
Configurational changes of heme followed by cytochrome c folding reaction†
Jungkweon
Choi‡
*a,
Dae Won
Cho
b,
Sachiko
Tojo
a,
Mamoru
Fujitsuka
a and
Tetsuro
Majima
*a
aThe Institute of Scientific and Industrial Research (SANKEN), Osaka University, Mihogaoka 8-1, Ibaraki, Osaka 567-0047, Japan. E-mail: jkchoi@sanken.osaka-u.ac.jp; majima@sanken.osaka-u.ac.jp
bDepartment of Advanced Materials Chemistry, Korea University, Sejong Campus, Sejong 339-700, Korea
First published on 22nd October 2014
Abstract
Although the folding kinetics of cytochrome c (Cyt-c), ferric or ferrous Cyt-c, has been extensively investigated as a paradigm for a protein folding reaction using various time-resolved spectroscopic techniques, the configurational change of heme associated with the folding reaction from a ferric Cyt-c to a ferrous Cyt-c induced by one-electron reduction has not been elucidated. To address this issue, we investigated the configurational change of heme in the Cyt-c folding process induced by one-electron reduction using a combination of time-resolved resonance Raman spectroscopy and pulse radiolysis. The results presented herein reveal that the reduction of ferric Cyt-c and the ligation of Met80 occur simultaneously within a timescale of approximately 2 μs, and that the ligand binding and exchange of heme depend on the initial configuration of the heme. The rapid ligation of Met80 observed in this study may be attributed to the intramolecular diffusion of Met80 into ferrous Cyt-c with a 5-coordinated high-spin configuration. Conversely, the ligand exchange of a ferrous Cyt-c with a 6-coordinated low-spin configuration was significantly slower.
Introduction
Conformational changes play an essential role in the biological functions of proteins, in vivo. Especially, the dynamics of protein folding are of particular importance in biomedical science, as accumulation of misfolded proteins can cause a variety of fatal diseases. As a result, numerous experimental and theoretical efforts have focused on understanding protein folding–unfolding processes, as summarized in recent reviews.1–5In vivo, conformational changes during protein folding occur over a wide timescale (fs–s). Therefore, various time-resolved spectroscopic techniques such as time-resolved circular dichroism,6–8 infrared and Raman spectroscopic techniques,9–17 the fast mixing stopped-flow method,14,18 transient absorption,19–21 transient grating,22–25 and pulse radiolysis26,27 have been applied to study the conformational changes that occur during protein folding reactions. Among these spectroscopic techniques, pulse radiolysis is a unique technique that has been used to trace real-time observations of the redox processes of various molecules, including protein and DNA.27–32 Indeed, using the pulse radiolysis technique we previously demonstrated that the oxidized form of cytochrome c (Cyt-c), ferric Cyt-c, is selectively reduced within a timescale of a few microseconds by electron injection, and that the resulting ferrous Cyt-c then initiates a folding reaction.26 Our results indicated that the folding dynamics of Cyt-c, triggered by one-electron reduction, exhibited a multistep rearrangement into the native conformation.26
Although various time-resolved spectroscopic techniques have been used to investigate the folding dynamics of Cyt-c in the presence of a denaturant such as guanidine hydrochloride (GdHCl), the dynamics of the heme coordination states accompanied by the electron transfer-triggered folding reaction of Cyt-c are not yet clear. To our knowledge, here, we present the first study on the configurational changes of heme accompanied by Cyt-c folding induced by one-electron reduction. For this study, we utilized time-resolved resonance Raman (TR3) spectroscopy combined with pulse radiolysis. TR3 spectroscopy is a sensitive technique that can accurately trace the tertiary structures and structural changes of various biomolecules over a wide timescale.10,12–14,33
Our findings indicate that although the reduction dynamics of ferric Cyt-c occur within a time range of a few microseconds, the ligand binding and exchange of heme, accompanied by one-electron reduction, depend on the initial configuration of the heme. In this report, we discuss the detailed configurational changes of heme in the Cyt-c folding process induced by one-electron reduction.
Results and discussion
In native Cyt-c, the Fe ion of the heme coordinates axially with the His18 and Met80 residues of the Cyt-c protein (Scheme 1). However, upon addition of GdHCl, the bond between Met80 and the Fe ion is disrupted, resulting in denaturation of the protein. In denatured ferric Cyt-c, the coordination site of met80 is occupied by a histidine (His-33 or His-26) to form a 6cLS configuration. Interestingly, previous studies have shown that while ferric Cyt-c is denatured in the presence of ∼4 M GdHCl, ferrous Cyt-c remains folded under identical conditions.20,25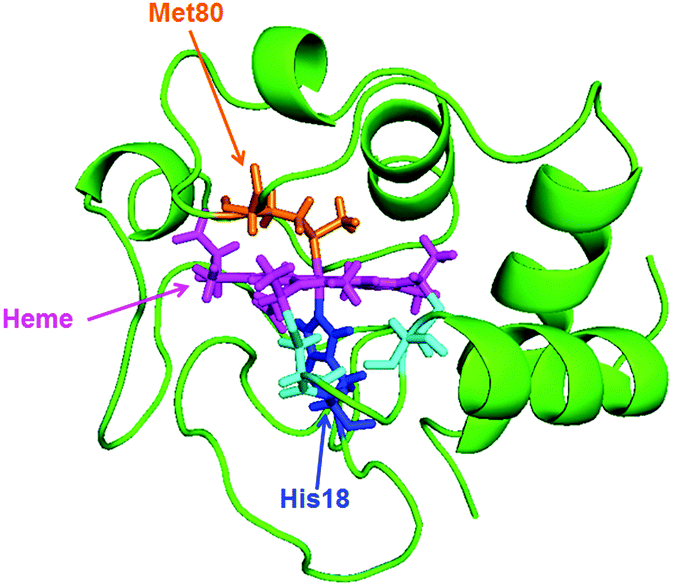 | ||
| Scheme 1 Solution structures of equine heart Cyt-c (1GIW) retrieved from the Protein Data Bank. | ||
To determine whether we could reproduce these findings, we measured the steady-state Raman spectra of ferric and ferrous Cyt-c in the presence of ∼4 M GdHCl by using an excitation wavelength of 514.5 nm (Fig. 1). Based on the assignments of resonance Raman spectra of Cyt-c, as reported by Spiro et al.15,16 and Oellerich et al.,13 Raman bands for ferric and ferrous Cyt-c in the presence of GdHCl are assigned to those of the heme molecule, as depicted in Fig. 1. The denatured ferric Cyt-c exhibited the characteristic ν4 (ν(pyrrole half-ring)sys) mode at 1375 cm−1, corresponding to the porphyrin breathing mode, and the ν10 (ν(CαCm)asys) mode at 1639 cm−1. In addition, the ν2 (ν(CβCβ)sys) mode, a spin-state marker band, was observed at 1565 and 1588 cm−1, corresponding to 6-coordinated low-spin (6cLS) and 5-coordinated high-spin (5cHS) configurations, respectively.13,14,17 Consistent with the data from previous studies,12–14,17 these data indicate that the heme moiety has a mixed configuration under denaturing conditions.
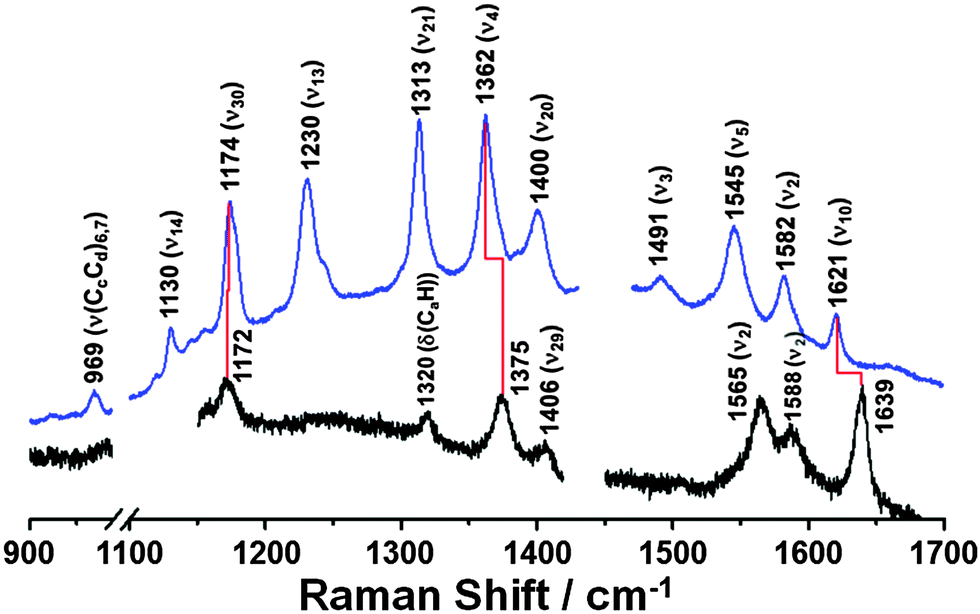 | ||
| Fig. 1 Steady-state Raman spectra of ferric Cyt-c (black line) and ferrous Cyt-c (blue line) in ∼4 M GdHCl solution, respectively. ([Cyt-c] = 50 μM, λExcitation = 514.5 nm). The Raman spectra show that ferric Cyt-c is unfolded in the ∼4 M GdHCl solution, whereas ferrous Cyt-c is folded under the same experimental conditions. | ||
In comparison to the Raman spectrum of unfolded ferric Cyt-c, ferrous Cyt-c exhibits significantly down-shifted ν4 and ν10 vibrational modes at 1362 and 1621 cm−1, respectively, which is characteristic of the properly folded ferrous Cyt-c.15 Indeed, the Raman spectrum of ferrous Cyt-c measured in ∼4 M GdHCl solution is consistent with that measured in 0.5 M GdHCl solution, where both ferric and ferrous Cyt-c are properly folded (Fig. S1, ESI†). Therefore, these data support previous findings that ferrous Cyt-c remains folded in the presence of 3.2–4.0 M GdHCl.20,26 This result also implies that the reduction of ferric Cyt-c in the presence of 3.2–4.0 M GdHCl results in a conformational change to form a folded structure. Generally, the ν4 mode is considered an indicator of the oxidation state of heme because of its sensitivity to the electron density of iron porphyrin (heme). Meanwhile, the ν10 mode is sensitive to structural changes in a protein. Thus, the down-shifts in both the ν4 and ν10 modes, observed in the spectrum of ferrous Cyt-c, can likely be attributed to changes in both the oxidation states and in the structures of ferric and ferrous Cyt-c in the presence of ∼4 M GdHCl. It is also noteworthy that the ν3 (ν(CαCm)sys) mode is more pronounced in the spectrum of ferrous Cyt-c than in that of ferric Cyt-c, suggesting that this mode can also be used as an indicator of the folding state of Cyt-c.
To investigate the configurational changes of heme during the reduction-induced folding reaction of ferric Cyt-c, we measured the TR3 spectra during pulse radiolysis of ferric Cyt-c in 3.5 M GdHCl. Ferric Cyt-c, in the presence of GdHCl, shows a strong absorption band at 409 nm (Soret band) and a weak broad band at approximately 530 nm (Q band). In contrast, the ferrous Cyt-c displays a strong Soret band at 417 nm and a slightly structured Q band at 500–570 nm.26 Therefore, to measure the TR3 spectra at the resonance wavelength, a 416 nm pulse of light was used as an excitation source. Fig. 2 shows the TR3 spectra obtained after pulse radiolysis of unfolded ferric Cyt-c in 3.5 M GdHCl. The Raman spectrum of the unfolded ferric Cyt-c in 3.5 M GdHCl exhibits ν4, ν3, ν2, and ν10 modes at 1373, 1505, 1587, and 1639 cm−1, respectively (Fig. 2). With the exception of the ν2 mode, this spectrum is similar to the steady-state Raman spectrum for unfolded ferric Cyt-c. However, the intensity of some Raman signals was altered by the use of different excitation wavelengths. Furthermore, the ν2 mode at ∼1585 cm−1 had a very broad feature. However, peak analysis revealed that this mode actually consisted of two peaks with centre frequencies of 1564 and 1587 cm−1 (Fig. S2, ESI†). This is consistent with the steady-state Raman spectrum of unfolded ferric Cyt-c. Thus, these results provide further evidence that the 6cLS state of heme coexists with the 5cHS state under denaturing conditions, as mentioned above. According to previous studies,13,17 formation of the unfolded ferric Cyt-c with 6cLS configuration is mediated by His33 (or His26) and His18, whereas formation of the unfolded ferric Cyt-c with 5cHS configuration is coordinated by the His18 residue alone.
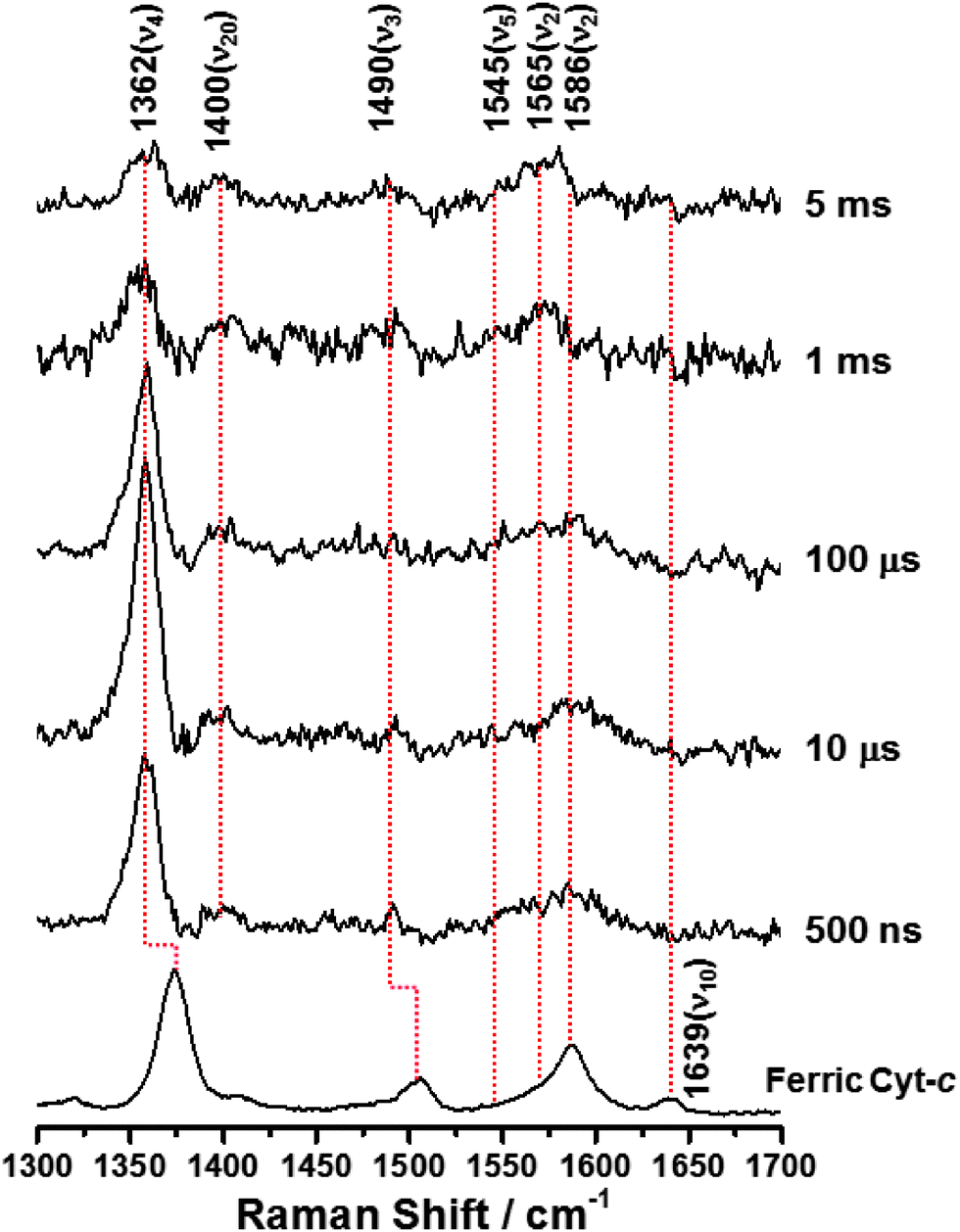 | ||
| Fig. 2 Time-resolved resonance Raman spectra obtained during pulse radiolysis of ferric Cyt-c in 3.5 M GdHCl. ([Cyt-c] = 50 μM, λExcitation = 416 nm). | ||
As depicted in Fig. 2, on the other hand, the TR3 spectra measured during pulse radiolysis of unfolded ferric Cyt-c in 3.5 M GdHCl also exhibited ν4 and ν20 modes at 1362 and 1400 cm−1, respectively, even in the early time-delay (e.g. at 500 ns). These ν4 and ν20 vibrations are characteristic Raman modes of ferrous Cyt-c. This result is therefore consistent with the proposal that we put forth in our previous study.26 The unfolded ferric Cyt-c undergoes rapid reduction via an electron transfer from the guanidine radical formed by the fast interaction of eaq− with GdHCl. The reduced form of Cyt-c then structurally relaxes into an unfolded ferrous Cyt-c molecule. For the reduction dynamics of an unfolded ferric Cyt-c, we observed a time-dependent signal intensity of the ν4 mode at 1362 cm−1 (Fig. 3). The temporal profile depicted in Fig. 3 could be well fitted by a tetra-exponential function with relaxation times of 1.4 ± 0.9 μs, 5.4 ± 4.5 μs, 296 ± 30 μs, and >5 ms (constant). These observed dynamics are similar to those reported in our previous study,26 where transient absorption (TA) spectroscopy was utilized to demonstrate that the reduction of ferric Cyt-c and the binding of the Met-80 residue to heme iron occur with rate constants of 1.3 × 105 s−1 (7.7 μs) and 4.3 × 105 s−1 (2.3 μs), respectively. These findings indicated that the reduction dynamics of ferric Cyt-c is slower than the ligation of Met-80 to heme.26 TA spectroscopy is a useful technique for obtaining information on local protein structural changes around a chromophore. TR3 spectroscopy, however, can provide detailed information about molecular vibrations by which a molecule can be chemically and structurally identified. From this point of view, the fast component of 1.4 μs, determined by the TR3 analysis, may be due to the reduction dynamics of ferric Cyt-c because the ν4 mode is very sensitive to the oxidation state of heme. Consequently, one interpretation of this observation is that the reduction of ferric Cyt-c occurs either more rapidly than or simultaneously with the ligation of Met-80. While this interpretation is slightly different from the proposal made in our previous study,26 Chen et al. found that the reduction of oxidized Cyt-c and the ligation of Met80 to the heme iron (Fe2+) occur simultaneously within a 5 μs timescale.34 Several other studies have reported that binding of methionine residues (Met-80 or Met-65) to the heme iron (Fe2+), followed by photodissociation of the CO ligand, takes place within a timescale of a few microseconds (∼2 μs).6,7,24 Furthermore, as shown in Fig. 2, a new ν3 vibrational mode at 1490 cm−1 due to the presence of ferrous Cyt-c with the 6cLS heme configuration was observed in the early delay time (500 ns); however, the intensity of this mode was weak. Therefore, we propose that the reduction of ferric Cyt-c and the ligation of Met80 occur simultaneously within a timescale of approximately 2 μs. Meanwhile, the two decay components of 5.4 and 296 μs probably stem from conformational changes triggered by the reduction of a denatured ferric Cyt-c and the ligation of Met80. The slow dynamics of >5 ms (constant) may be due to the rearrangement to the native conformation, as proposed by our prior study.26
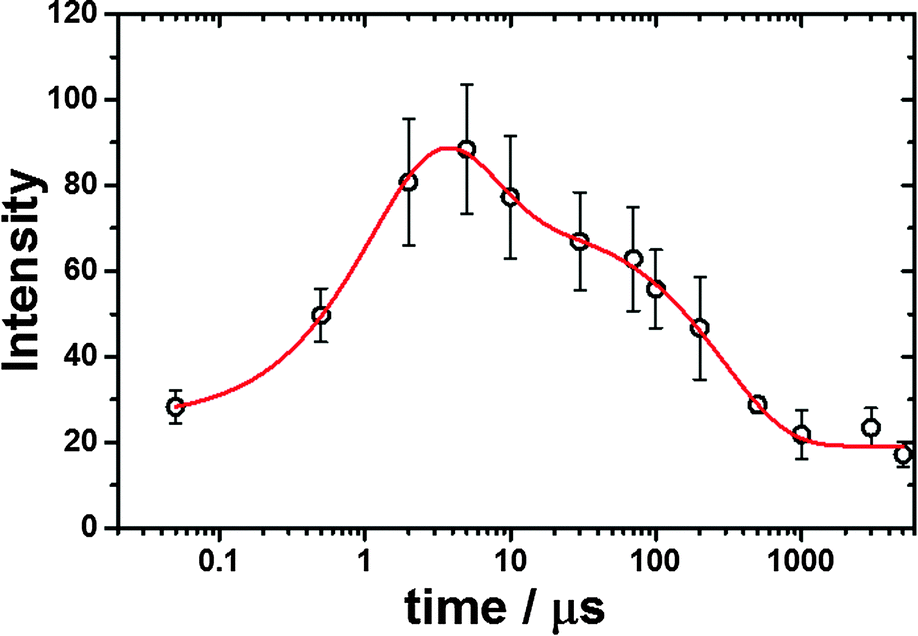 | ||
| Fig. 3 Intensity changes of the ν4 vibrational mode at 1362 cm−1 as a function of delay times. The theoretical fitting curve is shown in a red straight line. | ||
During the folding of ferric Cyt-c, the ligand exchange from His18-Fe3+-His33 (or His18-Fe3+-His26; 6cLS) to His18-Fe3+-Met80 (6cLS) occurs via a 6-coordinate intermediate linked to a water molecule as a distal ligand. Consequently the dynamics of this reaction are very slow.11,14 Here, we consider an explanation for the rapid ligation of Met80 observed in this system. The data presented herein show that ferric Cyt-c, under denaturing conditions, exists in both the 6cLS and 5cHS forms, as shown in Fig. 1 and 2. Oellerich et al. suggested that, similar to the formation of an unfolded ferric Cyt-c, the formation of an unfolded ferrous Cyt-c with a 6cLS heme might also be mediated by His-18 and His-33.13 Given the folding dynamics of ferric Cyt-c, one would predict that the ligand exchange from His18-Fe2+-His33 (or His18-Fe2+-His26) to His18-Fe2+-Met80 during the protein folding reaction would be slow. Indeed, the ν2 mode at 1565 cm−1 of ferrous Cyt-c, corresponding to the 6cLS configuration, was measured with a weak intensity even in the late delay time of 5 ms, as shown in Fig. 2. In contrast, the ν2 mode of 1586 cm−1, corresponding to the 5cHS configuration of ferrous Cyt-c, was undetectable, suggesting a rapid ligand binding process. This result indicates that the ligand binding and exchange of heme depend on the initial coordination state of the heme molecule even though the reduction dynamics of ferric Cyt-c occur within a timescale of a few microseconds. Consequently, our data demonstrate that the ligand binding of ferrous Cyt-c with the 5cHS configuration, generated by pulse radiolysis, is relatively fast, whereas ferrous Cyt-c with the 6cLS configuration exhibits slow reaction dynamics. With regard to ligand exchange of unfolded ferrous Cyt-c with the 5cHS configuration, the ligation dynamics of Met80 are governed by the intramolecular diffusion of Met80. Abel et al. investigated the ligand rebinding dynamics that occurred after photodissociation of the CO ligand from an unfolded ferrous Cyt-c and proposed that the mean intramolecular diffusion time of a methionine or a histidine residue into the heme iron was 3 ± 2 μs,6 which is close to the speed limit of protein folding (∼1 μs).2,23,35,36 This value is consistent with the results presented in this study. Thus, the fast ligation of Met80 can likely be attributed to the intramolecular diffusion of Met80 into a reduced ferrous Cyt-c with a 5cHS configuration.
Conclusions
Using TR3 spectroscopy combined with pulse radiolysis, we analysed the conformational changes that occurred during the Cyt-c folding process induced by one-electron reduction. This technique provided a vibrational level understanding of these protein folding and reduction processes. The reduction dynamics of ferric Cyt-c occurred within a timescale of a few microseconds. We also observed that the ligand binding and exchange of heme depended on its initial configuration, such as 5cHS and 6cLS. Specifically, the ligation of Met80 to a reduced ferrous Cyt-c with a 5cHS configuration occurs within a timescale of a few microseconds, which is close to the intramolecular diffusional time of Met80 under denaturing conditions. In contrast, a ferrous Cyt-c with a 6cLS configuration exhibited slow ligand-exchange dynamics.Our results demonstrate that TR3 combined with pulse radiolysis can be easily applied to study the redox reactions of various biomolecules, including protein and DNA, and can help elucidate the relationship between their structure and biological functions.
Experimental section
Equine heart Cyt-c was purchased from Sigma-Aldrich and used without further purification.Pulse radiolysis
Pulse radiolysis experiments were performed using an electron pulse (27 MeV, 11 A, 8 ns, 0.8 kGy per pulse) from a linear accelerator at Osaka University.Steady-state Raman spectroscopy
In order to clearly measure Steady-state Raman spectra, we used a 514.5 nm laser line of Ar ion laser (Spectra Physics, BeamLok 2060L-S4W, 10 mW), which is a resonance wavelength. Raman signals were detected using a CCD detector (Andor Technology, DU940P-BU) equipped with a polychromator (Andor Technology, SR750-B1E).TR3 spectroscopy
All Cyt-c solutions (100 mM phosphate buffer, pH 7.0, 50 μM protein, and an appropriate amount of GdHCl, 3.0–4.0 M) were purged with Ar gas during the TR3 measurements. The Raman experiments were performed by the flow of sample solution through a quartz capillary tube at a rate sufficient to ensure that each electron beam and laser pulse encounter a fresh sample volume. When we need accumulation to improve the signal-to-noise ratio, the sample was frequently replaced by a fresh sample. The TR3 spectra of Cyt-c after pulse radiolysis were obtained by photoexcitation with a 416 nm pulse. The excitation light of 416 nm pulse is generated by the hydrogen Raman shifter of the third harmonic (355 nm) from a nanosecond Q-switched Nd:YAG laser (5 ns fwhm, Brilliant, Quantel; Les Ulis, France). The laser excitation light is synchronized with the electron pulse. The sample solution was passed through a quartz capillary tube at a slow rate sufficient to ensure that each laser pulse encountered a fresh volume of the sample. The TR3 spectra were collected using a polychromator (Acton, SP2500i; Trenton, NJ, USA) equipped with a charge-coupled device (CCD) camera (Princeton Instruments, PI-MAX3; Trenton, NJ, USA).Acknowledgements
We thank the members of the Research Laboratory for Quantum Beam Science of ISIR, Osaka University, for running the linear accelerator. This work has been partly supported by a Grant-in-Aid for Scientific Research (Project 24550188, 25220806, and 25288035) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japanese Government.Notes and references
- J. R. Winkler, Curr. Opin. Chem. Biol., 2004, 8, 169–174 CrossRef CAS PubMed.
- J. Kubelka, J. Hofrichter and W. A. Eaton, Curr. Opin. Struct. Biol., 2004, 14, 76–88 CrossRef CAS PubMed.
- R. L. Baldwin and G. D. Rose, Curr. Opin. Struct. Biol., 2013, 23, 4–10 CrossRef CAS PubMed.
- B. Schuler and H. Hofmann, Curr. Opin. Struct. Biol., 2013, 23, 36–47 CrossRef CAS PubMed.
- D. Thirumalai, Z. Liu, E. P. O'Brien and G. Reddy, Curr. Opin. Struct. Biol., 2013, 23, 22–29 CrossRef CAS PubMed.
- C. J. Abel, R. A. Goldbeck, R. F. Latypov, H. Roder and D. S. Kliger, Biochemistry, 2007, 46, 4090–4099 CrossRef CAS PubMed.
- E. F. Chen, M. J. Wood, A. L. Fink and D. S. Kliger, Biochemistry, 1998, 37, 5589–5598 CrossRef CAS PubMed.
- T. Konno, Protein Sci., 1998, 7, 975–982 CrossRef CAS PubMed.
- J. Kim, J. Park, T. Lee and M. Lim, J. Phys. Chem. B, 2008, 113, 260–266 CrossRef PubMed.
- M. Negrerie, S. Cianetti, M. H. Vos, J. L. Martin and S. G. Kruglik, J. Phys. Chem. B, 2006, 110, 12766–12781 CrossRef CAS PubMed.
- S. R. Yeh, S. W. Han and D. L. Rousseau, Acc. Chem. Res., 1998, 31, 727–736 CrossRef CAS.
- T. Jordan, J. C. Eads and T. G. Spiro, Protein Sci., 1995, 4, 716–728 CrossRef CAS PubMed.
- S. Oellerich, H. Wackerbarth and P. Hildebrandt, J. Phys. Chem. B, 2002, 106, 6566–6580 CrossRef CAS.
- S. Takahashi, S. R. Yeh, T. K. Das, C. K. Chan, D. S. Gottfried and D. L. Rousseau, Nat. Struct. Biol., 1997, 4, 44–50 CrossRef CAS PubMed.
- S. Z. Hu, I. K. Morris, J. P. Singh, K. M. Smith and T. G. Spiro, J. Am. Chem. Soc., 1993, 115, 12446–12458 CrossRef CAS.
- S. Z. Hu and T. G. Spiro, Biophys. J., 1993, 64, A157 CrossRef.
- E. Droghetti, S. Oellerich, P. Hildebrandt and G. Smulevich, Biophys. J., 2006, 91, 3022–3031 CrossRef CAS PubMed.
- C. K. Chan, Y. Hu, S. Takahashi, D. L. Rousseau, W. A. Eaton and J. Hofrichter, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1779–1784 CrossRef CAS.
- J. A. Stevens, J. J. Link, Y. T. Kao, C. Zang, L. Wang and D. Zhong, J. Phys. Chem. B, 2010, 114, 1498–1505 CrossRef CAS PubMed.
- T. Pascher, J. P. Chesick, J. R. Winkler and H. B. Gray, Science, 1996, 271, 1558–1560 CAS.
- S. J. Hagen, J. Hofrichter, A. Szabo and W. A. Eaton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11615–11617 CrossRef CAS.
- J. Choi, Y. O. Jung, J. H. Lee, C. Yang, B. Kim and H. Ihee, ChemPhysChem, 2008, 9, 2708–2714 CrossRef CAS PubMed.
- J. Choi, S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, Phys. Chem. Chem. Phys., 2011, 13, 5651–5658 RSC.
- J. Choi, C. Yang, J. Kim and H. Ihee, J. Phys. Chem. B, 2011, 115, 3127–3135 CrossRef CAS PubMed.
- T. Nada and M. Terazima, Biophys. J., 2003, 85, 1876–1881 CrossRef CAS.
- J. Choi, M. Fujitsuka, S. Tojo and T. Majima, J. Am. Chem. Soc., 2012, 134, 13430–13435 CrossRef CAS PubMed.
- K. Kobayashi, H. Une and K. Hayashi, J. Biol. Chem., 1989, 264, 7976–7980 CAS.
- M. Fujikawa, K. Kobayashi and T. Kozawa, J. Biol. Chem., 2012, 287, 35702–35708 CrossRef CAS PubMed.
- D. C. Grills, J. A. Farrington, B. H. Layne, S. V. Lymar, B. A. Mello, J. M. Preses and J. F. Wishart, J. Am. Chem. Soc., 2014, 136, 5563–5566 CrossRef CAS PubMed.
- K. Kawai, T. Kimura, K. Kawabata, S. Tojo and T. Majima, J. Phys. Chem. B, 2003, 107, 12838–12841 CrossRef CAS.
- K. Kawai, A. Yokoohji, S. Tojo and T. Majima, Chem. Commun., 2003, 2840–2841 RSC.
- K. Kawai, T. Takada, S. Tojo and T. Majima, Tetrahedron Lett., 2002, 43, 89 CrossRef CAS.
- S. R. Yeh and D. L. Rousseau, Biophys. J., 1998, 74, A128 Search PubMed.
- E. F. Chen, P. Wittung-Stafshede and D. S. Kliger, J. Am. Chem. Soc., 1999, 121, 3811–3817 CrossRef CAS.
- I. J. Chang, J. C. Lee, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3838–3840 CrossRef CAS PubMed.
- K. Chattopadhyay, E. L. Elson and C. Frieden, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2385–2389 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and supporting figures. See DOI: 10.1039/c4mb00551a |
| ‡ Present address: Center for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon 305-701, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2015 |