
CIP2A regulates cancer metabolism and CREB phosphorylation in non-small cell lung cancer†
Bo
Peng
a,
Ningjing
Lei
a,
Yurong
Chai
a,
Edward K. L.
Chan
b and
Jian-Ying
Zhang
*a
aBorder Biomedical Research Center & Department of Biological Sciences, The University of Texas at El Paso, El Paso, Texas 79968, USA. E-mail: jzhang@utep.edu; Fax: +1-915-747-5808; Tel: +1-915-747-6995
bDepartment of Oral Biology, The University of Florida, Gainesville, Florida, USA
First published on 10th October 2014
Abstract
The cancerous inhibitor of protein phosphatase 2A (CIP2A) is a recently characterized endogenous inhibitor of the phosphatase activity of protein phosphatase 2A (PP2A), which extends the half-life of oncogenic protein c-myc and promotes in vivo tumor growth. The function of CIP2A in cancer progression is still poorly understood. To uncover the underlying mechanism of CIP2A-mediated cell proliferation, we implemented a two-dimensional electrophoresis (2DE)-based proteomic approach to examine lung cancer cell H1299 with and without CIP2A. We found 47 proteins differentially expressed where 19 proteins were upregulated and 28 proteins were downregulated. These were categorized into functional groups such as metabolism (25%), transcriptional and translational control (23%), and the signaling pathway and protein degradation (20%). On one hand, we validate our proteomic work by measuring the metabolic change. The knockdown of CIP2A decreased the expression of LDH-A as well as the enzymatic activity, accompanying with a decreased lactate production, an increased NADH/NAD+ ratio and ROS production. On the other hand, we found that CIP2A may regulate CREB activity through bioinformatics analysis. Our following experiments showed that, CIP2A positively regulated the phosphorylation of CREB in response to the serum treatment. Therefore, our proteomic study suggested that CIP2A mediates cancer progression through the metabolic pathway and intracellular signaling cascade.
1. Introduction
Lung cancer is one of the most common cancer types, and is the major cause of cancer-related deaths throughout the world in recent years. According to the latest data from American Cancer Society, there were 221![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 130 new cases of lung cancer (both small cell and non-small cell) in the United States in 2011 and 156940 people died from it in the same year. Although the etiological factors causing lung cancer have not been well defined, like many other types of cancer, some common genetic lesions have been identified as important triggers for lung cancer formation. Oncogenes like the epidermal growth factor receptor (EGFR), V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (K-ras), c-myc, and tumor suppressors, APC, LKB1, p53, and protein phosphatase 2A (PP2A) are the most commonly mutated genes in lung cancer.1–4
130 new cases of lung cancer (both small cell and non-small cell) in the United States in 2011 and 156940 people died from it in the same year. Although the etiological factors causing lung cancer have not been well defined, like many other types of cancer, some common genetic lesions have been identified as important triggers for lung cancer formation. Oncogenes like the epidermal growth factor receptor (EGFR), V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (K-ras), c-myc, and tumor suppressors, APC, LKB1, p53, and protein phosphatase 2A (PP2A) are the most commonly mutated genes in lung cancer.1–4
PP2A, a large protein family with intrinsic serine/threonine phosphatase activity, is a known tumor suppressor by targeting cancer-related molecules or pathways like AKT, mitogen-activated protein kinase (MAPK), and beta-catenin.5 Inhibition of PP2A phosphatase activity has been found to be a prerequisite for cellular transformation in most epithelial cells.6 PP2A can be inactivated by exogenous or endogenous factors. Chemicals like okadaic acid, viral proteins like small t antigen and cellular proteins like I1PP2A and I2PP2A have been identified as PP2A inhibitors and therefore promote cellular transformation and cancer progression.4 However, few endogenous cellular PP2A inhibitors have been identified so far.
CIP2A was a recently identified endogenous PP2A inhibitor in human malignancies.7 It was previously identified as a tumor-associated autoantigen in gastric cancer.8 Although this protein was tightly regulated during development, its function was largely unknown due to the lack of homologies to any other function-known proteins at the time of identification. In search of PP2A interacting proteins in HeLa cells, Junttila et al. used the PP2A A subunit as the bait in tandem-affinity purification and identified p90 as the endogenous interacting proteins in cancer cells. The knockdown of this protein impaired cell proliferation and in vivo tumor growth. CIP2A exerts tumor-promoting function by inhibiting c-myc-associated PP2A phosphatase activity, thus named the cancerous inhibitor of PP2A (CIP2A). Following studies by examining the tissue specimen of different cancer types revealed that CIP2A was overexpressed in various cancer tissues but not in adjacent normal tissue. Although CIP2A is able to inhibit PP2A phosphatase activity, previous data strongly suggest that it has substrate selectivity since the phosphorylation of other PP2A substrates like p53 and MDM2 was unaffected.7 So far, only a few CIP2A-mediated PP2A targets have been identified. In addition to c-myc, UNC5B and AKT were found to be another two targets of CIP2A.9,10 Interestingly, both of these proteins linked the CIP2A to the role as anti-apoptotic function.
Despite the progress in recent years, our understanding in the role of CIP2A in human malignancies is incomplete. We still do not know the underlying mechanism by which CIP2A promotes cell proliferation, which is crucial to understand the role of CIP2A in cancer pathogenesis. Recently developed OMICs are expected to facilitate the understanding since the high throughput technologies not only identify a group of differentially expressed proteins for further investigation, but also integrate these targeted proteins with the database including all known pathways to uncover novel functions of certain proteins. The molecular mechanism of CIP2A in cancer has been detected by transcriptomic studies as a whole,11 but not at the proteome level, which does not necessarily correlate with mRNAs.12 In the present study, two-dimensional electrophoresis 2-DE based proteomics carrying cleared whole-cell protein fraction of H1299 cells have been developed to investigate the possible mechanisms of CIP2A in promoting lung cancer cell proliferation by identifying differential proteins in abundance in CIP2A knockdown cells. We quantitatively detected 49 differentially expressed protein spots in the cell line with the knockdown of CIP2A using comparative 2-DE based proteomics, and interestingly predicted that CIP2A might be involved in the regulation of c-myc and CREB transcriptional factors by gene interaction networks. At last, we documented CIP2A regulation of CREB transcription through the growth factor mediated pathway. These findings not only extended our understanding of the role of CIP2A in lung cancer progression by modulating the expression of cell-related behavior, more importantly, also indicated that CIP2A is able to regulate important transcriptional factors.
2. Materials and methods
2.1 Cell culture
The three lung cancer cell lines, A549, NCI-H838 and NCI-H1299, were purchased from American Type Culture Collection (ATCC, Manassas, VA). All cell lines except A549 were cultured in RPMI-1640 medium (Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum (Life Technologies, Carlsbad, CA) at 37 °C with 5% CO2. A549 was cultured under the same conditions as for other cell lines except that it was cultured in F12K medium (Life Technologies, Carlsbad, CA).2.2 Transient transfection
Transient transfection of pcDNA3.1 control or pcDNA3.1 containing CIP2A cDNA (general gift from Dr Westermarck, Finland) into the H1299 lung cancer cell line was performed with Lipofectamine LTX (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol.2.3 Production of lentivirus encoding CIP2A short hairpin RNA
Five CIP2A short hairpin RNAs, ligated in pLKO.1 vector, were obtained from Open Biosystems (Open Biosystems, Huntsville, AL). The knockdown efficiency of these shRNAs was evaluated in H1299 cells by Western blot and two of them showing the higher knockdown efficiency were chosen to produce lentivirus. Lentivirus was produced by co-transfection of pLKO.1 control (ligated with scramble sequence) or other pLKO.1-derived vector with pMD2.G (Addgene, Cambridge, MA) and pCMV-VSVG (Addgene, Cambridge, MA) into HEK293T packaging cell lines. The supernatants containing lentivirus of HEK293T were harvested at 36 and 72 h post transfection. Supernatants were pooled, centrifuged to remove cells and then filtered through a 0.45 μm low protein binding filter. Cells were plated in a monolayer at different densities and infected with lentivirus constructs using 8 ng mL−1 polybrene (Sigma, St. Louis, MO). The stable cell lines were selected in the presence of 1 μg mL−1 puromycin (Sigma, St. Louis, MO) for two weeks.2.4 Cell proliferation, clonogenic assay, and anchorage-independent cell growth assay
To compare cell proliferation in cells with CIP2A knockdown, the MTT assay was performed. The shRNA for CIP2A lentivirus-transduced cells and control shRNA-transduced cells was plated quadruplicate in 96 wells plates at a density of 3 × 103/100 μL medium for each well. After 3 day culture, 15 μL of the dye solution was added to each well and incubated at 37 °C for another 4 h. At the end of the incubation, 100 μL stop solution was added to each well and colorimetric absorbance was read in the SpectraMax Plus (Molecular Devices, Sunnyvale, CA) at 570 nm. Cell free medium was used as the mock control.To measure the effects of knockdown on tumor cell clonogenic expansion, CIP2A shRNA-transduced cells and control shRNA transduced cells were trypsinized and resuspended in complete growth medium to form single cells. For each group of cells, 5 × 103 cells were plated in 100 mm petri dish (BD Bioscience, Bedford, CA) and incubated for 15 days. Medium was changed every three days. At the end of incubation, colonies were fixed with 10% formalin and stained with crystal violet. The number of colonies was counted using ImageJ software (NIH, Bethesda, MA).
Colony formation ability of soft agar was evaluated as described.13 Briefly, a 2.5 mL base layer consisting of 0.4% Nobel agar medium was prepared in 100 mm petri dish (BD biosciences, Bedford, MA). CIP2A shRNA-transduced and control lentivirus-transduced cells were trypsinized, centrifuged, resuspended in 0.4% agarose medium (equal volumes of 0.8% agarose and 2× RPMI1640 containing 20% FBS culture medium), and plated onto the top agar at 12500 cells per well. The cells were kept wet by adding a 2.5 mL complete RPMI 1640 growth medium with 10% FBS and incubated for 3 week at 37 °C. Colonies were stained with crystal violet and counted under the microscope.
2.5 PP2A phosphatase activity assay
Measurement of PP2A phosphatase activity was performed by using the RediPlate™ 96 EnzChek® Serine/Threonine Phosphatase Assay Kit (Life Technologies, Carlsbad, CA). The substrate incorporated in this kit is 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP), which generates DiFMU. DiFMUP in this kit can be used to assay a variety of Ser/Thr PPases including PP-1, PP-2A and PP-2B by supplementing different additives (e.g. NiCl2 specifically for PP2A phosphatase activity). Briefly, cell lysates were prepared by using low detergent buffer (1% Nonidet P-40, 10 mM HEPES, 150 mM NaCl, 10% glycerol, 1 mM PMSF, and complete protease inhibitor cocktail). A total of 50 μL cell lysates were incubated with 1× PP2A phosphatase reaction buffer supplemented with NiCl2 at a final concentration of 1 mM for 30 min at 37 °C. Fluorescence intensity was measured using excitation at 355 nm and emission at 485 nm. The fluorescence intensity was normalized to the expression level of the PP2A catalytic domain.2.6 Western blot analysis
Cells were plated in 6-well tissue culture plates at 80% confluence and incubated overnight. Cell lysates were obtained from transduced cells using cold radioimmunoprecipitation assay buffer [20 mmol L−1 Tris-HCl (pH 8.0), 100 mmol L−1 NaCl, 10% glycerol, 1% NP40, 0.5% sodium deoxycholate]. Twenty micrograms of protein mixture were separated on 10% SDS-PAGE gels and wet transferred to nitrocellulose membranes (GE Healthcare Life Sciences, Pittsburgh, PA) and then blocked for 1 h at room temperature in TBS-T buffer [50 mmol L−1 Tris-HCl (pH 7.5), 150 mmol L−1 NaCl, 0.1% Tween 20] containing 5% nonfat milk. Membranes were then incubated overnight at 4 °C or 1 h at room temperature with the respective primary antibodies: anti-CIP2A (2G10-3B5, 1:500), anti-c-myc (9E10, 1:1000), anti-PP2A catalytic domain (4B7, 1:500), anti-LDH (H-10, 1:500) and anti-actin (N-21, 1:1000), (Santa Cruz Biotechnology, Santa Cruz, CA), pCREB (87G3, 1:1000), CREB (48H2, 1:1000) (Cell Signaling, Danvers, MA). The anti-mouse or the anti-rabbit secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA) was used to visualize the stained bands with an enhanced chemiluminescence visualization kit (Santa Cruz Biotechnology, Santa Cruz, CA).
2.7 Sample preparation, 2-dimensional electrophoresis and image analysis
Cells transduced with either control lentivirus or CIP2AshRNA1 were directly lysed in 2-DE rehydration sample buffer (7 M urea, 2 M thiourea, 4% CHAPS, 80 mM DTT, and 1% carrier ampholyte pH 3–10).14 Protein concentration was measured with Bradford assay (Bio-rad, Hercules, CA). One hundred and twenty five micrograms of proteins were loaded to a 11 cm, pH 3–10 non-linear isoelectric focusing (IEF) gel strip and IEF was carried out for 8000 Vh. IEF gels are equilibrated in equilibration buffer containing 2% DTT for 15 min, followed by equilibration in equilibration buffer containing 2.5% iodoacetamide for another 15 min.15 The gels were transferred to the second-dimension electrophoresis using 10% acrylamide gel. Protein spots were visualized by staining with freshly prepared 0.1% Coomassie blue R250 (prepared in 40% ethanol and 10% acetic acid at room temperature for 20 min, followed by destaining in 40% ethanol and 10% acetic acid) until the background of the gel was fully destained (Bio-rad, Hercules, CA).Each protein sample was run triplicate by 2DE, and a total of six gels were generated. Gel images were analyzed with PDQuest 5.0 (Bio-rad Hercules, CA). Spot detection and quantification were performed by both automatic and manual modes. Well-resolved spots in all the gels were fixed as landmarks. After matching and normalization, gels were grouped together with the “Replicate group” function. Non-expression-related differences in spot intensity were corrected by using the PDQuest “total quantity in valid spots” normalization mode. Only spots whose staining intensity was significantly different between the groups and exhibiting a fold change minimum of 1.5 were considered differentially expressed. Statistical evaluation of PDQuest-normalized spot intensity values was performed in MiniTab statistic software. p < 0.05 was considered as significant.
2.8 Protein digestion
The stained protein spots were cut from the gel and destained three times with 100 μL of 50 mM ammonium bicarbonate/50% acetonitrile (ACN) for 30 min at ambient temperature. The destained gel pieces were completely dried in a SpeedVac vacuum concentrator (Savant Instruments, Farmingdale, NY). Gel pieces were reswollen with 8 μL of 25 mM ammonium bicarbonate containing 10 ng of trypsin at 4 °C for 1 h. After 13.5 h of incubation at 37 °C, the gel pieces were vacuum-dried, 8 μL of 50 mM ammonium bicarbonate was added, and the samples were incubated at 37 °C for 1 h. The supernatant was transferred into another micro tube, and 8 μL of 2.5% trifluoroacetyl (TFA)/50% ACN was used for the second step of extraction at 30 °C for 1 h, and the supernatant was combined with the first one. Finally, to extract hydrophobic peptides, 8 μL of 100% ACN was used at room temperature for 1 h, and the supernatant was also combined with the previous supernatants. The combined supernatants were then dried in a SpeedVac and redissolved with 2 μL of 0.5% TFA.2.9 MALDI-TOF/MS analysis
As previously described 0.5 μL of the sample solution, along with equivalent matrix solution (α-cyano-4-hydroxycinnamic acid), was mixed and applied onto the MALDI-TOF target for MALDI-TOF/MS analysis. MALDI-TOF spectra were calibrated using trypsin autodigestion peptide signals and matrix ion signals. MALDI analysis was performed using a fuzzy logic feedback control system (Reflex III MALDI-TOF system Bruker, Karlsruhe, Germany) equipped with delayed ion extraction. For MS/MS spectra, the 5 most abundant precursor ions per sample were selected for subsequent fragmentation and 1000–1200 Da laser shots were accumulated per precursor ion. The criterion for precursor selection was a minimum S/N of 50. Both the MS and MS/MS data were interpreted and processed by using Flexanalysis 3.0 (Bruker Daltonics, Billerica, MA), and then the obtained MS and MS/MS spectra per spot were combined and submitted to the MASCOT search engine (V2.3, Matrix Science, London) by Biotools 3.1 (Bruker Daltonics, Billerica, MA) and searched with the following parameters: the NCBI in SwissProt (http://www.matrixscience.com), one missed cleavage site, carbamidomethyl as fixed modification of cysteine, oxidation of methionine as a variable modification, and tolerance of 100 ppm for MS and 0.6 Da for MS/MS. Known contaminant ions (keratin) were excluded.162.10 Metabolic measurements
000 × g for 5 min to remove insoluble material. And LDH activity was determined using an LDH enzyme assay kit according to the manufacturer's instructions (Sigma, St. Louis, MO). The test results were obtained in SpectraMax Plus (Molecular Devices, Sunnyvale, CA) at 450 nm.
000 × g for 5 min. The test results were obtained in SpectraMax Plus (Molecular Devices, Sunnyvale, CA) at 565 nm.
2.11 Bioinformatics analysis
The gene interaction network was built in Pathway Studio 4.0 (Adriadne Genomics, Rockville, MD) using the latest ResNet Mammalian Database. Pathway Studio contained an automated text-mining tool that enables the software to generate pathways from the PubMed database and public resources. The identified interaction was confirmed through the PubMed/Medline hyperlink embedded in each node. All the differentially expressed proteins were entered into the database and set filter as “Promoter Binding” to create the shortest pathways, which gave predictions on transcription factor candidates in this study.3. Results
3.1 Establishment of the stable cell line expressing CIP2A shRNA
The H1299 cell line, expressing a relatively higher CIP2A level than another three lung cancer cell lines A549, H838 and H460 (unpublished data), was used in this study to generate cell lines stably expressing CIP2A shRNA by lentiviral-mediated transduction which achieved >90% knockdown effect as determined by western blot (Fig. 1). According to the previous report showing that the loss of CIP2A induced the degradation of c-myc by restoring c-myc associated PP2A phosphatase activity in HeLa cells, the expression level of c-myc and PP2A phosphatase activity was also measured. Consistently, the knockdown of CIP2A in H1299 with two independent CIP2A shRNAs led to increased PP2A phosphatase activity in the total cell lysates (Fig. 1) and a reduced level of c-myc (Fig. 1). Therefore, these data confirmed the role of CIP2A in regulating PP2A phosphatase activity and c-myc stability in lung cancer cells. | ||
| Fig. 1 CIP2A knockdown in lung cancer cell H1299 increased PP2A phosphatase activity. PP2A phosphatase activity assay. Total cell lysates extracted from transduced cells by control shRNA, CIP2A shRNA1, or CIP2A shRNA2 were loaded to measure the PP2A phosphatase activity by using DiMUF as the substrates (upper panel) in PP2A phosphatase assay buffer supplemented with NiCl2 (1 mM). Fluorescence readings were normalized to the expression level of the PP2Ac catalytic subunit (lower panel). Data represent three independent experiments. Statistics were performed with Student's t test and bars represent mean ± SD. p value < 0.05 was considered as significant. | ||
3.2 Depletion of CIP2A impairs cell proliferation, clonogenic ability, and anchorage-independent growth of the H1299 cell line
Knockdown of CIP2A in the H1299 cell line reduced the cell proliferation rate to as low as 69% compared to cells transduced with the scramble shRNA sequence (Fig. 2A), which implied the positive regulation of CIP2A in lung cancer cell proliferation. To further support the role of CIP2A in tumorigenesis of lung cancer, clonogenic expansion assay and anchorage-dependent cell growth assay were performed. The H1299 cell line CIP2A showed fewer colony formation both in clonogenic expansion assay (52%, Fig. 2B) and anchorage-dependent cell growth assay (62%, Fig. 2C) than those formed by the control cell line. These data therefore suggested that CIP2A was important for the lung cancer cell proliferation in vitro. | ||
| Fig. 2 Depletion of CIP2A induced decreased cell proliferation, clonogenic expansion, soft agar growth, and PP2A phosphatase activity. (A) Knockdown of CIP2A in H1299 cells reduced cell proliferation in MTT assay. (B) Knockdown of CIP2A in H1299 cells significantly lowered the number of colonies in the clonogenic expansion assay. (C) Knockdown of CIP2A in H1299 cells significantly reduced anchorage-independent cell growth. 1.25 × 104 cells were resuspended in 0.4% agarose, poured to base agar containing 0.8% Nobel agar and incubated for three weeks. Colonies were stained with crystal violet and counted under a microscope. Statistics were performed with Student's t test and bars represent mean ± SD. p value < 0.05 was considered as significant. | ||
3.3 Comparative proteomic analysis of proteins associated with CIP2A knockdown
To gain insight into how CIP2A regulates tumor growth, 2-DE and mass spectrometry-based proteomic approach were utilized to systematically investigate the change in proteome between the H1299 control cell line and the same cell line with CIP2A knockdown. By comparing the 2-D profile from these two lines, 49 differentially expressed protein spots were detected (Fig. 3A).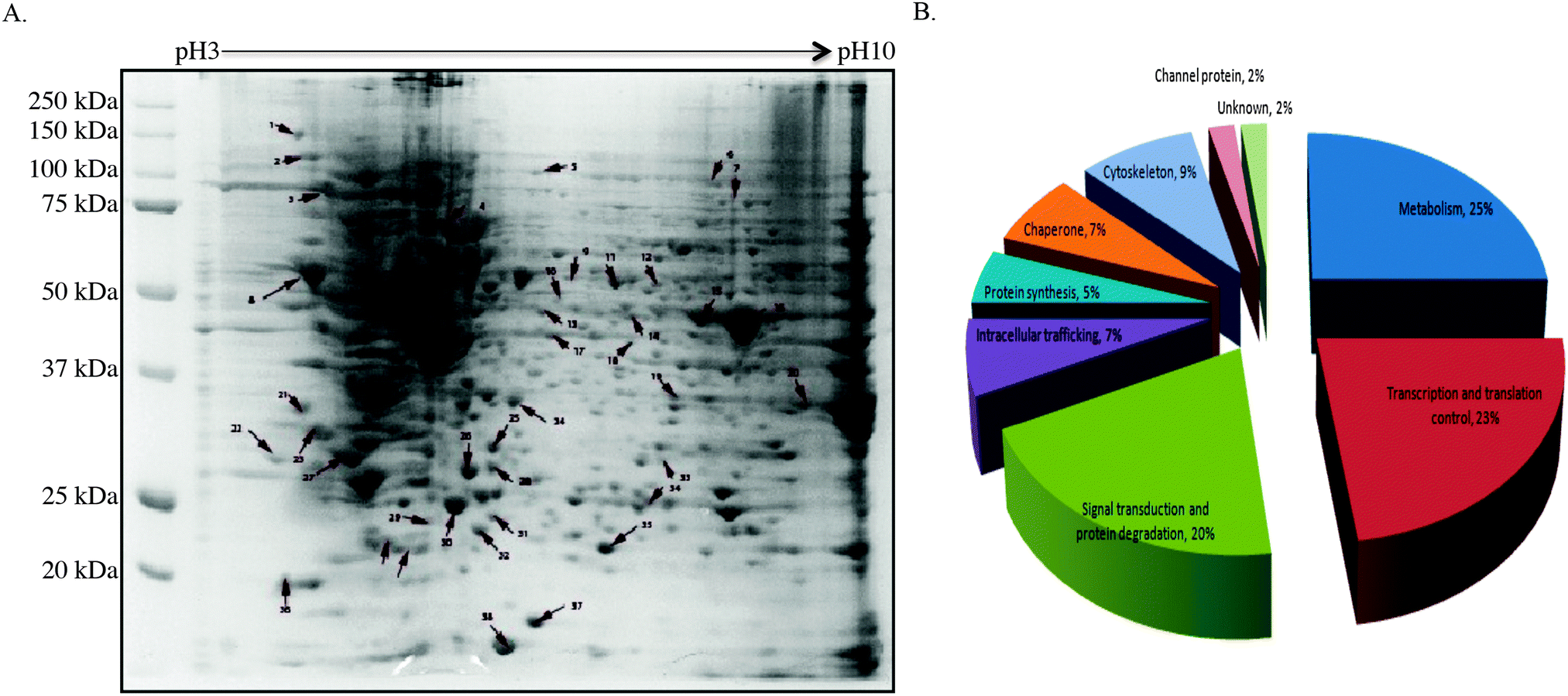 | ||
| Fig. 3 Two-dimensional electrophoresis and the functional category of differentially expressed proteins. (A) Differentially expressed proteins were obtained by comparing the 2DE expression profile between H1299 cells stably expressing control shRNA and CIP2A shRNA1. Arrows indicate differentially expressed proteins between these two groups. Data were obtained from three independent experiments. (B) The differentially expressed proteins are classified by their reported function. The gene function was obtained from the Gene Ontology (GO) database. | ||
According to the reported function of these proteins, we found that a majority of altered proteins belong to the functional classes of metabolism (25%), signal transduction (20%), and transcriptional and translational control (23%) (Fig. 3B). Proteins like phosphoglycerate mutase 1 (PGM), α-enolase, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are involved in different steps of glycolysis by catalyzing the chemical reactions (Table S1, ESI†).17–19 Tumor cells often exhibited increased glycolysis to generate ATP to support their growth under aerobic conditions, known as the Warburg effect.20 The loss of CIP2A impaired the expression of these proteins and implied that CIP2A indirectly affects their expression through c-myc since c-myc is known to promote the aerobic glycolysis of cancer cells. Other metabolism-related proteins like nucleoside diphosphate kinase (NDK) play roles in catalyzing the exchange of phosphate groups between different nucleoside diphosphates.21 Nucleophosmin, encoded by nm23-H1 gene, has been reported to overexpress in many different types of malignancies. The clinical study showed that its overexpression is strongly associated with tumor cell motility, invasion, and metastasis.22
Twenty four percent of the differentially expressed proteins are associated with transcriptional and translational control. Among these proteins, SAP-domain containing ribonucleoprotein (CIP29) and heterogeneous nuclear ribonucleoprotein H1 (hnRNP H1) are known to be able to bind nucleotide and regulate RNA stability and localization (Table S1, ESI†).23,24 CIP29, like CIP2A, is a fetal protein which only expressed during the embryonic stage and is silenced in adult tissue. This protein is overexpressed in breast and pancreatic tumors.23 hnRNP H1 is a mRNA binding protein and its interaction with target mRNA can determine its subcellular localization. The function of this protein in cancer is not well understood, but it has been reported to involve in anti-cancer drug resistance.24 Protein DJ-1 and far upstream element-binding protein 2 (FUBP-2) can facilitate the gene transcription.25,26 For example, protein DJ-1 can positively regulate the transcription of the androgen receptor (AR) in the nucleus.25 FUBP-2 and its family protein FUBP-1 are known to be involved in the transcriptional control of c-myc. The knockdown of this protein will lead to decreased cell proliferation and reduced xenographt tumor growth.26 Other proteins like elongation factor 2 (eEF2), acidic leucine-rich nuclear phosphoprotein 32 (ANAP32), ruvB-like 1 participate in diverse cellular functions such as translation elongation, nucleosome assembly and transcription.27–29
Another major functional class is related to signal transduction and protein degradation (Table S1, ESI†). This functional class protein consists of several cancer-related genes. Annexin A5 and 14-3-3 epsilon are proteins of great importance in regulating cancer cell proliferation.30,31 Proteins like Annexin A1 and EBP1 have anti-proliferative effects in cancer. EBP1 is an ErbB-3 binding protein, which inhibits cell proliferation and induces cell differentiation in breast cancer cells. Although how this protein contributes to inhibiting cell proliferation is unknown, it can act as a transcription factor or a transcription co-regulator, which is normally sequestered in the cytoplasm as inactive form until it has been stimulated with proper ligands.32,33
Some proteins related to cytoskeleton are also identified including septin, F-actin-capping protein subunit-beta, stathamin-1 and microtubule-associated protein RP/EB family member (MAPRE1). These proteins participate in chromosome stability, cell polarity and cytokinesis.34,35 Except F-actin-capping protein subunit-beta, all of the other three proteins have been confirmed related to cancer.
3.4 Network analysis of differentially expressed proteins in CIP2A knockdown
Since proteins normally work together with other molecules to perform certain functions rather than work as a single entity, the possible pathways or interactions connected to these differentially expressed proteins were analyzed using the Pathway Studio software. All of the 47 differentially expressed proteins were used for this pathway analysis. Although these proteins were not connecting to any known signaling pathways, five of the differentially expressed proteins (CALR, ENO1, PHB, STMN1 and GAPDH) were identified directly or indirectly regulated by either c-myc or cAMP responsive element binding protein (CREB) which had the most number of connections (Fig. 4). Since c-myc is one of the proteins which has been shown to be a direct target of CIP2A, the bioinformatics predicted the possibility that CREB might be another protein targeted by CIP2A.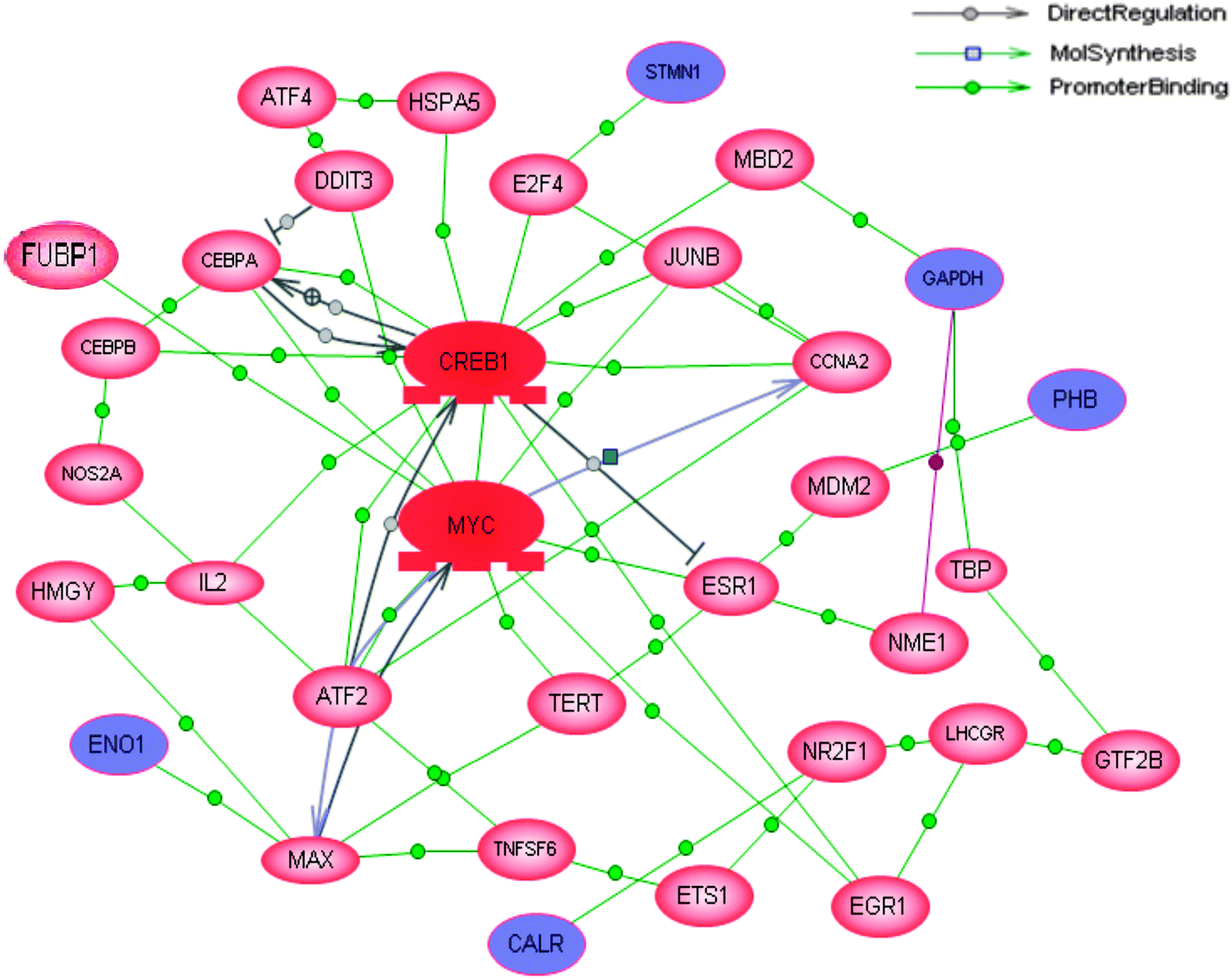 | ||
| Fig. 4 The gene interaction network of differentially expressed proteins in CIP2A knockdown cells. The differentially expressed proteins were searched against potential transcription factors in the Pathway Studio database using the “Promoter binding” filter. The two highlighted proteins, MYC and CREB (dark red), are the nexus proteins, which have the most connections to other proteins including the identified differentially expressed proteins: enolase 1 (ENO1), prohibitin (PHB), calreticulin (CALR), stathmin (STMN), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) found in the interaction network. | ||
3.5 CIP2A regulated CREB transcription through the serum-mediated pathway
Both c-myc and CREB are transcription factors which have been demonstrated to promote cancer cell proliferation including lung cancer.36 We therefore tested whether CIP2A can regulate the activity of CREB in the phosphorylation at Serine 133, a critical event for the transcriptional activity of CREB. Phosphorylation of CREB can be triggered by various stimuli like growth factors, calcium influx and increased intracellular concentration of cAMP. After being starved for 24 h, H1299 (control shRNA) or H1299 (CIP2A shRNA1) cells were treated with either KCl (depolarization of cell membrane and induces calcium influx), forskolin (increase intracellular cAMP concentration) or serum (10% FBS). Interestingly, the treatment with KCl did not alter the phosphorylation of CREB (Fig. 5A). In contrast, the serum treated CIP2AshRNA H1299 cells showed large reduction of phosphorylated CREB (33% of the control shRNA sample) (Fig. 5A). Noticeably, the knockdown of CIP2A also showed reduced phosphorylated CREB (80%) compared to the control shRNA treated group but not as significant as the serum-treated sample. In addition, the loss of CIP2A reduced CREB phosphorylation in another two lung cancer cell lines, H838 and A549 (Fig. 5B). | ||
| Fig. 5 CIP2A regulates CREB phosphorylation. (A) CIP2A knockdown decreased serum-induced CREB phosphorylation. H1299 transduced with either control shRNA or CIP2A shRNA1 were serum starved for 16 h and then treated with either KCl (30 mM), forskolin (10 μM) or complete growth medium containing 10% FBS. (B) CIP2A regulated CREB phosphorylation in another two lung cancer cell lines. CREB phosphorylation was examined in another two lung cancer cells, H838 and A549, with depleted CIP2A. Cells were starved for 24 h, treated with complete growth serum for 30 min and analyzed for CREB phosphorylation by Western blot analysis. | ||
3.6 CIP2A regulates L-lactate dehydrogenase A activity and ROS production
LDH-A, which catalyzes the final step of glycolysis by interconversion of lactate and pyruvate, plays critical roles in cellular transformation and cancer progression.37 Our proteomic analysis identified LDH-A is the protein associated with the expression change of CIP2A. Therefore, we further confirmed the proteomic results by Western-blot. The knock down of CIP2A by two independent shRNAs reduced the expression of LDH-A to 63% to 54% of the control shRNA, respectively, which is consistent with our 2D findings (Fig. 6A). The decreased expression of LDH-A is also accompanied by reduced LDH-A activity in both of the knockdown cell lines (Fig. 6B). Furthermore, the production of lactate in the two CIP2A knockdown cell lines (2.59 nM and 2.64 nM, respectively) was lower than that of the control shRNA cell line (3.31 nM) (Fig. 6C). In contrast, the ratio of NADH/NAD+ and ROS production was increased 34% and 29%, and 41% and 39% after CIP2A knock down, respectively, indicating the reduced glycolytic rate and increased ROS production (Fig. 6D and E). These data suggested that the overexpression of CIP2A may reprogram the metabolic pathway to drive tumor growth. | ||
| Fig. 6 CIP2A regulates L-lactate dehydrogenase A activity and ROS production. (A) Expression of LDH-A was measured in CIP2A knock down cells by Western blot analysis. Fold of change was measured by densitometry. (B) LDH-A enzymatic assay. The enzymatic change was measured in control shRNA, CIP2A shRNA1 and CIP2A shRNA2 cells by the colorimetric method. The enzymatic activity was normalized to the number of cells. (C) Lactate assay. The production of lactate in the three cells was measured by the colorimetric method. The cell supernatant was centrifuged and deproteinized before assay. The lactate concentration was calculated from the standard curve according to manufacture's instructions. (D) NADH/NAD+ redox homeostasis. The status of NADH/NAD+ was measured in the three cell lines by the colorimetric method. The knock down of CIP2A increases the ratio of NADH to NAD+. (E) ROS production assay. The production of H2O2 was measured by a fluorometric method using H2DCFDA as the substrate. The ROS production was normalized with the number of cells. The statistical analysis for the above tests was Students' t test. P value < 0.05 was considered as significant. | ||
4. Discussion
In our study, the phenotypic analysis of CIP2A in lung cancer cell revealed the role of CIP2A in cancer cell proliferation by showing the decreased cell proliferation, clonogenic ability and anchorage-dependent cell growth along with the knockdown of CIP2A. The functional analysis of CIP2A by combinatorial use of the proteomic approach and bioinformatics analysis helped us to identify CIP2A as a positive regulator of the CREB signaling pathway. However, one question needs to be established is that how CIP2A can exert an effect on CREB. According to the previous findings that CREB is mainly localized in the nucleus, but CIP2A is localized in the cytoplasm. The distinct subcellular localization of these two proteins seems that they are not able to interact with each other in a direct way. Therefore, this event may occur through the regulation of upstream kinases like AKT, p90RSK, p38 α/β, CamK-II, CamK-IV and protein kinase A.38 These kinases activate CREB through phosphorylation at Serine 133, which is dependent on the type of stimuli. In this study, we used FBS (including different types of growth factors, which activate AKT, p90RSK and p38 α/β), KCl (depolarize cell membranes to induce calcium influx, which activates CamK-II and CamK-IV), and forskolin (activates PKA). We found that serum had the highest effect on CREB phosphorylation, and forskolin had modest effect. This indicated AKT, p90RSK, p38 α/β and PKA might be the upstream kinases targeted by CIP2A to regulate CREB phosphorylation. This is not astonishing because several of the kinases, AKT, p90RSK and p38 α/β are the targets of PP2A. Although CIP2A modulates AKT-associated PP2A phosphatase activity, the possibility of CIP2A has similar effects on p90RSK and p38 α/β cannot be excluded. PKA is not a substrate of PP2A. Instead, PKA phosphorylated the catalytic subunit of PP2A, which increased the PP2A phosphatase activity.39 However, the increased PKA activation only leads to limited restoration PP2A phosphatase activity (evidenced by the CREB phosphorylation) in CIP2A knockdown cells, indicating the possible interplay between CIP2A and PKA, which requires further investigation.In our study, the knockdown of CIP2A showed differential abundance of proteins participating in cell metabolism, transcriptional and translational control, and protein signaling pathways. Twenty-five percent of the altered proteins were associated with metabolism. Although the relationship between metabolism and cancer has been described a long time ago since the discovery of cells undergo glycolysis instead of oxidative phosphorylation in cancer and proliferating cells, only until recently it is shown that metabolism-associated genes are active in cell transformation and tumorigenesis rather than a by-stander. Oncogenic factors like Ras, c-myc, and HIF1 have been reported to reprogram cellular metabolism pathways to favor cancer cell growth. In the current study, we showed that the knockdown of CIP2A decreased the expression of LDH-A, enzymatic activity as well as the production of L-lactate. These results not only validated our proteomic results but also established the relationship between CIP2A and cancer metabolism. In addition, the knockdown of CIP2A affects the redox homeostasis and ROS production. Although the proteins like peroxidase, reductase or oxidase were not identified in our study, our results on the ratio of NADH/NAD+ and ROS production strongly suggested the participation of CIP2A in response to oxidative stress. However, the mechanism of how CIP2A interacts with redox homeostasis should be further investigated.
Aberrant transcription and translation have been reported in cancer. Besides the transcription factors, other factors in transcription and translation machinery have also been found to be altered in cancer, which can be triggered by abnormal activation of upstream biochemical pathways.40 Although the mechanism of how CIP2A contributed to such alterations is still unknown, it is speculated that CIP2A may exert such effects by regulating the upstream biochemical pathways like AKT. AKT is a central regulator, which integrates different stimuli and then transmits the signal to appropriate substrates through phosphorylation.41,42 Upon the stimulation of growth factors, AKT is able to promote protein synthesis by regulating the phosphorylation of ribosomal protein p70 S6 kinase, and eukaryotic initiation factor 4E binding protein. Noticeably, we also found that CIP2A is able to regulate the phosphorylation status of AKT, which thus may partially explain the resulted protein changes of transcription and translation machinery from CIP2A knockdown.42,43
In conclusion, by using comparative proteomics, we found that CIP2A is able to regulate the metabolism, translational and transcriptional control, and signaling transduction of cancer cells. On one hand, we validated our proteomic results by studying LDH-A-associated metabolism. On the other hand, we also identify a novel CIP2A target, CREB, by bioinformatics analysis. Further functional studies showed that CIP2A is able to regulate the phosphorylation of CREB under serum stimulated conditions in a c-myc-independent manner, which implied a novel mechanism by which CIP2A promotes cancer cell progression. Therefore, our study not only extended our understanding of the role of CIP2A in lung cancer progression by modulating the expression of cell-related behavior but also proposed CIP2A as a valuable target for cancer therapeutic design.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Dr Eng M. Tan (Scripps Research Institute, La Jolla, CA) for his comments on this manuscript. This work was supported by grants from NIH (SC1CA166016). We also thank the Biomolecule Analysis Core Facility, Cell Culture Core Facility, DNA Sequencing Core Facility, and Analytical Cytology Core Facility of Border Biological Research Center (BBRC) at The University of Texas at El Paso (UTEP), funded by NIH grant (5G12MD007592) for their support.References
- H. J. Burstein and R. S. Schwartz, N. Engl. J. Med., 2008, 358, 527 CrossRef CAS PubMed.
- X. Wang, D. C. Christiani, J. K. Wiencke, M. Fischbein, X. Xu, T. J. Cheng, E. Mark, J. C. Wain and K. T. Kelsey, Cancer Epidemiol., Biomarkers Prev., 1995, 4, 543–548 CAS.
- M. Sanchez-Cespedes, Oncogene, 2007, 26, 7825–7832 CrossRef CAS PubMed.
- J. Westermarck and W. C. Hahn, Trends Mol. Med., 2008, 14, 152–160 CrossRef CAS PubMed.
- A. A. Sablina and W. C. Hahn, Cancer Metastasis Rev., 2008, 27, 137–146 CrossRef CAS PubMed.
- W. Chen, R. Possemato, K. T. Campbell, C. A. Plattner, D. C. Pallas and W. C. Hahn, Cancer Cell, 2004, 5, 127–136 CrossRef CAS.
- M. R. Junttila, P. Puustinen, M. Niemela, R. Ahola, H. Arnold, T. Bottzauw, R. Ala-aho, C. Nielsen, J. Ivaska, Y. Taya, S. L. Lu, S. Lin, E. K. Chan, X. J. Wang, R. Grenman, J. Kast, T. Kallunki, R. Sears, V. M. Kahari and J. Westermarck, Cell, 2007, 130, 51–62 CrossRef CAS PubMed.
- L. Soo Hoo, J. Y. Zhang and E. K. Chan, Oncogene, 2002, 21, 5006–5015 CrossRef PubMed.
- K. F. Chen, C. Y. Liu, Y. C. Lin, H. C. Yu, T. H. Liu, D. R. Hou, P. J. Chen and A. L. Cheng, Oncogene, 2010, 29, 6257–6266 CrossRef CAS PubMed.
- C. Guenebeaud, D. Goldschneider, M. Castets, C. Guix, G. Chazot, C. Delloye-Bourgeois, A. Eisenberg-Lerner, G. Shohat, M. Zhang, V. Laudet, A. Kimchi, A. Bernet and P. Mehlen, Mol. Cell, 2010, 40, 863–876 CrossRef CAS PubMed.
- M. Niemela, O. Kauko, H. Sihto, J. P. Mpindi, D. Nicorici, P. Pernila, O. P. Kallioniemi, H. Joensuu, S. Hautaniemi and J. Westermarck, Oncogene, 2012, 31, 4266–4278 CrossRef CAS PubMed.
- N. L. Anderson and N. G. Anderson, Electrophoresis, 1998, 19, 1853–1861 CrossRef CAS PubMed.
- N. A. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS PubMed.
- T. Rabilloud, Electrophoresis, 1998, 19, 758–760 CrossRef CAS PubMed.
- A. Gorg, W. Postel and S. Gunther, Electrophoresis, 1988, 9, 531–546 CrossRef CAS PubMed.
- Y. Ma, C. Guo, H. Li and X. X. Peng, J. Proteomics, 2013, 87, 78–88 CrossRef CAS PubMed.
- A. Kimura, T. Sakurai, A. Koumura, M. Yamada, Y. Hayashi, Y. Tanaka, I. Hozumi, R. Tanaka, M. Takemura, M. Seishima and T. Inuzuka, J. Neuroimmunol., 2010, 219, 105–108 CrossRef CAS PubMed.
- E. Zhang, J. M. Brewer, W. Minor, L. A. Carreira and L. Lebioda, Biochemistry, 1997, 36, 12526–12534 CrossRef CAS PubMed.
- M. E. Campanella, H. Chu and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2402–2407 CrossRef CAS PubMed.
- A. J. Levine and A. M. Puzio-Kuter, Science, 2010, 330, 1340–1344 CrossRef CAS PubMed.
- M. H. Bosnar, R. Bago and H. Cetkovic, Mol. Cell. Biochem., 2009, 329, 63–71 CrossRef CAS PubMed.
- A. Bilitou, J. Watson, A. Gartner and S. Ohnuma, Mol. Cell. Biochem., 2009, 329, 17–33 CrossRef CAS PubMed.
- K. Maruyama and S. Sugano, Gene, 1994, 138, 171–174 CrossRef CAS.
- M. Stark, E. E. Bram, M. Akerman, Y. Mandel-Gutfreund and Y. G. Assaraf, J. Biol. Chem., 2011, 286, 3741–3754 CrossRef CAS PubMed.
- J. Xu, N. Zhong, H. Wang, J. E. Elias, C. Y. Kim, I. Woldman, C. Pifl, S. P. Gygi, C. Geula and B. A. Yankner, Hum. Mol. Genet., 2005, 14, 1231–1241 CrossRef CAS PubMed.
- R. Duncan, L. Bazar, G. Michelotti, T. Tomonaga, H. Krutzsch, M. Avigan and D. Levens, Genes Dev., 1994, 8, 465–480 CrossRef CAS.
- A. G. Ryazanov, M. D. Ward, C. E. Mendola, K. S. Pavur, M. V. Dorovkov, M. Wiedmann, H. Erdjument-Bromage, P. Tempst, T. G. Parmer, C. R. Prostko, F. J. Germino and W. N. Hait, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4884–4889 CrossRef CAS.
- N. Tochio, T. Umehara, Y. Munemasa, T. Suzuki, S. Sato, K. Tsuda, S. Koshiba, T. Kigawa, R. Nagai and S. Yokoyama, J. Mol. Biol., 2010, 401, 97–114 CrossRef CAS PubMed.
- T. Puri, P. Wendler, B. Sigala, H. Saibil and I. R. Tsaneva, J. Mol. Biol., 2007, 366, 179–192 CrossRef CAS PubMed.
- Y. Wen, J. L. Edelman, T. Kang and G. Sachs, Biochem. Biophys. Res. Commun., 1999, 258, 713–721 CrossRef CAS PubMed.
- A. Craparo, R. Freund and T. A. Gustafson, J. Biol. Chem., 1997, 272, 11663–11669 CrossRef CAS PubMed.
- X. Xia, A. Cheng, T. Lessor, Y. Zhang and A. W. Hamburger, J. Cell. Physiol., 2001, 187, 209–217 CrossRef CAS PubMed.
- Y. Zhang, J. D. Fondell, Q. Wang, X. Xia, A. Cheng, M. L. Lu and A. W. Hamburger, Oncogene, 2002, 21, 5609–5618 CrossRef CAS PubMed.
- L. K. Su, M. Burrell, D. E. Hill, J. Gyuris, R. Brent, R. Wiltshire, J. Trent, B. Vogelstein and K. W. Kinzler, Cancer Res., 1995, 55, 2972–2977 CAS.
- P. A. Kuhlman and V. M. Fowler, Biochemistry, 1997, 36, 13461–13472 CrossRef CAS PubMed.
- M. D. Conkright and M. Montminy, Trends Cell Biol., 2005, 15, 457–459 CrossRef CAS PubMed.
- V. R. Fantin, J. St-Pierre and P. Leder, Cancer Cell, 2006, 9, 425–434 CrossRef CAS PubMed.
- B. Mayr and M. Montminy, Nat. Rev. Mol. Cell Biol., 2001, 2, 599–609 CrossRef CAS PubMed.
- J. H. Ahn, T. McAvoy, S. V. Rakhilin, A. Nishi, P. Greengard and A. C. Nairn, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2979–2984 CrossRef CAS PubMed.
- F. Meric and K. K. Hunt, Mol. Cancer Ther., 2002, 1, 971–979 CAS.
- J. Faridi, J. Fawcett, L. Wang and R. A. Roth, Am. J. Physiol.: Endocrinol. Metab., 2003, 285, E964–E972 CAS.
- J. W. Watters and C. J. Roberts, Mol. Cancer Ther., 2006, 5, 2444–2449 CrossRef CAS PubMed.
- N. J. Lei, B. Peng and J. Y. Zhang, Oncol. Rep., 2014, 4, 1689–1694 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00513a |
| This journal is © The Royal Society of Chemistry 2015 |