section for more details. | ||
Fragment-based drug discovery (FBDD)9–11 is a new paradigm in drug discovery that utilizes small molecules (molecular weight ≤300 Dalton, heavy atoms ≤22)12–14 as starting points for hit optimization. Within the context of FBDD, fragment-based screening is a more efficient way to sample chemical space and generally yields higher hit rates than classical high-throughput screening (HTS) campaigns of larger, drug-like compound.11,14,15 Biochemical and biophysical fragment screening studies of small chemical libraries (circa 25–1010 compounds) against different GPCRs have been reported with 0.4–14% hit rates yielding several new chemical starting points for fragment-based GPCR ligand optimization.14,16–22 So far only few experimental screens of the same fragment library against multiple GPCR targets have been reported14,16 that can provide information about the molecular determinants of GPCR-ligand selectivity by fragment-based chemogenomics analyses.14,23
Virtual fragment screening approaches, the in silico prediction of fragment binding to protein binding sites that has the potential to explore protein–ligand space more extensively, have been successfully applied to identify new fragment-like ligands for several GPCR targets, with 20–73% hit rates (% of experimentally tested in silico hits with detectable binding affinity).18,20,22,24–27 While ligand-based virtual screening methods often only allow the identification of chemically similar ligands, protein-based virtual screening approaches potentially offer the possibility of scaffold hopping, the discovery of ligands with new chemical functional groups.24,28–30 GPCR structure-based virtual fragment screening (SBVFS),18,22,24–26 the identification of smaller fragment-like molecules by docking simulations31 of large chemical databases in GPCR 3D structures, is however, still challenging and several problems have been identified including: (i) Conformational sampling problem: proper consideration of protein flexibility in docking simulations is difficult and small differences between experimentally-determined crystal structures, as well as structural inaccuracies in protein homology models, can affect sampling and scoring of different GPCR-ligand conformations.32–35 In particular, docking small fragments in a large binding pocket may result in multiple distinct binding modes with similar docking scores.36–38 (ii) Scoring problem: the ability of docking scoring functions to rank ligand docking poses in order to predict ligand binding modes and discriminate ligands from inactive molecules depends on the properties of the GPCR binding site and fine details of GPCR-ligand interactions.27,30,34,39,40 Moreover, docking scoring functions are generally not optimized for discriminating docking poses of small fragment-like molecules with comparable energy scores.36–38
(iii) Training problem: There are limited experimental data on true inactive fragment-like compounds that are required to optimize and validate structure-based virtual screening approaches.14,20,25 Furthermore, experimentally-determined crystal structures of the targeted protein in complex with fragment-like ligands are often lacking, and therefore cannot be used to validate structure-based virtual fragment screening approaches.
Several recent developments in the field of GPCR structural biology and FBDD can help to address these conformational sampling, scoring, and training problems associated with structure-based virtual fragment screening. In the past eight years crystal structures of 27 different GPCRs have been solved,41–43 including the adrenergic beta-2-receptor (β2R)44 and histamine H1 receptor (H1R).45 Structure-based virtual screening (SBVS) campaigns against GPCR crystal structures (in particular β2R,46adenosine A2A receptor (A2AR),47,48dopamine D3 receptor (D3R),395-hydroxytryptamine receptor 2B (5HT2BR),49 and H1R 24 have resulted in relatively high hit rates (24–73%) and yielded several small fragment-like ligands (≤22 heavy atoms).18,22,27 Although some successful SBVS studies with high hit rates (>20%) have also been reported based on GPCR homology models,39,50,51 the GPCR crystal structures seem to offer improved opportunities to push the limits of structure-based ligand discovery and design,18,20,22,24,27,30,52 including the application of virtual fragment screening to GPCRs.22,27,30,52 The increasing number of GPCR crystal structures for different GPCR subfamilies furthermore offer higher-resolution templates for modeling the structures of GPCRs for which crystal structures have not yet been solved.30,32,33,35,53 Three-dimensional H4R-ligand binding-mode models have been derived by (combining) ligand-based and protein-based modeling approaches, ligand structure–activity relationships, and site-directed mutagenesis studies.54–61 Experimentally validated homology models of human H4R have been constructed based on bovine Rhodopsin (bRho),54–56 β2R,58,59,62 and more recently the H1R crystal structure.26,60,61 Although H4R shares more ligands with H1R than with β2R,23 sequence identity between the H4R and H1R binding site (28%) is only slightly higher than between the H4R and β2R binding site (26%, ESI† Fig. S1).23 Interestingly, H4R models based on β2R and H1R crystal structure templates were equally successful in explaining H4R mutation data, while H1R-based H4R models could better explain ligand SAR than β2R-based H4R homology models.60
The challenges in structure-based virtual screening against GPCR homology models have been demonstrated by previous H4R virtual screening campaigns.26,56 In a large scale virtual screening study of more than 5 million compounds against a bRho-based H4R model (refined with histamine), 255 in silico hits were selected for experimentally testing, of which 11 had low affinity (>10 μM) and 5 had Ki values of 10 μM or lower (compounds 4–7, ESI† Fig. S2).56 Although the discovery of fragment-like molecules was not the aim of this study, most of the identified ligands were fragment-like.12–14 In another study retrospective virtual screening studies against MD simulation snapshots of a H1R-based H4R model (refined with JNJ-777120), allowed for the identification of representative H4R structures that gave optimal enrichments of known H4R ligands versus decoy molecules compared to the initial H4R homology model.34 This ensemble docking approach was subsequently applied in a prospective virtual screening campaign in which 50 in silico hits were selected for experimental testing. Nine of the fragment hits had low H4R affinity (>10 μM) and one fragment (compound 8, ESI† Fig. S2) had a Ki value of 8 μM.26
The aim of this study is to investigate the possibilities and limitations of structure-based virtual screening for the identification of new fragment-like ligands for H4R. Conformational sampling problems will be addressed by the construction of different three-dimensional receptor models of the human H4R with different ligands (compounds 1–3, Fig. 1),7,8,59,63 based on two different crystal structure templates (β2R 44 and H1R 45), and by the consideration of different molecular dynamics simulation snapshots. Although crystal structures of several aminergic GPCRs are available to construct H4R homology models (i.e. β1R, β2R, D3R, H1R, M2R, M3R, 5HT1BR, 5HT2BR),44,45,64–68 β2R and H1R are selected as H4R modelling templates because: i) this allows us to further build from our β2R-based and H1R-based H4R modelling studies;60 ii) H1R is the crystallized GPCR that shares the highest number of ligands with H4R,23 and iii) β2R has been a frequently used crystal structure target46,69–71 and GPCR modelling template26,39,72 in prospective structure-based virtual screening studies in the past few years.27Scoring and training problems will be addressed by: i) the use of a molecular interaction fingerprint (IFP) scoring method that considers protein–ligand interaction similarity to experimentally validated H4R-ligand binding-mode models consistent with H4R ligand structure–activity and structure–selectivity relationship and site-directed mutagenesis (SDM) studies. ii) the retrospective validation, comparison, and optimization of different virtual screening approaches based on a training set containing not only known H4R fragment-like ligands but also experimentally validated inactive fragments.13,17 The IFP scoring method has been shown to outperform energy-based scoring methods in previous GPCR structure-based virtual (fragment) screening studies36,73,74 and enable the identification of new chemical ligand scaffolds.18,24,25 Optimal structure-based virtual screening approaches identified in the current study will therefore be compared with two-dimensional (2D) and three dimensional (3D) ligand-based virtual screening methods. Although pharmacophore-based virtual screening techniques were not considered in the current study, it should be noted that scaffold hopping potential of pharmacophoric methods is also well recognized,75 as for example demonstrated in virtual screening studies for new histamine H3 receptor ligands.25 Systematic analysis and comparison of hit sets in both retrospective and prospective virtual screening studies will provide insights into the relative performance and complementarity of different virtual screening approaches in the identification of novel fragment-like H4R ligands. Fragment training sets of experimentally determined binders/actives and non-binders/inactives have been previously used for the optimization and validation of ligand-based and protein-based FLAP pharmacophore models for the discovery of new fragment-like H3R ligands25 and the evaluation of different consensus-scoring strategies for ligand-based virtual screening for fragment-like H1R and H4R ligands.76 In the current study, training sets of experimentally determined binders and non-binders have been used for the first time to optimize and validate protein structure-based virtual screening methods. The lessons learned from our comparative retrospective and prospective virtual fragment screening studies can be used as a blueprint for future structure-based virtual (fragment) screening studies.
Results
Retrospective evaluation ligand-based and structure-based virtual screening methods
A dedicated training set, containing 100 unique fragment-like H4R ligands from our in-house fragment library14,17 and the ChEMBL database77 and 959 fragments inactive at human H4R,14,17 was used for retrospective validation of different structure-based and ligand-based (LBVS) virtual screening approaches (Fig. 1). The chemical structures and binding modes of the H4R ligands histamine (1), JNJ7777120 (2), and VUF10497 (3) (Fig. 1) were used as reference compounds in the retrospective VS runs. These selected ligands represent different steps in H4R ligand-optimization strategies in the past years:4–6 (i) modification of the basic amine, and (ii) substitution of the imidazole ring with bioisosteres.4Histamine (1) is the endogenous ligand of H4R and JNJ7777120 (2) is the first published selective non-imidazole H4R ligand, a biased agonist for the β-arrestin pathway,78,79 and both have served as reference compounds in previous ligand-based and protein-based H4R virtual screening and ligand design studies.8,26,56,80 VUF10497 is a high affinity H4R inverse agonist and is representative for a series based on an in-house discovered H4R scaffold.8,63The binding modes of the reference compounds in the H4R binding pocket were derived following the information extracted from previous structure–activity relationship and mutation studies on H4R.4,59,61,82 All compounds display H-bond interactions to D943.32 and E1825.46.4,59 Mutagenesis studies have indicated that these residues are essential H-bond acceptor interaction points for H4R ligands54,55,60 and suggest that D943.32 and E1825.46 form H-bond interaction networks with Q3477.42 and N1474.57, respectively.58,59 The transmembrane (TM) binding pocket is very similar in both H4R models based on β2R and H1R (Fig. 2),4,59,82 but there are differences in the second extracellular loop (EL2).74,83,84 These differences slightly affect the binding orientation of JNJ7777120 (2) (Fig. 2),58 while the binding poses of histamine (1)58,85 and VUF10497 (3) remain very similar (Fig. 2). In the β2R-based H4R model the chlorine atom of JNJ7777120 (2) is located between EL2 (F168), TM5 (L1755.39), and TM6 (T3236.55), while in the H1R-based model the chlorine atom of JNJ7777120 is accommodated between TM5 (L1755.39 and T1785.42) and EL2 (F168) (Fig. 2).4,58 It should be noted that the functional effect of these ligands vary considerably, from inverse agonist (VUF10497 (3)) to biased agonist (JNJ-7777120 (2)) to full agonist (histamine). Clearly, the fragments to be identified in these VS efforts do not represent optimized compounds. It is apparent from literature that even the smallest structural changes of a ligand during hit or lead optimization (i.e., for fragments but also for drug-like molecules) can completely alter the functional activity from agonist to antagonist and vice versa.86,87 Furthermore, the functional activity is highly dependent on the species investigated and on the signalling pathways studied.58,78,79,88–90 On a protein molecular level this probably means that the differences between agonists and antagonists can be relatively small, but the effect these changes can have on the conformation (activation state) of the receptor can be big.27 For this fragment-based VS campaign, we have chosen not to focus on the functional effect of the ligands, but on the affinity for H4R.
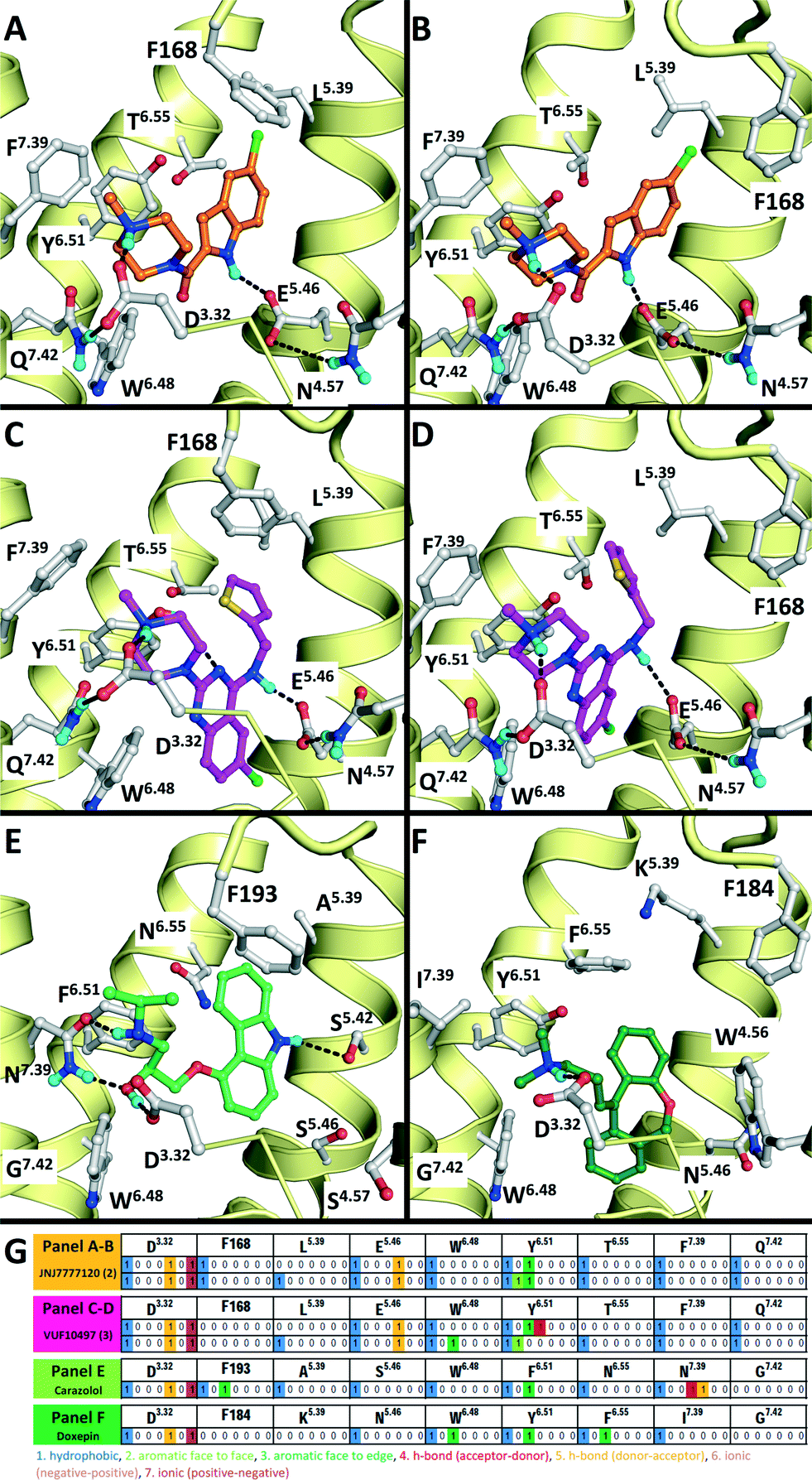 | ||
| Fig. 2 Comparison of β2R–carazolol (light green) crystal structure44 (E) based H4R models (A, C) and H1R–doxepin dark green) crystal structure (F) based H4R models45 (B, D). H4R models are constructed with JNJ7777120 (2, orange carbonatoms, A and B) and VUF10497 (3, magenta, C and D). Protein–ligand interaction fingerprints (IFPs) of the binding modes in A–F are presented in G. The backbone of TM helices 4, 5, 6, and 7 are represented by yellow tubes and part of TM3 is shown as ribbon (the top of the helix is not shown for clarity). Important binding residues identified previously4,55,56,59,61,82 are depicted in grey. | ||
The retrospective VS flowchart is presented in Fig. 1. 2D-LBVS and 3D-LBVS similarity searches of the test set of H4R binders and non-binders were ranked according to ECFP-4 (Tanimoto score)91 and ROCS-EON (Comboscore)92 similarity against reference ligands (1–3).24 In the SBVS runs, compounds were docked against molecular dynamics simulation snapshots of β2R-based44,58,59 and H1R-based45 H4R homology models. The resulting docking poses were ranked subsequently using PLANTSChemPLP93 and interaction fingerprint (IFP)36,74,94 similarity scores to reference binding modes of ligands 1–3 (Fig. 2). ROC plots (% true positives (TP) versus % false positives (FP) in the ranked database)74 of the retrospective analysis of 2D-LBVS, 3D-LBVS and SBVS hit lists are presented in Fig. 3. The enrichment factor 1% (EF1%) of the VS protocols together with the area under ROC curves are summarized in Table 1.95–97
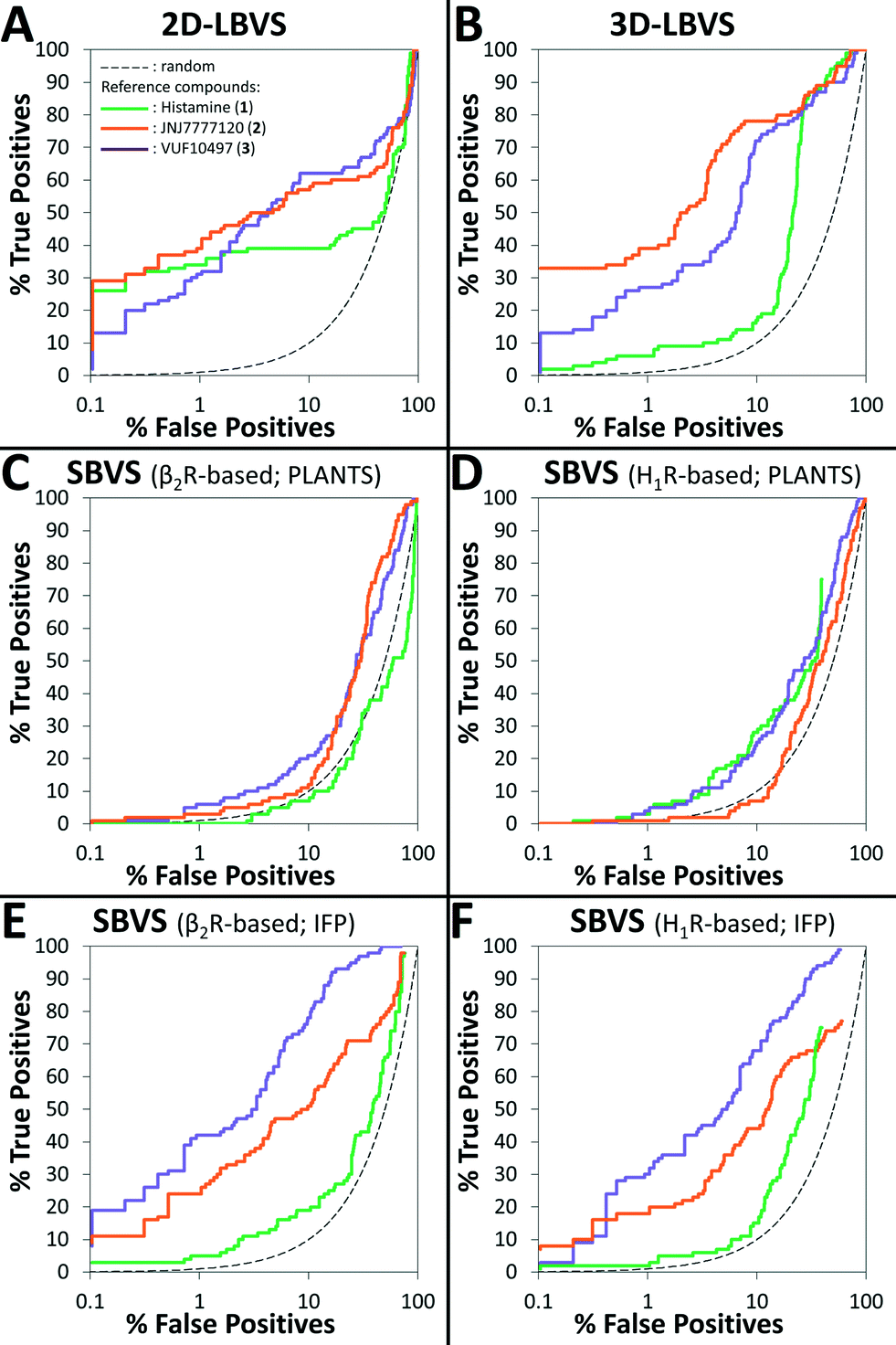 | ||
| Fig. 3 Graphs of % true positives (fragment-like H4R ligands) vs. % false positives (fragments that do not bind H4R) in the ranked database resulted from 2D-LBVS (A), 3D-LBVS (B), β2R-based SBVS ranked by PLANTSChemPLP score (C), H1R-based SBVS ranked by PLANTSChemPLP score (D), β2R-based SBVS re-ranked by Tc-IFP score (E), and H1R-based SBVS re-ranked by Tc-IFP score (F) based on histamine (1, green), JNJ-7777120 (2, orange), and VUF10497 (3, purple) reference ligands (A–B) and ligand-H4R IFPs (C–F). | ||
| VS protocol | References | |||||
|---|---|---|---|---|---|---|
| Histamine (1) | JNJ777120 (2) | VUF10497 (3) | ||||
| EF1% | AUC (CIa) | EF1% | AUC (CIa) | EF1% | AUC (CIa) | |
| a Confidence interval of the AUC with level of confidence of 95% calculated using pROC packages in R statistical computing software.95 b Fig 3A. c Fig 3B. d Fig 3C. e Fig 3D. f Fig 3E. g Fig 3F. | ||||||
| Ligand-based VS | ||||||
| 2Db | 32.6 | 0.61 (0.54–0.98) | 37.4 | 0.71 (0.64–0.78) | 29.7 | 0.73 (0.66–0.80) |
| 3Dc | 5.7 | 0.77 (0.74–0.81) | 37.4 | 0.89 (0.85–0.93) | 25.9 | 0.85 (0.81–0.90) |
| β2R-based SBVS | ||||||
| PLANTSd | 0.0 | 0.58 (0.52–0.65) | 2.9 | 0.69 (0.64–0.73) | 5.7 | 0.66 (0.61–0.71) |
| Tc-IFPe | 4.8 | 0.62 (0.57–0.67) | 23.0 | 0.78 (0.73–0.84) | 40.3 | 0.94 (0.92–0.97) |
| H1R-based SBVS | ||||||
| PLANTSf | 2.9 | 0.67 (0.62–0.72) | 1.0 | 0.58 (0.52–0.63) | 3.8 | 0.68 (0.63–0.73) |
| Tc-IFPg | 1.9 | 0.68 (0.63–0.73) | 17.3 | 0.73 (0.66–0.79) | 28.8 | 0.89 (0.86–0.93) |
Table 1 and Fig. 3 indicate that 2D-LBVS, 3D-LBVS and SBVS-IFP can give a good early enrichment. SBVS using PLANTSChemPLP scoring on the other hand resulted in significantly lower enrichments. The 2D-LBVS runs result in a lower global virtual screening accuracy (reflected by the area under ROC curves) compared to the 3D approaches. On the other hand, low early enrichments as well as global virtual screening accuracies were obtained with the 3D approaches using histamine (1) as the reference compound. JNJ7777120 (2) appeared to be the best reference for 2D- and 3D-LBVS runs, while the VUF10497 (3) binding mode performed as the best reference for post-processing SBVS docking simulations. Interestingly, β2R-based SBVS resulted slightly higher enrichments compared to H1R-based SBVS.
We furthermore evaluated the ability of the different methods to identify “novel” fragment-like molecules (Fig. 4). In our retrospective virtual screening studies “novel” fragments were defined as compounds that have an ECFP-4 Tanimoto similarity (Tc-ECFP4) score to any reference compounds of less than 0.26.98 β2R-based SBVS yielded the most novel hits (Fig. 4C), followed by H1R-based SBVS (Fig. 4D) and 3D-LBVS (Fig. 4B).
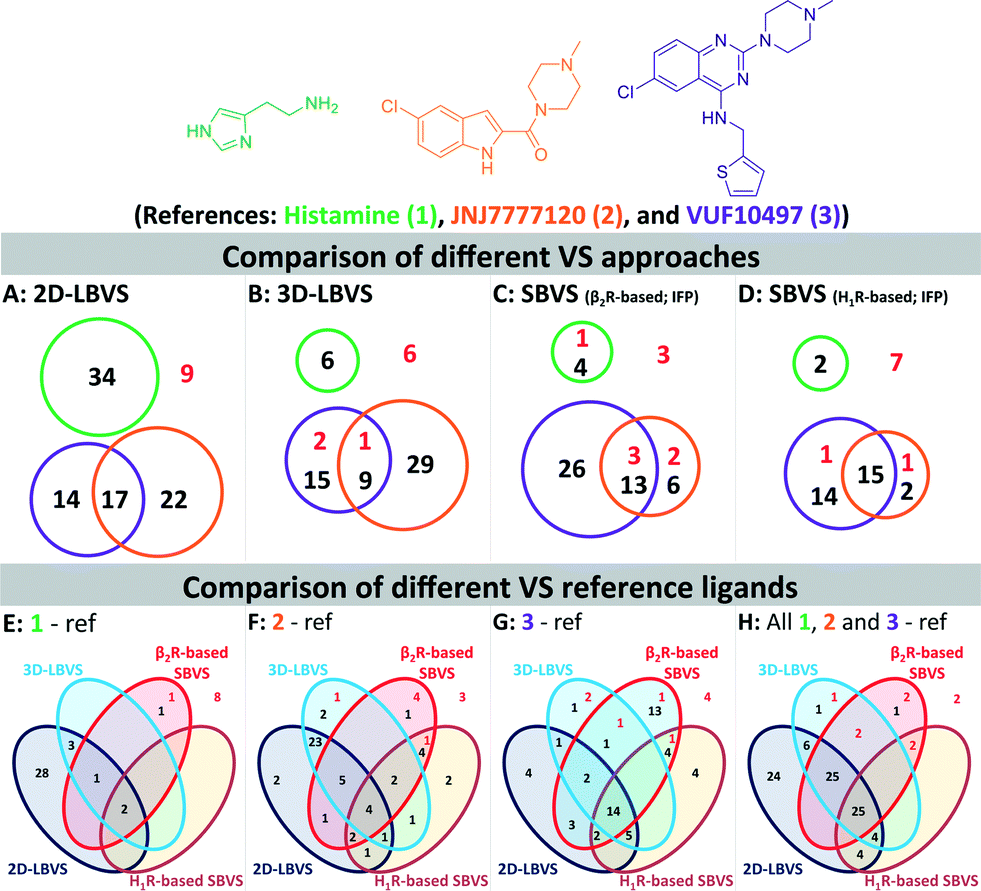 | ||
| Fig. 4 Venn diagrams of the number of actives at a 1% false positive rate resulted in 2D-LBVS (A), 3D-LBVS (B), β2R-based SBVS re-ranked by Tc-IFP score (C), and H1R-based SBVS re-ranked by Tc-IFP score (D). The circles proportionally indicate the number of actives at a 1% false positive rate based on histamine (1, green), JNJ777120 (2, blue), or VUF10497 (3, purple) references (Panels A–D). Venn diagrams of the number of actives at a 1% false positive rate resulted in all used VS methods using histamine (1, panel E), JNJ777120 (2, panel F), VUF10497 (3, panel G) or consensus references (panel H). Red numbers indicate active H4R fragments in the database that have an ECFP-4 Tanimoto similarity score of less than 0.26 to any of the references.98 | ||
Histamine (1) (and histamine-H4R binding modes) was only a suitable reference in 2D-LBVS runs (Fig. 4E), while both JNJ7777120 (2) and VUF10497 (3) were good references in different virtual screening protocols (Fig. 4F–G). Fig. 4E–H show clear overlaps between 2D- and 3D-LBVS by using JNJ7777120 (2) or VUF10497 (3) as the reference. Fig. 4F shows a high number of hits only identified by the LBVS studies, which were not identified in the SBVS studies by using JNJ7777120 (2) as the reference. Fig. 4F shows that the use of JNJ7777120 (2) in the SBVS studies increases the chance to retrieve “novel” H4R fragments. The highest number of shared hits between all SBVS methods were retrieved in the VUF10497-based SBVS studies (Fig. 4G). Collection of the hits that were retrieved at a 1% false positive rate using all references results in the Venn diagram presented in Fig. 4H. Although most active H4R fragments were retrieved using LBVS, SBVS provided a higher probability of retrieving “novel” H4R fragments.
Based on the results of our retrospective virtual screening studies, we performed the prospective SBVS campaigns using the β2R-based44,58,59 and H1R-based45 H4R homology models in complex with reference ligands 2 and 3 (Fig. 3 and 5).
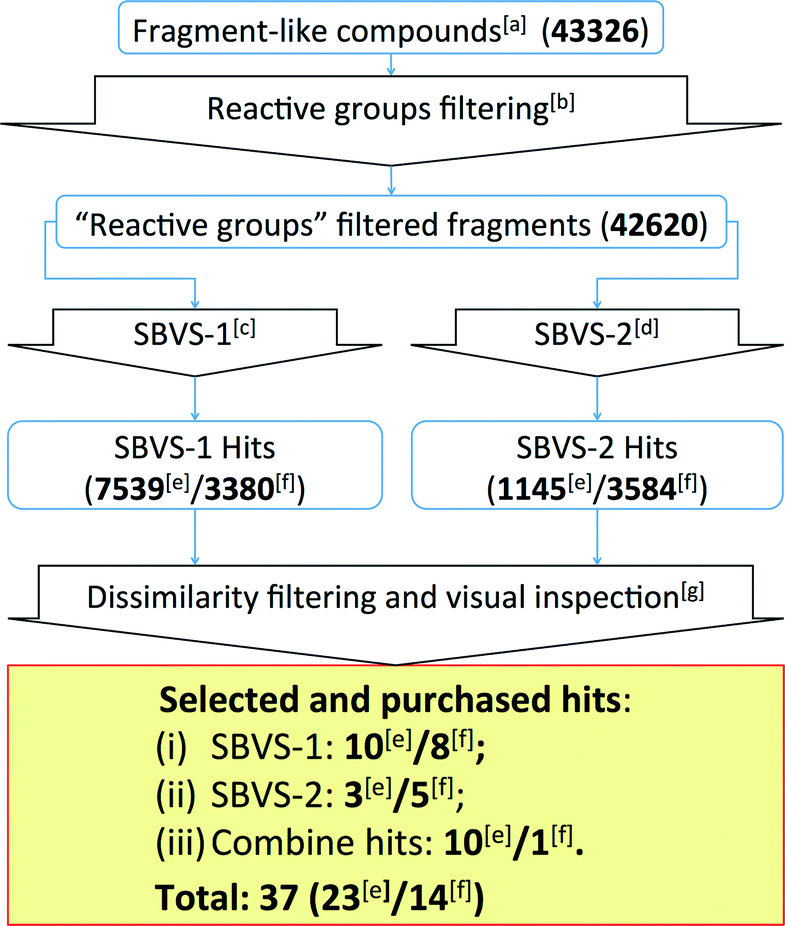 | ||
| Fig. 5 Flowchart of the β2R-based and H1R-based prospective SBVS campaigns. See Experimental section for more details. [a]Fragment-like compounds collected from the ZINC database.37,99[b]Filtering fragments containing plausible reactive/toxic groups.100[c]SBVS with JNJ7777120 (2) as the reference compound.7,56,80[d]SBVS with VUF10497 (3) as the reference compound.8,56,63[e]Results from the β2R-based prospective SBVS studies. [f]Results from the H1R-based prospective SBVS studies. [g]Fragments with ECFP-4 Tanimoto similarity score of less than 0.40 to any of the H4R ligands used in the retrospective VS.24 In the H1R-based prospective SBVS campaigns, H4R purchased hits from the β2R-based prospective SBVS studies were added as the reference compounds in the dissimilarity filtering. | ||
Prospective structure-based virtual screening studies to discover new H4R ligands
The SBVS approach against the β2R-based H4R model was determined to be the best method in identifying novel fragment-like ligands (Fig. 4). We therefore used this model in a prospective in silico screening study to discover new active H4R fragments from a subset of fragment-like (and commercially-available) molecules extracted from the ZINC database (Fig. 5, ESI† Table S1).99 Based on the results of the retrospective VS studies (Table 1, Fig. 4), two β2R-based H4R models and their corresponding IFP references were used in parallel: i) The first SBVS run was rescored using the Tanimoto coefficient-based IFP similarity (Tc-IFP) against the JNJ7777120 (2) customized H4R model (SBVS-1), and ii) A second in silico screen rescored using Tc-IFP against the VUF10497 (3) based H4R model (SBVS-2) (Fig. 4). In the retrospective VS studies, the re-scoring using Tc-IFP clearly shows the increase of SBVS quality compared to the ones using PLANTSChemPLP scores (Table 1, Fig 3). Our retrospective validation studies showed that many of the “novel” ligands (i.e., not identified with 2D ligand-based VS) were identified in the SBVS-1/SBVS-2 consensus hit list and the SBVS-1 hit list (Fig. 4E).We therefore especially selected compounds from these lists in our prospective VS campaign: 23 fragments were selected (ESI† Fig. S3 and ESI† Table S2) and purchased, from which 6 were experimentally confirmed as H4R ligands (Table 2), including three piperazine-benzofuropyrimidines with submicromolar affinity (9a–c), and three pyrimido-indole containing fragments with micromolar affinity (10a–c). None of the validated hits had detectable binding affinity for β2R, demonstrating that the SBVS was not biased towards the structural template39 used to construct the H4R homology models (Table 2). Following the successful efforts in the discovery of novel H4R fragments by using the β2R-based H4R model, we subsequently performed similar SBVS campaigns employing H1R-based H4R model (Fig. 5). In order to increase our chances of finding ligands with a different chemotype we added the newly discovered H4R hit ligands as references compounds in the dissimilarity filter (Fig. 5). Fourteen fragments were selected and purchased (ESI† Fig. S4 and ESI† Table S3). Three of the hits from the H1R-based prospective SBVS (11–13) were experimentally confirmed as H4R ligands (Table 3). In total, 37 fragments were selected and purchased. Nine out of these 37 purchased fragments were confirmed as H4R ligands with binding affinities between 0.14 and 6.9 μM (Tables 2–3; Fig. 6). The hits represent five different scaffolds: the isosteric benzofuropyrimidines 9a–c and pyrimido-indoles 10a–c scaffolds identified in the β2R-based H4R model, and bezo-imidazole 11, triazoloquinoxaline 12, and morpholinopyrimidine 13 identified in the H1R-based H4R model.
| Compounds | Biological activities (pKi ± SEM)a | Tc-IFP scored (#rank) | PLANTSChemPLP scoree (#rank) | ECFP4 similarityf (#rank) | Reference ROCS-EONg (#rank) | ECFP4 similarityh | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H4Rb | β2Rc | SBVS-1 | SBVS-2 | SBVS-1 | SBVS-2 | JNJ (2) | VUF (3) | JNJ (2) | VUF (3) | ChEMBL | |
| a pKi values are calculated from at least three independent measurements as the mean ± SEM. b Measured by displacement of [3H]-histamine binding using membranes of HEK293T cells transiently expressing the human H4R. c Measured by displacement of [3H]-dihydroalprenolol binding using membranes of HEK293T cells transiently expressing the human β2R. d IFP Tanimoto similarity with the pose of either JNJ7777120 (SBVS-1) or VUF10497 (SBVS-2) in the H4R model. Tc-IFP ranking is given between brackets. e PLANTSChemPLP docking score using H4R model bound to JNJ7777120 (SBVS-1) or VUF10497 (SBVS-2). The ranking is given between brackets. f ECFP-4 2D Tanimoto similarity to JNJ7777120 (2), or VUF10497 (3). A similarity higher than 0.40 is considered as significant.24 ECFP-4 ranking is given between brackets. g ROCS/EON shape/electrostatic-based 3D similarity to JNJ7777120 (2), or VUF10497 (3) based on Comboscore. A similarity higher than 1.30 is considered as significant. Comboscore ranking is given between brackets. h ECFP-4 circular fingerprint Tanimoto similarity to the closest known H4R-active fragment used in the retrospective VS.17,101 A similarity higher than 0.40 is considered as significant.24 | |||||||||||
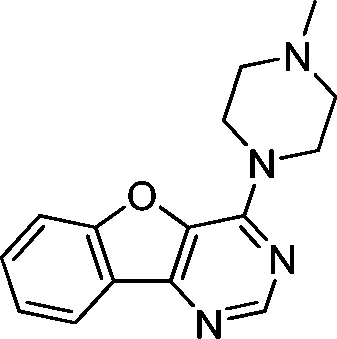 VUF13682 (9a) VUF13682 (9a) |
6.84 ± 0.12 | <5 | — | 0.909 (#14) | −90.759 (#4653) | −97.161 (#3550) | 0.121 (#8124) | 0.178 (#1790) | 1.513 (#3945) | 1.506 (#408) | 0.254 |
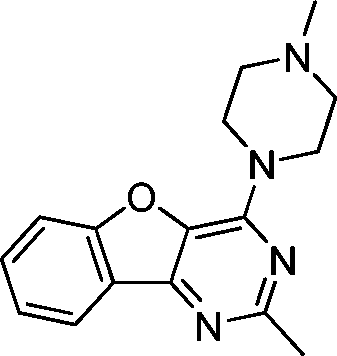 VUF13686 (9b) VUF13686 (9b) |
6.83 ± 0.08 | <5 | 0.750 (#4513) | 0.909 (#10) | −76.260 (#25 334) | −99.754 (#2232) | 0.119 (#8566) | 0.178 (#1805) | 1.519 (#3829) | 1.513 (#352) | 0.294 |
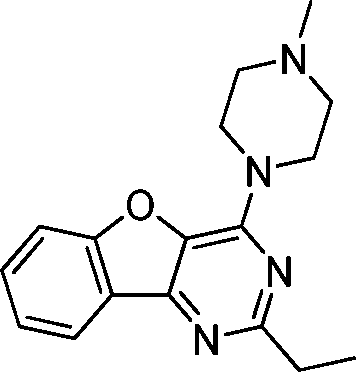 VUF13687 (9c) VUF13687 (9c) |
6.57 ± 0.06 | <5 | 0.750 (#4821) | 0.909 (#6) | −68.907 (#36 530) | −101.757 (#1448) | 0.113 (#10 241) | 0.184 (#1462) | 1.528 (#3651) | 1.529 (#244) | 0.268 |
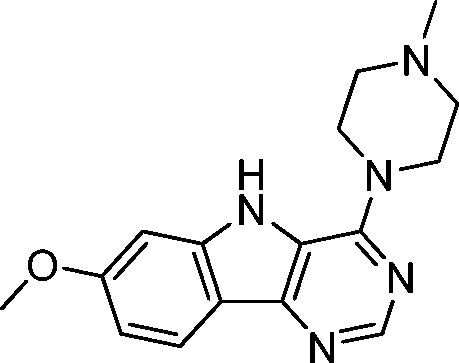 VUF13690 (10a) VUF13690 (10a) |
5.20 ± 0.06 | <5 | 1.000 (#1) | 0.864 (#103) | −92.650 (#3285) | −95.021 (#4986) | 0.139 (#5171) | 0.192 (#1038) | 1.565 (#2886) | 1.527 (#257) | 0.261 |
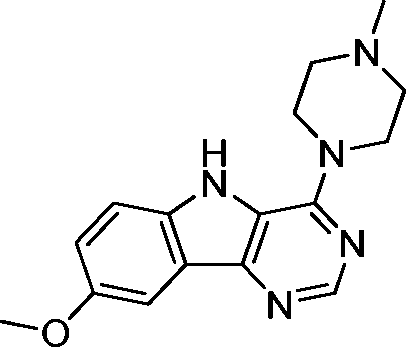
 VUF13694 (10b) VUF13694 (10b) |
5.23 ± 0.06 | <5 | 0.842 (#418) | 0.955 (#1) | −83.531 (#12 968) | −97.851 (#3129) | 0.159 (#3191) | 0.223 (#306) | 1.653 (#1455) | 1529 (#243) | 0.319 |
 VUF13695 (10c) VUF13695 (10c) |
5.23 ± 0.03 | <5 | 0.810 (#777) | 0.909 (#9) | −78.414 (#21 595) | −100.022 (#2097) | 0.141 (#4888) | 0.179 (#1721) | 1.554 (#3102) | 1.588 (#62) | 0.286 |
| Compounds | Biological activities (pKi ± SEM)a | Tc-IFP scored (#rank) | PLANTSChemPLP scoree (#rank) | ECFP4 similarityf (#rank) | Reference ROCS-EONg (#rank) | ECFP4 similarityh | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H4Rb | H1Rc | SBVS-1 | SBVS-2 | SBVS-1 | SBVS-2 | JNJ (2) | VUF (3) | JNJ (2) | VUF (3) | ChEMBL | |
| a pKi values are calculated from at least three independent measurements as the mean ± SEM. b Measured by displacement of [3H]-histamine binding using membranes of HEK293T cells transiently expressing the human H4R. c Measured by displacement of [3H]-mepyramine binding using membranes of HEK293T cells transiently expressing the human H1R. d IFP Tanimoto similarity with the pose of either JNJ7777120 (SBVS-1) or VUF10497 (SBVS-2) in the H4R model. Tc-IFP ranking is given between brackets. e PLANTSChemPLP docking score using H4R model bound to JNJ7777120 (SBVS-1) or VUF10497 (SBVS-2). The ranking is given between brackets. f ECFP-4 2D Tanimoto similarity to JNJ7777120 (2), or VUF10497 (3). A similarity higher than 0.40 is considered as significant.24 ECFP-4 ranking is given between brackets. g ROCS/EON shape/electrostatic-based 3D similarity to JNJ7777120 (2), or VUF10497 (3) based on Comboscore. A similarity higher than 1.30 is considered as significant.102 Comboscore ranking is given between brackets. h ECFP-4 circular fingerprint Tanimoto similarity to the closest known H4R active fragments used in the retrospective VS and the purchased hits based on β2R-based prospective VS.17,101 A similarity of higher than 0.40 is considered as significant.24 i At 10 μM, the compounds showed [3H]-mepyramine displacement of less than 50%. j At 10 μM, the compounds showed [3H]-mepyramine displacement of more than 50%. | |||||||||||
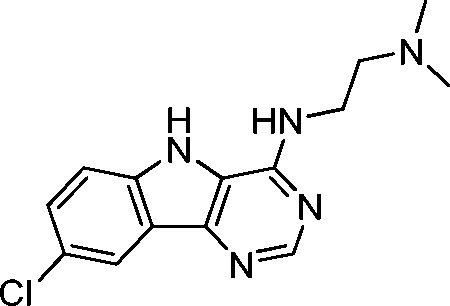
 VUF13848 (11) VUF13848 (11) |
5.16 ± 0.03 | <5i | 0.800 (#413) | — | −83.836 (#2764) | −89.297 (#10 026) | 0.125 (#71 765) | 0.120 (#87 147) | 1.635 (#6966) | 1.380 (#5019) | 0.200 |
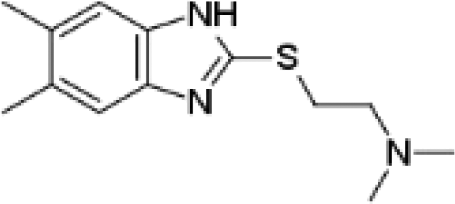
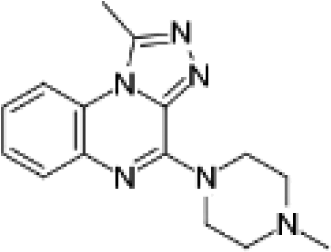 VUF13860 (12) VUF13860 (12) |
5.19 ± 0.07 | >5j | 0.727 (#3374) | — | −41.868 (#34 454) | −78.980 (#26 862) | 0.215 (#12 441) | 0.300 (#600) | 1.624 (#8066) | 1.563 (#142) | 0.396 |
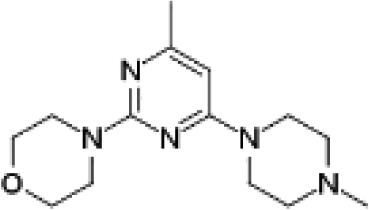 VUF13867 (13) VUF13867 (13) |
5.16 ± 0.04 | >5j | — | 0.739 (#626) | −74.071 (#12 599) | −93.292 (#1051) | 0.213 (#13 059) | 0.323 (#277) | 1.643 (#6170) | 1.472 (#1100) | 0.264 |
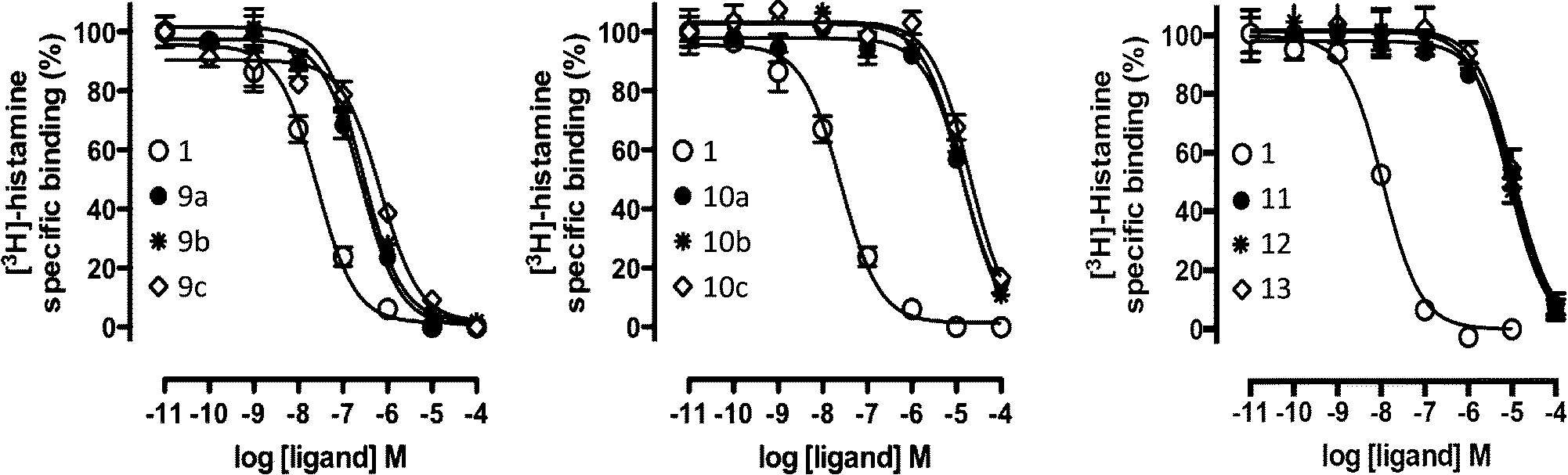 | ||
| Fig. 6 [3H]-histamine binding displacement by reference compound 1 and the nine virtual screening hits (9a–c, 10a–c, 11, 12, 13). Data shown are representative binding curves of at least three experiments performed in triplicate. Error bars indicate SEM values. | ||
Discussion
Ligand- and protein-based virtual screening methods are complementary
One of the challenges in SBVFS is the limited experimental data on true inactive (fragment-like) compounds to properly validate and optimize virtual screening approaches. Our in-house fragment screening against H4R provided invaluable data that enabled us to construct a balanced training set of true active and true inactive H4R fragments.14,17 The H4R active fragments from our in-house screens14,17 were appended by active fragment-like H4R ligands from the ChEMBL database77,101 to further increase the number of true H4R active fragments. This dedicated training set of fragment-like compounds enabled us to retrospectively validate different ligand- and protein-based virtual fragment screening protocols (Fig. 1).Both (2D and 3D) ligand-based and structure-based (IFP) virtual screening approaches gave good early enrichment in our retrospective virtual screening studies (Fig. 3, Table 1). SBVS using PLANTS scoring on the other hand resulted in significantly lower enrichments (Fig. 3, Table 1). Although 2D-LBVS gave high early enrichments, the global virtual screening accuracies (AUC values) of the ECFP-4 2D similarity searches were relatively low, indicating that this method is, as expected, rather dependent on the reference ligand. 3D-LBVS runs also gave high enrichments for the relatively larger reference ligands (Fig. 3, Table 1), and retrieved ligands that were not identified in 2D-LBVS simulations (Fig. 5). Enrichments obtained by 3D-similarity screens based on the relatively small histamine reference ligand (only 8 heavy atoms) are, however, significantly lower than 2D-similarity screens based on the same ligand (5-fold lower EF1%). This suggests that very small fragments are less distinguishable based on shape and electrostatic/pharmacophore similarity. Indeed, histamine-based runs give relatively higher ROCS-EON scores (% of compounds within the database with a score ≥1.3 = 32%), than when using JNJ7777120 (26%) and VUF10497 (15%) as a reference. Another explanation could be that only disconnected groups are common between the reference and the target compound (maximum common edge (MCE) subgraph).103 This is supported by the fact that 41% of the actives and 21% of the inactives share an imidazole ring with histamine. It should be noted that in a 3D-LBVS campaign, using JNJ7777120 (2) as the reference compound, new H4R ligands were discovered80 that are similar to the experimentally confirmed hits 9a–c that were independently identified in our prospective SBVS study. These hit compounds from Cramp et al. were, however, not yet published when we performed our virtual screening and were therefore also not yet included in the ChEMBL database77 version (downloaded on August 19, 2010) used in our study for the novelty assessment.
Structure-based virtual screening with a molecular interaction fingerprint (IFP) scoring method to rank PLANTS docking poses in JNJ7777120- and VUF10497-customized H4R homology models (based on β2R and H1R crystal structure templates) gave comparably high enrichments as 2D and 3D ligand-based virtual screening methods (Fig. 3, Table 1). In fact, SBVS against the β2R-based H4R model refined with VUF10497 (3) gave the best early enrichment of all methods used in the retrospective evaluation. For both β2R-based H4R models higher retrospective virtual screening enrichments were obtained with VUF10497 (3) reference IFPs than with JNJ7777120 (2) or histamine (1) reference IFPs. A possible explanation for this result could be that the larger VUF10497 (3) ligand binds a larger part of the hydrophobic pocket between TM helices 3–6 (between W3166.48, Y3196.51, and L1755.39) than JNJ777712 and histamine (Fig. 2). As a result the VUF10497 reference interaction fingerprint allows the retrieval of H4R ligand docking poses that target the subpocket between W3166.48 and Y3196.51 and/or the subpocket between Y3196.51 and L1755.39, which may lead to a better enrichment in the retrospective SBVS studies.36 It should furthermore be noted that VUF10497 (3) has the highest affinity compared to histamine (1) and JNJ7777120 (2).7,8 Comparison of the overlap of ligands retrieved by the different methods furthermore shows that SBVS methods are capable of identifying of novel ligands that are chemically dissimilar from the ligands used to define the reference IFP that are not retrieved by 2D or 3D LBVS methods (Fig. 4). This indicates that ligand- and structure-based virtual screening methods are complementary as previously shown by Krüger and Evers.29 Our results furthermore highlight the scaffold-hopping potential of SBVS in combination with IFP, as shown in previous studies.24
Structure-based virtual screening is an efficient way to identify novel fragment-like H4R ligands
Based on the results of the retrospective VS studies (Table 1, Fig. 2–3), two β2R-based H4R models and their corresponding IFP references were used in prospective in silico screening runs to discover new active H4R fragments from a subset of fragment-like and commercially-available molecules extracted from the ZINC database (Fig. 5). Six out of 23 purchased in silico hits were experimentally confirmed as active H4R fragments with pKi values of 5.2–6.8 (Table 2 and Fig. 7). By re-ranking the docking poses according to their IFP similarity the rank of the confirmed hits increased from #1448–#3550 for PLANTS to #1–#14 using IFP (Table 2). It should be noted that compounds 9a–c (Table 2 and Fig. 7) is similar to the H4R scaffold discovered by Cramp et al.80 in an independent ligand-based virtual screening campaign. It should be emphasized however that the compounds discovered in this ligand-based screening were not yet included in the ChEMBLdb database77 version used in our study,101 and therefore were identified after completion of our own prospective structure-based virtual screening study.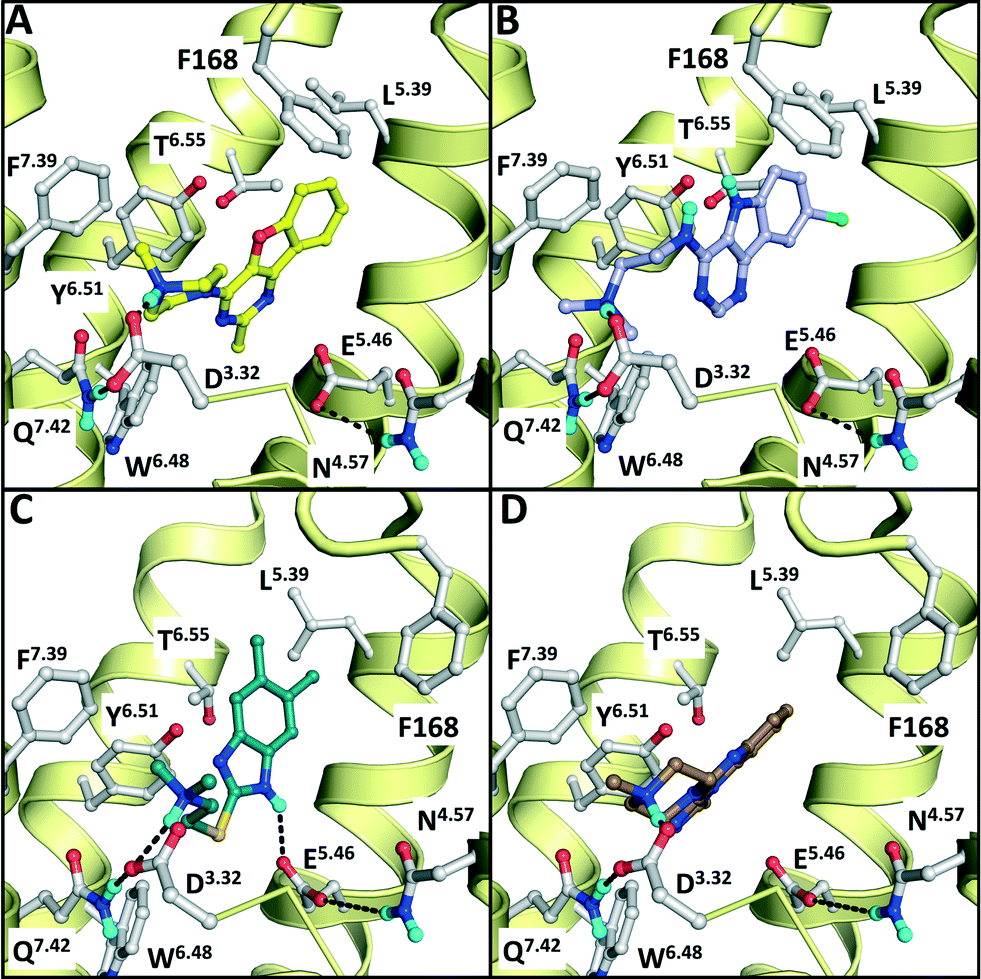 | ||
| Fig. 7 Binding modes of the validated hits VUF13686 (9b, yellow, panel A) and VUF13694 (10b, purple, B) in the β2R-based H4R model, compared to the binding modes of VUF13848 (11, darkgreen, C), and VUF13860 (12, brown, D) in the H1R-based H4R model. Rendering is similar to Fig. 2. | ||
We subsequently performed the prospective SBVS campaigns studies against the H4R models that were built based on the H1R crystal structure45 (Fig. 3 and 5). The SBVS runs against the H1R-based H4R models identified three experimentally confirmed H4R ligands (11–13) (Table 3, Fig. 6 and 7). These three additional ligands combine a basic amine moiety with scaffolds (bezo-imidazole 11, triazoloquinoxaline 12, morpholinopyrimidine 13) that are different from the two isosteric H4R ligand scaffolds (benzofuropyrimidines 9a–c and pyrimido-indoles 10a–c) discovered in the prospective SBVS campaigns against β2R-based H4R models (Tables 2–3, ESI† Fig. S5). Although the TM fold of the β2R and H1R crystal structure templates are similar,44,45 the different EL2 loop conformations (in particular the orientation of F168) results in different H4R models. As a result, the reference ligands (2–3) and the novel ligands identified in prospective virtual screening studies (9–13) have similar binding modes in β2R based and H4R models, including H-bond conserved H-bond interactions with D943.32 and E1825.46, but adopt slightly different orientations in the EL2 region (Fig. 2 and 7). These subtle differences in both binding pocket structure and reference ligand binding mode result in relatively small differences in retrospective VS accuracies between H4R homology models based on β2R and H1R crystal structure templates. Both modeling templates yield H4R models with good early enrichments, but the retrospective virtual screening accuracy of the β2R-based models is somewhat higher than the H1R-based H4R models (Table 1 and Fig. 2–3). Although the sequence identity between the H4R and H1R binding site is only slightly higher than between the H4R and β2R binding site,23 H1R is expected be a better template to model the EL2 region downstream from the conserved C164EL2 that forms a disulphide bridge with C773.25 (including F168, ESI† Fig. S1). While H1R-based H4R models have previously been shown to better explain ligand SAR than β2R-based H4R homology models,60 the current study indicates that the differences between H1R and β2R crystal structure templates does not significantly affect structure-based virtual screening accuracy of H4R homology models. Moreover, prospective virtual screening studies against β2R-based and H1R-based H4R models resulted both in the discovery of different new ligand chemotypes (Tables 2–3, Fig. 7–8). Our results are in line with recent comparative GPCR modeling studies which showed that comparable virtual screening results can be obtained with GPCR homology models and crystal structures.25,27,39,40,104
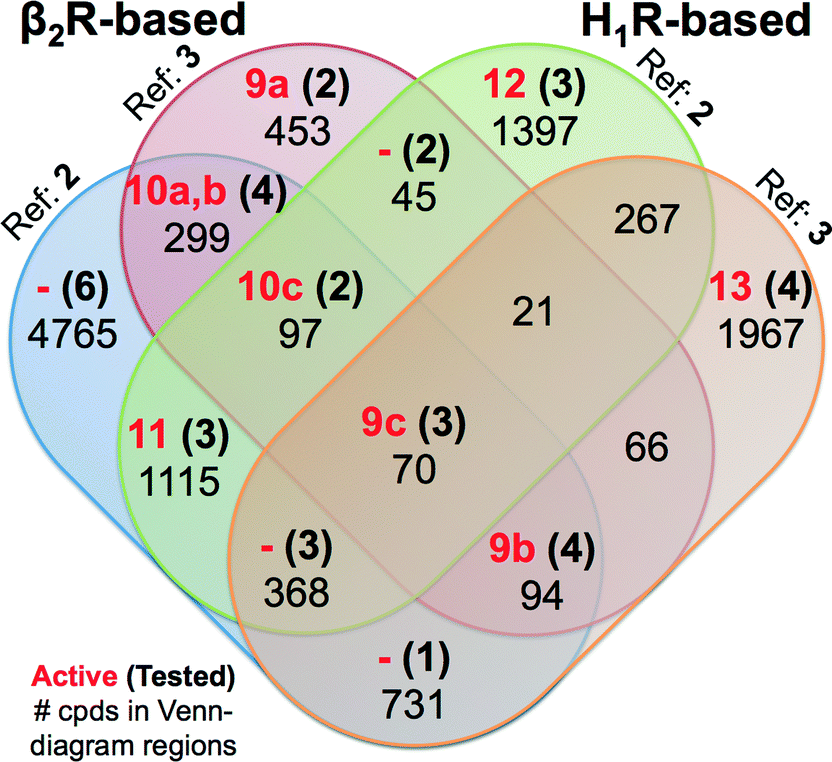 | ||
| Fig. 8 Venn diagram showing the SBVS hit overlap for all prospectively applied homology models (see Fig. 5). The red bold numbers, the black bold numbers and the black regular numbers indicate the number of confirmed active hits (Tables 2 and 3), purchased hits (ESI† Figs. S4 and S5), and the number of hits after applying the IFP cutoffs, respectively. | ||
Notably, in contrast to the ligands found using β2R-based H4R models the confirmed hits discovered in the prospective SBVS campaigns against H1R-based H4R models were not the highest ranking compounds. Re-ranking the docking poses by their IFP similarity after the docking increased the rank of the confirmed hits from #1051–#26 862 using PLANTS to #413–#3374 using IFP (Table 3). This indicates that the post SBVS campaign steps presented in Fig. 5 (dissimilarity filter and visual inspection) has led to diverse sets of selected and purchased hits. The Venn Diagram of Fig. 8 shows the overlap between the hit lists obtained for each of the homology models (after applying the retrospectively identified IFP score cutoffs, see Methods). The amount of unique ligands for each approach shows the complementarity of employing different modelling templates and different reference ligands to refine homology models. The purchased hits cover 12 out of 15 possible Venn-diagram regions, i.e. overlap combinations between homology-model hit lists (Fig. 8). Most of the confirmed active H4R fragments were present in the hit lists of the VUF10497 models (7 out of 9). The remaining 2 confirmed H4R fragments were identified in the SBVS using the H1R-based model with JNJ7777120. None of the confirmed hits were unique for β2R-based model with JNJ7777120, although one confirmed hit (VUF13848 (11)) was identified in both JNJ7777120 homology models.
Most of previous ligands designs for H4R were highly inspired from the structure of JNJ7777120 (2)4,59 since it was the first published non-imidazole antagonist for H4R.7 This might be the cause that the JNJ7777120 H4R models resulted in more diverse selected active hits compared to the SBVS campaigns using VUF10497 (3) as the reference (Fig. 8). The PLANTSChemPLP scores of the active H4R fragments were higher (≤−90)24 in the SBVS with VUF10497 (3) as the reference compound (ESI† Fig. S6). The scoring distributions (Fig. S5) also show that with a lower PLANTS score the IFP score is increasing for only the VUF10497 models, which is in line with the finding that 7 out of the 9 confirmed hits were present in hit lists of these models. Hence, employing PLANTSChemPLP score and Tc-IFP as a combined scoring function in SBVS campaigns could increase the SBVS quality (as it did for the aforementioned H1R crystal structure-based VS),24 but it depends on the structure models and the IFP references. Cut-offs optimization is therefore required to build SBVS protocols with optimized quality.
Similar to the SBVS on the H1R crystal structure,24 the combined approaches can lead in to a good hit rate (9 out of 36) of H4R small fragments. Although the hit rate is lower than the SBVS on the H1R crystal structure,24 these results of the prospective virtual screening exercise validate our structure-based virtual fragment screening method.
Conclusions
We have investigated the possibilities of structure-based virtual fragment screening against optimized homology models of the histamine H4R receptor. Structure- and ligand-based methods performed equally well in retrospective virtual screening studies, but structure-based virtual fragment screening using an interaction fingerprint scoring method enabled the identification of H4R ligands that were not identified in ligand-based VS runs. Surprisingly, retrospective virtual screening validation studies against H4R homology models based on the H1R crystal structure did not give higher VS enrichments than H4R models based on the crystal structure of the more distantly-related β2R. Optimized SBVFS methods were successfully used to find two new series of fragment-like H4R ligands. Nine out of the 37 tested compounds (representing five different scaffolds) had binding affinities between 0.14 and 6.9 μM at the H4R. The results of our comparative study can be used to guide future structure-based virtual fragment screening campaigns.Experimental section
Retrospective virtual screening
Prospective virtual screening
Pharmacological assays
HEK293T cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 50 IU ml−1penicillin and 50 μg ml−1streptomycin at 37 °C and 5% CO2. Approximately 4 × 10 6 cells in 10 cm dishes were transiently transfected with 5 μg receptor DNA using 25 kDa linear polyethylenimine (PEI; Polysciences, Warrington, USA) as transfection reagent (1 : 4 DNA/PEI ratio). The cells were harvested 2 days after transfection and homogenized in 50 mM Tris-HCl binding buffer (pH 7.4). Cell homogenates were co-incubated with indicated concentrations of compounds and ~3 nM [3H]-mepyramine (human H1R), ~10 nM [3H]-histamine (human H4R), or ~2 nM [3H]-dihydroalprenolol (human β2R) in a total volume of 100 μl per well. All radioligands were purchased from PerkinElmer Life Sciences. The reaction mixtures were incubated for 1–1.5 h at 25 °C on a microtiter shaker (750 rpm). Incubations were terminated by rapid filtration through Unifilter glass fiber C plates (PerkinElmer Life Sciences) that were presoaked in 0.3% polyethylenimine and subsequently washed three times with ice-cold binding buffer (pH 7.4 at 4 °C). Retained radioactivity was measured by liquid scintillation using a MicroBeta Trilux (PerkinElmer Life Sciences). Nonlinear curve fitting was performed using GraphPad Prism version 6.00 for Windows/Mac OSX, GraphPad Software, La Jolla California USA, www.graphpad.com. The Ki values were calculated using the Cheng–Prusoff equation:118Ki = IC50/(1 + [radioligand]/Kd).Acknowledgements
We thank Mitchell K. L. Han for assistance with radioligand displacement and Antoni R. Blaazer for performing the quality control of the compounds. This research was financially supported by The Netherlands Organization for Scientific Research (NWO VENI Grant 700.59.408 to C. d. G. and NWO TOP PUNT Grant to R. L.), Indonesian Directorate General of Higher Education (Fundamental Research Block Grant 1320/K5/KM/2014 to E. P. I.) and Institute for Research and Community Services, Sanata Dharma University (Research Grant 083/Panel/LPPM-USD/SP/X/2013 to E. P. I). A. J. K., H. F. V., S. N., I. J. P. d. E., R. L., and C. d. G. participate in the European Cooperation in Science and Technology Action CM1207 [GPCR-Ligand Interactions, Structures, and Transmembrane Signalling: A European Research Network (GLISTEN)].Notes and references
- M. C. Lagerström and H. B. Schiöth, Nat. Rev. Drug Discovery, 2008, 7, 339–357 CrossRef PubMed.
- R. Leurs, H. F. Vischer, M. Wijtmans and I. J. P. de Esch, Trends Pharmacol. Sci., 2011, 32, 250–257 CrossRef CAS PubMed.
- H. D. Lim, R. A. Smits, R. Leurs and I. J. P. de Esch, Curr. Top. Med. Chem., 2006, 6, 1365–1373 CrossRef CAS.
- E. P. Istyastono, C. de Graaf, I. J. P. de Esch and R. Leurs, Curr. Top. Med. Chem., 2011, 11, 661–679 CrossRef CAS.
- R. A. Smits, R. Leurs and I. J. P. de Esch, Drug Discovery Today, 2009, 14, 745–753 CrossRef CAS PubMed.
- H. Engelhardt, R. A. Smits, R. Leurs, E. Haaksma and I. J. P. de Esch, Curr. Opin. Drug Discovery Dev., 2009, 12, 628–643 CAS.
- J. A. Jablonowski, C. A. Grice, W. Chai, C. A. Dvorak, J. D. Venable, A. K. Kwok, K. S. Ly, J. Wei, S. M. Baker, P. J. Desai, W. Jiang, S. J. Wilson, R. L. Thurmond, L. Karlsson, J. P. Edwards, T. W. Lovenberg and N. I. Carruthers, J. Med. Chem., 2003, 46, 3957–3960 CrossRef CAS PubMed.
- R. A. Smits, I. J. P. de Esch, O. P. Zuiderveld, J. Broeker, K. Sansuk, E. Guaita, G. Coruzzi, M. Adami, E. Haaksma and R. Leurs, J. Med. Chem., 2008, 51, 7855–7865 CrossRef CAS PubMed.
- D. A. Erlanson, R. S. McDowell and T. O'Brien, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS PubMed.
- G. E. de Kloe, D. Bailey, R. Leurs and I. J. P. de Esch, Drug Discovery Today, 2009, 14, 630–646 CrossRef PubMed.
- C. W. Murray, M. L. Verdonk and D. C. Rees, Trends Pharmacol. Sci., 2012, 33, 224–232 CrossRef CAS PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef.
- S. M. Boyd and G. E. de Kloe, Drug Discovery Today: Technol., 2010, 7, e173–e180 CrossRef CAS PubMed.
- C. de Graaf, H. F. Vischer, G. E. de Kloe, A. J. Kooistra, S. Nijmeijer, M. Kuijer, M. H. Verheij, P. J. England, J. E. van Muijlwijk-Koezen, R. Leurs and I. J. de Esch, Drug Discovery Today, 2013, 18, 323–330 CrossRef CAS PubMed.
- A. R. Leach and M. M. Hann, Curr. Opin. Chem. Biol., 2011, 15, 489–496 CrossRef CAS PubMed.
- M. Congreve, R. L. Rich, D. G. Myszka, F. Figaroa, G. Siegal and F. H. Marshall, Methods Enzymol., 2011, 493, 115–136 CAS.
- M. H. P. Verheij, C. de Graaf, G. E. de Kloe, S. Nijmeijer, H. F. Vischer, R. A. Smits, O. P. Zuiderveld, S. Hulscher, L. Silvestri, A. J. Thompson, J. E. van Muijlwijk-Koezen, S. C. R. Lummis, R. Leurs and I. J. P. de Esch, Bioorg. Med. Chem. Lett., 2011, 21, 5460–5464 CrossRef CAS PubMed.
- A. Visegrady and G. M. Keseru, Expert Opin. Drug Discovery, 2013, 8, 811–820 CrossRef CAS PubMed.
- D. Chen, J. C. Errey, L. H. Heitman, F.H. Marshall, A. P. Ijzerman and G. Siegal, ACS Chem. Biol., 2012, 7, 2064–2073 CrossRef CAS PubMed.
- D. Chen, A. Ranganathan, I. J. AP, G. Siegal and J. Carlsson, J. Chem. Inf. Model., 2013, 53, 2701–2714 CrossRef CAS PubMed.
- J. A. Christopher, J. Brown, A. S. Dore, J. C. Errey, M. Koglin, F. H. Marshall, D. G. Myszka, R. L. Rich, C. G. Tate, B. Tehan, T. Warne and M. Congreve, J. Med. Chem., 2013, 56, 3446–3455 CrossRef CAS PubMed.
- S. P. Andrews, G. A. Brown and J. A. Christopher, ChemMedChem, 2014, 9, 256–275 CrossRef CAS PubMed.
- A. J. Kooistra, S. Kuhne, I. J. de Esch, R. Leurs and C. de Graaf, Br. J. Pharmacol., 2013, 170, 101–126 CrossRef CAS PubMed.
- C. de Graaf, A. J. Kooistra, H. F. Vischer, V. Katritch, M. Kuijer, M. Shiroishi, S. Iwata, T. Shimamura, R. C. Stevens, I. J. de Esch and R. Leurs, J. Med. Chem., 2011, 54, 8195–8206 CrossRef CAS PubMed.
- F. Sirci, E. P. Istyastono, H. F. Vischer, A. J. Kooistra, S. Nijmeijer, M. Kuijer, M. Wijtmans, R. Mannhold, R. Leurs, I. J. P. de Esch and C. de Graaf, J. Chem. Inf. Model., 2012, 52, 3308–3324 CrossRef CAS PubMed.
- M. Vass, É. Schmidt, F. Horti and G. M. Keserű, Eur. J. Med. Chem., 2014, 77, 38–46 CrossRef CAS PubMed.
- A. J. Kooistra, R. Leurs, I. J. P. de Esch and C. de Graaf, Adv. Exp. Med. Biol., 2014, 796, 129–157 CrossRef CAS.
- J. Venhorst, S. Nunez, J. W. Terpstra and C. G. Kruse, J. Med. Chem., 2008, 51, 3222–3229 CrossRef CAS PubMed.
- D. M. Krüger and A. Evers, ChemMedChem, 2010, 5, 148–158 CrossRef PubMed.
- A. J. Kooistra, L. Roumen, R. Leurs, I. J. P. de Esch and C. de Graaf, Methods Enzymol., 2013, 522, 279–336 CAS.
- N. Moitessier, P. Englebienne, D. Lee, J. Lawandi and C. R. Corbeil, Br. J. Pharmacol., 2008, 153(Suppl 1), S7–26 CAS.
- M. Michino, E. Abola, GPCR Dock 2008 participants, C. L. Brooks 3rd, J. S. Dixon, J. Moult and R. C. Stevens, Nat. Rev. Drug Discovery, 2009, 8, 455–463 CrossRef CAS PubMed.
- I. Kufareva, M. Rueda, V. Katritch, R. C. Stevens, R. Abagyan and GPCR Dock 2010 participants, Structure, 2011, 19, 1108–1126 CrossRef CAS PubMed.
- A. Tarcsay, G. Paragi, M. Vass, B. Jójárt, F. Bogár and G. M. Keserű, J. Chem. Inf. Model., 2013, 53, 2990–2999 CrossRef CAS PubMed.
- I. Kufareva, V. Katritch, GPCR Dock 2013 participants, R. C. Stevens and R. Abagyan, Structure, 2014, 22, 1120–1139 CrossRef CAS PubMed.
- G. Marcou and D. Rognan, J. Chem. Inf. Model., 2007, 47, 195–207 CrossRef CAS PubMed.
- K. Loving, I. Alberts and W. Sherman, Curr. Top. Med. Chem., 2010, 10, 14–32 CrossRef CAS.
- M. L. Verdonk, I. Giangreco, R. J. Hall, O. Korb, P. N. Mortenson and C. W. Murray, J. Med. Chem., 2011, 54, 5422–5431 CrossRef CAS PubMed.
- J. Carlsson, R. G. Coleman, V. Setola, J. J. Irwin, H. Fan, A. Schlessinger, A. Sali, B. L. Roth and B. K. Shoichet, Nat. Chem. Biol., 2011, 7, 769–778 CrossRef CAS PubMed.
- S. Vilar, G. Ferino, S. S. Phatak, B. Berk, C. N. Cavasotto and S. Costanzi, J. Mol. Graphics Modell., 2011, 29, 614–623 CrossRef CAS PubMed.
- V. Katritch, V. Cherezov and R. C. Stevens, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 531–556 CrossRef CAS PubMed.
- A. J. Venkatakrishnan, X. Deupi, G. Lebon, C. G. Tate, G. F. Schertler and M. M. Babu, Nature, 2013, 494, 185–194 CrossRef CAS PubMed.
- A. J. Kooistra, C. de Graaf and H. Timmerman, Neurochem. Res., 2014, 39, 1850–1861 CrossRef CAS PubMed.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS PubMed.
- T. Shimamura, M. Shiroishi, S. Weyand, H. Tsujimoto, G. Winter, V. Katritch, R. Abagyan, V. Cherezov, W. Liu, G. W. Han, T. Kobayashi, R. C. Stevens and S. Iwata, Nature, 2011, 475, 65–70 CrossRef CAS PubMed.
- P. Kolb, D. M. Rosenbaum, J. J. Irwin, J. J. Fung, B. K. Kobilka and B. K. Shoichet, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6843–6848 CrossRef CAS PubMed.
- V. Katritch, V. P. Jaakola, J. R. Lane, J. Lin, A. P. Ijzerman, M. Yeager, I. Kufareva, R. C. Stevens and R. Abagyan, J. Med. Chem., 2010, 53, 1799–1809 CrossRef CAS PubMed.
- J. Carlsson, L. Yoo, Z. Gao, J. J. Irwin, B. K. Shoichet and K. A. Jacobson, J. Med. Chem., 2010, 53, 3748–3755 CrossRef CAS PubMed.
- D. Rodriguez, J. Brea, M. I. Loza and J. Carlsson, Structure, 2014, 22, 1140–1151 CrossRef CAS PubMed.
- J. Varady, X. Wu, X. Fang, J. Min, Z. Hu, B. Levant and S. Wang, J. Med. Chem., 2003, 46, 4377–4392 CrossRef CAS PubMed.
- A. Evers and T. Klabunde, J. Med. Chem., 2005, 48, 1088–1097 CrossRef CAS PubMed.
- M. Congreve, J. M. Dias and F. H. Marshall, Prog. Med. Chem., 2014, 53, 1–63 CAS.
- K. A. Jacobson and S. Costanzi, Mol. Pharmacol., 2012, 82, 361–371 CrossRef CAS PubMed.
- N. Shin, E. Coates, N. J. Murgolo, K. L. Morse, M. Bayne, C. D. Strader and F. J. Monsma, Mol. Pharmacol., 2002, 62, 38–47 CrossRef CAS.
- A. Jongejan, H. D. Lim, R. A. Smits, I. J. de Esch, E. Haaksma and R. Leurs, J. Chem. Inf. Model., 2008, 48, 1455–1463 CrossRef CAS PubMed.
- R. Kiss, B. Kiss, A. Konczol, F. Szalai, I. Jelinek, V. Laszlo, B. Noszal, A. Falus and G. M. Keseru, J. Med. Chem., 2008, 51, 3145–3153 CrossRef CAS PubMed.
- Y. Tanrikulu, E. Proschak, T. Werner, T. Geppert, N. Todoroff, A. Klenner, T. Kottke, K. Sander, E. Schneider, R. Seifert, H. Stark, T. Clark and G. Schneider, ChemMedChem, 2009, 4, 820–827 CrossRef CAS PubMed.
- H. D. Lim, C. de Graaf, W. Jiang, P. Sadek, P. M. McGovern, E. P. Istyastono, R. A. Bakker, I. J. de Esch, R. L. Thurmond and R. Leurs, Mol. Pharmacol., 2010, 77, 734–743 CrossRef CAS PubMed.
- E. P. Istyastono, S. Nijmeijer, H. D. Lim, A. van de Stolpe, L. Roumen, A. J. Kooistra, H. F. Vischer, I. J. de Esch, R. Leurs and C. de Graaf, J. Med. Chem., 2011, 54, 8136–8147 CrossRef CAS PubMed.
- S. Schultes, S. Nijmeijer, H. Engelhardt, A. J. Kooistra, H. F. Vischer, I. J. P. de Esch, E. E. J. Haaksma, R. Leurs and C. de Graaf, Med. Chem. Commun., 2013, 4, 193 RSC.
- S. Schultes, H. Engelhardt, L. Roumen, O. P. Zuiderveld, E. E. Haaksma, I. J. de Esch, R. Leurs and C. de Graaf, ChemMedChem, 2013, 8, 49–53 CrossRef CAS PubMed.
- M. Wijtmans, C. de Graaf, G. de Kloe, E. P. Istyastono, J. Smit, H. Lim, R. Boonnak, S. Nijmeijer, R. A. Smits, A. Jongejan, O. Zuiderveld, I. J. P. de Esch and R. Leurs, J. Med. Chem., 2011, 54, 1693–1703 CrossRef CAS PubMed.
- R. A. Smits, M. Adami, E. P. Istyastono, O. P. Zuiderveld, C. M. van Dam, F. J. de Kanter, A. Jongejan, G. Coruzzi, R. Leurs and I. J. de Esch, J. Med. Chem., 2010, 53, 2390–2400 CrossRef CAS PubMed.
- T. Warne, M. J. Serrano-Vega, J. G. Baker, R. Moukhametzianov, P. C. Edwards, R. Henderson, A. G. Leslie, C. G. Tate and G. F. Schertler, Nature, 2008, 454, 486–491 CrossRef CAS PubMed.
- E. Y. Chien, W. Liu, Q. Zhao, V. Katritch, G. W. Han, M. A. Hanson, L. Shi, A. H. Newman, J. A. Javitch, V. Cherezov and R. C. Stevens, Science, 2010, 330, 1091–1095 CrossRef CAS PubMed.
- K. Haga, A. C. Kruse, H. Asada, T. Yurugi-Kobayashi, M. Shiroishi, C. Zhang, W. I. Weis, T. Okada, B. K. Kobilka, T. Haga and T. Kobayashi, Nature, 2012, 482, 547–551 CrossRef CAS PubMed.
- A. C. Kruse, J. Hu, A. C. Pan, D. H. Arlow, D. M. Rosenbaum, E. Rosemond, H. F. Green, T. Liu, P. S. Chae, R. O. Dror, D. E. Shaw, W. I. Weis, J. Wess and B. K. Kobilka, Nature, 2012, 482, 552–556 CrossRef CAS PubMed.
- C. Wang, Y. Jiang, J. Ma, H. Wu, D. Wacker, V. Katritch, G. W. Han, W. Liu, X. P. Huang, E. Vardy, J. D. McCorvy, X. Gao, X. E. Zhou, K. Melcher, C. Zhang, F. Bai, H. Yang, L. Yang, H. Jiang, B. L. Roth, V. Cherezov, R. C. Stevens and H. E. Xu, Science, 2013, 340, 610–614 CrossRef CAS PubMed.
- S. Topiol and M. Sabio, Bioorg. Med. Chem. Lett., 2008, 18, 1598–1602 CrossRef CAS PubMed.
- D. R. Weiss, S. Ahn, M. F. Sassano, A. Kleist, X. Zhu, R. Strachan, B. L. Roth, R. J. Lefkowitz and B. K. Shoichet, ACS Chem. Biol., 2013, 8, 1018–1026 CrossRef CAS PubMed.
- A. J. Kooistra, I. J. de Esch, R. Leurs and C. de Graaf, PhD Thesis, VU University Amsterdam, 2015 Search PubMed.
- M. M. Mysinger, D. R. Weiss, J. J. Ziarek, S. Gravel, A. K. Doak, J. Karpiak, N. Heveker, B. K. Shoichet and B. F. Volkman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5517–5522 CrossRef CAS PubMed.
- D. Rognan, Top. Curr. Chem., 2012, 317, 201–222 CrossRef CAS.
- C. de Graaf, N. Foata, O. Engkvist and D. Rognan, Proteins, 2008, 71, 599–620 CrossRef CAS PubMed.
- G. Hessler and K.-H. Baringhaus, Drug Discovery Today: Technol., 2010, 7, e263–e269 CrossRef CAS PubMed.
- S. Schultes, A. J. Kooistra, H. F. Vischer, S. Nijmeijer, E. Haaksma, R. Leurs, I. J. P. de Esch and C. de Graaf, J. Chem. Inf. Model., 2015 DOI:10.1021/ci500694c.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–1107 CrossRef CAS PubMed.
- S. Nijmeijer, H. F. Vischer, E. M. Rosethorne, S. J. Charlton and R. Leurs, Mol. Pharmacol., 2012, 82, 1174–1182 CrossRef CAS PubMed.
- S. Nijmeijer, H. F. Vischer, F. Sirci, S. Schultes, H. Engelhardt, C. de Graaf, E. M. Rosethorne, S. J. Charlton and R. Leurs, Br. J. Pharmacol., 2013, 170, 78–88 CrossRef CAS PubMed.
- S. Cramp, H. J. Dyke, C. Higgs, D. E. Clark, M. Gill, P. Savy, N. Jennings, S. Price, P. M. Lockey, D. Norman, S. Porres, F. Wilson, A. Jones, N. Ramsden, R. Mangano, D. Leggate, M. Andersson and R. Hale, Bioorg. Med. Chem. Lett., 2010, 20, 2516–2519 CrossRef CAS PubMed.
- H. D. Lim, R. M. V. Rijn, P. Ling, R. A. Bakker, R. L. Thurmond and R. Leurs, J. Pharmacol. Exp. Ther., 2005, 314, 1310–1321 CrossRef CAS PubMed.
- S. Nijmeijer, H. Engelhardt, S. Schultes, A. C. van de Stolpe, V. Lusink, C. de Graaf, M. Wijtmans, E. E. Haaksma, I. J. de Esch, K. Stachurski, H. F. Vischer and R. Leurs, Br. J. Pharmacol., 2013, 170, 89–100 CrossRef CAS PubMed.
- J. A. Salon, D. T. Lodowski and K. Palczewski, Pharmacol. Rev., 2011, 63, 901–937 CrossRef CAS PubMed.
- D. Wacker, G. Fenalti, M. A. Brown, V. Katritch, R. Abagyan, V. Cherezov and R. C. Stevens, J. Am. Chem. Soc., 2010, 132, 11443–11445 CrossRef CAS PubMed.
- H. D. Lim, A. Jongejan, R. A. Bakker, E. Haaksma, I. J. de Esch and R. Leurs, J. Pharmacol. Exp. Ther., 2008, 327, 88–96 CrossRef CAS PubMed.
- M. Govoni, H. D. Lim, D. El-Atmioui, W. M. P. B. Menge, H. Timmerman, R. A. Bakker, R. Leurs and I. J. P. De Esch, J. Med. Chem., 2006, 49, 2549–2557 CrossRef CAS PubMed.
- B. M. Savall, J. P. Edwards, J. D. Venable, D. J. Buzard, R. Thurmond, M. Hack and P. McGovern, Bioorg. Med. Chem. Lett., 2010, 20, 3367–3371 CrossRef CAS PubMed.
- E. M. Rosethorne and S. J. Charlton, Mol. Pharmacol., 2011, 79, 749–757 CrossRef CAS PubMed.
- R. Seifert, E. H. Schneider, S. Dove, I. Brunskole, D. Neumann, A. Strasser and A. Buschauer, Mol. Pharmacol., 2011, 79, 631–638 CrossRef CAS PubMed.
- D. Wifling, K. Löffel, U. Nordemann, A. Strasser, G. Bernhardt, S. Dove, R. Seifert and A. Buschauer, Br. J. Pharmacol., 2015, 172, 785–798 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- OpenEye Scientific Software Inc., 2009, ROCS version 2.3.1, Santa Fe. http://www.eyesopen.com..
- O. Korb, T. Stützle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS PubMed.
- C. de Graaf and D. Rognan, Curr. Pharm. Des., 2009, 15, 4026–4048 CrossRef CAS.
- R Development Core Team, 2008, R: A Language and Environment for Statistical Computing, Vienna. http://www.r-project.org..
- M. H. Zweig and G. Campbell, Clin. Chem., 1993, 39, 561–577 CAS.
- X. Robin, N. Turck, A. Hainard, N. Tiberti, F. Lisacek, J.-C. Sanchez and M. Müller, BMC Bioinf., 2011, 12, 77 CrossRef PubMed.
- A. Steffen, T. Kogej, C. Tyrchan and O. Engkvist, J. Chem. Inf. Model., 2009, 49, 338–347 CrossRef CAS PubMed.
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed.
- T. I. Oprea, J. Comput. Mol. Des., 2000, 14, 251–264 CrossRef CAS.
- European Bioinformatics Institute, ChEMBLdb, 2010, Cambridgeshire. https://www.ebi.ac.uk/chembldb. Accessed 19 August 2010.
- A. B. Yongye, J. R. Appel, M. A. Giulianotti, C. T. Dooley, J. L. Medina-Franco, A. Nefzi, R. A. Houghten and K. Martínez-Mayorga, Bioorg. Med. Chem., 2009, 17, 5583–5597 CrossRef CAS PubMed.
- J. W. Raymond, E. J. Gardiner and P. Willett, J. Chem. Inf. Comput. Sci., 2002, 42, 305–316 CrossRef CAS PubMed.
- J. C. Mobarec, R. Sanchez and M. Filizola, J. Med. Chem., 2009, 52, 5207–5216 CrossRef CAS PubMed.
- J. A. Ballesteros and H. Weinstein, Methods Neurosci., 1995, 25, 366–428 CAS.
- C. H. Wu, R. Apweiler, A. Bairoch, D. A. Natale, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H. Huang, R. Lopez, M. Magrane, M. J. Martin, R. Mazumder, C. O'Donovan, N. Redaschi and B. Suzek, Nucleic Acids Res., 2006, 34, D187–D191 CrossRef CAS PubMed.
- ChemAxon, 2009, MarvinBeans 5.2.5.1, Budapest.
- Molecular Networks GmbH., 2008, Corina version 3.46, Erlangen, http://www.molecular-networks.com/products/corina..
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, Amber 10, University of California, San Francisco, 2008 Search PubMed.
- E. Urizar, S. Claeysen, X. Deupí, C. Govaerts, S. Costagliola, G. Vassart and L. Pardo, J. Biol. Chem., 2005, 280, 17135–17141 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- O. Korb, T. Stützle and T. E. Exner, Proc. IEEE Swarm Intell. Symp., 2007, 1, 115–134 CrossRef PubMed.
- OpenEye Scientific Software Inc., 2008, OMEGA version 2.3.2, Santa Fe. http://www.eyesopen.com.
- Accelrys, 2007, Pipeline Pilot version 6.1.5,San Diego. http://accelrys.com/products/pipeline-pilot.
- A. N. Jain and A. Nicholls, J. Comput.-Aided Mol. Des., 2008, 22, 133–139 CrossRef CAS PubMed.
- OpenEye Scientific Software Inc., 2009, FILTER version 2.1.1, Santa Fe. http://www.eyesopen.com.
- J. Sadowski, J. Gasteiger and G. Klebe, J. Chem. Inf. Model., 1994, 34, 1000–1008 CrossRef CAS.
- Y. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00022j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |