
Permethrin and its metabolites affect Cu/Zn superoxide conformation: fluorescence and in silico evidences
Gabbianelli
Rosita
*a,
Carloni
Manuel
a,
Marmocchi
Franco
a,
Nasuti
Cinzia
a,
Fedeli
Donatella
a,
Laudadio
Emiliano
b,
Massaccesi
Luca
b and
Galeazzi
Roberta
b
aScuola del Farmaco e dei Prodotti della Salute Università di Camerino, Via Gentile III da Varano, Camerino, Italy. E-mail: rosita.gabbianelli@unicam.it
bDi.S.VA, Università Politecnica delle Marche, via Brecce Bianche, Ancona, Italy. E-mail: r.galeazzi@univpm.it; Tel: +39 0712204724
First published on 21st October 2014
Abstract
The proclivity of permethrin and its metabolites to affect the structure and activity of Cu/Zn superoxide dismutase (SOD) has been investigated by using intrinsic fluorescence and 8-ANS fluorescence techniques. In silico molecular docking investigations were carried out in order to assess the means of interaction at a molecular level between SOD and the considered ligands. Results show that both, permethrin and its metabolites are able to induce conformational variation on SOD. Permethrin and 3-phenoxybenzyl alcohol metabolite induce a blue shift toward the hydrophobic amino acids Leu-101, Ile-102, Leu-104, Ile-110 and Ile-111, with a significant peak increase. An opposite effect was shown by 3-phenoxy benzaldehyde and 3-phenoxybenzoic acid with a progressive reduction of tyrosine fluorescence emission, without any shift. Computational findings confirm that all the molecules considered have more than one allosteric binding site but none of them interact with SOD at its catalytic Cu/Zn cleft. Moreover, all the binding poses found are very close in binding energy thus demonstrating that there is not only a preferred interaction site but most of them are important due to their relative energy in equilibrium with a population strictly connected to the ligand concentration. In the obtained complexes, all the ligands are involved in many hydrogen bonds through their polar oxygen moieties but due to the presence of a common aromatic hydrophobic core, many hydrophobic interactions are due to the SOD nature rich in apolar amino acids. Furthermore, for each ligand it can be pointed out the presence of a highly populated docked structure with a specific interaction of permethrin and its metabolites with Tyr-108, responsible for changes in fluorescence emission.
Introduction
Permethrin (PERM) was the first synthetic pyrethroid photostable enough to be used as a pesticide in agriculture, in health applications to treat tropical scabies, and on mosquito nets as an antimalarial strategy in tropical areas.1,2 Generally PERM, both in its two isomeric forms cis and trans, as well as the other members of the pyrethroid family, is considered the insecticide with the lowest mammalian toxicity because mammals have a high level of enzymes that detoxify pyrethroids and their dermal layer, thus the fast metabolism rapidly produces pyrethroid metabolites. Permethrin metabolites, formed by enzymatic cleavage due to oxidases in the cis isomer and to esterase in the trans one, are 3-phenoxybenzyl alcohol (3-PBA), 3-phenoxybenzaldehyde (3-PBALD) and 3-phenoxybenzoic acid (3-PBACID) (Fig. 1).3–5 After cleavage, both isomers are hydrolysed by an esterase and finally oxidized.6 Following oral intake of permethrin, the largest part of the urinary metabolites (93%) is excreted within 24 hours as glucurono-conjugated.7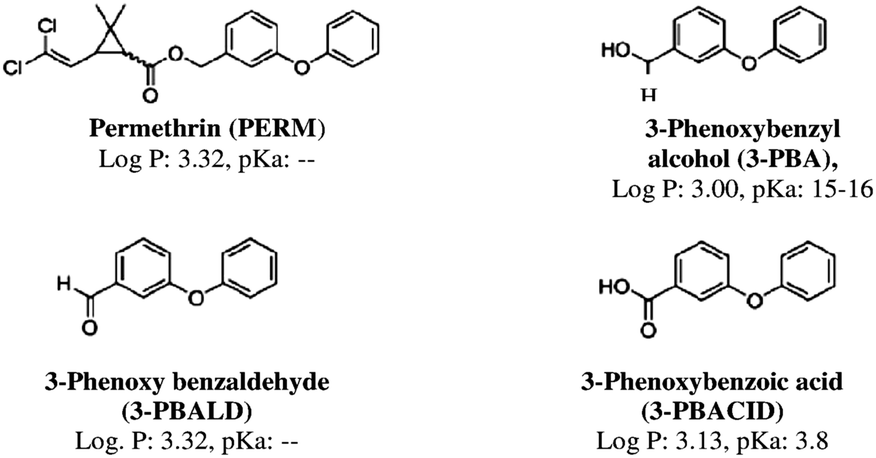 | ||
Fig. 1 Chemical structure, lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and acid dissociation constant (pKa) values of permethrin (PERM) and its main metabolites. P) and acid dissociation constant (pKa) values of permethrin (PERM) and its main metabolites. | ||
Some studies indicated that hydrolysed products of permethrin are more cytotoxic than the parent compound, pointing out carcinogenic effects and interactions with the steroid hormone system.8–12 A noticeable pro-oxidant activity on several cells (erythrocytes lymphocyte, neurons, etc.) and on various cellular targets (DNA, protein and lipids) has been reported following PERM treatment.13–21 Particularly, a significant impairment of the enzymatic antioxidant system (i.e. superoxide dismutase, catalase and glutathione peroxidase activities) resulted in an animal model exposed to PERM.11–16,18,20
In particular, superoxide dismutases are metalloenzymes that can be classified depending on their metal selectivity (Cu–Zn, Fe, Ni, Mn-SOD). They are present in both prokaryotic and eukaryotic species and catalyze the formation of oxygen and hydrogen peroxide from superoxide through a one-electron redox cycle.22,23 Metal ions act as an intermediary for electronic transfer between two superoxide anions. At first, the enzyme is reduced by O2−, then it acts as a reducing factor by transferring the electron received just before to another O2−˙ and being ready for another cycle. The importance of SOD on cellular protection against aging and cancer has been demonstrated,24,25 as well as age-related diseases such as cataracts, neurodegenerative diseases and atherosclerosis, are strictly related with low levels of SOD.20,21 Besides, Muller et al. verified that the longevity of the knock out mice is directly related with the activity of Mn-SOD and Cu–Zn-SOD, while the inactivation of other antioxidant enzymes as catalase and glutathione peroxidase did not affect their life span.26
Taking into account that the overall conformation influences protein function, the aim of this study is to investigate the interaction between the Cu/ZnSOD and PERM or its metabolites, the 3-phenoxybenzyl alcohol (3-PBA), the 3-phenoxy benzaldehyde (3-PBALD), and the 3-phenoxybenzoic acid (3-PBACID) (Fig. 1) using intrinsic and 8-ANS fluorescence. Moreover the capability of PERM and its metabolites to affect the SOD enzyme activity was measured.
Finally, a computational investigation by using molecular docking protocols, was carried out in order to assess the molecular basis of interaction between SOD and permethrin or its metabolites.
Materials and methods
Materials
All reagents were of pure and analytical grade. 3-PBA, 3-PBALD, 3-PBACID and 8-anilino-1-naphthalene sulfonic acid (8-ANS) and Horse heart cytochrome-c (type IV) were purchased from Sigma Chemical Co. Technical grade (75:25, trans:cis; 94% purity) 3-phenoxybenzyl-(1R,S)-cis,trans-3-(2,2-dichlorovinyl)-2,2-dimethylcyclopropanecarboxylate (PERM) (NRDC 143) was generously donated by Dr A. Stefanini of ACTIVA, Milan, Italy.
P, were calculated by using CambridgeSoft ChemDraw Suite 2010.
:3) and stirred for at least 15 min. The precipitate was removed by centrifugation at 2500 rpm. The supernatant was collected and 50 ml l−1 of saturated lead acetate (450 g l−1) was added. The precipitate was removed by centrifugation at 9000 rpm and homogenized with about 1.5 l of 0.33 M KH2PO4, pH 6. The supernatant was dialyzed against 5 mM Tris-HCl buffer, pH 9, and then applied to a QAE-Sephadex A-50 column equilibrated with the same buffer. Elution of Cu/Zn SOD was performed with a linear pH gradient between 5 mM Tris-HCl buffer, pH 9 and 20 mM sodium cacodylate/HCl, pH 5. A single peak of a bluish-green protein was collected, concentrated and applied to a Sephadex G-75 column equilibrated with 0.1 M potassium phosphate buffer pH 7.4; Cu/Zn SOD was eluted with the same buffer and separated from minor contaminating proteins. The preparation showed a single band in sodium dodecylsulfate-polyacrylamide gel electrophoresis. Cu,Zn SOD activity was measured according to the method of McCord and Fridovich27 Polyacrylamide gel electrophoresis and activity staining of gels were carried out as previously described.28 Protein concentration was measured by the method of Lowry et al.29 Prussian Blue was precipitated following the procedure previously described.30 In this instance, a weighted amount of protein, wetted with 40 μl of supporting electrolyte, was deposited directly on the surface of the electrode. Horse heart cytochrome-c (type IV) was used without further purification as reference.
PERM, 3-PBA, 3-PBALD and 3-PBACID were dissolved in DMSO. A solution of 0.33 mg ml−1 SOD was incubated for 180 min at 37 °C in 1 ml of 10 mM Tris pH 7.4 in the presence of dimethylsulfoxide (10 μl DMSO) (control) or PERM or its metabolites (10 μl) at the final concentration of 10, 30, 50 or 70 μM. Data are presented as percentage (%) of the maximum absorbance peak (A) of the sample containing the compound with respect to its control.
Measurements of intrinsic SOD steady-state fluorescence were performed using a spectrofluorometer Hitachi 4500, at an excitation wavelength of 277 nm. Emission spectra were recorded from 290 to 370 nm every 30 min until 180 min of incubation.
In experiments with 8-ANS (1 μM final concentration) the excitation wavelength was 277 nm and the emission was recorded from 370 to 510 nm (ref. 31) every 30 min until 180 min of incubation.
| %SOD Inhibition = 100 − (ΔODs/ΔODb) |
Computational methods
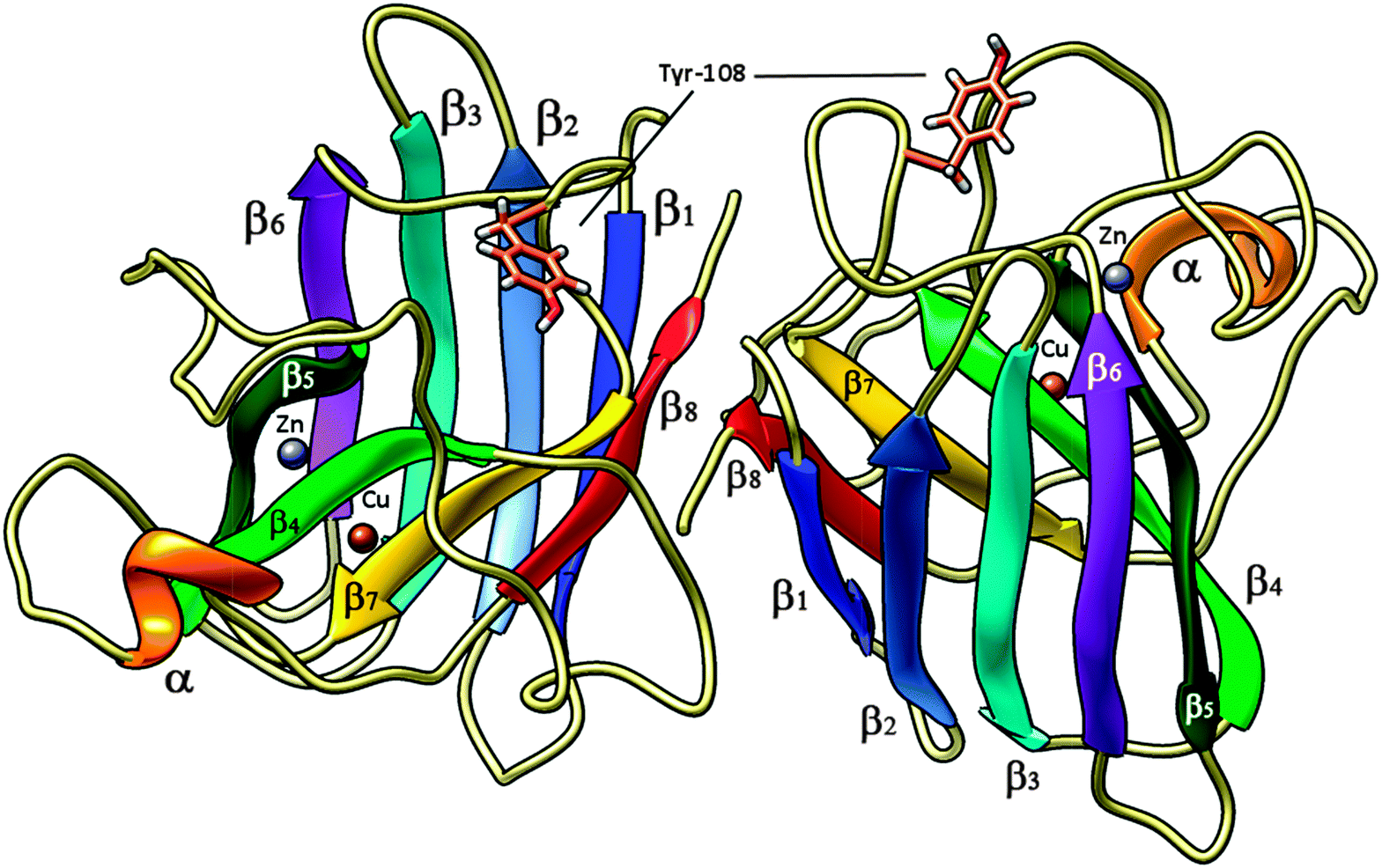 | ||
| Fig. 2 High pressure (0.57 Gpa) crystal structure of bovine copper,zinc superoxide dismutase at 2.0 Å (pdb code 2SOD) in its dimeric form; Tyr-108 side chain is shown in tubes – light brown, Cu and Zn ions are reported as spheres, brown and violet, respectively. α-helices are shown in orange, β1-sheets in blue, β2 in light blue, β3 in cyan, β4 in light green, β5 in dark green, β6 in purple, β7 in yellow and β8 in red. | ||
Thus, the chosen set of ligands were docked into minimized SOD with AutoDock Suite 4.2.38 At first, in order to assess the correct position of the ligand own binding pocket and to assess the presence of allosteric binding sites, we performed a full flexible Blind Docking centred on the target macromolecule. Its graphical front-end, AUTODOCKTOOLS,38 was used to add polar hydrogen atoms and partial charges for proteins, ligand charges were obtained at AM1 level and added manually. Atomic solvation parameters and fragmental volumes for the proteins were assigned using the ADDSOL tool (included in the program package). Flexible torsions in the ligands were assigned with the AUTOTORS module and all dihedral angles were allowed to rotate freely. In general, these were all acyclic, non-terminal single bonds (excluding amide bonds) in a given ligand molecule. Affinity grid fields were generated using the auxiliary program AUTOGRID.
The docking process was performed in two steps. In the first, the docking procedure was applied to the whole protein target, without imposing the binding site (Blind Docking).41,42
In this step, the grid field was formed by a grid box of (126 × 126 × 126) Å3, which includes the entire macromolecule, with spacing of 0.375 Å cent modified at the middle of the protein. The powerful genetic algorithm method implemented in the program Auto Dock 4.2 was employed.40
Particularly, the genetic algorithm-local search (GA-LS) hybrid was used to perform an automated molecular docking. Default parameters were used, except for the number of generations, energy evaluations, and docking runs, which were set to 200, 25000000, 150 respectively.
In the second step (Refined Focused Docking), after cluster analysis we started from the representative structure of each corresponding low energy cluster to go further with a focused docking to better positioning the ligand into its binding pocket using a Lamarckian genetic algorithm GA41 with a flexible ligand and a rigid receptor, a population size of 300, 10000000 evaluations, and a maximum of 27000 generations for 100 GA runs. This time, the grid field was a (44 × 44 × 44) Å3 cube with grid points separated by 0.34 Å centred on the best scored conformation obtained in the first step. Lennard-Jones parameters 12_10 and 12_6 (supplied with the program package) were used for modelling H-bonds and van der Waals interactions, respectively.
The resulting docked conformations were clustered into families of similar binding modes, with a root mean square deviation (RMSD) clustering tolerance of both 0.5 and 2 Å. In all cases the lowest docking-energy conformations were included in the largest cluster found (which usually contains 80–100% of total conformations). Otherwise, the lowest docking-energy conformations were considered as the most stable orientations. The docking energy represents the sum of the intermolecular energy and the internal energy of the ligand while the free-binding energy is the sum of the intermolecular energy and the torsional free energy.43 The VMD program was used for graphical interpretation and representation of results.44 The ligand–receptor binding interactions at the orthosteric binding sites were analyzed with CHIMERA software.45
The docked complexes binding energies were calculated by an empirical free energy force field with a Lamarckian genetic algorithm (LGA), which provides a fast prediction of conformation and free energy. In addition, the force field of AutoDock has been calibrated by using a database containing 188 various protein–ligand complexes and has exhibited a low standard error in estimating the experimental binding free energy which became lower if AM1 instead of Gasteiger–Marsili charges are used [GM]. The representative structure of each low energy cluster has been further minimized and stabilized by using full atoms micro molecular dynamics simulations (1 ns) in order to better refine the obtained docked complexes.33–35
These calculated free binding energies can be related to the Ki through the known thermodynamic law ΔG = −RTlnKi.
Data of Table 1 and Fig. 4 are from one representative set of tracings from at least three experiments.
| Sample | DMSO | 10 μM | 30 μM | 50 μM | 70 μM |
|---|---|---|---|---|---|
| PERM | 312 | 312 | 309 | 305 | 303 |
| 3-PBA | 317 | 308 | 304 | 303 | 300 |
| 3-PBALD | 317 | 316 | 317 | 312 | 326 |
| 3-PBACID | 315 | 315 | 312 | 315 | 316 |
The statistical analysis was performed using StatSoft Statistica 8.0 software by applying 1-way ANOVA followed by the Student–Newman–Keuls test. A P value <0.05 was considered statistically significant.
Results
Steady state fluorescence of SOD' tyrosines
Fig. 3A–D shows the changes of the fluorescence emission peak of SOD samples incubated with PERM and its metabolites versus their control. The presence of 10 μM PERM decreases the peak intensity, while an increase in the emission maxima is measured in the presence of 30, 50 and 70 μM PERM (P < 0.05) (Fig. 3A). A dose dependent blue shift from about 312 to 303 nm is measured as shown in Table 1. The change towards lower wavelengths means that PERM induces a conformational change of SOD and that tyrosine residues move near hydrophobic domains. Moreover the decrease of peak intensity at low PERM concentration, could be related to proton transfer from Tyr to near carbonyl or amino groups before the folding process observed at higher concentrations of PERM accordingly with the increase of the peak values. This outcome remains unchanged for all incubation times (data not shown). Fig. 3B shows the fluorescence emission spectra of SOD in the presence of 3-PBA. This metabolite increases significantly in a dose dependent manner from 10 μM to 50 μM in the peak emission spectra (P < 0.05). There is a significant progressive blue shift from 317 nm to 300 nm (Table 1). When samples were incubated for 180 min a similar behaviour was observed for the peak values (data not shown). These data are associated with the shift towards a more apolar environment and to the folding of the protein due to 3-PBA.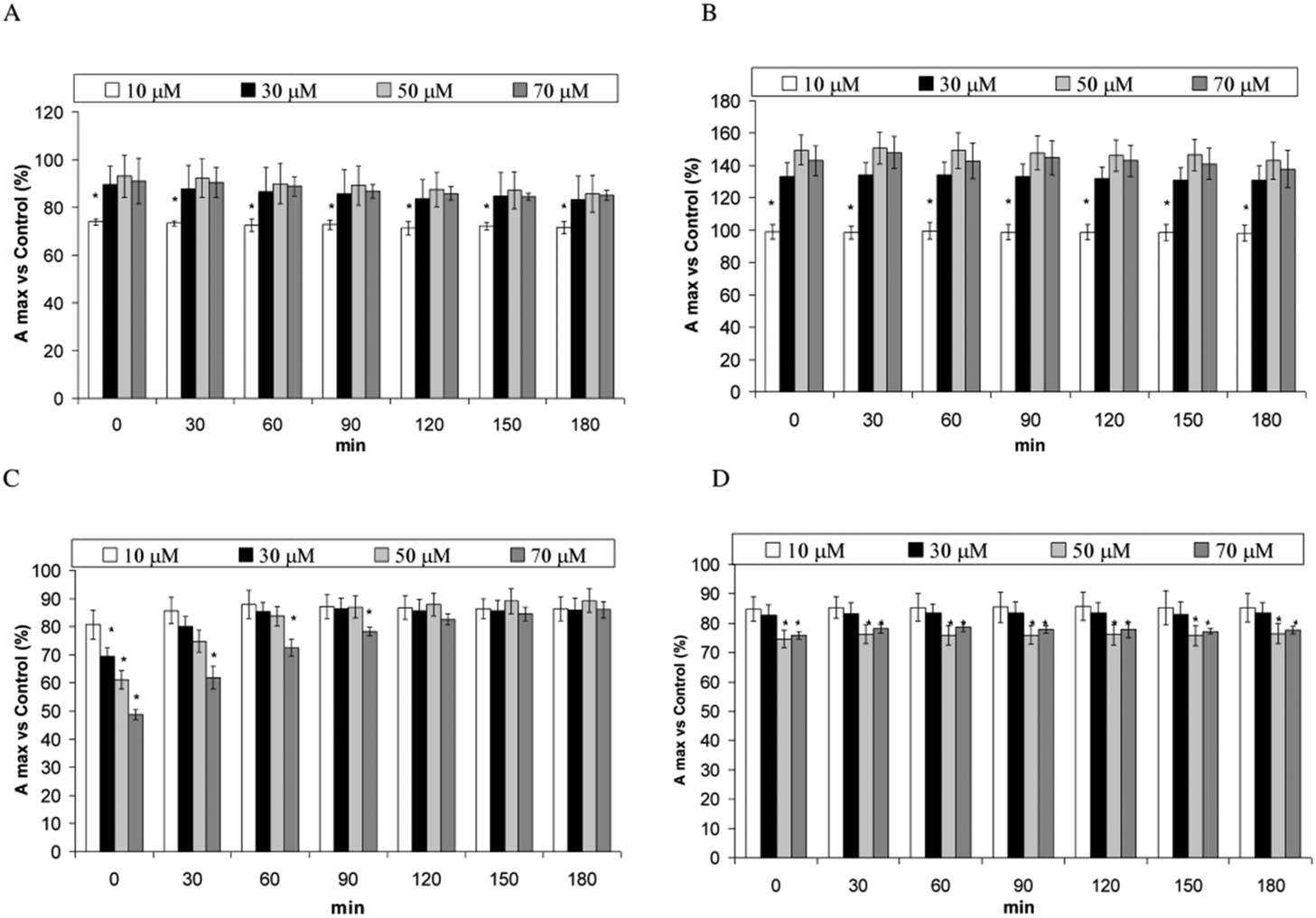 | ||
| Fig. 3 Fluorescence emission spectra of SOD in the presence of 10, 30, 50 and 70 μM PERM (A), 3-PBA (B), 3-PBALD (C) and 3-PBACID (D) measured at 37 °C, and the changes of fluorescent emission peak of the SOD samples incubated at 37 °C with 10, 30, 50 and 70 μM of PERM admits metabolites vs. control measured each 30 min for 3 h. *P < 0.05 vs. other concentrations. | ||
The presence of 3-PBALD induces the opposite effect with a dose dependent decrease of emission spectra of Tyr fluorescence (Fig. 3C); this effect disappears over time, showing no significant differences after 90 min of incubation. During the experiment no shift in the maximum peak was observed until 50 μM, and the increase reported at 70 μM should be considered in accordance with the plateau observed in the emission spectra (Table 1). These results indicate that 3-PBALD could lead to a more unfolded protein in a domain with the same polarity. However the presence of amino acid residues like Asp-25 could also facilitate the proton transfer to this residue decreasing peak intensity.
A similar folding can be observed in the presence of 3-PBACID that induces a dose dependent weak decrease on the Tyr emission spectra during the entire incubation time at low 3-PBACID concentrations (Fig. 3D) and a 3 nm shift was observed only in the 30 μM sample (Table 1). However in this case no proton transfer can be suggested.
Since pH is a key factor that influences the structure and function of enzymes, we verified that the addition of various amounts of PERM or its metabolites to the sample containing SOD, did not modify the pH where fluorescence measurements were performed. pH data remained constant in all samples at each time (data not shown), indicating that the observed changes cannot be due to any pH variations of buffer where experiments were performed.
Spectra at 0 and 180 min of incubation of SOD with PERM or its metabolites show that only the metabolite 3-PBA induces an increase of 8-ANS fluorescence emission after 180 min of incubation (Fig. 4B). No changes were reported in the presence of the PERM metabolites (data not shown).
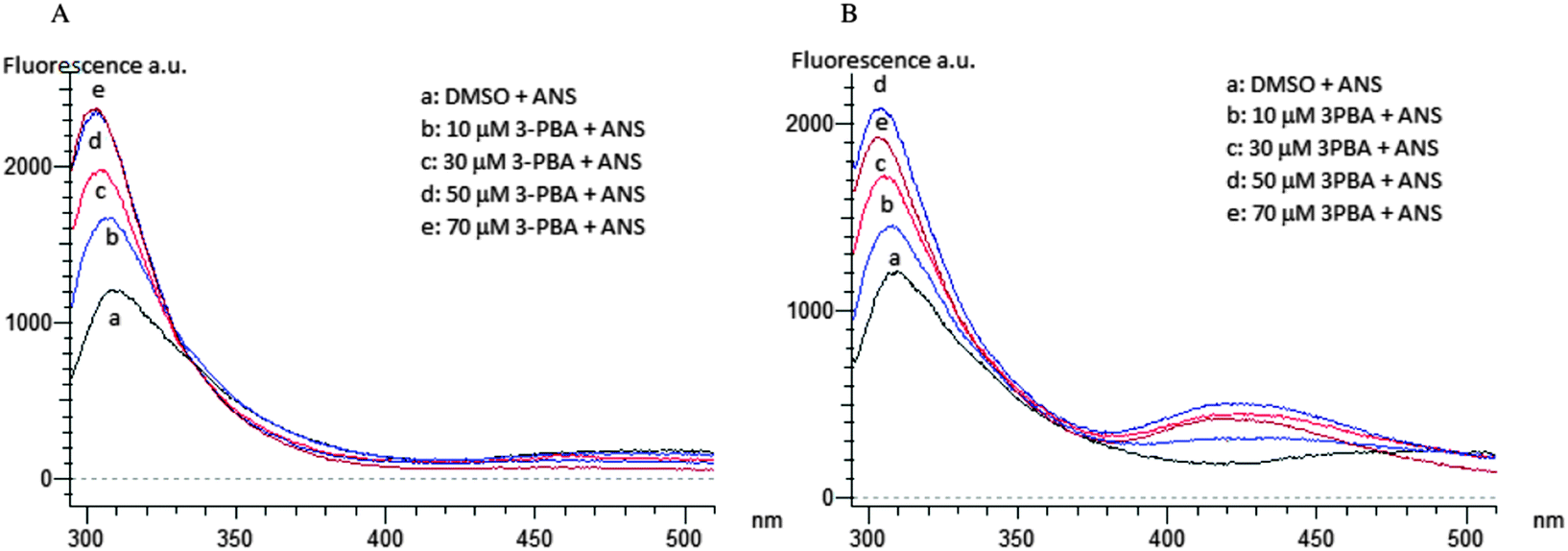 | ||
| Fig. 4 ANS fluorescence emission spectra of SOD incubated for 0 min (A) and 180 min (B) at 37 °C with 10, 30, 50 and 70 μM of 3-PBA. | ||
Computational and docking results
At first in order to assess the means of interaction between SOD and permethrin or its metabolites, we performed an initial screening using the Blind Docking protocol (see Computational methods section). The ligands, i.e. permethrin (PERM), 3-PBA, 3-PBACID and 3-PBALD, were treated separately. For each ligand we pointed out four allosteric areas of interaction. The peculiar feature is that each representative structure of the obtained clusters has a very close binding energy. Anyway, as a regular computational protocol, we investigated further, proceeding with the described focused docking method and we finally localized the exact poses and conformations for all the ligands considered. As a result, it is clear that all the molecules considered have more than one allosteric binding site but none of them interacts with SOD at the catalytic Cu,Zn cleft. This fact explains perfectly the experimental data activity which remains unchanged in the presence of all of the compounds.Moreover, all these binding poses are very close in binding energy and these results led us to conclude that there is not only a preferred binding site but that most of them (those with energy within 0.5–0.7 kcal mol−1) are important due to their relative energy in equilibrium with a population strictly connected with the ligand concentration. In particular, PERM interacts with SOD in seven different zones spread through all the enzyme surface (Fig. 5) (Ebinding between −7.04/−5.33 kcal mol−1, in which five are within 0.6 kcal mol−1).
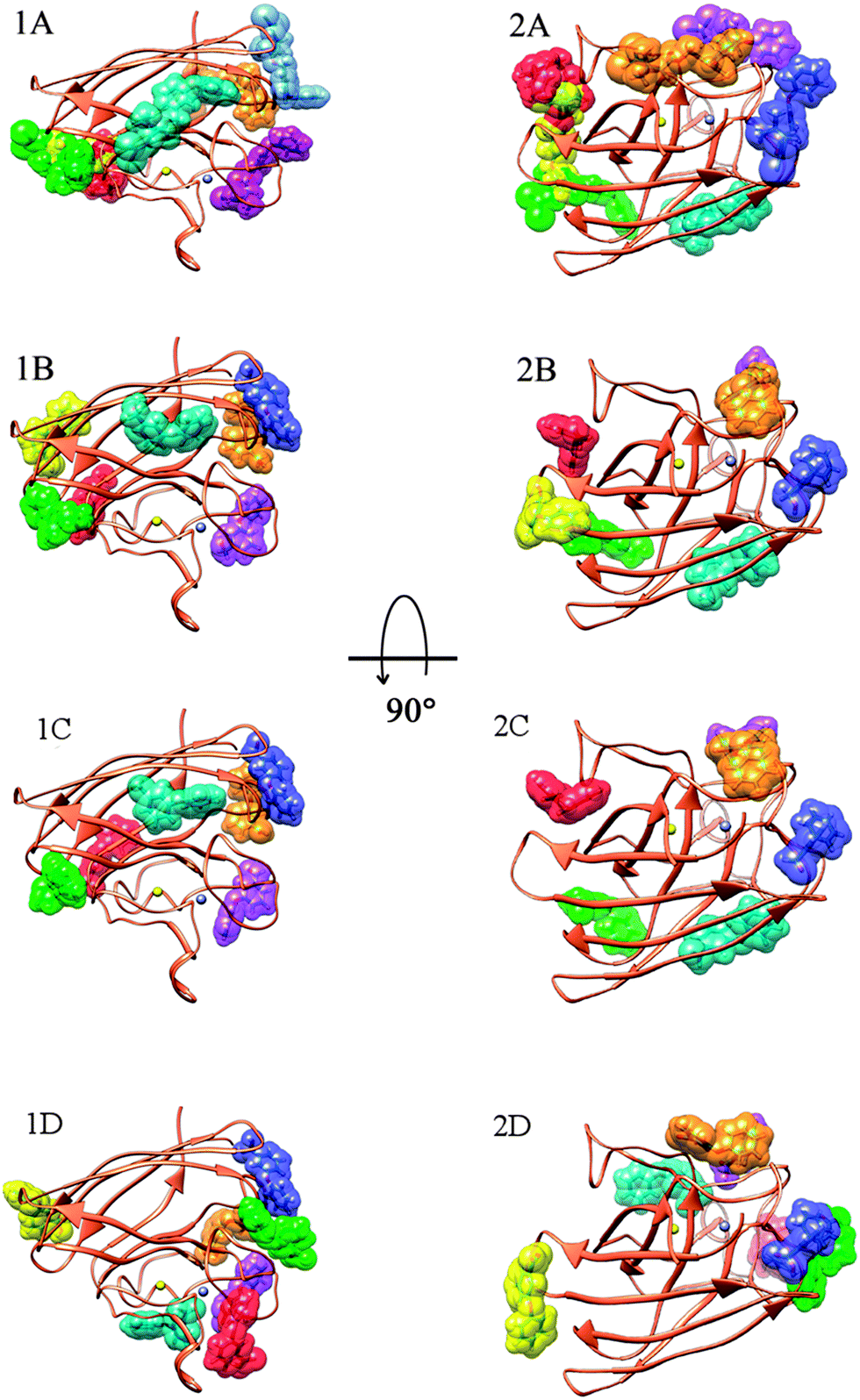 | ||
| Fig. 5 1A and 2A: SOD enzyme, in coral, and permethrin PERM in its allosteric sites of interaction. The interaction sites are highlighted representing the ligand in van der Waals spheres colored differently and show how permethrin is able to encircle the enzyme. Only energy accessible sites of interactions are reported (ΔE within 0.7 kcal mol−1). Zn and Cu ions, are represented as spheres in violet and yellow, respectively; 1B and 2B: SOD enzyme, in coral, and 3-PBA its allosteric sites of interaction. The interaction sites are highlighted representing the ligands in van der Waals spheres colored differently and show how the ligand is able to encircle the enzyme. Only energy accessible sites of interactions are reported (ΔE within 0.7 kcal mol−1). Zn and Cu ions, are reported as spheres in violet and yellow, respectively; 1C and 2C: SOD enzyme, in coral, and for 3-PBALD its allosteric sites of interaction. The interaction sites are highlighted representing the ligand in van der Waals spheres colored differently and show how the ligand is able to encircle the enzyme (ΔE within 0.7 kcal mol−1). Only energy accessible sites of interactions are reported. Zn and Cu ions, are reported in violet and yellow, respectively; 1D and 2D: SOD enzyme, in coral, and 3-PBACID its allosteric sites of interaction. The interaction sites are highlighted representing the ligand in van der Waals spheres colored differently and show how the ligand is able to encircle the enzyme. Only energy accessible sites of interaction are reported (ΔE within 0.7 kcal mol−1). Zn and Cu ions, are reported in violet and yellow, respectively. | ||
Analyzing in detail the interactions of ligands with SOD amino acids, it should be pointed out that in every populated pose, PERM and its metabolites are all involved in many hydrogen bonds through their polar oxygen moieties but due to the presence of a common aromatic hydrophobic core and to the SOD nature rich in apolar amino acids, many hydrophobic interaction contribute to the complex stabilization. In more detail, we focused on the specific binding interaction involved in the allosteric site near the Tyr-108 residues. This, in particular, is a key point since experimental evidence of changes arise from the fluorescent emission of the aromatic Tyr-108 and they can be connected to a shift in both conformation and position of this amino acid.
The same docking results have been obtained for 3-PBA and 3-PBACID, pointing out seven energy accessible poses while for 3-PBALD we found out only six binding sites for SOD association; for all the PERM metabolites we can point out a very small energy ranges (ΔE) between sites thus corresponding to an almost equal population (−5.67/−4.71 kcal mol−1) (Table 2).
| Compound | E b cl1 | E b cl2 | E b cl3 | E b cl4 | E b cl5 | E b cl6 | E b cl7 |
|---|---|---|---|---|---|---|---|
| PERM | −6.92 | −7.03 | −5.96 | −6.95 | −5.33 | −7.04 | −6.46 |
| 3-PBA | −5.35 | −4.99 | −5.1 | −5.06 | −5.09 | −4.75 | −4.8 |
| 3-PBALD | −5.4 | −4.84 | −4.71 | −5.01 | −4.86 | −4.74 | — |
| 3-PBACID | −5.67 | −5.05 | −5.11 | −5.18 | −4.74 | −4.77 | −4.73 |
In fact, for metabolites 3-PBALD and 3-PBACID, a decrease of the emission fluorescence signal is registered, instead for PERM and 3-PBA an increase of the same signal is observed. These results can be put into direct relation with the Tyr-108 side chain position which is different for each molecule considered. As can be seen in Fig. 2 and 6, this tyrosine residue is positioned within the enzyme in a highly hydrophobic region where residues Ile-101, Ile-102, Leu-104, Ile-110 and Ile-111 can be found. Despite this fact, in the free enzyme, its side chain aromatic ring is not involved in any intramolecular interaction but is instead free and oriented externally (Fig. 7). Thus, it is highly accessible both to solvent and to the approaching ligands. As a result for each compound considered we can point out a low energy cluster corresponding to the ligand positioning in this allosteric site which corresponds to cluster 6 for PERM, cluster 2 for 3-PBA, cluster 2 for 3-PBALD and cluster 1 for 3-PBACID; see Table 2.
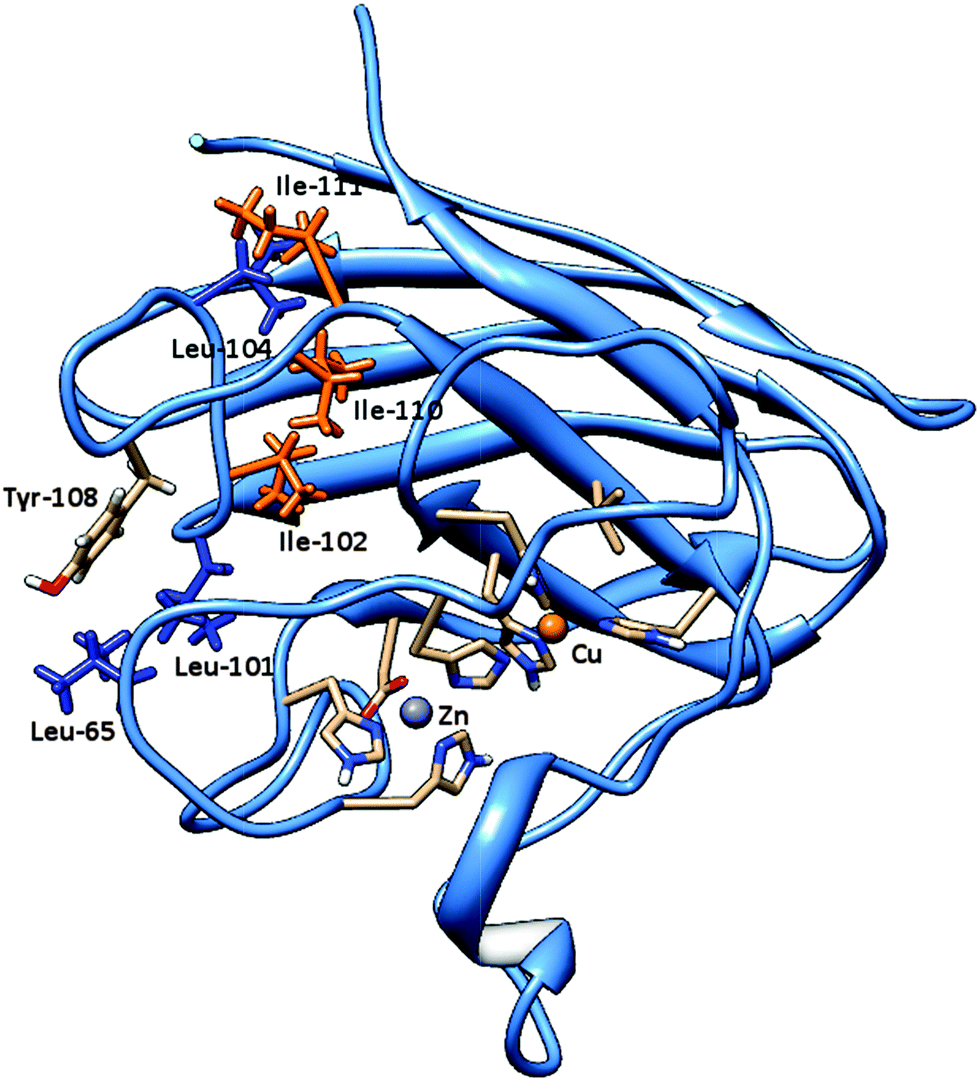 | ||
| Fig. 6 The SOD 3D structure is shown focusing onto the catalytic site position (Zn ion in violet, Cu ion in coral) with respect to Tyr-118. Its aromatic side chain is visible in orange whilst in yellow the loop backbone in which it can be found together with the hydrophobic residues Leu-101, Ile-102, Leu-104, Ile-110 and Ile-111. | ||
 | ||
| Fig. 7 Tyr-108-site Docked structures of SOD with PERM (cyan), 3-PBA (blue), 3-PBALD (violet) and 3-PBACID (yellow); the ligand binding interaction is represented in detail. Zn and Cu ions are represented as spheres in violet and brown, respectively. | ||
Observing in depth these binding regions, we can point out that both 3-PBALD and 3-PBACID (downside in Fig. 5C and D) interact with Tyr-108 on the opposite orientation and side with respect to PERM and 3-PBA (uppersite in Fig. 5A and B).
This can be ascribed to the presence of different preferred non-bonding interactions of the two groups of compounds, which are herein described in detail.
Considering first PERM and 3-PBA, we can point out that both share common interactions: they are involved in hydrogen bonding with Asn-63 (as acceptors) and hydrophobic interactions with Phe-62 and, worthy of more notice, with Tyr-108.
In greater detail, for PERM, the ligand makes two H-bonds with SOD: the first occurs between its ether-aromatic oxygen (acceptor) and amino group of Arg-113 residue (in β7) (donor) (d = 2.619 Å); the second, instead, occurs between chetonic oxygen of permethrin (acceptor) and the amino group of Asn-63 (located between β4 and β5) (donor) (d = 1.961 Å). Furthermore, two strong π-stacking interactions can be observed, the first face-to-face with the Phe-62 side chain (located between β4 and β5), second face-to-edge as expected with Tyr-108 residue.
For 3-PBA, we can point out the presence of three H-bonds with SOD, two of them occur between the hydrogen of the alcoholic group of 3-PBA (as donor) and the first with the carbonylic oxygen of His-61 residue (located between β4 and β5) (acceptor) (d = 1.925 Å), the second with the amino group of Asn-63 residue (located between β4 and β5) (as acceptor, d = 2.523 Å); the third involves ether-aromatic oxygen of the ligand (acceptor) and backbone N–H of Asn-63 (donor) (d = 2.055 Å). Beside, 3-PBA also makes important hydrophobic interactions; more precisely, CH–π interaction between the aromatic moiety of 3-PBA and the CH2 group of Asn-63, and two edge-to-face π-stacking interactions, the first with Phe-62 side chain (located between β4 and β5), the second, once again, with Tyr-108 (located between β6 and β7).
Besides, considering the intermolecular interactions of the second group, 3-PBACID and 3-PBALD in the correspondent cluster close to Tyr-108, we can immediately notice that both compounds are less involved in hydrogen bonding interactions, while sharing only a hydrophobic interaction, the first with Leu-101 and the second with the aromatic moiety of Tyrosine, but in the opposite site with respect to the PERM and 3-PBA orientation.
Analyzing in more detail the ligands' interactions (Fig. 7), for 3-PBACID, we observe the presence of a H-bond, between its carboxylic moiety and Thr-26 (located between β2 and β3) (2.019 Å), and two hydrophobic interactions. The latter is a CH–π interaction with Leu-101, and an edge-to-face π-stacking interaction with Tyr-108 (notice that both these residues are located between β6 and β7). Finally, in 3-PBALD's cluster, the ligand, as already observed for 3-PBACID, makes one H-bond, involving a carboxylic oxygen of the metabolite and the backbone N–H moiety of Asp-25 (located between β2 and β3), (d = 1.905 Å). Also the hydrophobic interactions present are the same as for 3-PBACID, resulting in a CH–π interaction with Leu-101, and an edge-to face π-stacking interaction with Tyr-108 residue.
From the overall data analysis, we can evince that PERM and 3-PBA make a greater number of H-bonds compared to 3-PBACID and 3-PBALD. This propensity induces the binding of the first two compounds in a more polar region with respect to the last two, which are more deeply inserted into the “hydrophobic pocket” (Fig. 6) of SOD. Moreover, all of them are involved in π-stacking interaction with Tyr-108 aromatic moiety, but while both permethrin and 3-PBA (that are correlated to fluorescence increase) strongly interact with Phe-62, Asn-63 residues (located between β4 and β5), 3-PBACID and 3-PBALD (that correlate to fluorescence decrease) interact with Leu-101 and the nearby apolar residues (located in another secondary domain between region β6 and β7). Thus, the overall protein conformational changes induced from Tyr-108-mediated-hydrophobic interactions in different zones of the enzymatic domains (β4 and β5vs. β6 and β7) is confirmed by ligand–SOD complex energy minimization followed by atomistic molecular dynamics simulation33–35 carried out by using AMBER force field46 and including the solvent water effect explicitly (TIP3P model).47
Discussion
Bovine superoxide dismutase is a Cu/Zn SOD type with a relative molecular mass of 31200, composed by two dimers of 151 amino acidic residues (Fig. 2). The two identical active sites containing the metal ions are located 3.4 nm apart.48–50 Copper ion is situated at the bottom of a deep channel bound to three histidines (His-44, His-46, and His-118) and its role is strictly related with the catalytic activity of the enzyme, while zinc ion, important for enzyme stability, is completely buried and bound to two histidines (His-69 and His-78) and to an aspartic acid residue (Asp-81). These two metal ions share an imidazolate ligand (His 61).50–52 Each dimer contains a tyrosine residue localized in 108 position (Fig. 6), strictly related with the intrinsic fluorescence, while tryptophan is missing in both chains. The absence of Trp and the presence of one Tyr in each subunit makes this protein a good model for fluorescence studies in the particular region around the aromatic amino acid.53,54 Tyrosine residues play a special role in several biochemical processes such as the phosphorylation carried out by tyrosine phosphorylase; besides, conformational change of this amino acid in an active site is often related to activity and kinetic alterations of enzymes.55 Conformational changes of the enzyme are related to a variation in the fluorescence emission intensity of aromatic amino acids in the protein. In this study we used Tyr-108 as intrinsic probe to investigate the dynamics of bovine Cu/Zn-SOD following incubation with permethrin and its metabolites.
Results show that both, PERM and metabolites are capable of inducing conformational variation on SOD and those changes are present during all the experimental time. Tyr fluorescence intensity was decreased by 10 μM PERM and in a dose dependent manner in the presence of 3-PBALD and 3-PBACID. This data could be related to different preferred non-bonding interactions between these compounds and the protein surface residues which are observed from in silico results (see Computational results section). The decrease of emission signal measured in samples incubated with 3-PBALD and 3-PACID metabolites (Fig. 3), indicates a protein–unfolding process which can be related not only to π-stacking interaction with Tyr-108 side-chain but also to H-bonding with a backbone N–H in the β2–β3 region and other hydrophobic interaction in the β6–β7 region.
On the other hand a dose-dependent increase of fluorescence emission intensity due to PERM and 3-PBA (Fig. 3) could be related to a protein folding process.56 The wrapping, also confirmed by the blue shift proclivity, is more evident in 3-PBA than in permethrin although the first compound is less hydrophilic than the second one (Table 1). This SOD conformational change could be related to the shift of the two Tyr-108 to a more hydrophobic region of the molecule, where more hydrophobic residues, such as Leu-101, Ile-102, Leu-104, Ile-110 and Ile-111, are present.57 This conformational change is indeed mediated by PERM and 3-PBA strong hydrophobic interactions in the β4–β5 domain as results from docking analysis. Moreover the conformational variation due to 3-PBA, is in addition evidenced also by a moderate increase of 8-ANS fluorescence emission due to interaction of the probe with protein hydrophobic amino acidic residues after 180 min of incubation (Fig. 3).
Thus, having observed the presence of important conformational changes occurring in the presence of all the considered compounds, we went further in studying a possible change in enzyme activity being the overall protein conformation related to it.
Indeed, SOD is characterized by catalytic efficiency and remarkable stability due to several factors such as the prosthetic metal ions,58 the intrasubunit disulfide bond,48 and the close packing of the hydrophobic interface between the subunits and the two halves of the β-barrel core.59 The Greek key eight-stranded β-barrel is very important for the biological function of SOD and structural changes in β1, β4, β7 and β8, might induce the reduction or lose of activity of SOD (thermal dissociation and conformational lock of superoxide dismutase). The upon cited, His-44, His-46, and His-118, situated in the active site and very important for the catalytic function, are located in β4 (His-44, His-46) and β7 (His-118), so spatial structure modifications of β4, β7 cause changes in the active site that can lead to the reduction or loss of activity. It has been reported that the intra-subunit disulfide bond between Cys144 and Cys55 is very important for maintaining the spatial structure and compactness of SOD48 and Cys-144 is located on β8, for this reason spatial changes of β8 that affect the disulfide bond, cause the reduction of compactness and stability of SOD.
Thus, in order to evaluate the eventual influence of the conformational modification on enzyme function, we assessed the SOD activity and no changes in its activity were measured. This outcome is due to the spatial position of Tyr-108, whose movement is not able to affect the accessibility of the catalytic cleft related with the enzyme activity in the experimental conditions used nor can PERM and its metabolites. In fact, computational results confirm that all the molecules considered have more than one allosteric binding site but none of them interacts with SOD at the catalytic Cu/Zn cleft. Besides although the Ile-111 is one of the residues involved in the contact areas between the two subunits, and 3-PBA and PERM induce a SOD blue shift of Tyr-108 toward the Ile-111, no effect on enzyme activity can be measured because this residue is not determinant for catalytic activity of SOD. This fact explains perfectly the experimental data on SOD activity which remains unchanged in the presence of all compounds.
Since human SOD is characterized by several similarities with bovine SOD, structural information obtained on bovine SOD can be extended also to human enzymes, because they do not depend on the specific nature and on the position of the intrinsic fluorescence probe (Tyr-108 in bovine SOD versus Trp 32 in human SOD).56 This fluorescence technique together with computational and docking results, could be very useful for increasing our knowledge of protein dynamic events in the presence of different exogenous molecules.53,54
Conclusions
PERM and its metabolites are able to interact with SOD in 6/7 allosteric sites very close in binding energy. PERM and 3-PBA interact with Tyr-108 in different regions compared to 3-PBALD and 3-PBACID, which explains the differences in the fluorescence measured.Although these residues have no influence on the catalytic site activity, the conformational changes observed should be considered as an insight of “in vivo” longer and constant exposition to insecticides, where even other factors could contribute to affect the conformation and consequently the SOD functionality.
Notes and references
- M. Bjørling-Poulsen, H. R. Andersen and P. Grandjean, Environ. Health, 2008, 7, 50 CrossRef PubMed.
- S. M. Bradberry, S. A. Cage, A. T. Proudfoot and J. A. Vale, Toxicol. Rev., 2005, 24, 93 CrossRef CAS PubMed.
- S. Hussain, T. Masud and K. Ahad, Pak. J. Nutr., 2002, 1, 41 CrossRef.
- M. C. Fortin, G. Carrier and M. Bouchard, Environ. Health, 2008, 7, 55 CrossRef PubMed.
- G. Leng, W. Gries and S. Selim, Toxicol. Lett., 2006, 162, 195 CrossRef CAS PubMed.
- A. R. McCarthy, B. M. Thomson, I. C. Shaw and A. D. Abell, J. Environ. Monit., 2006, 8, 197 RSC.
- D. Noort, A. van Zuylen, A. Fidder, B. van Ommen and A. G. Hulst, Chem. Res. Toxicol., 2008, 21, 1396 CrossRef CAS PubMed.
- J. S. Taylor, B. M. Thomson, C. N. Lang, F. Y. T. Sin and E. Podivinsky, J. Toxicol. Environ. Health, Part A, 2010, 73, 1075 CrossRef CAS PubMed.
- H. Bouwman and H. Kylin, Environ. Health Perspect., 2009, 117, 1477 CAS.
- D. M. S. Vadhana, C. Nasuti and R. Gabbianelli, Cardiovasc. Toxicol., 2010, 10, 199 CrossRef PubMed.
- R. Gabbianelli, M. L. Falcioni, C. Nasuti, F. Cantalamessa, I. Imada and M. Inoue, Chem.-Biol. Interact., 2009, 182, 245 CrossRef CAS PubMed.
- C. Parker, D. R. Patterson, G. A. van Gelder, E. B. Gordon, M. G. Valerio and W. C. Hall, J. Toxicol. Environ. Health, 1984, 13, 83 CrossRef CAS PubMed.
- C. Nasuti, F. Cantalamessa, G. Falcioni and R. Gabbianelli, Toxicology, 2003, 191, 233 CrossRef CAS.
- C. Nasuti, M. L. Falcioni, I. E. Nwankwo, F. Cantalamessa and R. Gabbianelli, Toxicology, 2008, 251, 45 CrossRef CAS PubMed.
- R. Gabbianelli, G. Falcioni, C. Nasuti and F. Cantalamessa, Toxicology, 2002, 175, 91 CrossRef CAS.
- R. Gabbianelli, C. Nasuti, G. Falcioni and F. Cantalamessa, Toxicology, 2004, 203, 17 CrossRef CAS PubMed.
- R. Gabbianelli, M. L. Falcioni, F. Cantalamessa and C. Nasuti, J. Appl. Toxicol., 2009, 29, 317 CrossRef CAS PubMed.
- I. Chargui, M. L. Falcioni, H. B. Cheikh and R. Gabbianelli, Environ. Toxicol. Pharmacol., 2010, 30, 116 CrossRef CAS PubMed.
- M. L. Falcioni, C. Nasuti, C. Bergamini, R. Fato, G. Lenaz and R. Gabbianelli, Neuroscience, 2010, 168, 2 CrossRef CAS PubMed.
- D. M. S. Vadhana, M. Carloni, C. Nasuti, D. Fedeli and R. Gabbianelli, Exp. Gerontol., 2011, 46, 731 CrossRef PubMed.
- D. M. S. Vadhana, M. Carloni, C. Nasuti and R. Gabbianelli, Cardiovasc. Toxicol., 2011, 11, 226 CrossRef CAS PubMed.
- M. B. Yim, P. B. Chock and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5006 CrossRef CAS.
- A. L. Brioukhanov and A. I. Netrusov, Biochemistry, 2004, 69, 949 CAS.
- B. Halliwell and J. M. Gutteridge, Hum. Exp. Toxicol., 1988, 7, 7 CAS.
- B. S. Berlett and E. R. Stadtmani, J. Biol. Chem., 1997, 272, 20313 CrossRef CAS PubMed.
- F. L. Muller, M. S. Lustgarten, Y. Jang, A. Richardson and H. Van Remmen, Trends in oxidative aging theories, Free Radical Biol. Med., 2007, 43, 477 CrossRef CAS PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049 CAS.
- F. Marmocchi, G. Venardi, G. Caulini and G. Rotilio, FEBS Lett., 1974, 44, 337 CrossRef CAS.
- O. H. Lowry, N. J. Rosenbrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265 CAS.
- P. J. Kulesza, M. A. Malik, S. Zamponi, M. Berrettoni and R. Marassi, J. Electroanal. Chem., 1995, 397, 287 CrossRef.
- O. K. Gasymov and B. J. Glasgow, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 403 CrossRef CAS PubMed.
- H. P. Misra and I. Fridovich, J. Biol. Chem., 1972, 247, 6960 CAS.
- Suite 2011: MacroModel, Version 9.9., Schrödinger, LLC, New York, 2011.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- J. C. Gilbert and J. Nocedal, SIAM J. Optim., 1992, 2, 21 CrossRef.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef CAS.
- G. B. Rocha, R. O. Freire, A. M. Simas and J. J. Stewart, J. Comput. Chem., 2006, 27, 1101 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. R. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- O. Trott and A. J. Olsen, J. Comput. Chem., 2010, 31, 455 CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- R. Galeazzi, L. Massaccesi, F. Piva, G. Principato and E. Laudadio, J. Mol. Model., 2014, 20, 2120 CrossRef PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- W. K. den Otter and W. J. Briels, J. Chem. Phys., 1998, 109, 4139 CrossRef CAS PubMed.
- VMD visualization software: W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- W. Junmei, M. Romain, J. W. Wolf, P. A. Caldwell and D. A. Kollman, J. Comput. Chem., 2004, 25, 1157 CrossRef PubMed.
- W. L. Jorgensen, J. Comput. Chem., 1998, 19, 1179 CrossRef CAS.
- J. L. Abernethy, H. M. Steinman and R. L. Hill, J. Biol. Chem., 1974, 249, 7339 CAS.
- H. M. Steinman, V. R. Naik, J. L. Abernethy and R. L. Hill, J. Biol. Chem., 1974, 249, 7326 CAS.
- J. Richardson, K. A. Thomas, B. H. Rubin and D. C. Richardson, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1349 CrossRef CAS.
- K. G. Strothkamp and S. J. Lippard, Acc. Chem. Res., 1982, 15, 318 CrossRef CAS.
- J. A. Tainer, E. D. Getzoff, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 284 CrossRef CAS.
- S. T. Ferreira, L. Stella and E. Gratton, Biophys. J., 1994, 66, 1185 CrossRef CAS.
- I. Ascone, C. Savino, R. Kahn and R. Fourme, Acta Crystallogr., 2010, 66, 654 CAS.
- P. Limphong, N. E. Adams, M. F. Rouhier, R. M. McKinney, M. Naylor, B. Bennett, C. A. Makaroff and M. W. Crowder, Biochemistry, 2010, 49, 8228 CrossRef CAS PubMed.
- L. J. Libertini and E. W. Small, Biophys. J., 1985, 47, 765 CrossRef CAS.
- S. Chen, X. Li and H. Ma, ChemBioChem, 2009, 10, 1200 CrossRef CAS PubMed.
- H. S. Forman and I. Fridovich, J. Biol. Chem., 1973, 248, 2645 CAS.
- E. D. Getzoff, J. A. Tainer, M. M. Stempien, G. I. Bell and R. A. Hallewell, Proteins: Struct., Funct., Genet., 1989, 5, 322 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |