
The status of the lysophosphatidic acid receptor type 1 (LPA1R)
Inés
González-Gil
,
Debora
Zian
,
Henar
Vázquez-Villa
,
Silvia
Ortega-Gutiérrez
* and
María L.
López-Rodríguez
*
Departamento de Química Orgánica I, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, E-28040 Madrid, Spain. E-mail: mluzlr@ucm.es; siortega@ucm.es
First published on 22nd September 2014
Abstract
Lysophospholipids are lipid molecules that are receiving growing attention because, in addition to their structural function in the cell membrane, they are now regarded as important regulators for diverse biological functions through activation of specific receptors. These receptors have been characterized during the last two decades as G protein-coupled receptors (GPCRs) and, among them, two families stand out: lysophosphatidic acid (LPA1–6) and sphingosine 1-phoshate (S1P1–5) receptors. Despite their interest, the high structural similarity between them has restrained the development of selective and high affinity ligands and therefore the elucidation of the role of these receptors in the central nervous system (CNS). This review provides an overview about the different LPA receptors with a special focus on the LPA1 subtype from a medicinal chemistry perspective. It summarizes the most recent developments in the search for selective and specific agonists and antagonists of the LPA1 receptor and highlights their current status in the drug development pipeline.
Introduction
The phospholipid superfamily has been traditionally linked to structural roles, as key constituents of biological membranes. Nevertheless, research from the last decades has associated some phospholipids with diverse signalling functions, reporting their action as extracellular signals and, moreover, their involvement in many physiological and pathological processes.1,2Phospholipids are usually divided into two broad families: (i) glycerophospholipids,3 which are structurally based on the glycerol scaffold (Fig. 1A) and (ii) sphingophospholipids, which are derivatives of the amino alcohol sphingosine (Fig. 1B). They present a polar head bearing a phosphate group (–OPO3Y−, Y = H, choline, ethanolamine, Fig. 1A and B) and two hydrophobic chains (R, X, Fig. 1A and B). When one of the fatty acid chains is missing (X = H, Fig. 1A and B), the resulting derivatives are denominated lysophospholipids. These molecules are quantitatively minor lipid species compared to their parent compounds, the phospholipids – which have a major presence in cell membranes. Despite their low concentration, lysophospholipids are important because of their ability to signal through G protein-coupled receptors (GPCRs). Lysophosphatidic acid (LPA, 1-acyl-sn-glycero-3-phosphate, Fig. 1C) and sphingosine 1-phosphate3 (S1P, Fig. 1D) are the two most prominent molecules of this family, which are being extensively studied and their biological activities have been shown to be extremely relevant.4
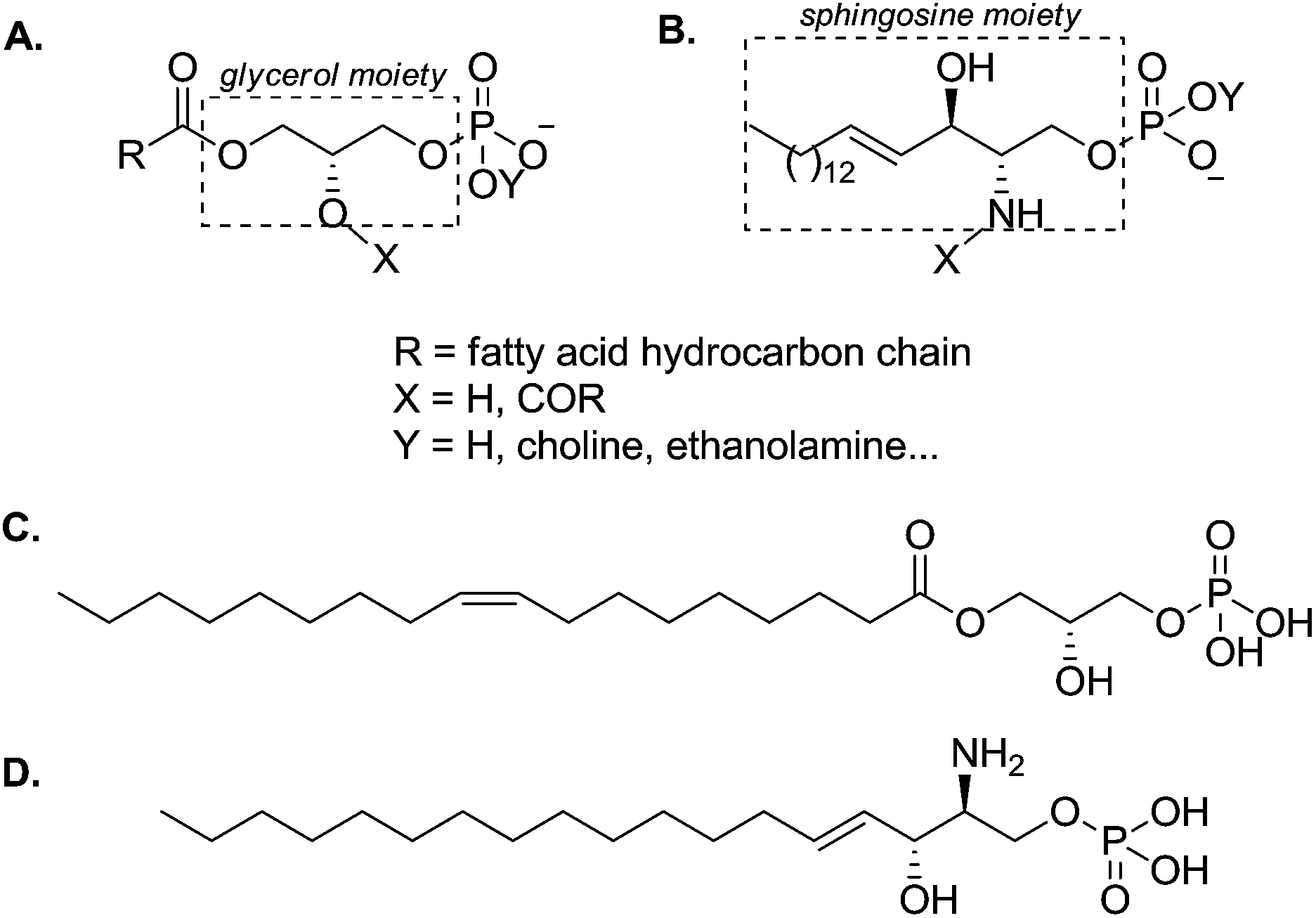 | ||
| Fig. 1 General structure of common glycerophospholipids (A) and sphingophospholipids (B). Structures of LPA (C) and S1P (D). | ||
Although they belong to distinct signalling systems, similarities between these two lipids extend to their tissue distribution and concentration, homology and effector pathways of their cognate receptors, and the broad range of their biological roles. In contrast, the actions of other lysophospholipids have not been elucidated to such a high degree and very little is known about their endogenous receptors. However, recent in vitro studies suggest that they can induce various and unique cellular responses.5
Among the bioactive phospholipids, LPA stands out as a molecule that elicits a plethora of biological effects, both in the central nervous system (CNS) and in the periphery, by acting on at least six different receptors. Nonetheless, its therapeutic potential is still far from being established given the complexity of the system and the lack of specific ligands, agonists and antagonists, that enable the elucidation of the (patho)physiological roles played by a particular LPA receptor subtype. This review summarizes the most important aspects of the LPA signaling system with a special focus on the LPA1 receptor, its ligands and their potential for drug development. In the first part, we provide an overview of the different LPA receptors with particular attention to the LPA1 subtype, its endogenous ligand LPA, and the main (patho)physiological functions regulated by the LPA1 receptor. Although more exhaustive reviews have been published on the molecular, biochemical and pharmacological aspects of the complex LPA system (see references), this introduction will allow us to proceed to the second part of the review, in which we address, from a medicinal chemistry perspective, the most essential advances reported so far regarding the development of selective and specific ligands of the LPA1 receptor. Finally, we will summarize the current status and the clinical perspectives of the compounds that have progressed most in the drug development pipeline.
Lysophosphatidic acid receptors
LPA has a well-known structural function as a precursor and a metabolite in the biosynthesis of membrane phospholipids. However, it was not until the 1960s that several groups started to report biological actions mediated by LPA, such as smooth muscle contraction and platelet aggregation.6 Nevertheless, the specific function of this intriguing molecule was still unknown.During the mid-1980s, proliferative LPA-dependent effects in fibroblasts were described. These responses were completely inhibited with pertussis toxin pre-treatment, which specifically inactivates Gαi/o-type G proteins.7 This was followed by the description in the early 1990s of several morphological cell changes attributed to LPA, such as cell growth, cell rounding/neurite retraction8,9 and actin stress fiber formation.10 At the same time, S1P was reported to evoke cellular responses similar to those induced by LPA, suggesting that they might even share the same GPCRs.11
Growing evidence was making clear that LPA was acting through a GPCR, as it was finally demonstrated by van Blitterswijk12 through photoaffinity labeling experiments, which revealed [32P] LPA-binding membrane proteins of 38–40 kDa present in various LPA-responsive cell types and in the brain. This binding protein met all the pharmacological criteria for a specific, high-affinity LPA receptor since its labeling was competitively and specifically inhibited by unlabeled LPA with an IC50 as low as 10 nM. In addition to the LPA responses, LPA binding was not detectable in LPA-unresponsive cells such as human neutrophils, and was blocked by suramin, a known inhibitor of LPA actions. Although similar evidences were shown independently by Clark,13 the biophysical properties of LPA or the possibility of second messenger activities were also proposed as alternative mechanisms for LPA actions, and this ambiguity persisted in the absence of molecularly identified receptors.
Finally, in 1996, Chun and coworkers reported the discovery of the first lysophospholipid receptor gene, ventricular zone gene 1 (vzg-1),14 during their studies on mammalian neurogenesis. Vzg-1 encoded a GPCR that had the properties of a high-affinity LPA receptor. Identification of this gene as encoding an LPA receptor was independently demonstrated by Goetzl15 and Kiefer.16 Definitive confirmation about the identity of this receptor was achieved by heterologous expression in mammalian cells17 and genetic deletion of the receptor.18
Similar approaches allowed the identification of new receptors, like the first receptor for S1P, which was independently reported by two groups in 1998.19,20 Since then, several members of the orphan GPCR receptor family called “endothelial differentiation genes” (Edg) were identified as GPCRs for both LPA and S1P, including Edg4 (LPA2),21,22 Edg7 (LPA3),23 Edg5 (S1P2), Edg3 (S1P3), Edg6 (S1P4)24 and Edg8 (S1P5).25 Regarding the LPA receptors, another group of less similar GPCR genes have also been identified, which are GPR23 (LPA4),26,27 GPR92 (LPA5),28,29 and P2Y5 (LPA6).30,31 This latter group is more closely related to the family of P2Y purinergic receptor genes, indicating that LPA receptors have evolved via two distinct lineages in the rhodopsin GPCR family. To date, a total of eleven receptors have been described, six for LPA (LPA1–6) and five for S1P (S1P1–5). The current nomenclature includes the cognate ligand and the chronological order of identification. All LPA receptors are type I, rhodopsin-like GPCRs that differ in their tissue distribution and downstream signalling pathways32 (see Fig. 2 and Table 1 for a summary of some relevant features).
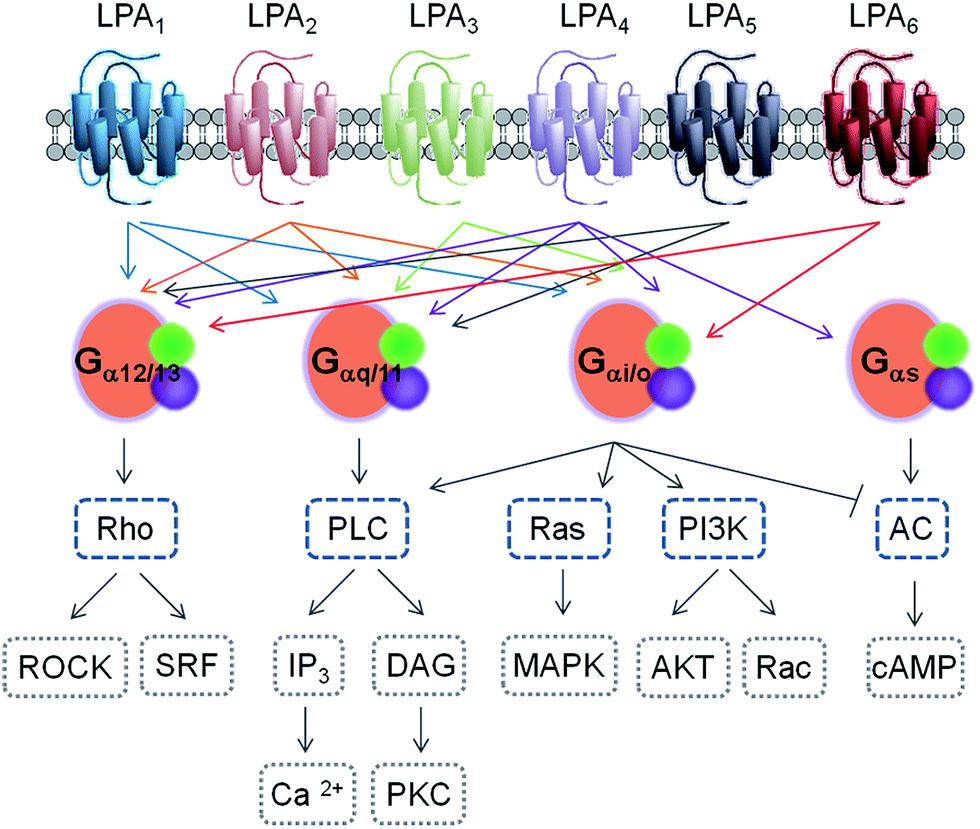 | ||
| Fig. 2 Schematic representation of the signalling pathways activated by the LPA1–6 receptors. The heterotrimeric G proteins are defined here by their α subunits in orange (green and purple circles represent the β and γ subunits). | ||
| Namea | Gene symbol (human) | Chromosomal location (human) | Number of amino acids (human) | Similarity to LPA1 (%) |
|---|---|---|---|---|
| a Nomenclature of the International Union of Basic and Clinical Pharmacology (IUPHAR). | ||||
| LPA1 | LPAR1 | 9q31.3 | 364 | |
| LPA2 | LPAR2 | 19p12 | 348 | 60 |
| LPA3 | LPAR3 | 1p22.3 p31.1 | 353 | 50 |
| LPA4 | LPAR4 | Xq13–q21.1 | 370 | 10 |
| LPA5 | LPAR5 | 12p 13.31 | 372 | 12 |
| LPA6 | LPAR6 | 13q14 | 344 | 13 |
LPA1 receptor
The mammalian LPAR1 gene encodes a ∼41 kDa protein of 364 amino acids with seven putative transmembrane domains.Human LPAR1 is widely expressed in the heart, brain, placenta, skeletal muscle, kidney, pancreas, spleen, prostate, testis, ovary, small intestine and colon.33 A similar distribution is observed for the Lpar1 mouse gene, although it is more spatially restricted during embryogenesis, where it is mainly found in the ventricular zone (VZ), a major site for neuroprogenitor cell proliferation during prenatal developmental stages. VZ disappears prior to birth, reappearing during the postnatal life within oligodendrocytes and Schwann cells that may influence myelination in the central and peripheral nervous system, indicating roles for LPA signalling in cortical development.34
Signalling through LPA1 receptor induces a range of cellular responses: cell proliferation and survival, cell migration and cytoskeletal changes. At the molecular level, LPA1 receptor activation can be transduced through three types of G proteins: Gαi/o, Gαq/11, and Gα12/13, that are responsible for Ca2+ mobilization, adenylyl cyclase inhibition and activation of phospholipase C, Akt, Rho and mitogen-activated protein kinase pathways.
The targeted disruption of Lpar1 in mice revealed unanticipated in vivo functions of this receptor. Lpar1−/− mice show 50% perinatal lethality and survivors have a reduced body size, craniofacial dysmorphism, and increased apoptosis in sciatic nerve Schwann cells.18
Other LPA receptors
Together with the LPA1 receptor, LPA2 and LPA3 have been the most thoroughly studied ones. The expression pattern of the LPA2 receptor is more spatiotemporally restricted compared to the LPA1 receptor. Lpar2 is found in the embryonic brain, but its expression strongly attenuates one week after birth. In humans, LPAR2 is found in the testis, leukocytes, prostate, spleen, thymus and pancreas.32 LPA2 null mice were born normally and showed no obvious behavioural, anatomical or histological abnormalities, in contrast to LPA1 null mice.35 When LPA1/LPA2 double-null mice were generated, aggravation of the phenotypic abnormalities was expected, as LPA2 is coexpressed with LPA1 in several organs and cells and thus a major loss of LPA signalling would be achieved. However, no qualitative differences in phenotypes, compared to LPA1 null mice, were observed. Thus, LPA1 and LPA2 receptors may have redundant functions in LPA signalling.LPAR3 encodes an ∼40 kDa GPCR broadly expressed in humans. This receptor shows a strong preference for unsaturated chains and has a relatively high affinity for 2-acyl-LPA containing unsaturated fatty acids. Despite the fact that LPA3 is expressed in the frontal cortex, hippocampus, and amygdala, no phenotypes related to LPA3 loss in the nervous system have been reported to date.32
The LPA4 receptor is structurally distinct from classical LPA1–3 and S1P receptors that share significant homology, and is more closely related to P2Y purinergic receptors. It does not, however, respond to any nucleotide or nucleoside tested. LPA4 is ubiquitously expressed in both humans and mice and it is specifically abundant in the ovary. LPA5 (GPR92) was identified by two independent groups in 2005 from the receptor gene data bank. This receptor is structurally different to LPA1–3, but shares 35% homology with LPA4. Lpar5 is relatively broadly expressed in murine and human tissues.32
The orphan receptor P2Y5, closely related to the purinergic family and sharing high homology with LPA4 receptor, has been recently classified as the LPA6 receptor.31 This receptor has been found to be essential for the maintenance of hair growth.36
Recently other receptors have been proposed. GPR87 and P2Y10 are orphan GPCRs that have been described to be responsive either to LPA or to both LPA and S1P, respectively. They belong to the P2Y family and are similar to LPA4 and LPA5 receptors.37
Among all LPA actions, those elicited through LPA1–3 receptors have been the most studied to date, revealing crucial roles in the nervous, vascular, immune and reproductive systems. Focusing on the CNS, LPA1 is described as the receptor with a major expression and, even though, the information is very scarce, there is evidence enough to suggest that it can contribute to the pathogenesis of several diseases and, accordingly, could be endowed with therapeutic relevance for the treatment of CNS disorders.
Lysophosphatidic acid receptor structure
Currently, no crystal structures have been elucidated for any native LPA receptor. The only phospholipid GPCR crystal structure available is the structure of S1P1,38 which has provided valuable information about the receptor–ligand interaction of this type of receptor.39 This is especially useful for molecular modelling of LPA receptors, because they share much higher sequence homology with this receptor than with any of the other currently available GPCR crystal structures, a fact that will enable the construction of homology models of LPA1–3 receptors using the structure of the S1P1 receptor as the template.40Mutagenesis studies combined with computational analysis identified several important residues in LPA1–3 receptors, most of them located in transmembrane domains.41,42 In this regard, Arg3.28 is important for efficacy and potency for all three receptors, as it forms a salt bridge with the phosphate group while Gln3.29 interacts with the hydroxy group of LPA. Thus, this latter position is responsible for LPA/S1P selectivity, as S1P receptors bear glutamic acid instead. These two residues are conserved over the LPA1–3 receptors, together with Trp4.64, which, in contrast, is only implicated in LPA3 activation. Other amino acids important for ligand recognition and selectivity among the LPA1–3 and S1P receptors are found in positions 5.38 (Asp in LPA1) and 7.36 (Lys in LPA1), though their function is still not clear. LPA4 and LPA5 share less amino acid identity with LPA1–3, and detailed models of their interaction with LPA are not available. Most of the residues described above are not present in LPA4 and LPA5, suggesting that these receptors have different ligand binding characteristics. Further research is needed to identify the critical residues for these receptors, and this information will need to be re-evaluated once crystal structure data become available.43
Biosynthesis and degradation of LPA
LPA is generally known as a mixture of various lysophospholipids with both saturated (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 18:0) and unsaturated (16:1, 18:1, 18:2, 20:4) fatty acid chains. It must be noted that in the context of LPA as a signalling molecule, and thus throughout this review, LPA refers to 1-oleoyl-sn-glycero-3-phosphate. It is found in almost all eukaryotic tissues and biological fluids, including blood.32 Among them, serum is the best characterized source of LPA, where it is bound to albumin and other proteins, probably preventing the molecule from rapid degradation.44
:0, 18:0) and unsaturated (16:1, 18:1, 18:2, 20:4) fatty acid chains. It must be noted that in the context of LPA as a signalling molecule, and thus throughout this review, LPA refers to 1-oleoyl-sn-glycero-3-phosphate. It is found in almost all eukaryotic tissues and biological fluids, including blood.32 Among them, serum is the best characterized source of LPA, where it is bound to albumin and other proteins, probably preventing the molecule from rapid degradation.44
Autotaxin (ATX),45,46 a secreted glycoprotein with lysophospholipase D activity, is the primary enzyme responsible for LPA production in blood. In fact, ATX heterozygote knockout mice have a 50% reduction of circulating LPA compared to wild type mice47 and negligible levels of LPA are detected after treatment with ATX inhibitors.48 Outside the cell, the enzyme ATX converts lysophosphatidylcholine (LPC), produced from different membrane phospholipids via phospholipase A2 (PLA2), into LPA (Fig. 3).
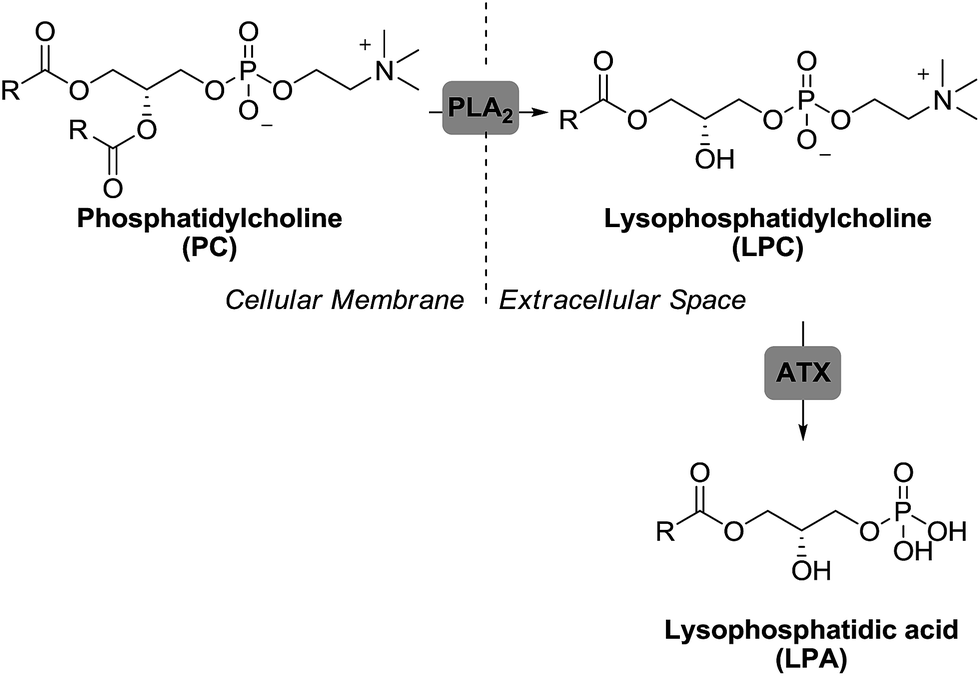 | ||
| Fig. 3 Pathway for LPA production. | ||
Degradation of LPA can occur through two main routes. In the first one, LPA is irreversibly dephosphorylated to monacylglycerol by lipid phosphate phosphohydrolases, presumably LPP1. In the second route, LPA is reversibly esterified to phosphatidic acid (PA) by the enzyme LPA-acyltransferase (LPAAT) (Fig. 4).49
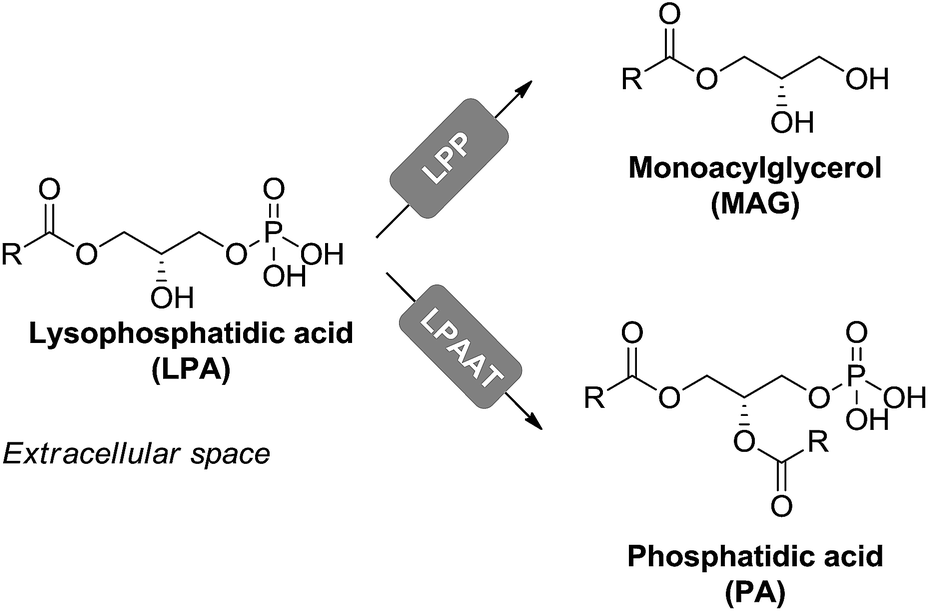 | ||
| Fig. 4 LPA degradation pathways. | ||
Physiological roles of the LPA1 receptor and therapeutic potential
LPA displays a wide range of cellular effects through its receptors. Among the most important actions of LPA, those mediated by the LPA1 receptor in the CNS stand out, a fact that immediately suggests a potential for the treatment of related diseases.Nervous system
As highlighted before, the nervous system is one of the major locations for LPA receptors,34 as they are expressed in most of its cell types under physiological and pathological conditions. In addition, LPA can be found in the brain in high concentrations, influencing many developmental processes and neurological disorders.Among the different LPA receptors, the LPA1 subtype is the most abundant one in the brain,32 where it plays a major role in the development of the embryonic brain and thus in neurogenesis, due to its main expression in the VZ of the embryonic brain. Neural progenitor cells (NPCs) – the differentiable cells responsible for neurogenesis – express LPA1–3 receptors and are found in this area, where they proliferate and sequentially differentiate into various cell types, such as neurons, astrocytes or oligodendrocytes.
Several in vitro and in vivo studies have demonstrated that LPA controls proliferation and differentiation of NPCs via LPA1. Furthermore, LPA1 null NPCs do not present the ability to achieve LPA-dependent neurogenesis-related changes, confirming LPA1 as a modulator of neurogenesis. In adult neurons, LPA is known to influence neuronal survival/death processes. Moreover, it has been described that LPA1 is related to neuroprotection, as apoptotic cell death is described in LPA1 null mouse brains.34
Neuropsychiatric disorders, like schizophrenia, anxiety, memory impairment or Alzheimer's disease, have been recently linked to LPA signalling. LPA1 null mutants share schizophrenia-type defects, such as pre-pulse inhibition, serotonin synthesis alteration or cranial dysmorphism. In addition, LPA signalling through LPA1 in the hippocampus modulates neurogenesis, which is related to learning and emotional behaviour; and memory impairments have been reported in LPA1 deficient mice.34
Two important developmental disorders linked to the LPA1 receptor are: fetal hypoxia and fetal hydrocephalus. It was shown that mouse brains exposed to LPA develop fetal hydrocephalus, and that treatment with an LPA1 antagonist blocked this response, demonstrating the implication of the receptor.50 Regarding fetal hypoxia, it has been described that the absence of the adequate supply of oxygen causes cortical disorganization throughout NPCs via over-activation of the LPA1 receptor.51 Since both diseases are associated with later development of CNS disorders, such as epilepsy, schizophrenia or autism, it is clear that LPA1 signalling needs to be tightly regulated to ensure unaltered brain functions.
LPA1 has also been associated with myelination because its expression in oligodendrocytes (CNS myelinating cells) correlates spatiotemporally with their maturation and myelination, and it has been shown that LPA influences several of its cellular responses. Moreover, a recent study has shown that LPA, acting through the LPA1 receptor, promotes Schwann cell migration, which precedes myelination and remyelination in the peripheral nervous system.52
It has been suggested that LPA plays a key role in the initiation of neuropathic pain, a form of chronic pain which accounts for almost 20% of its diagnosed cases in the U.S.A. Neuropathic pain is the result of a combination of multiple factors, but a direct link with fiber demyelination has been reported.53 LPA produces nerve injury via LPA1-mediated demyelination with subsequent loss of the structural and functional integrity of neurons. In further support of this, LPA1 deficient mice do not show neuropathic pain behaviour or demyelination in response to intrathecal LPA injection or nerve injury. LPA5 null mice are also protected from developing neurophatic pain, although the mechanisms involved are different from those mediated by LPA1.54
Peripheral roles of the LPA1 receptor
The main peripheral roles of LPA characterized so far are related to the ability of this molecule to influence cellular proliferation and differentiation in several tissues and systems. In this regard, LPA performs an important role in the vascular system, where it modulates different effects in vascular smooth muscle cells (VSMCs) and vascular endothelial cells (VECs), which are involved in processes like angiogenesis (the formation of new capillary networks from pre-existing vasculature by sprouting and/or splitting of capillaries) or vascular maturation. Angiogenesis involves coordinated proliferation, migration, adhesion, differentiation, and assembly of both VECs and their surrounding VSMCs, and its dysregulation can lead to diverse pathological conditions, such as atherosclerosis,55 cardiovascular disease, or development of tumours.Similarly, and related to the ability of LPA to promote cell proliferation, the LPA1 receptor is gaining attention as a druggable target for fibrosis.56,57 This disease involves the formation of excessive connective tissue, and it has been found to be strongly influenced by receptor-mediated LPA signalling in lung, kidney and skin. Hence, increased epithelial cell apoptosis, migration and proliferation of lung fibroblasts, together with enhanced fibroblast resistance to apoptosis are LPA1-mediated processes directly linked to the development of pulmonary, dermal and kidney fibrosis. In addition, the results obtained with a dual LPA1/LPA3 antagonist suggest a possible implication of the LPA3 receptor. Supporting these data, one LPA1 antagonist has entered phase II clinical trials for idiopathic pulmonary fibrosis (IPF)58 and another one is in preclinical stages, indicated for the treatment of liver, lung and kidney fibrosis.59
Recent research has also associated the LPA1 receptor with the initiation and development of rheumatoid arthritis (RA). It is known that synovial fibroblasts (SFs), implicated in the beginning and perpetuation of RA, express all LPA receptors and that LPA stimulates proliferation, adhesion and migration of SFs. Accordingly, the LPA1 receptor has been suggested as a possible therapeutic target in the treatment of this disease.60,61
LPA1 is also the most widely expressed lysophospholipid receptor in adipose tissue, a fact that makes it an interesting pharmacological target for the treatment of obesity-associated metabolic diseases. Obesity, one of the key factors leading to type II diabetes, is accompanied by an increased ATX-mediated synthesis of LPA by adipocytes, where LPA exerts different biological actions through the activation of the LPA1 receptor.62
Finally, it is well known that LPA signalling influences cancer-related processes,63 especially via LPA2. Nevertheless, there is also evidence of LPA1 implication in cancer progression, specifically in ovarian, breast and gastrointestinal ones.
LPA1 receptor ligands
Given the importance of the LPA1 receptor in a variety of pathologies, the need for potent and selective ligands is crucial to unravel its potential as a therapeutic target, but up to this moment there are no drugs in the market targeting any of the LPA receptors.Although much research is ongoing in this field,64 the lack of potent and selective ligands is still an issue. Lipid-resembling molecules encounter solubility problems and show very high protein binding with only a small percentage within plasma available to interact with receptors. Moreover, the abundant cell surface lipid phosphate phosphohydrolases may rapidly degrade them. Regarding non-lipid structures, some advances have been done in the field of antagonists, as two of them have currently reached clinical trials.58,65,66 Still, small-molecule agonists structurally different from LPA have not been described yet.
Agonists of the LPA1 receptor
Detailed studies have been carried out on the search for the essential patterns required to obtain selective agonism at the LPA1 receptor.40 The information available so far comes from LPA analogues, as no structurally different synthetic agonists have been described yet.The first LPA-based agonist was N-acyl ethanolamide phosphoric acid (2-[(9Z)-octadec-9-enoylamino]ethyl dihydrogen phosphate or NAEPA, 1) described by Sugiura in 1994 (ref. 67) as an LPA mimetic and later confirmed as a dual LPA1/LPA2 agonist.68 Several changes in its structure have led to ligands with improved activity and, in some cases, selectivity over the LPA1 receptor. Initial modifications included the introduction of different substituents in the β-carbon atom (Fig. 5, left panel) and revealed a strong enantiomer preference, as well as a decrease in agonist potency when bulky substituents were introduced. Among all the synthesized compounds, 2–4 stand out as potent dual LPA1/LPA3 agonists, with a stronger preference for LPA1 and activity values similar to LPA [EC50 (LPA1) = 7.9, 4.9 and 3.4 nM; EC50 (LPA3) = 321.8, 683.7 and 112.6 nM, respectively].69
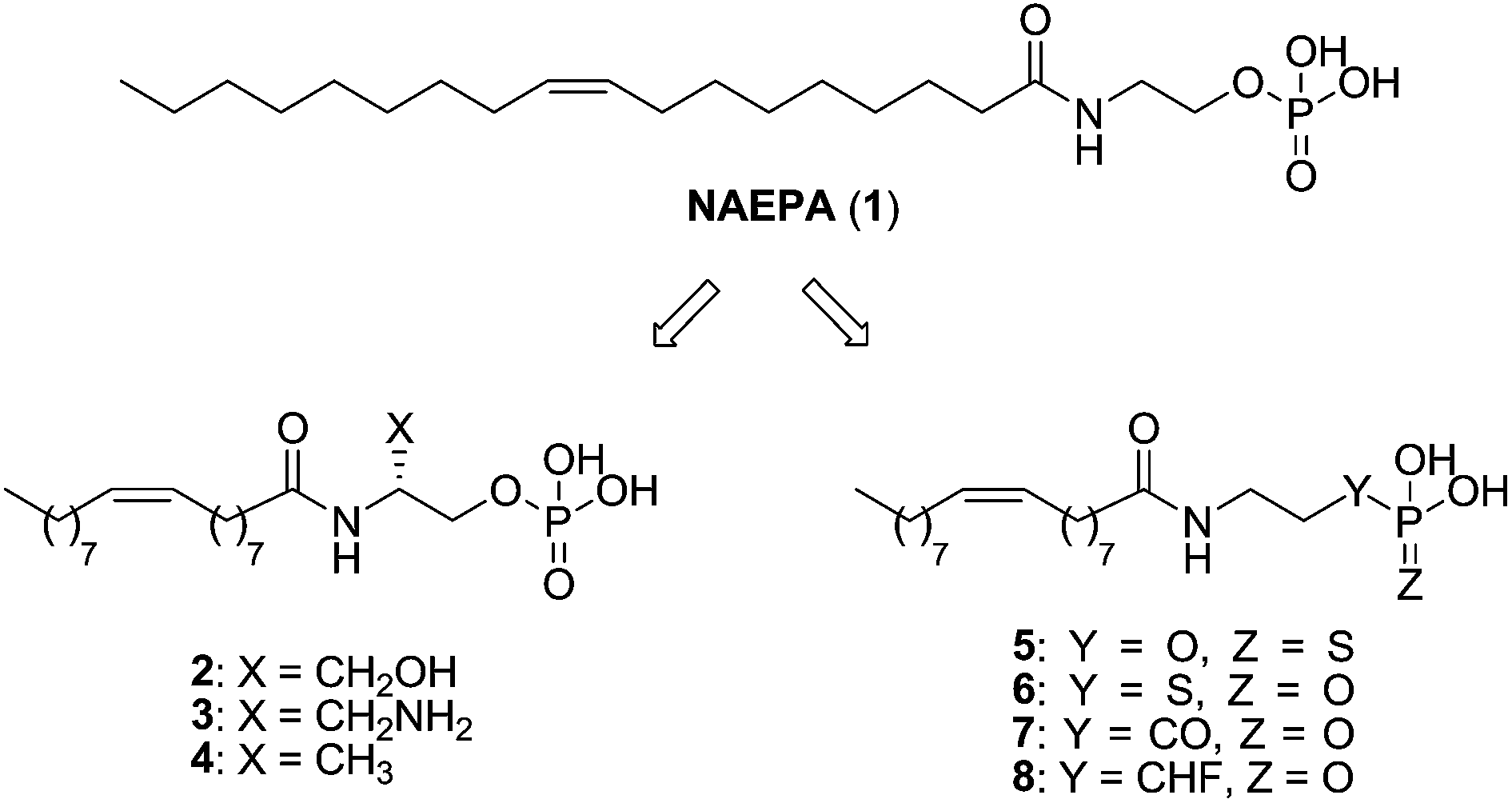 | ||
| Fig. 5 Structure of NAEPA and derivatives. | ||
Further replacements of the phosphate group by its mimetics thiophosphate (Y = S, Z = O, Fig. 5), and the metabolically stabilized phosphorothioate (Y = O, Z = S, Fig. 5) and phosphonate groups (Y = CO, CHF, Z = O, Fig. 5) were carried out. These groups, especially phosphonates, had higher pKa values than LPA, so α-substituted phosphonates with electronegative groups at the α-carbon were also prepared in order to maintain acidity (Fig. 5, right panel). Among them, selective compounds 6 [EC50 (LPA1) = 318 nM] and 7 [EC50 (LPA1) = 221 nM] exhibit an activity similar to NAEPA at the LPA1 receptor (EC50 = 197 nM), and compound 5 [EC50 (LPA1) = 40 nM; EC50 (LPA2) = 108 nM] improved it. It must be noted that analogue 8, bearing an α-fluorophosphonate moiety, more acid than compound 5, was inactive at the LPA1 receptor, indicating that acidity is not the only requirement for receptor activation when modifying the phosphate moiety.70 In fact, other LPA-derived phosphonates and analogues bearing fluoro or difluoro moieties in the α-carbon act as good LPA2 or LPA3 agonists, but are inactive at LPA1.71
The LPA analogue (2S)-2-methoxy-3-(thiophosphonooxy)propyl (9Z)-octadec-9-enoate or OMPT (9) was one of the first selective LPA3 agonists, with an EC50 value of 276 nM.72 Its modification led to diverse structures, such as the enantiomers 10 [EC50 (LPA1) = 790 nM; EC50 (LPA3) = 62 nM] and 11 [EC50 (LPA1) = 571 nM; EC50 (LPA3) = 80 nM], which turned out to be good LPA3 agonists but also present modest activity at LPA1 (Fig. 6).73 In order to prevent acyl chain migration, other metabolically stabilizing modifications were carried out, leading to phosphorothioate analogues of sn-2-acyl LPA (compounds 12–14, Fig. 6). These three compounds displayed weak LPA1 agonism, but they stand out as potent LPA3, LPA5 and LPA6 agonists.74
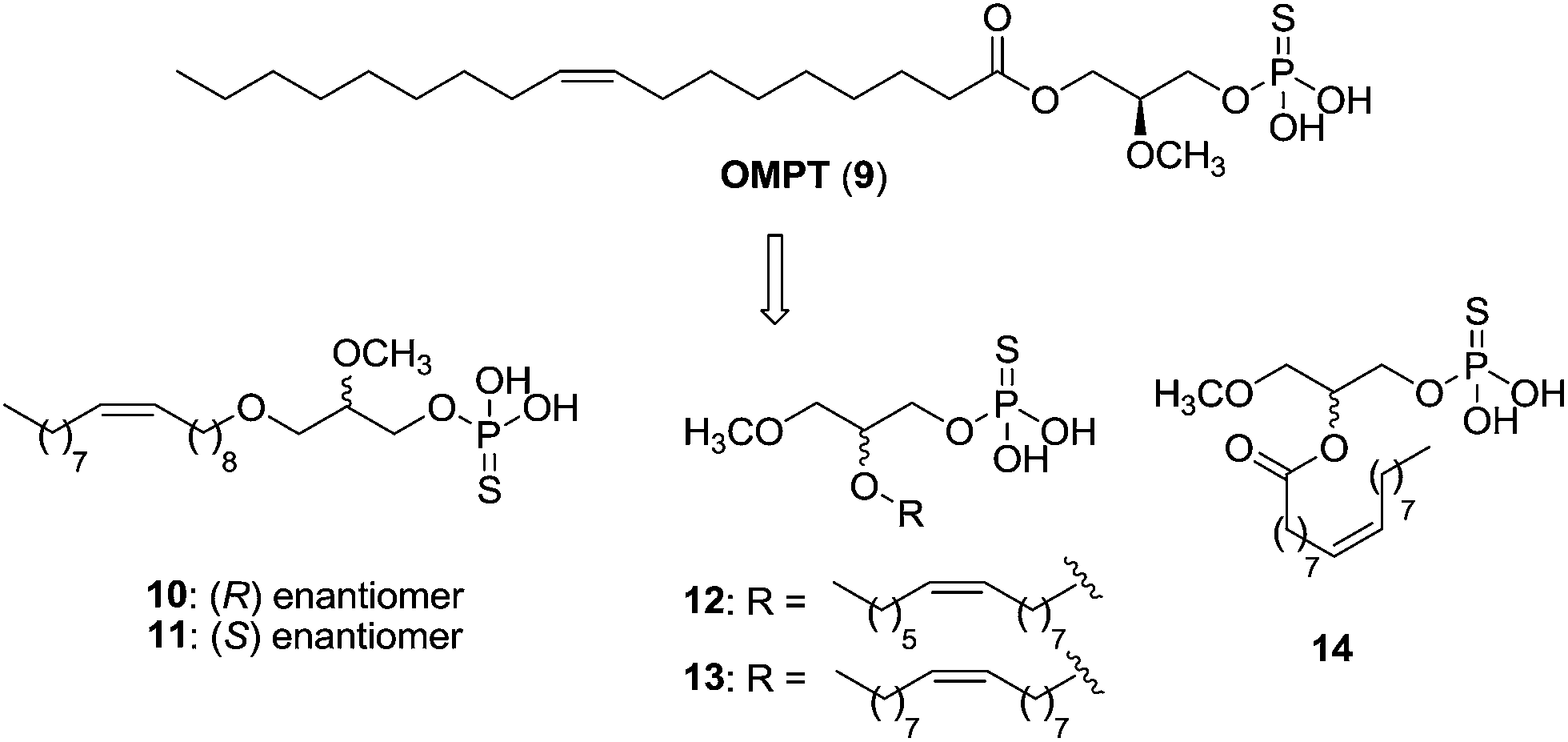 | ||
| Fig. 6 Structure of OMPT and derivatives. | ||
The influence of the position of the acyl chain has also been studied. For example, sn-2 LPA derivatives resistant to acyl migration such as 1,1-difluorinated phosphates,75 difluoromethyl phosphates76 or α-fluorinated phosphonates77 were synthesized. Unfortunately, none of these compounds was active at the LPA1 receptor,71 though LPA1 and LPA2 receptors were reported to show no regioisomeric preference between sn-1 and sn-2 positions.
Cyclic phosphate analogues have also been described as LPA1 agonists (Fig. 7). The cyclic difluorophosphate 15 was reported as a weak LPA1–3 agonist [EC50 (LPA1) > 1940 nM; EC50 (LPA2) > 9460 nM; EC50 (LPA3) > 7030 nM].78 In addition, some acetal phosphatidates, also known as Darmstoff analogues, have been reported as LPA mimetics. Some of these compounds are LPA pan-agonists (16–19), though with activity in the low micromolar range at the LPA1 receptor.79 Again, small structural modifications turn the compounds into antagonists.
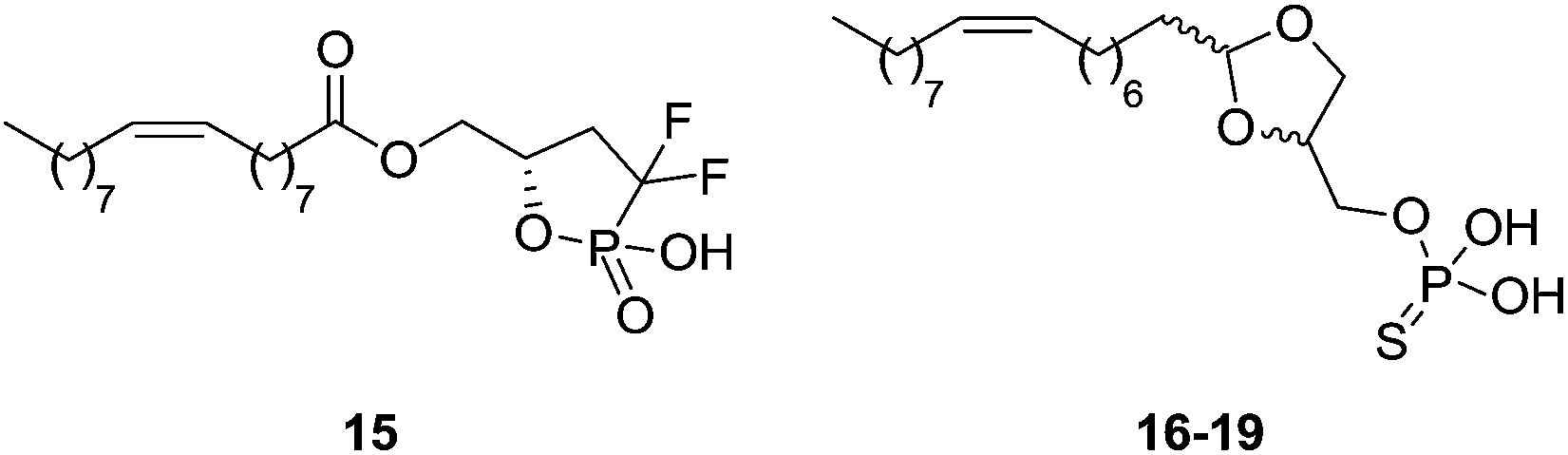 | ||
| Fig. 7 Structure of cyclic phosphate agonists. | ||
In summary, around 20 years after the discovery of the first LPA1 ligands, there is still a lack of potent and selective agonists. Nowadays, the knowledge about the features needed for the activity has been somehow disclosed, but despite that, the complete puzzle of the structural requirements for activating this receptor is not yet fully understood.
Antagonists of LPA1 receptor
The LPA1 antagonist field is a current focus of pharmaceutical companies. Structurally, LPA1 antagonists can be classified into two broad classes: a family closely related to LPA and a second group formed by compounds whose structures widely differ from LPA.Starting with LPA analogues, modification of the agonist NAEPA (1, Fig. 5) with a bulky substituent in the β-carbon atom led to compound 20, which turned out to be a dual LPA1/3 antagonist [IC50 (LPA1) = 5210 nM; IC50 (LPA3) = 6450 nM],69 which has been used in vivo in a model of lung fibrosis.80 An exhaustive SAR of this structure yielded compounds 21, a selective LPA1 ligand with moderate activity [IC50 (LPA1) = 2490 nM], and 22, which showed increased potency [IC50 (LPA1) = 109 nM; IC50 (LPA3) = 175 nM] and which is five times more active than its (S)-enantiomer.81 Further optimizations led to 23, a dual LPA1/3 antagonist with nanomolar potency [IC50 (LPA1) = 84 nM; IC50 (LPA3) = 48 nM] (Fig. 8).82
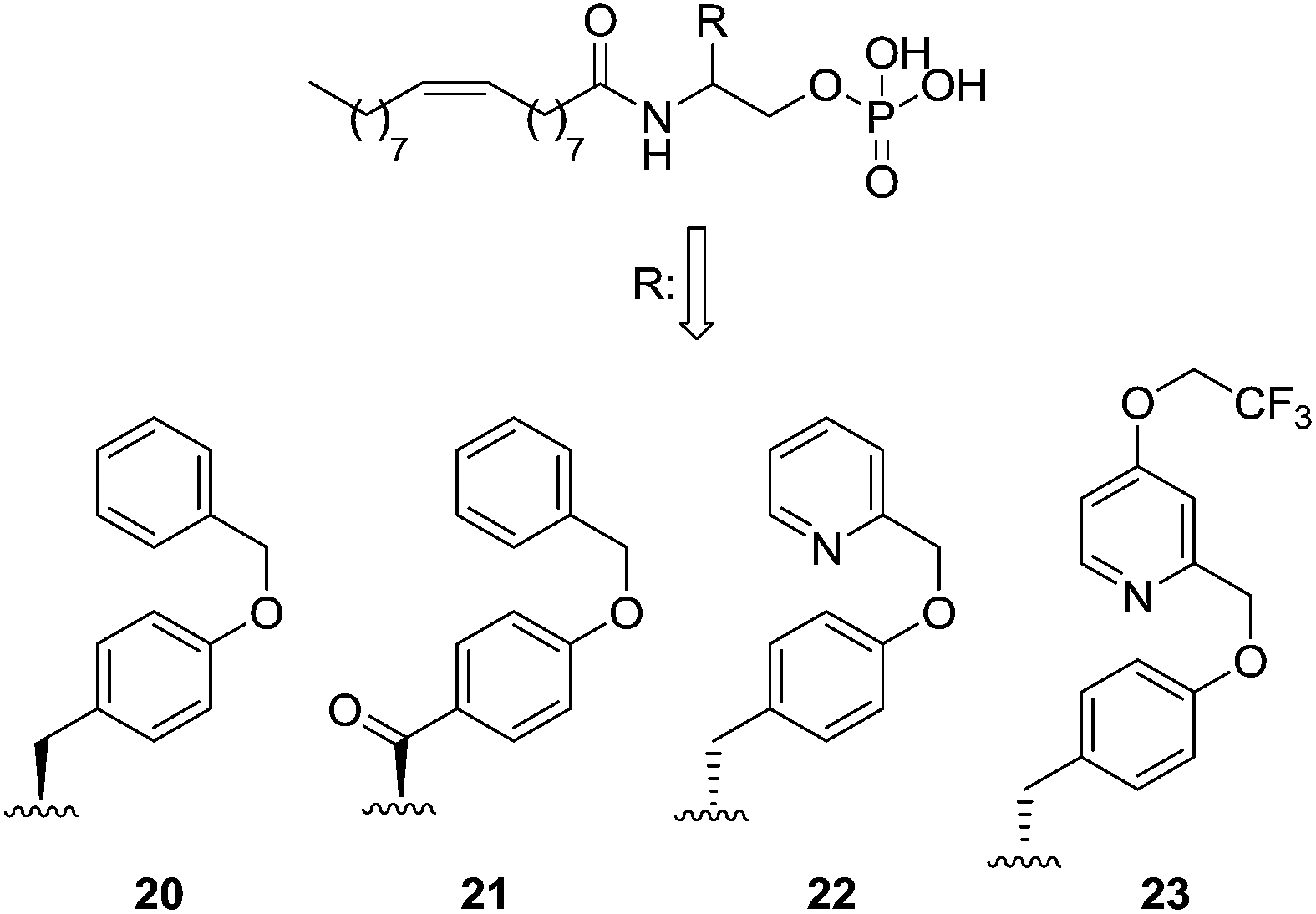 | ||
| Fig. 8 Structure of NAEPA-derived antagonists. | ||
Another important LPA analogue is bromophosphonate 24 (Fig. 9), also known as BrP-LPA, an LPA pan-antagonist [IC50 (LPA1) = 1500 nM; IC50 (LPA2) = 1420 nM; IC50 (LPA3) = 1160 nM; IC50 (LPA4) = 266 nM] and ATX inhibitor with in vivo activity.83 This molecule has contributed to elucidate the involvement of LPA receptors in the inhibition of tumour growth84 and in the attenuation of arthritis in animal models.85
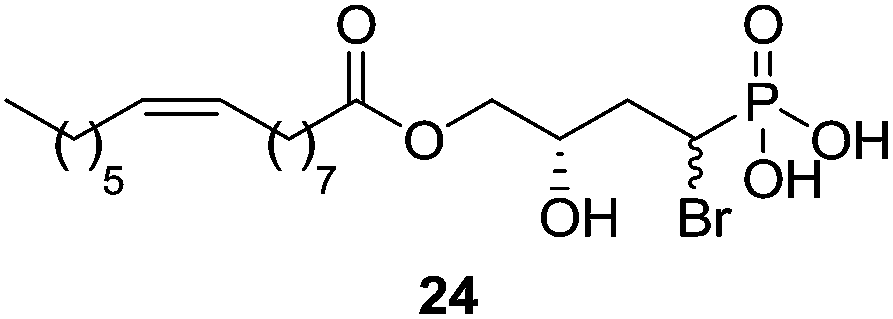 | ||
| Fig. 9 Structure of the pan-antagonist bromophosphonate BrP-LPA (24). | ||
Cyclic LPA derivatives have also been described as LPA1 antagonists (Fig. 10). Compound 25 and cyclic phosphorothioates 26 and 27 show activity as partial LPA1/LPA3 antagonists with moderate potencies [IC50 (LPA1) = 106–941 nM; IC50 (LPA3) = 1270–7720 nM].78
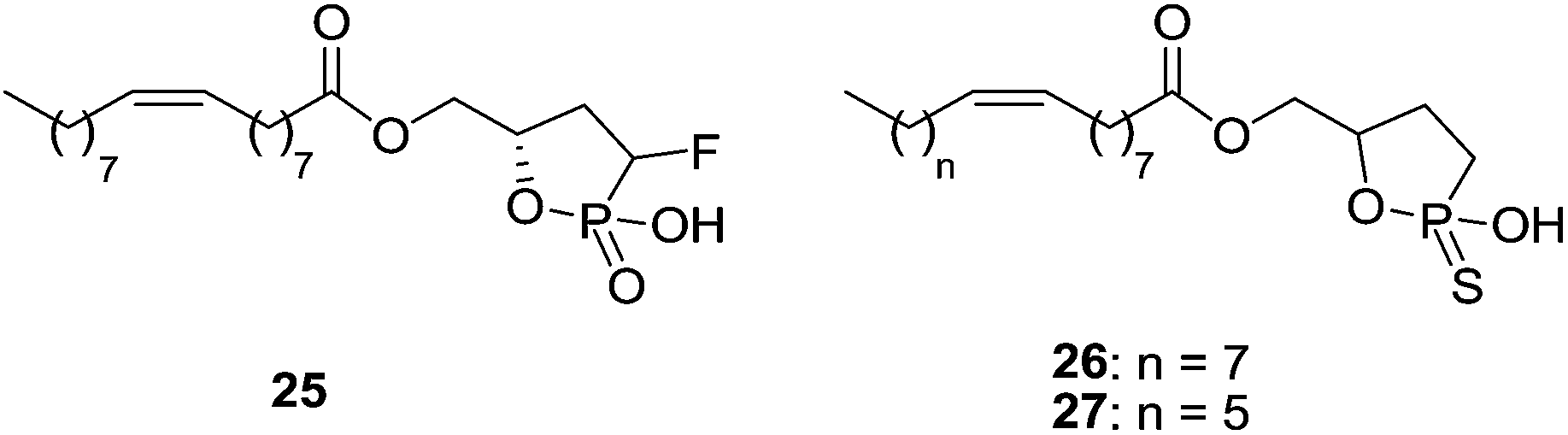 | ||
| Fig. 10 Structure of cyclic phosphate antagonists. | ||
Overall, these series of compounds show the difficulty of discovering the requirements needed to regulate the pharmacology of LPA-derived ligands, as subtle changes in their structures convert agonists into antagonists and cause drastic changes in activity. In addition, the coexistence of a polar head and a long hydrophobic tail becomes a problem in order to obtain orally active compounds. Thus, high-throughput screening was used to discover novel hits, structurally different from LPA, followed by hit to lead processes to improve their pharmacology.
The first reported non-lipid dual LPA1/3 antagonist was compound 28 (Ki16425, Fig. 11)86 [IC50 (LPA1) = 130 nM; IC50 (LPA3) = 2300 nM] which has been widely used as a tool compound, as it displays in vivo activity.50,61 Several modifications of its structure by different academic groups and pharmaceutical companies have led to more potent and selective compounds, some of them even achieving clinical trials.
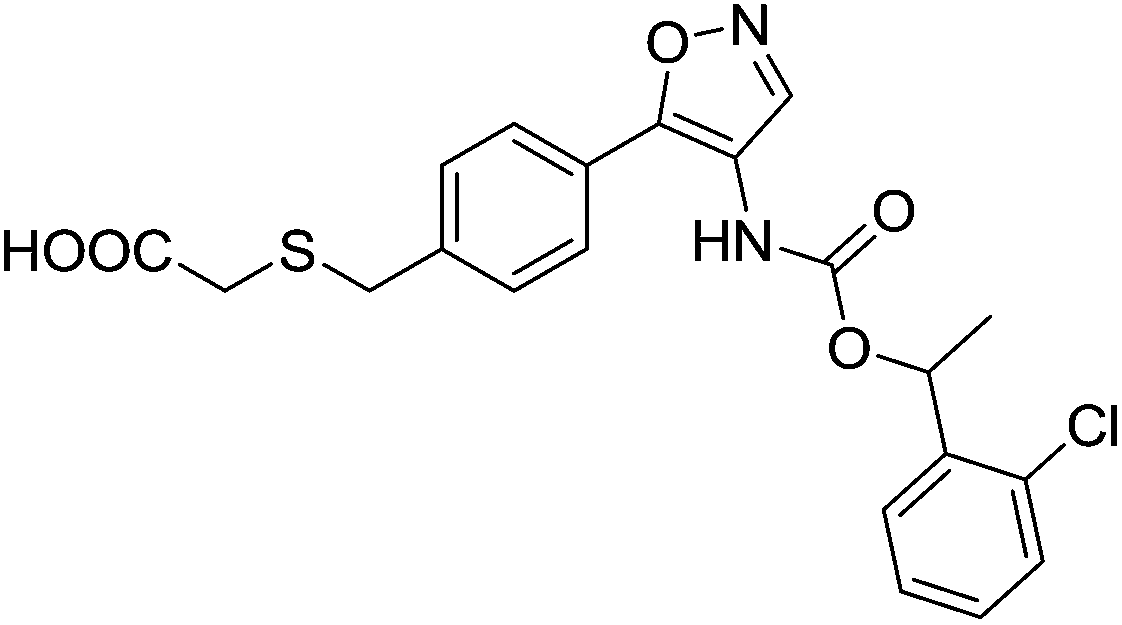 | ||
| Fig. 11 Structure of the dual LPA1/3 antagonist Ki16425 (28). | ||
Based on this scaffold, Amira Pharmaceuticals (currently Bristol-Myers Squibb) developed a series of isoxazole derivatives. Among them, compounds 29 (AM966),87 [IC50 (LPA1) = 17 nM; IC50 (LPA2) = 1700 nM; IC50 (LPA3) = 1600 nM] and 30 (AM095)88,89 [IC50 (LPA1) = 25 nM; IC50 (LPA2–5) > 8000 nM] (Fig. 12) stand out as potent LPA1 antagonists with good oral bioavailability and antifibrotic in vivo activity. Moreover, a compound coming from this series, BMS-986020, whose structure has not been disclosed yet, is currently facing phase II trials for the treatment of IPF.58
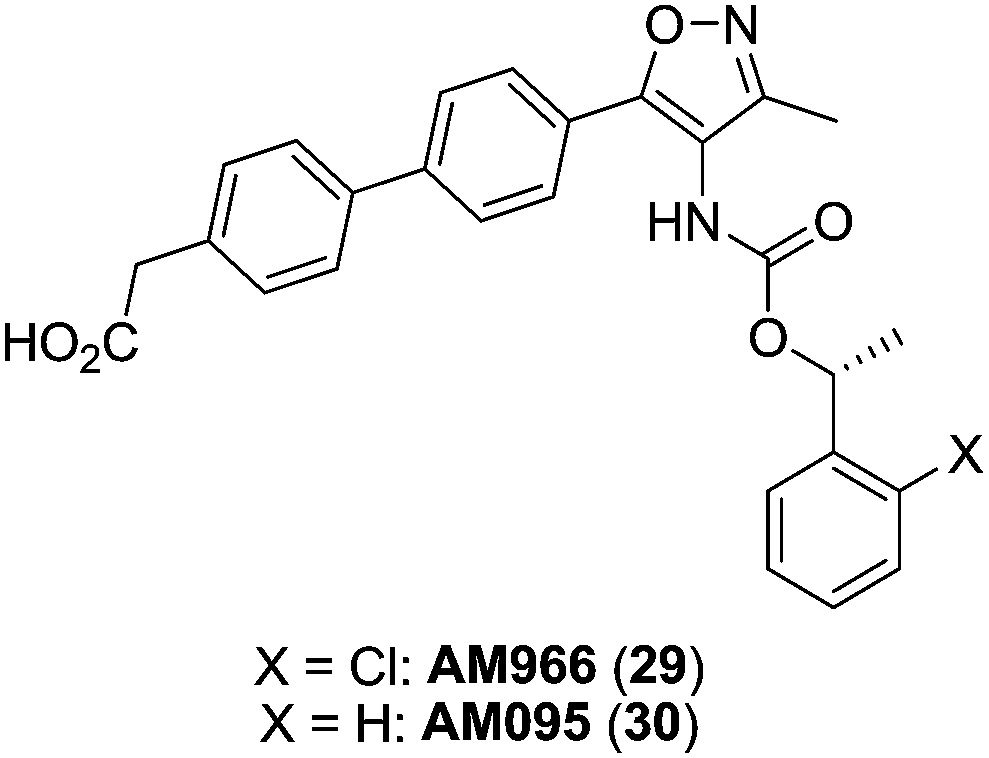 | ||
| Fig. 12 Isoxazole LPA1 receptor antagonists developed by Amira Pharmaceuticals. | ||
Hoffman-La Roche's modifications of Ki16425 involved changes in the carboxylic acid and the heterocyclic core, replacing the isoxazole moiety with pyrazole and triazole rings. The best compounds were 31, with low nanomolar activity and good selectivity values [IC50 (LPA1) = 25 nM; IC50 (LPA3) > 30000 nM], and 32, a dual LPA1/LPA3 antagonist [IC50 (LPA1) = 24 nM; IC50 (LPA3) = 65 nM] (Fig. 13).90
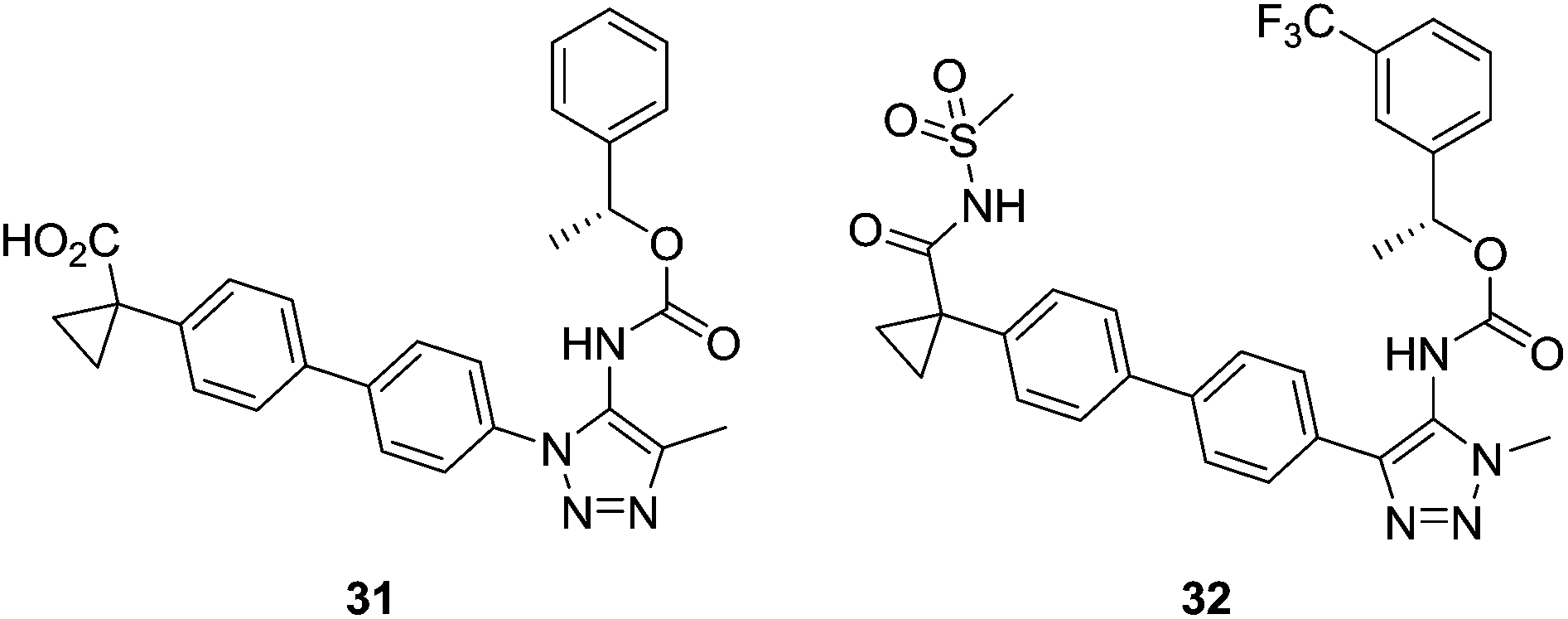 | ||
| Fig. 13 Triazole LPA1 receptor antagonists developed by Hoffman-La Roche. | ||
Further exploration of the central heterocycle ring by other pharmaceutical companies has led to potent antagonists of the LPA1 receptor, with IC50 values in the low nanomolar range, such as compound 33, which is a selective LPA1 receptor antagonist [IC50 (LPA1) < 50 nM; IC50 (LPA3) > 500 nM].91 Introduction of different sulfonamide groups led to LPA1 antagonists with activities in the low nanomolar scale.92,93 For example, compound 34 (Fig. 14) is an LPA1 antagonist with an IC50 value of 6.6 nM.
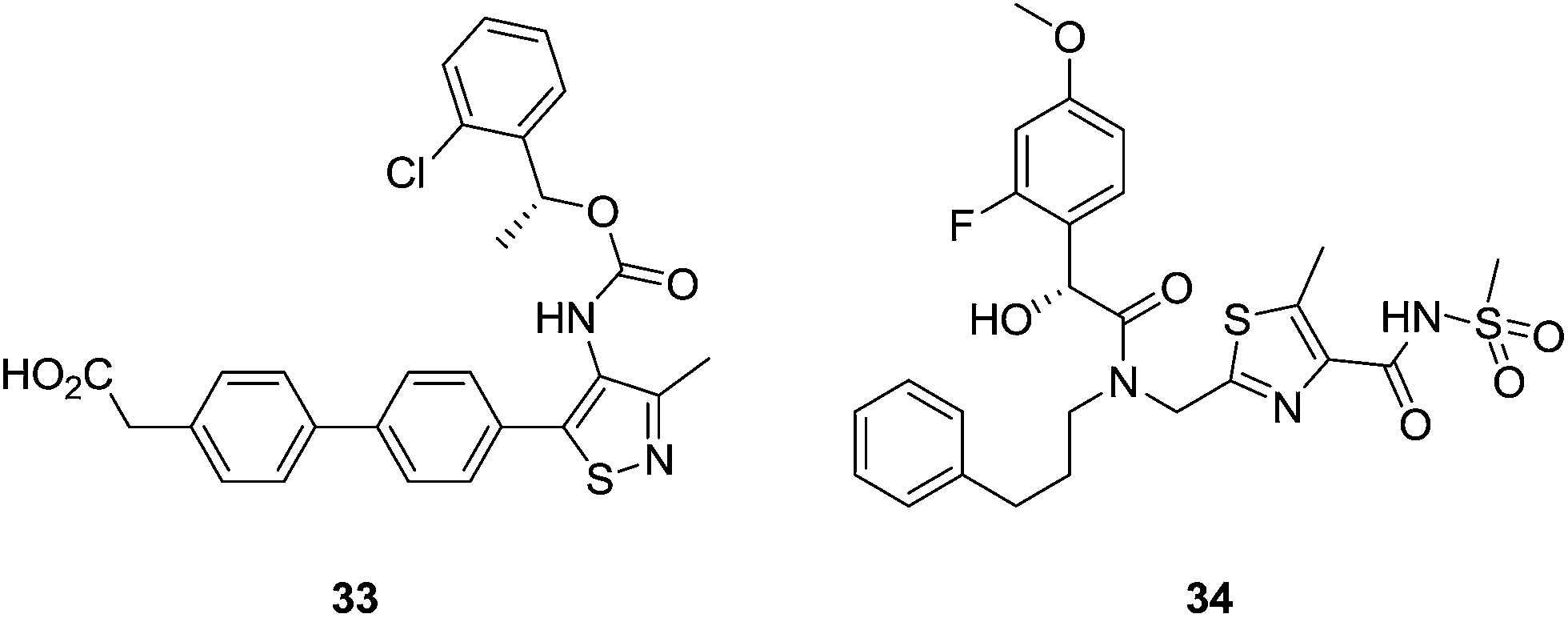 | ||
| Fig. 14 Thiazole LPA1 receptor antagonists. | ||
Initially inspired by Ki16425, Sanofi-Aventis synthesized a series of non-natural amino acids, such as compound 35 (Fig. 15), with IC50 values lower than 100 nM. It must be highlighted that SAR100842 (structure not yet disclosed) is an LPA1/LPA3 antagonist from this set of compounds, which has completed phase II clinical trials for systemic sclerosis.66
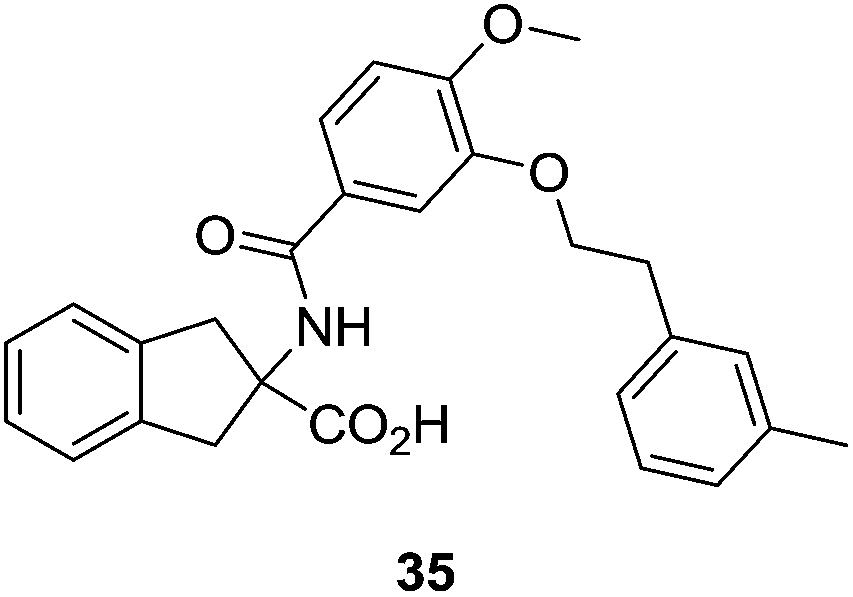 | ||
| Fig. 15 Sanofi-Aventis antagonist. | ||
In conclusion, it is clear that the LPA1 receptor has an outstanding but yet intriguing role under physiological and pathological conditions. Thus, the discovery of potent and selective agonists and antagonists is nowadays a crucial need to achieve the validation of this receptor as a therapeutic target.
LPA1 receptor ligands under clinical development
The implication of LPA in multiple diseases has attracted research interest from both academia and pharmaceutical companies in order to validate the LPA pathway as a source of novel druggable targets. Focusing on the LPA1 receptor, preclinical results obtained from LPA1 knockout mice and the use of specific antagonists in animal disease models have demonstrated the key role of this receptor in mediating the pro-fibrotic effects of LPA in fibroblasts. Therefore, the challenge remains to prove that LPA1 antagonists could be developed as effective therapeutics for the treatment of fibrotic disorders, such as IPF, hepatic fibrosis and systemic sclerosis. To date, two LPA1 antagonists, BMS-986020 and SAR100842, have advanced into clinical investigation for the treatment of IPF58 and systemic sclerosis.66 Furthermore, an orally bioactive small-molecule LPA1 antagonist developed by Angion is at a preclinical stage for the treatment of liver, lung and kidney fibrosis.59BMS-986020, initially developed by Amira and then by Bristol-Myers Squibb, has recently completed a phase I study to assess the pharmacokinetics, metabolism and excretion, as well as safety and tolerability of a single oral dose. Moreover, a single sequence study has also been conducted to evaluate the effect of concomitant administration of BMS-986020 on the pharmacokinetics of rosuvastatin. Currently, two new clinical trials are recruiting participants: a phase I study to evaluate the relationship between plasma drug levels and receptor binding in the lungs using positron emission tomography (PET); and a phase II trial to determine if once or twice daily administration of 600 mg of BMS-986020 will reduce the decline in the forced vital capacity and will be tolerated well in subjects with IPF. Additionally, a phase I study to assess the drug–drug interaction in healthy volunteers is announced to start in September 2014. This study will determine the effect of BMS-986020 on the pharmacokinetics of montelukast, flurbiprofen, and digoxin.65
Sanofi-Aventis has conducted a phase II trial with the dual LPA1/LPA3 antagonist SAR100842 to evaluate its safety and tolerability in an 8 week study in patients with diffuse, cutaneous systemic sclerosis.66 This clinical study was completed in April 2014 and the results have not been reported yet.
In summary, the progression of LPA1 antagonists into clinical trials will hopefully help to ascertain the therapeutic utility of the LPA1 receptor as a novel target for the treatment of disorders with high unmet medical need such as IPF.
Outlook and future perspectives
Lipid-binding GPCRs are potential drug targets for many diseases including neuropshychiatric and neurodegenerative disorders, multiple sclerosis, pain, inflammation-related diseases, and cancer. In particular the LPA1 receptor plays fundamental roles in both the central and the peripheral nervous system. However, the paucity of currently available potent and selective (ant)agonists for the different LPA receptors is hampering the validation of this receptor as a therapeutically useful target. In this regard, some advances have been made in terms of the development of antagonists, some of which are currently undergoing clinical trials. However, the field of agonists is still clearly lagging behind as not really potent and selective agents structurally different from LPA have been disclosed. In addition, it is likely that the progress in structural determination of GPCRs will extend also to LPA receptors, and structures of these receptors in complex with different ligands can be elucidated in the upcoming future. These advances should also consider the importance of biased and allosteric ligands, since they can help to unravel the biology behind these receptors and to provide new therapeutic solutions for important diseases that lack adequate clinical treatments today.Acknowledgements
This work was supported by grants from MINECO (SAF2010-22198-C02, SAF2013-48271-C2) and Comunidad de Madrid (S2011/BMD2353). I. G.-G. and D. Z. are grateful to MINECO for predoctoral fellowships.References
- H. A. Brown and L. J. Marnett, Chem. Rev., 2011, 111, 5817–5820 CrossRef CAS PubMed.
- P. M. Musille, J. A. Kohn and E. A. Ortlund, FEBS Lett., 2013, 587, 1238–1246 CrossRef CAS PubMed.
- S. A. Scott, T. P. Mathews, P. T. Ivanova, C. W. Lindsley and H. A. Brown, Biochim. Biophys. Acta Mol. Cell Biol. Lipids, 2014, 1841, 1060–1084 CrossRef CAS PubMed.
- (a) T. Mutoh, R. Rivera and J. Chun, Br. J. Pharmacol., 2012, 165, 829–844 CrossRef CAS PubMed; (b) Lysophospholipid Receptors, ed. J. Chun, T. Hla, W. Moolenaar and S. Spiegel, John Wiley & Sons, Inc., Hoboken, New Jersey, 2013 Search PubMed.
- A. Grzelczyk and E. Gendaszewska-Darmach, Biochimie, 2013, 95, 667–679 CrossRef CAS PubMed.
- W. Vogt, Biochem. Pharmacol., 1963, 12, 415–420 CrossRef CAS.
- E. J. van Corven, A. Groenink, K. Jalink, T. Eichholtz and W. H. Moolenaar, Cell, 1989, 59, 45–54 CrossRef CAS.
- D. Dyer, G. Tigyi and R. Miledi, Mol. Brain Res., 1992, 14, 293–301 CrossRef CAS.
- K. Jalink and W. H. Moolenaar, J. Cell Biol., 1992, 118, 411–419 CrossRef CAS.
- A. J. Ridley and A. Hall, Cell, 1992, 70, 389–399 CrossRef CAS.
- S. Yamamura, Y. Yatomi, F. Ruan, E. A. Sweeney, S. Hakomori and Y. Igarashi, Biochemistry, 1997, 36, 10751–10759 CrossRef CAS PubMed.
- R. L. van der Bend, J. Brunner, K. Jalink, E. J. van Corven, W. H. Moolenaar and W. J. van Blitterswijk, EMBO J., 1992, 11, 2495–2501 CAS.
- F. J. Thomson, L. Perkins, D. Ahern and M. Clark, Mol. Pharmacol., 1994, 45, 718–723 CAS.
- J. H. Hecht, J. A. Weiner and J. Chun, J. Cell Biol., 1996, 83, 1071–1083 CrossRef.
- S. An, M. A. Dickens, T. Bleu, O. G. Hallmark and E. J. Goetzl, Biochem. Biophys. Res. Commun., 1997, 231, 619–622 CrossRef CAS PubMed.
- J. R. Erickson, J. J. Wu, J. G. Goddard, G. Tigyi, K. Kawanishi, L. D. Tomei and M. C. Kiefer, J. Biol. Chem., 1998, 273, 1506–1510 CrossRef CAS PubMed.
- N. Fukushima, Y. Kimura and J. Chun, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6151–6156 CrossRef CAS.
- J. J. A. Contos, N. Fukushima, J. A. Weiner, D. Kaushal and J. Chun, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13384–13389 CrossRef CAS PubMed.
- G. C. Zondag, F. R. Postma, I. V. Etten, I. Verlaan and W. H. Moolenaar, Biochem. J., 1998, 330, 605–609 CAS.
- M. J. Lee, J. R. Van Brocklyn, S. Thangada, C. H. Liu, A. R. Hand, R. Menzeleev, S. Spiegel and T. Hla, Science, 1998, 279, 1552–1555 CrossRef CAS.
- S. An, T. Bleu, O. G. Hallmark and E. J. Goetzl, J. Biol. Chem., 1998, 273, 7906–7910 CrossRef CAS PubMed.
- J. J. A. Contos and J. Chun, Genomics, 2000, 64, 155–169 CrossRef CAS PubMed.
- K. Bandoh, J. Aoki, H. Hosono, S. Kobayashi, T. Kobayashi, K. Murakami-Murofushi, M. Tsujimoto, H. Arai and K. Inoue, J. Biol. Chem., 1999, 274, 27776–27785 CrossRef CAS PubMed.
- J. R. Van Brocklyn, M. H. Gräler, G. Bernhardt, J. P. Hobson, M. Lipp and S. Spiegel, Blood, 2000, 95, 2624–2629 CAS.
- D.-S. Im, C. E. Heise, N. Ancellin, B. F. O'Dowd, G. Shei, R. P. Heavens, M. R. Rigby, T. Hla, S. Mandala, G. McAllister, S. R. George and K. R. Lynch, J. Biol. Chem., 2000, 275, 14281–14286 CrossRef CAS PubMed.
- K. Noguchi, S. Ishii and T. Shimizu, J. Biol. Chem., 2003, 278, 25600–25606 CrossRef CAS PubMed.
- C.-W. Lee, R. Rivera, A. E. Dubin and J. Chun, J. Biol. Chem., 2007, 282, 4310–4317 CrossRef CAS PubMed.
- C.-W. Lee, R. Rivera, S. Gardell, A. E. Dubin and J. Chun, J. Biol. Chem., 2006, 281, 23589–23597 CrossRef CAS PubMed.
- K. Kotarsky, Å. Boketoft, J. Bristulf, N. E. Nilsson, Å. Norberg, S. Hansson, C. Owman, R. Sillard, L. M. Leeb-Lundberg and B. Olde, J. Pharmacol. Exp. Ther., 2006, 318, 619–628 CrossRef CAS PubMed.
- S. M. Pasternack, I. von Kugelgen, K. Al Aboud, Y. A. Lee, F. Ruschendorf, K. Voss, A. M. Hillmer, G. J. Molderings, T. Franz, A. Ramirez, P. Nurnberg, M. M. Nothen and R. C. Betz, Nat. Genet., 2008, 40, 329–334 CrossRef CAS PubMed.
- K. Yanagida, K. Masago, H. Nakanishi, Y. Kihara, F. Hamano, Y. Tajima, R. Taguchi, T. Shimizu and S. Ishii, J. Biol. Chem., 2009, 284, 17731–17741 CrossRef CAS PubMed.
- (a) J. W. Choi, D. R. Herr, K. Noguchi, Y. C. Yung, C.-W. Lee, T. Mutoh, M. E. Lin, S. T. Teo, K. E. Park, A. N. Mosley and J. Chun, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 157–186 CrossRef CAS PubMed; (b) Y. Kihara, M. Maceyka, S. Spiegel and J. Chun, Br. J. Pharmacol., 2014, 171, 3575–3594 CrossRef CAS PubMed.
- Y. C. Yung, N. C. Stoddard and J. Chun, J. Lipid Res., 2014, 55, 1192–1214 CrossRef CAS PubMed.
- J. W. Choi and J. Chun, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 20–32 CrossRef CAS PubMed.
- J. J. A. Contos, I. Ishii, N. Fukushima, M. A. Kingsbury, X. Ye, S. Kawamura, J. H. Brown and J. Chun, Mol. Cell. Biol., 2002, 22, 6921–6929 CrossRef CAS.
- A. M. Christiano, Int. Patent, WO2009089380A2, 2009 Search PubMed.
- (a) A. P. Davenport, S. P. Alexander, J. L. Sharman, A. J. Pawson, H. E. Benson, A. E. Monaghan, W. C. Liew, C. P. Mpamhanga, T. I. Bonner, R. R. Neubig, J. P. Pin, M. Spedding and A. J. Harmar, Pharmacol. Rev., 2013, 65, 967–986 CrossRef CAS PubMed; (b) K. Makide, A. Uwamizu, Y. Shinjo, J. Ishiguro, M. Okutani, A. Inoue and J. Aoki, J. Lipid Res., 2014, 55, 1986–1995 CrossRef CAS PubMed.
- M. A. Hanson, C. B. Roth, E. Jo, M. T. Griffith, F. L. Scott, G. Reinhart, H. Desale, B. Clemons, S. M. Cahalan, S. C. Schuerer, M. G. Sanna, G. W. Han, P. Kuhn, H. Rosen and R. C. Stevens, Science, 2012, 335, 851–855 CrossRef CAS PubMed.
- C. O'Sullivan and K. K. Dev, Trends Pharmacol. Sci., 2013, 34, 401–412 CrossRef PubMed.
- A. L. Parrill and G. Tigyi, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 2–12 CrossRef CAS PubMed.
- Y. Fujiwara, V. Sardar, A. Tokumura, D. Baker, K. Murakami-Murofushi, A. Parrill and G. Tigyi, J. Biol. Chem., 2005, 280, 35038–35050 CrossRef CAS PubMed.
- W. J. Valentine, J. I. Fells, D. H. Perygin, S. Mujahid, K. Yokoyama, Y. Fujiwara, R. Tsukahara, J. R. Van Brocklyn, A. L. Parrill and G. Tigyi, J. Biol. Chem., 2008, 283, 12175–12187 CrossRef CAS PubMed.
- J. K. Archbold, J. L. Martin and M. J. Sweet, Trends Pharmacol. Sci., 2014, 35, 219–226 CrossRef CAS PubMed.
- C. Pages, M. F. Simon, P. Valet and J. S. Saulnier-Blache, Prostaglandins Other Lipid Mediators, 2001, 64, 1–10 CrossRef CAS.
- A. Tabchy, G. Tigyi and G. B. Mills, Nat. Struct. Mol. Biol., 2011, 18, 117–118 CAS.
- H. M. Albers and H. Ovaa, Chem. Rev., 2012, 112, 2593–2603 CrossRef CAS PubMed.
- L. A. van Meeteren, P. Ruurs, C. Stortelers, P. Bouwman, M. A. van Rooijen, J. P. Pradere, T. R. Pettit, M. J. Wakelam, J. S. Saulnier-Blache, C. L. Mummery, W. H. Moolenaar and J. Jonkers, Mol. Cell. Biol., 2006, 26, 5015–5022 CrossRef CAS PubMed.
- J. Gierse, A. Thorarensen, K. Beltey, E. Bradshaw-Pierce, L. Cortes-Burgos, T. Hall, A. Johnston, M. Murphy, O. Nemirovskiy, S. Ogawa, L. Pegg, M. Pelc, M. Prinsen, M. Schnute, J. Wendling, S. Wene, R. Weinberg, A. Wittwer, B. Zweifel and J. Masferrer, J. Pharmacol. Exp. Ther., 2010, 334, 310–317 CrossRef CAS PubMed.
- V. A. Blaho and T. Hla, Chem. Rev., 2011, 111, 6299–6320 CrossRef CAS PubMed.
- Y. C. Yung, T. Mutoh, M. E. Lin, K. Noguchi, R. R. Rivera, J. W. Choi, M. A. Kingsbury and J. Chun, Sci. Transl. Med., 2011, 3, 99ra87 Search PubMed.
- K. J. Herr, D. H. Herr, C.-W. Lee, K. Noguchi and J. Chun, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15444–15449 CrossRef CAS PubMed.
- B. Anliker, J. W. Choi, M. E. Lin, S. E. Gardell, R. R. Rivera, G. Kennedy and J. Chun, Glia, 2013, 61, 2009–2022 CrossRef PubMed.
- H. Ueda, H. Matsunaga, O. I. Olaposi and J. Nagai, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 61–73 CrossRef CAS PubMed.
- M. E. Lin, R. R. Rivera and J. Chun, J. Biol. Chem., 2012, 287, 17608–17617 CrossRef CAS PubMed.
- A. Schober and W. Siess, Br. J. Pharmacol., 2012, 167, 465–482 CrossRef CAS PubMed.
- I. Sevastou, E. Kaffe, M. A. Mouratis and V. Aidinis, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 42–60 CrossRef CAS PubMed.
- D. C. Budd and Y. Qian, Future Med. Chem., 2013, 5, 1935–1952 CrossRef CAS PubMed.
- Safety and Efficacy of a Lysophosphatidic Acid Receptor Antagonist in Idiopathic Pulmonary Fibrosis, http://clinicaltrials.gov/ct2/show/NCT01766817?term=lysophosphatidic+acid%26rank=2, accessed 2nd September, 2014.
- Angion Biomedica Corp. Pipeline,http://angion.com/products.asp, accessed 2nd September, 2014.
- Y. Miyabe, C. Miyabe, Y. Iwai, A. Takayasu, S. Fukuda, W. Yokoyama, J. Nagai, M. Jona, Y. Tokuhara, R. Ohkawa, H. M. Albers, H. Ovaa, J. Aoki, J. Chun, Y. Yatomi, H. Ueda, M. Miyasaka, N. Miyasaka and T. Nanki, Arthritis Rheum., 2013, 65, 2037–2047 CrossRef CAS PubMed.
- B. Orosa, S. García, P. Martínez, A. González, J. J. Gómez-Reino and C. Conde, Ann. Rheum. Dis., 2014, 73, 298–305 CrossRef CAS PubMed.
- C. Rancoule, R. Dusaulcy, K. Treguer, S. Gres, C. Attane and J. S. Saulnier-Blache, Biochimie, 2014, 96, 140–143 CrossRef CAS PubMed.
- M. Gotoh, Y. Fujiwara, J. Yue, J. Liu, S. Lee, J. Fells, A. Uchiyama, K. Murakami-Murofushi, S. Kennel, J. Wall, R. Patil, R. Gupte, L. Balazs, D. D. Miller and G. J. Tigyi, Biochem. Soc. Trans., 2012, 40, 31–36 CrossRef CAS PubMed.
- A. L. Parrill, Future Med. Chem., 2014, 8, 871–883 CrossRef PubMed.
- Studies found in clinical trials for BMS-986020,http://www.clinicaltrials.gov/ct2/results?term=BMS-986020%26Search=Search, accessed 2nd September, 2014.
- Proof of Biological Activity of SAR100842 in Systemic Sclerosis, http://clinicaltrials.gov/ct2/show/NCT01651143?term=lysophosphatidic+acid%26rank=6, accessed 2nd September, 2014.
- T. Sugiura, A. Tokumura, L. Gregory, T. Nouchi, S. T. Weintraub and D. J. Hanahan, Arch. Biochem. Biophys., 1994, 311, 358–368 CrossRef CAS PubMed.
- K. R. Lynch, D. W. Hopper, S. J. Carlisle, J. G. Catalano, M. Zhang and T. L. MacDonald, Mol. Pharmacol., 1997, 52, 75–81 CAS.
- C. E. Heise, W. L. Santos, A. M. Schreihofer, B. H. Heasley, Y. V. Mukhin, T. L. Macdonald and K. R. Lynch, Mol. Pharmacol., 2001, 60, 1173–1180 CAS.
- W. L. Santos, B. H. Heasley, R. Jarosz, K. M. Carter, K. R. Lynch and T. L. Macdonald, Bioorg. Med. Chem. Lett., 2004, 14, 3473–3476 CrossRef CAS PubMed.
- Y. Xu, J. Aoki, K. Shimizu, M. Umezu-Goto, K. Hama, Y. Takanezawa, S. Yu, G. B. Mills, H. Arai, L. Qian and G. D. Prestwich, J. Med. Chem., 2005, 48, 3319–3327 CrossRef CAS PubMed.
- Y. Hasegawa, J. R. Erickson, G. J. Goddard, S. Yu, S. Liu, K. W. Cheng, A. Eder, K. Bandoh, J. Aoki, R. Jarosz, A. D. Schrier, K. R. Lynch, G. B. Mills and X. Fang, J. Biol. Chem., 2003, 278, 11962–11969 CrossRef CAS PubMed.
- L. Qian, Y. Xu, T. Simper, G. Jiang, J. Aoki, M. Umezu-Goto, H. Arai, S. Yu, G. B. Mills, R. Tsukahara, N. Makarova, Y. Fujiwara, G. Tigyi and G. D. Prestwich, ChemMedChem, 2006, 1, 376–383 CrossRef CAS PubMed.
- G. Jiang, A. Inoue, J. Aoki and G. D. Prestwich, Bioorg. Med. Chem. Lett., 2013, 23, 1865–1869 CrossRef CAS PubMed.
- Y. Xu and G. D. Prestwich, J. Org. Chem., 2002, 67, 7158–7161 CrossRef CAS PubMed.
- Y. Xu, L. Qian, A. V. Pontsler, T. M. McIntyre and G. D. Prestwich, Tetrahedron, 2004, 60, 43–49 CrossRef CAS PubMed.
- Y. Xu, L. Qian and G. D. Prestwich, Org. Lett., 2003, 5, 2267–2270 CrossRef CAS PubMed.
- Y. Xu, G. Jiang, R. Tsukahara, Y. Fujiwara, G. Tigyi and G. D. Prestwich, J. Med. Chem., 2006, 49, 5309–5315 CrossRef CAS PubMed.
- V. Gududuru, K. Zeng, R. Tsukahara, N. Makarova, Y. Fujiwara, K. R. Pigg, D. L. Baker, G. Tigyi and D. D. Miller, Bioorg. Med. Chem. Lett., 2006, 16, 451–456 CrossRef CAS PubMed.
- L. Gan, J. X. Xue, X. Li, D. S. Liu, Y. Ge, P. Y. Ni, L. Deng, Y. Lu and W. Jiang, Biochem. Biophys. Res. Commun., 2011, 409, 7–13 CrossRef CAS PubMed.
- B. H. Heasley, R. Jarosz, K. R. Lynch and T. L. Macdonald, Bioorg. Med. Chem. Lett., 2004, 14, 2735–2740 CrossRef CAS PubMed.
- J. E. East, K. M. Carter, P. C. Kennedy, N. A. Schulte, M. L. Toews, K. R. Lynch and T. L. Macdonald, MedChemComm, 2011, 2, 325–330 RSC.
- G. Jiang, Y. Xu, Y. Fujiwara, T. Tsukahara, R. Tsukahara, J. Gajewiak, G. Tigyi and G. D. Prestwich, ChemMedChem, 2007, 2, 679–690 CrossRef CAS PubMed.
- X. Xu and G. D. Prestwich, Cancer, 2010, 116, 1739–1750 CrossRef CAS PubMed.
- I. Nikitopoulou, E. Kaffe, I. Sevastou, I. Sirioti, M. Samiotaki, D. Madan, G. D. Prestwich and V. Aidinis, PLoS One, 2013, 8, e70941 CAS.
- H. Ohta, K. Sato, N. Murata, A. Damirin, E. Malchinkhuu, J. Kon, T. Kimura, M. Tobo, Y. Yamazaki, T. Watanabe, M. Yagi, M. Sato, R. Suzuki, H. Murooka, T. Sakai, T. Nishitoba, D. S. Im, H. Nochi, K. Tamoto and H. Tomura, et al., Mol. Pharmacol., 2003, 64, 994–1005 CrossRef CAS PubMed.
- J. S. Swaney, C. Chapman, L. D. Correa, K. J. Stebbins, R. A. Bundey, P. C. Prodanovich, P. Fagan, C. S. Baccei, A. M. Santini, J. H. Hutchinson, T. J. Seiders, T. A. Parr, P. Prasit, J. F. Evans and D. S. Lorrain, Br. J. Pharmacol., 2010, 160, 1699–1713 CrossRef CAS PubMed.
- F. V. Castelino, J. Seiders, G. Bain, S. F. Brooks, C. D. King, J. S. Swaney, D. S. Lorrain, J. Chun, A. D. Luster and A. M. Tager, Arthritis Rheum., 2011, 63, 1405–1415 CrossRef CAS PubMed.
- J. S. Swaney, C. Chapman, L. D. Correa, K. J. Stebbins, A. R. Broadhead, G. Bain, A. M. Santini, J. Darlington, C. D. King, C. S. Baccei, C. Lee, T. A. Parr, J. R. Roppe, T. J. Seiders, J. Ziff, P. Prasit, J. H. Hutchinson, J. F. Evans and D. S. Lorrain, J. Pharmacol. Exp. Ther., 2011, 336, 693–700 CrossRef CAS PubMed.
- Y. Qian, M. Hamilton, A. Sidduri, S. Gabriel, Y. Ren, R. Peng, R. Kondru, A. Narayanan, T. Truitt, R. Hamid, Y. Chen, L. Zhang, A. J. Fretland, R. A. Sanchez, K. C. Chang, M. Lucas, R. C. Schoenfeld, D. Laine, M. E. Fuentes, C. S. Stevenson and D. C. Budd, J. Med. Chem., 2012, 55, 7920–7939 CrossRef CAS PubMed.
- B. O. Buckman, J. B. Nicholas, K. Emayan and S. D. Seiwert, Int. Patent, WO2013025733, 2013 Search PubMed.
- E. Kawaminami, T. Takahashi, T. Kanayama and H. Kaizawa, Int. Patent, WO2011037192, 2011 Search PubMed.
- E. Kawaminami, T. Takahashi, T. Kanayama and K. Sakamoto, Int. Patent, WO2012039460, 2012 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |