
Molecular dynamics-based discovery of novel phosphodiesterase-9A inhibitors with non-pyrazolopyrimidinone scaffolds†
Zhe
Li‡
a,
Xiao
Lu‡
a,
Ling-Jun
Feng
a,
Ying
Gu
b,
Xingshu
Li
a,
Yinuo
Wu
*a and
Hai-Bin
Luo
*a
aSchool of Pharmaceutical Sciences, SunYat-Sen University, Guangzhou 510006, P. R. China. E-mail: luohb77@mail.sysu.edu.cn; wyinuo3@mail.sysu.edu.cn
bSchool of Information and Software Engineering, University of Electronic Science and Technology of China, Chengdu 610054, P. R. China
First published on 10th October 2014
Abstract
Phosphodiesterase-9A (PDE9A) is a promising therapeutic target for the treatment of diabetes and Alzheimer's disease (AD). The Pfizer PDE9A inhibitor PF-04447943 has completed Phase II clinical trials in subjects with mild to moderate AD in 2013. However, most of the reported PDE9A inhibitors share the same scaffold as pyrazolopyrimidinone, which lacks structural diversity and is unfavorable for the development of novel PDE9A inhibitors. In the present study, a combinatorial method including pharmacophores, molecular docking, molecular dynamics simulations, binding free energy calculations, and bioassay was used to discover novel PDE9A inhibitors with new scaffolds rather than pyrazolopyrimidinones from the SPECS database containing about 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds. As a result, 15 hits out of 29 molecules (a hit rate of 52%) with five novel scaffolds were identified to be PDE9A inhibitors with inhibitory affinities no more than 50 μM to enrich the structural diversity, different from the pyrazolopyrimidinone-derived family. The high hit ratio of 52% for this virtual screening method indicated that the combinatorial method is a good compromise between computational cost and accuracy. Binding pattern analyses indicate that those hits with non-pyrazolopyrimidinone scaffolds can bind the same active site pocket of PDE9A as classical PDE9A inhibitors. In addition, structural modification of compound AG-690/40135604 (IC50 = 8.0 μM) led to a new one, 16, with an improved inhibitory affinity of 2.1 μM as expected. The five novel scaffolds discovered in the present study can be used for the rational design of PDE9A inhibitors with higher affinities.
000 compounds. As a result, 15 hits out of 29 molecules (a hit rate of 52%) with five novel scaffolds were identified to be PDE9A inhibitors with inhibitory affinities no more than 50 μM to enrich the structural diversity, different from the pyrazolopyrimidinone-derived family. The high hit ratio of 52% for this virtual screening method indicated that the combinatorial method is a good compromise between computational cost and accuracy. Binding pattern analyses indicate that those hits with non-pyrazolopyrimidinone scaffolds can bind the same active site pocket of PDE9A as classical PDE9A inhibitors. In addition, structural modification of compound AG-690/40135604 (IC50 = 8.0 μM) led to a new one, 16, with an improved inhibitory affinity of 2.1 μM as expected. The five novel scaffolds discovered in the present study can be used for the rational design of PDE9A inhibitors with higher affinities.
1 Introduction
Both 3′-5′-cyclic adenosine monophosphate (cAMP) and 3′-5′-cyclic guanosine monophosphate (cGMP), as second messengers, play very important roles in intracellular signaling cascades. They are involved in many physiological processes, such as inflammation, smooth muscle contraction, steroid hormone function, and memory.1–7 Phosphodiesterases (PDEs) are the sole enzymes that hydrolyze cAMP and cGMP, and are used as therapeutic targets for many diseases.8,9 PDEs consist of 11 families, among which PDE4, PDE7, and PDE8 are cAMP-specific enzymes, whereas PDE5, PDE6, and PDE9 are cGMP-specific enzymes.PDE9 has attracted much attention in recent years since it has the highest affinity towards cGMP among those PDE families with a Km of 170 nM.10–13 It is distributed widely in the human body, especially in the central nervous system. A recent study found that in the aged rat brain there is an increase in PDE9 expression and activity and a decrease in cGMP concentration,14 which may cause Alzheimer's disease (AD). Until now, several PDE9 inhibitors have been developed for the treatment of diabetes, insulin-resistance syndrome, cardiovascular diseases, and CNS diseases such as AD.15–18 BAY 73-6691, a selective PDE9 inhibitor, is currently under preclinical development for the treatment of AD and tends to enhance long-term memory in an object recognition task.19 In addition, the Pfizer PDE9 inhibitor PF-04447943 has just completed Phase II clinical trials (Pfizer, ClinicalTrials.gov Identifier: NCT00930059) in subjects with mild to moderate AD in 2013. Interestingly, these inhibitors may serve as enhancers of memory and synaptic plasticity, reverse memory deficits, and attenuate forgetfulness.20
Sildenafil (Viagra and Revatio), a PDE5 inhibitor, has been widely used for the treatment of male erectile dysfunction and pulmonary hypertension.21 However, with a similar scaffold to cGMP, it has shown several notable side effects since it is related to the insufficient selectivity versus other PDEs, especially reported incidences of visual disturbances may be linked to the unexpected inhibition against PDE6,22 whereas another PDE5 inhibitor tadalafil with a different scaffold from both cGMP and sildenafil gives a high selectivity of 1020-fold against PDE6.23 The structures of PDE9A inhibitors reported nowadays are rather homogeneous, most of which exhibited a “pyrazolopyrimidinone”-like core of the cyclic nucleotides,13,18,24–28 as shown in Fig. S1 (ESI†). This scaffold could interact with Gln 543 and Phe 456, both of which are key residues in PDE9. It is obvious that lack of variety in the structure is unfavorable for the development of novel PDE9 inhibitors. Thus, a particular area of focus has been the discovery of new agents that have different scaffolds from the current PDE9 inhibitors. Thus, the search for novel scaffolds for PDE9 inhibitors continues unabated.
Reported in this paper is the discovery of novel PDE9A inhibitors with non-pyrazolopyrimidinone scaffolds to enrich the structural diversity, different from the current pyrimidinone-derived family, which were prescreened by molecular dynamics-based virtual modeling and validated by bioassay. During our attempt to enrich the structural diversity, 15 hits out of 29 molecules (a hit rate of 52%) with 5 novel scaffolds were identified to be PDE9A inhibitors. Chemical modification of compound AG-690/40135604 led to a new one, 16, with an improved affinity as expected. Thus, the discovery of novel PDE9 inhibitors herein will provide not only more choices for new medical applications, but also compounds with new scaffolds which might hopefully have less side effects.
2 Methods
A MD-augmented virtual screening method has been applied in this study. The workflow of virtual screening is shown in Fig. 2. First, a pharmacophore model was used to rapidly screen the small molecule database and get an initial result. Then, a molecular docking method was used to predict the binding energy and binding mode between the protein and small molecules. At last, molecular dynamics (MD) simulations and the MM/PBSA method were used to obtain more accurate binding modes and binding energies. Based on the virtual screening results, molecules with appropriate binding modes and high binding energies were further subjected to bioassay.2.1 Database preparation
Considering structural diversity and commercial availability, the SPECS database (http://www.specs.net) containing about 200000 compounds was selected in this study. The SPECS database was firstly filtered by using Lipinski's rule in order to obtain the preliminary database DB0, which would be used in the pharmacophore screening. Before our pharmacophore screening, at most 250 conformations of each molecule were generated by using the modeling software Molecular Operating Environment (MOE 2008.10) with the conformation import method. When generating conformations, default filter rules were applied, and molecules with the following properties were removed: molecular weight > 600, logP < −4, logP > 8, H-bond donors + acceptors > 12, rotatable bonds > 7, single bond chain length > 6, chiral centers > 4, unconstrained chiral centers > 3, transition metals, rings > 8, and d-hybrids. After the preliminary filtration, all the compounds in DB0 can be used for the subsequent pharmacophore screening.
2.2 Pharmacophore model generation and screening
Five crystal structures of PDE9A at high resolutions were retrieved from Protein Data Bank (PDB ID: 3JSI, 3JSW, 3K3E, 4E90, and 4GH6)13,24,27,28 to build the pharmacophore model by using the same software MOE 2008.10. The five crystal structures were first superimposed by residues, and as a result, the ligands in the crystal structures were also superimposed on each other. Based on the superimposed ligands, the pharmacophore features were generated by the consensus strategy. Considering the similarity between some features, the unified Aro|PiR (aromatic center or Pi ring center) feature was used instead of the aromatic center feature; the unified Hyd|Aro (hydrophobic centroid or aromatic center) feature was used instead of the hydrophobic centroid feature. The excluded volume features were then generated by the protein residues near the binding site pocket. The five proteins share a similar shape of binding site pockets, so only 3JSW was used here for excluded volume generation.It is very important to evaluate the screening efficacy after a pharmacophore model was generated. Thus a test set, which contained 78 active compounds from the Binding Database and 674 inactive compounds randomly selected from the DUD database (http://dud.docking.org/),29 was constructed. The conformation generation method for the test set was the same as the one used for the SPECS database. The Goodness of hit (GH) test30–32 was used here to evaluate the efficacy of the generated pharmacophore model. Based on the GH test score, the pharmacophore model was modified several times to improve its screening efficacy. If the GH test score is greater than 0.5 or 0.6, it means that the pharmacophore model is a good or excellent screening model.30–32 In general, the GH test score of the resulting pharmacophore model for the purpose of screening must be greater than 0.5.
After the pharmacophore model was generated, all compounds in DB0 were filtered by this pharmacophore model. Compounds matching all the features were selected to generate the initial database DB1 which would be used for subsequent molecular docking studies.
2.3 Molecular docking
A crystal structure of PDE9A in the complex with a highly selective inhibitor (PDB ID: 4GH6) reported in our previous studies28 was used in this study, and Surflex-dock33 embedded in the software Tripos Sybyl 2.0 was used for docking-based virtual screening. There are two metal ions in this crystal structure which were crucial for the PDE's catalytic activity, and they are surrounded by amino acid residues and water molecules. Most of the water molecules in this crystal structure were removed except the ones which form coordination bonds with the two metal ions. Hydrogen atoms were added, and the ionizable residues were protonated at the neutral pH. The protomol, which represents a set of molecular fragments that characterize the active site, was generated using a ligand-based approach, and the bound ligand was used for protomol generation. The proto_thresh and proto_bloat parameters represent how much the protomol can be buried in the protein and how far the protomol extends outside the cavity, respectively. The proto_thresh was selected as 0.5 and proto_bloat was selected as 0.After the preparation of the protomol, docking-based virtual screening was performed. All molecules in DB1 were docked to the prepared structure of PDE9A, and 200 molecules with the highest docking scores and appropriate binding modes were selected as DB2, which were subjected to the subsequent MD simulations and binding free energy calculations.
2.4 MD simulations
After the docking-based virtual screening, MD simulations were used to equilibrate the whole system. AMBER 10.034 was used for all the MD simulations.Docking poses for the compounds in DB2 were used as the initial structures for MD simulations. Gaussian 0335 program was first used to calculate the partial atomic charges of all the ligands in DB2 by using the Hartree–Fock method and the 6-31G* basis set. Antechamber was then used to fit the restricted electrostatic potential (RESP) and assign general amber force field (GAFF) parameters.36 The protein was assigned the amber03 force field and the two metal ions in the binding site pocket were treated by the “nonbond model” method.37 For each receptor–ligand complex, Na+ ions were used to neutralize the whole system, and an 8 Å truncated octahedral box of TIP3P water molecules was added.
All the residues were protonated at the neutral pH. His256 and His292 were protonated at the δ position to form coordination bonds with Zn2+ and Mg2+ ions. As proposed by the previous study,38 the oxygen atom between the two metal ions was treated as the hydroxide ion, and the histidine nearest the hydroxide ion can capture a proton, and thus was treated as HIP (protonated histidine). For each system, 4 steps of minimization were performed before MD simulation runs. In the first step of minimization, both the receptor and ligand were restricted, and water molecules were minimized; in the second step, only the receptor was restricted; in the third step, only the ligand was restricted; and in the last step, no restriction was added and the whole system was minimized. During the first 3 steps of minimization, a restriction force of 500 kcal (mol−1 Å−2) was applied, and each minimization step contained 2500 cycles of steepest descent minimization, followed by 2500 cycles of conjugated gradient minimization. After minimization, the whole system was firstly heated from 0 to 300 K in 50 ps by using Langevin dynamics in an NVT ensemble, and then equilibrated for 100 ps in an NPT ensemble (P = 1 atm). During heating and equilibration, a weak constraint of 10 kcal (mol−1 Å−2) was applied to all the heavy atoms in the receptor–ligand complexes. Finally, 8 ns MD simulations were performed in the NPT ensemble at a constant pressure of 1 atm and a constant temperature of 300 K. During the MD simulations, periodic boundary conditions were applied, and the cutoff was set to 8 Å with long-range electrostatic interactions treated with the partial mesh Ewald (PME) method.39,40 All the bonds involving hydrogen atoms were restricted by the SHAKE algorithm,41,42 and the time step was set to 2 fs. The snapshots were extracted from the MD trajectories of the last 1 ns with a time interval of 10 ps; the resultant 100 snapshots were used to perform MM/PBSA binding free energy calculations.
2.5 MM-PBSA binding free energy calculations
According to the molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method,43–46 binding free energy (ΔGbind) is calculated from the difference between the free energies of the complex, the free receptor and the free inhibitor as per the following equation:| ΔGbind = Gcom − Grec − Glig | (1) |
G com/rec/lig was evaluated as the sum of the MM energy, the solvation free energy Gsolv, and the entropy contribution (−TS).
| Gcom/rec/lig = EMMcom/rec/lig + Gsolvcom/rec/lig − TScom/rec/lig | (2) |
Based on the two eqn (1) and (2) above, we get:
| ΔGbind = ΔEMM + ΔGsolv − TΔS | (3) |
ΔEMM is the gas phase interaction energy, which is partitioned into three terms: EMM(com), EMM(rec) and EMM(lig). Solvation free energy is calculated by the sum of electrostatic solvation free energy and nonpolar solvation free energy. The electrostatic solvation free energy was calculated by solving the Poisson Boltzmann (PB) equation (ΔGPB), and the nonpolar solvation free energy is proportional to the solvation accessible surface area (SASA).
| ΔGsolv = ΔGPB + ΔGnp | (4) |
| ΔGnp = γSASA + b | (5) |
Entropy contribution (−TΔS) can be calculated by using normal mode analysis. But the normal mode calculation is extremely time-consuming for large systems, so entropy contribution was not considered in our ΔGbind (3) calculations in order to save computational costs for the 200 PDE9A–ligand complexes. All the calculations were done using the MM-PBSA module embedded in AMBER 10.0 with the default γ = 0.0072 kcal Å−2 and b = 0 kcal mol−1.
In MM-PBSA calculations, the charges of both Mg2+ and Zn2+ were kept as 2.0 for PB calculations, and their bond radii were used for SA calculation.
About 35 compounds with the most negative ΔGbind and appropriate binding patterns via visual inspection were retained in the final database DB3. Totally, 29 compounds with non-pyrazolopyrimidinone scaffolds in DB3 were purchased from SPECS and their inhibitory activities against PDE9A were determined via bioassay.
2.6 Bioassay
000–30000 cpm per assay. The reaction was carried out at room temperature (25 °C) for 15 min and then terminated by addition of 0.2 M ZnSO4. The reaction product was precipitated by 0.2 N Ba(OH)2, whereas unreacted 3H-cGMP remained in the supernatant. The radioactivity in the supernatant was measured in 2.5 ml of Ultima Gold liquid scintillation cocktails (PerkinElmer) using a PerkinElmer 2910 liquid scintillation counter. At least eight concentrations of inhibitors were used for the measurement of IC50 of inhibitors. Each measurement was repeated at least three times, and IC50 values were calculated by nonlinear regression. The mean values of the measurements were considered as the final IC50 values with the SD values of the measurements. In this assay, Bay73-6691 purchased from SIGMA was used as the reference compound.
2.7 Structure modification based on new scaffolds of PDE9A inhibitors
Based on the bioassay results, 5 novel scaffolds of PDE9A inhibitors were identified (Fig. 4). Structure modification based on scaffold 2 was performed by chemical synthesis and evaluated by bioassay.1H NMR and 13C NMR spectra were recorded at room temperature on a Bruker BioSpin GmbH spectrometer at 400.132 and 100.614 MHz, respectively. Coupling constants are given in Hz. MS spectra were recorded on an Agilent LC-MS 6120 instrument with an ESI and APCI mass selective detector. The high resolution mass spectrum was run on a ThermoFisher LTQ Orbitrap Elite. The infrared spectrum was recorded on the PerkinElmer Lambda950. Elemental analyses were carried out using an Elementa Vario EL cube with errors of ±0.3% for CHN elements. Melting points were determined using a WRS-1B digital melting point apparatus and were not corrected. Flash column chromatography was performed using silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. Thin-layer chromatography was performed on precoated silica gel F-254 plates (0.25 mm, Qingdao Haiyang Chemical Co. Ltd) and was visualized with UV light. All the starting materials and reagents were purchased from commercial suppliers and used directly without further purification.
Syntheses of intermediates M1–M4 and compound 16 (Scheme 1) are described as follows.48,49 Cyanoacetic acid ethyl ester (25 mmol), elemental sulfur (25 mmol) and cyclohexanone (23.8 mmol) were suspended in EtOH (10 ml). Diethyl amine (23.8 mmol) was added drop-wise for 25 min and the mixture was stirred at room temperature for 16 h. Then, the solvent was removed and the residue was partitioned between 10 ml water and 10 ml ethyl acetate. The aqueous phase was extracted by ethyl acetate three times. The combined organic fractions were washed with saturated salt water, dried over anhydrous Na2SO4, and evaporated in a vacuum to give the crude product M1 (84%). M1 (19 mmol) was suspended in formamide (20 ml) and refluxed for 4 h. The dark solution was placed inside a fridge overnight, and the precipitated product (M2, 95%) was filtered and washed with a cold solution of 40% EtOH. M2 (18 mmol) was dissolved in POCl3 (15 ml) and refluxed for 3 h. Excess POCl3 was removed in vacuo and then ice water was poured into the residue. The precipitate was filtered and recrystallized in hot MeOH to give M3 as white crystals (72%). M3 (13 mmol) and hydrazine (51% in H2O, 2 ml) were added to MeOH (200 ml). The mixture was refluxed for 4 h. The precipitated yellow crystals (M4, 85%) were filtered, and washed with a cold solution of 40% EtOH. M4 (11 mmol) and 2-hydroxy-1-naphthaldehyde (13 mmol) were dissolved in EtOH (10–15 ml). The mixture was refluxed for 4 h. After cooling to room temperature, the mixture was placed in a fridge overnight and the yellow solid (16) was filtered, and recrystallized from EtOH (81%). Bioassay was then performed for compound 16 by using the protocol described above.
 | ||
| Scheme 1 Reagents and conditions: (a) diethyl amine, EtOH, rt, 16 h; (b) formamide, reflux, 4 h; (c) phosphorus oxychloride, reflux, 3 h; (d) hydrazine 51% in H2O, MeOH, reflux, 4 h; and (e) 2-hydroxy-1-naphthaldehyde, EtOH, reflux, 4 h. | ||
The characterization of compound 16 is as follows. (E)-1-(2-(3,4,5,6,7,8-Hexahydrobenzo[2,3-d]pyrimidin-4-yl) hydrazono)methyl)naphthalene-2-ol (16). Yellow solid, 81%, MS(ESI): m/z: 375.12([M + H]); 1H NMR (400 MHz, DMSO) δ 9.50 (s, 1H), 8.21 (d, J = 68.7 Hz, 1H), 7.87 (t, J = 9.0 Hz, 2H), 7.56 (t, J = 7.5 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.21 (d, J = 8.9 Hz, 1H), 2.98 (s, 1H), 2.75 (d, J = 33.5 Hz, 2H), 2.10–1.65 (m, 4H), 1.18 (d, J = 17.8 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 159.39, 157.57, 132.13, 131.83, 131.68, 129.50, 128.80, 128.28, 127.78, 127.62, 127.39, 126.94, 123.36, 120.91, 118.81, 48.57, 24.89, 22.19, 21.98. IR(Pure, cm−1): 3442, 2930, 2840, 2028, 1621, 1582, 1566, 1540, 1468, 1425, 1391, 1377, 1340, 1319, 1278, 1239, 1222, 1209, 1186, 1143, 778, 748. Anal. calcd for C21H18N4OS: C, 65.71, H, 5.218, N, 14.31; Found: C, 67.3, H, 4.8, N, 14.95. Mp. 248–249.5 °C.
3 Results and discussion
Before the pharmacophore screening, the SPECS database was prefiltered by Lipinski's rules. The default filtration rules implemented in the conformation import method in MOE 2008.10 were also used to further reduce the database size. As a result, 112755 molecules passed the filtration and were saved as DB0.
3.1 Pharmacophore-based virtual screening
Based on the superimposed PDE9A crystal structures, our pharmacophore model was generated by using the consensus strategy. After the pharmacophore model was generated, a test set was used to evaluate the efficacy of this model. The GH test is a well-accepted method for evaluating the efficacy of a resulting screening model. The pharmacophore model was modified several times based on the test set result to improve its screening efficacy.Here, a hit refers to what fits the derived pharmacophore model well. In general, GH test scores of ≥0.5 and ≥0.6 indicate a good screening model and an excellent screening model, respectively. After modifications, as shown in Table 1, 77 out of the 78 active compounds and 66 out of the 674 inactive compounds in the test set were selected by the pharmacophore model, and the final GH test score of the pharmacophore model was 0.58, which means our pharmacophore model has high efficacy and is acceptable.
| Parameters | Values |
|---|---|
| a EF = (Ha × D)/(Ht × A). b GH = {[Ha × (3A + Ht)]/(4Ht × A)} × [1 − (Ht − Ha)/(D − A)]. | |
| Total number of molecules in database (D) | 752 (78 + 674) |
| Total number of hit compounds in database (Ht) | 144 |
| Total number of known potent compounds in database (A) | 78 |
| Total number of known potent compounds in hit list (Ha) | 77 |
| False positive | 67 |
| False negative | 1 |
| True positive | 77 |
| True negative | 607 |
| Ratio of known potent compounds [(Ha/A) × 100]% | 98.7 |
| Yield of known potent compounds [(Ha/Ht) × 100]% | 53.5 |
| Enrichment Factora (EF) | 5.16 |
| Goodness of hit test scoreb (GH) | 0.58 |
This pharmacophore model contains 5 features as shown in Fig. 1. In this pharmacophore model, the Acc2 feature represents the acceptor projection, the Don2 feature represents the donor projection, and the Aro|PiR is a unified feature which represents the aromatic center or Pi ring center, while the Aro|Hyd feature represents the hydrophobic centroid or aromatic center. Aside from the features above, this model also contains the excluded volume features, which are not represented in this figure.
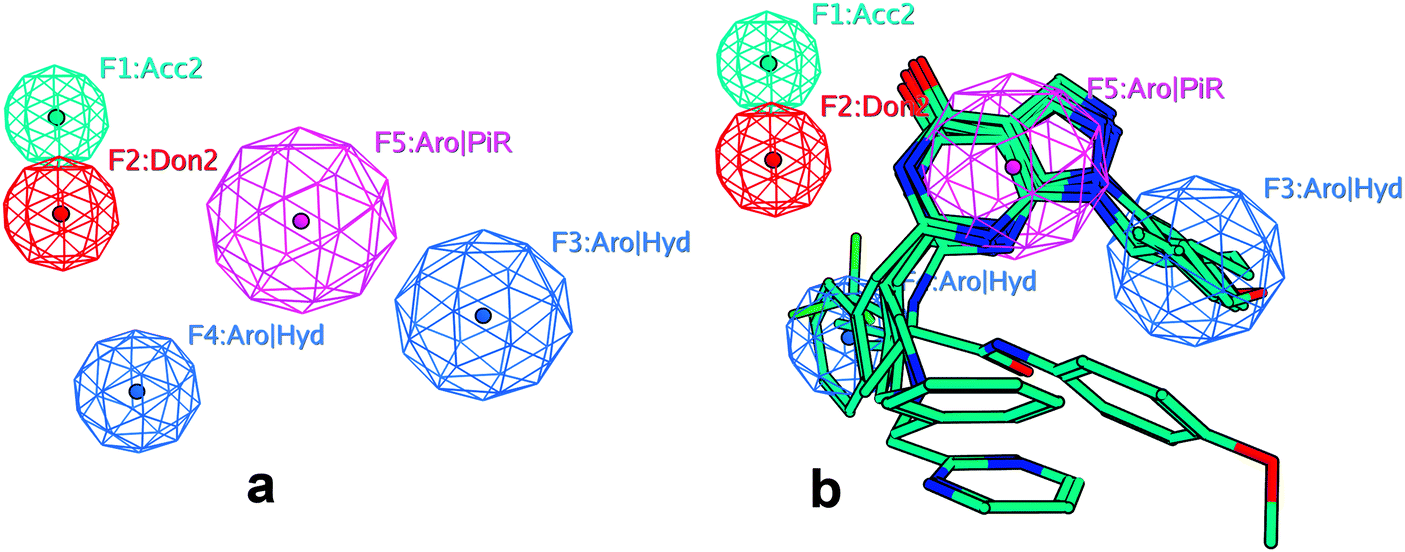 | ||
| Fig. 1 (a) The pharmacophore model used in the screening of PDE9A inhibitors. This model contains 5 features: the F1 feature is a hydrogen bond acceptor projection; the F2 feature is a hydrogen bond donor projection; the F5 feature is a unified aromatic ring center or pi ring center; and the F3 and F4 features are a unified aromatic ring center or hydrophobic centroid. (b) The pharmacophore model mapped with the ligands in the training set. | ||
This pharmacophore model was then used for the screening of DB0. A molecule must fit all the features in this model to pass the screening. Based on this screening strategy, 114745 out of 122755 molecules were filtered out, whereas 8010 molecules were retained and saved as DB1.
3.2 Molecular docking-based virtual screening
The 8010 compounds in DB1 were then screened by molecular docking. Before the screening, the docking method was evaluated, so the original ligand 28s in 4GH629 was extracted from the crystal structure and redocked back to the structure. There are several parameters controlling the docking procedures, and different combinations of the parameters were attempted. Before the screening, the ‘additional starting conformations per molecules’ parameter was set to 5, and the ‘density of search’ parameter was set to 6; each molecule was set to generate at most 50 conformations, and other parameters were set to the default values. In the docking procedure, the average RMSD for the ligand 28s between the original X-ray pose and the top 20 docking poses was 1.5 Å, which suggests that the Surflex-dock approach is suitable for the PDE9A system. Thus, identical parameters were used for subsequent database screening.In the binding site pocket of PDE9A, Gln453 and Phe456 are two conservative amino acid residues which play very important roles in the receptor–ligand interactions. In the present study, only molecules interacting with the two residues were retained as potent PDE9 inhibitors. Some of the molecules with appropriate binding poses are shown in Fig. S2 (ESI†). After the docking-based virtual screening, the binding mode of each molecule with PDE9A was carefully examined. As a result, 200 molecules with both appropriate binding modes and the highest docking scores were selected as DB2 for the further study.
3.3 MD-based virtual screening
To evaluate the reliability of the MD simulations method and selected parameters, crystal structure 4GH6 was used as the reference. During the MD simulation, the RMSD values of the backbone atoms for 4GH6 were monitored and are shown in Fig. S3 (ESI†). As seen from this figure, the RMSD curve became stable after 3 ns and the PDE9A/28s complex achieved equilibrium, which implied that this MD method and the relevant parameters were suitable for the PDE9A system. Identical conditions were used for the following MD-based virtual screening.8 ns MD simulations were performed for all the 200 PDE9A–ligand complexes from DB2. After all the complexes were stabilized, 100 snapshots were extracted from the last 1 ns of each trajectory with a time interval of 10 ps for the MM/PBSA binding energy calculations. Finally, 36 compounds with both appropriate binding modes and the highest predicted binding energies were selected as DB3. There are 7 purine-like and 29 non-pyrazolopyrimidinone compounds in DB3, and only the latter were ordered from SPECS for the final bioassay. RMSD curves of the selected 29 compounds with PDE9A during MD simulations are contained in the ESI.†
3.4 Bioassay validation
Here Bay73-6691 was used as the reference compound and its IC50 value was 48 nM. Under identical conditions, 15 of the 29 tested compounds were confirmed to be PDE9A inhibitors in vitro and their IC50 values were no more than 50 μM (Table 2). The best IC50 of compound 1 was 6.1 μM. Among the 15 compounds, 4 of them had IC50 values of less than 10 μM. The inhibitory curves of the four representative compounds are shown in Fig. S4 (ESI†), which shows an unambiguous dose-dependent effect. The structures of the 15 novel PDE9A inhibitors are shown in Fig. 2, along with their SPECS IDs and inhibitory activities.| No. | SPECS ID | IC50 | SD | ΔG(exp)1 | ΔG(cal)1 |
|---|---|---|---|---|---|
| Bay73-6691 serves as the reference compound for the bioassay (IC50 = 0.048 μM). ΔG(exp)1 = −RTln(IC50) and ΔG(cal)1 = −ΔG(MM/PBSA). |
|||||
| 1 | AG-690/12412006 | 6.5 | ±0.6 | 7.11 | 30.69 |
| 2 | AG-690/40135604 | 8.0 | ±2.3 | 6.99 | 28.5 |
| 3 | AB-323/13887077 | 9.4 | ±1.2 | 6.90 | 30.92 |
| 4 | AK-968/15363760 | 9.7 | ±0.8 | 6.88 | 30.95 |
| 5 | AG-690/12412005 | 13.1 | ±0.8 | 6.70 | 34.06 |
| 6 | AO-476/43380494 | 15.1 | ±2.3 | 6.61 | 26.22 |
| 7 | AN-970/40920752 | 15.1 | ±2.0 | 6.61 | 30.85 |
| 8 | AO-476/43362623 | 18.5 | ±0.3 | 6.49 | 25.18 |
| 9 | AG-205/11867117 | 20.9 | ±2.4 | 6.42 | 31.17 |
| 10 | AO-476/43250258 | 31.9 | ±4.6 | 6.17 | 27.07 |
| 11 | AK-968/41024707 | 34.6 | ±2.8 | 6.12 | 26.25 |
| 12 | AG-690/37029001 | ≈50 | — | — | |
| 13 | AN-329/40528720 | ≈50 | — | — | |
| 14 | AO-476/43362622 | ≈50 | — | — | |
| 15 | AG-205/37136077 | ≈50 | — | — | |
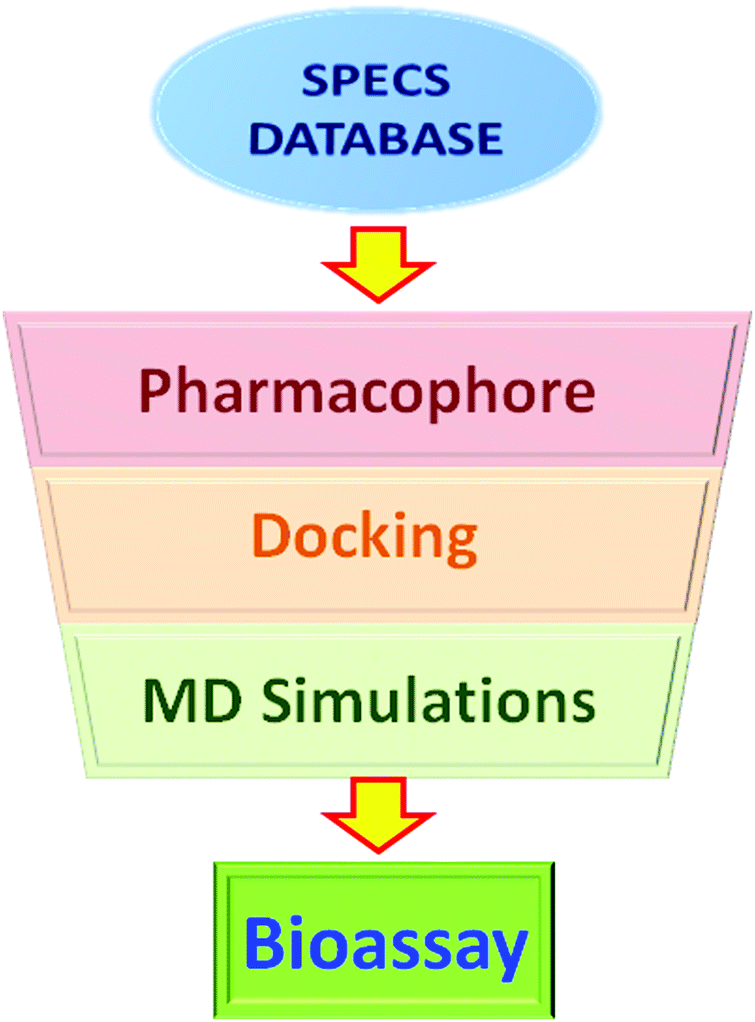 | ||
| Fig. 2 Workflow of the virtual screening process, containing the pharmacophore model, docking, and MD simulations. The selected molecules were submitted for further bioassay. | ||
3.5 Manual modification of the pharmacophore model is important
The pharmacophore model used in this screening was built based on several crystal structures of PDE9A that have purine-like inhibitors in their binding site pockets. Because of the similarity between the ligands, the initial pharmacophore model constructed automatically by MOE has 14 features that match all the ligands simultaneously, as shown in Fig. S5 (ESI†). This model has too many restraints and is obviously an over-fitting model, and as a result, almost no molecule can fit this model. However, there are no other kinds of PDE9A inhibitors reported, thus the only way to solve this problem is to analyze the receptor structure and get rid of the unimportant features. The key residues in the receptor are Phe456 and Gln453, which form π–π stacking and hydrogen bonds with the purine-like scaffolds of the ligands. So according to the interactions with the two key residues, all the features in the purine structure were abstracted to an Aro|PiR feature, an Acc2 feature, and a Don2 feature. Acc2 and Don2 are acceptor projection and donor projection features, which represent the location of the other end of hydrogen bonds according to the acceptors and donors, respectively. In this model, the two features represent the location of the side chain of Gln453. No Acc or Don features were used to in order to reduce the restraints of this screening.After manual modification, 14 features were abstracted to 5 features, and this allows enough compounds to pass the pharmacophore screening before molecular docking and MD studies.
3.6 Binding patterns after 8 ns MD simulations
Understanding the binding modes of the new PDE9 inhibitors is very important for further structural modification, so their binding patterns after 8 ns of MD simulations were analyzed. The binding structures of the 4 most representative inhibitors are illustrated in Fig. 3. The binding mode between AG-690/12412005 and PDE9A is shown in picture 1 (Fig. 3), from which we can see that the most obvious interactions are two hydrogen bonds, a π–π stacking, and a salt bridge formed by water molecules. The binding between AG-690/40145604 and PDE9A is shown in picture 2 (Fig. 3), and this ligand forms pi–pi stacking with Phe456, two hydrogen bonds with Gln453, and a hydrogen bond with Asn405. Picture 3 shows the binding mode between AB-323/13887077 and PDE9A. The molecule forms hydrogen bond interactions with Tyr424, Ala452, and Gln453, and also stacks against Phe456. The final picture shows the binding mode between AO-476/43380494 and PDE9A. This molecule forms two hydrogen bonds with Gln453, while the π–π stacking interaction with Phe456 also exists.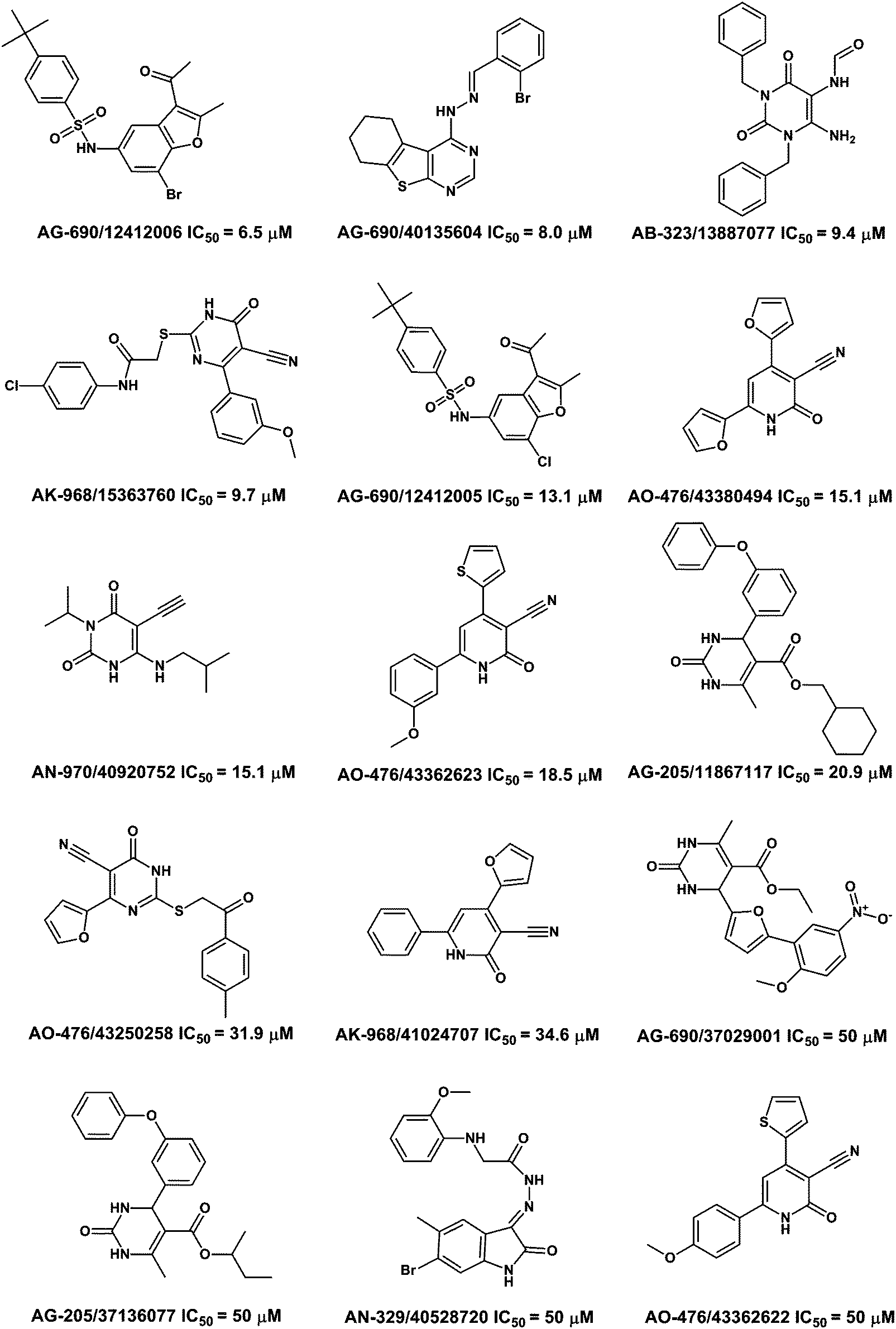 | ||
| Fig. 3 Chemical structures and IC50 values of the 15 PDE9A inhibitors along with their SPECS IDs. | ||
The four molecules described above have formed π–π stacking with Phe456 and most of them, except the molecule coded AG-690/12412005, have formed hydrogen bond(s) with Gln453. The two residues were also considered as conservative residues in PDE families. Based on the binding mode between AG-690/12412005 and PDE9A, it is clear that this molecule does not form a hydrogen bond with Gln453, which reveals that PDE inhibitors are not necessary to have specific interactions with Gln453. This binding mode may also indicate a new way to find novel PDE inhibitors.
3.7 New scaffolds for PDE9 inhibitors
As much as we know, most of the reported PDE9 inhibitors have a common purine-like nucleus (or with the same scaffold as pyrazolopyrimidinone, see Fig. S1, ESI†), and the lack of variety in structures is obviously very unfavorable for the development of PDE9 inhibitors. So the proposition of new scaffolds is very important. Based on our screening result and the binding mode analysis, several new scaffolds could be summarized. As shown in Fig. 4, AG-690/12412006 and AG-690/12412005 have a common scaffold 1; AG-690/40135604 has scaffold 2; AB-323/13887077 has scaffold 3; AN-970/40920752 has scaffold 4; and most of the other molecules have scaffold 5 (Fig. 5).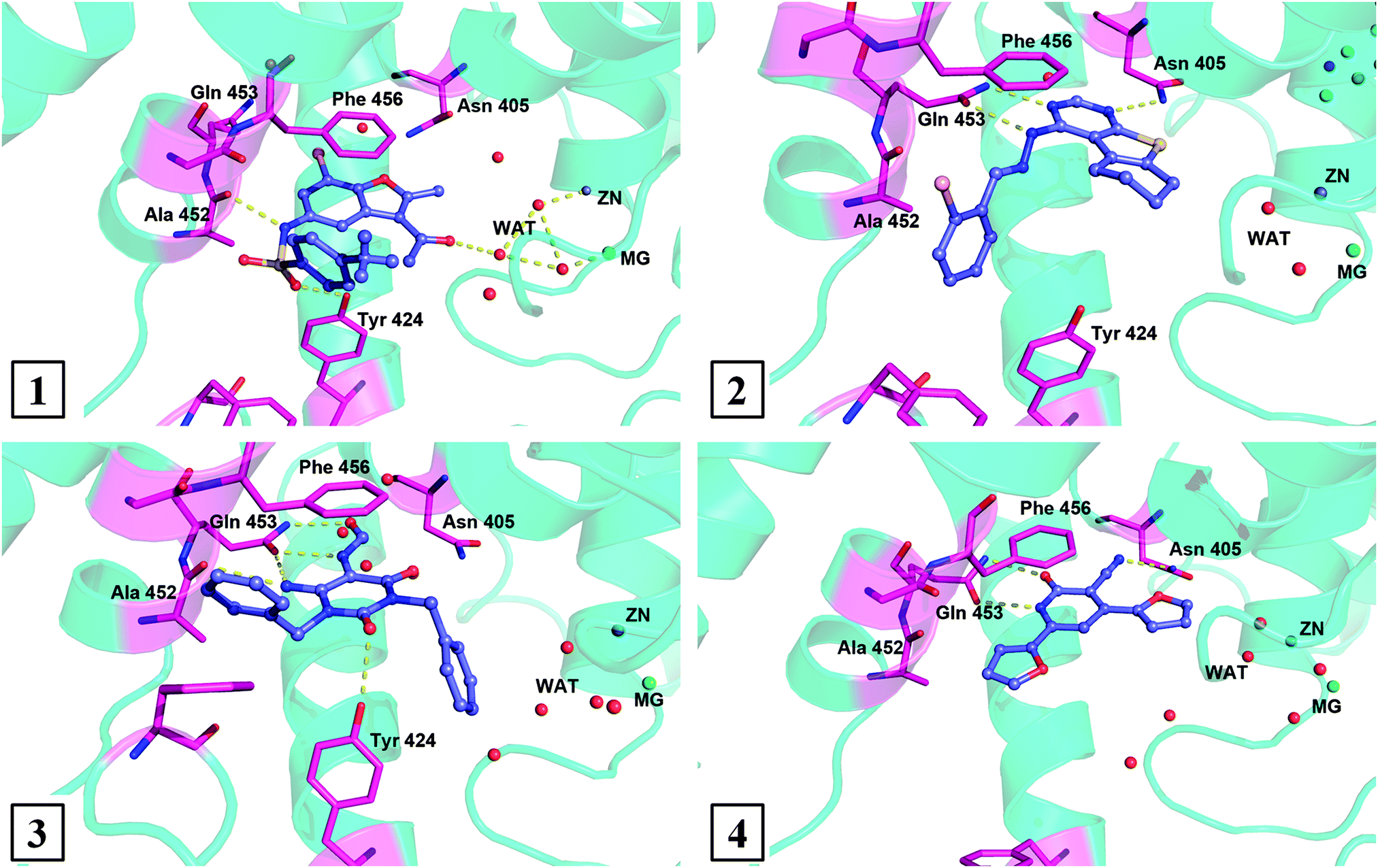 | ||
| Fig. 4 Binding modes of the most four representative inhibitors with PDE9A after 8 ns MD simulations. | ||
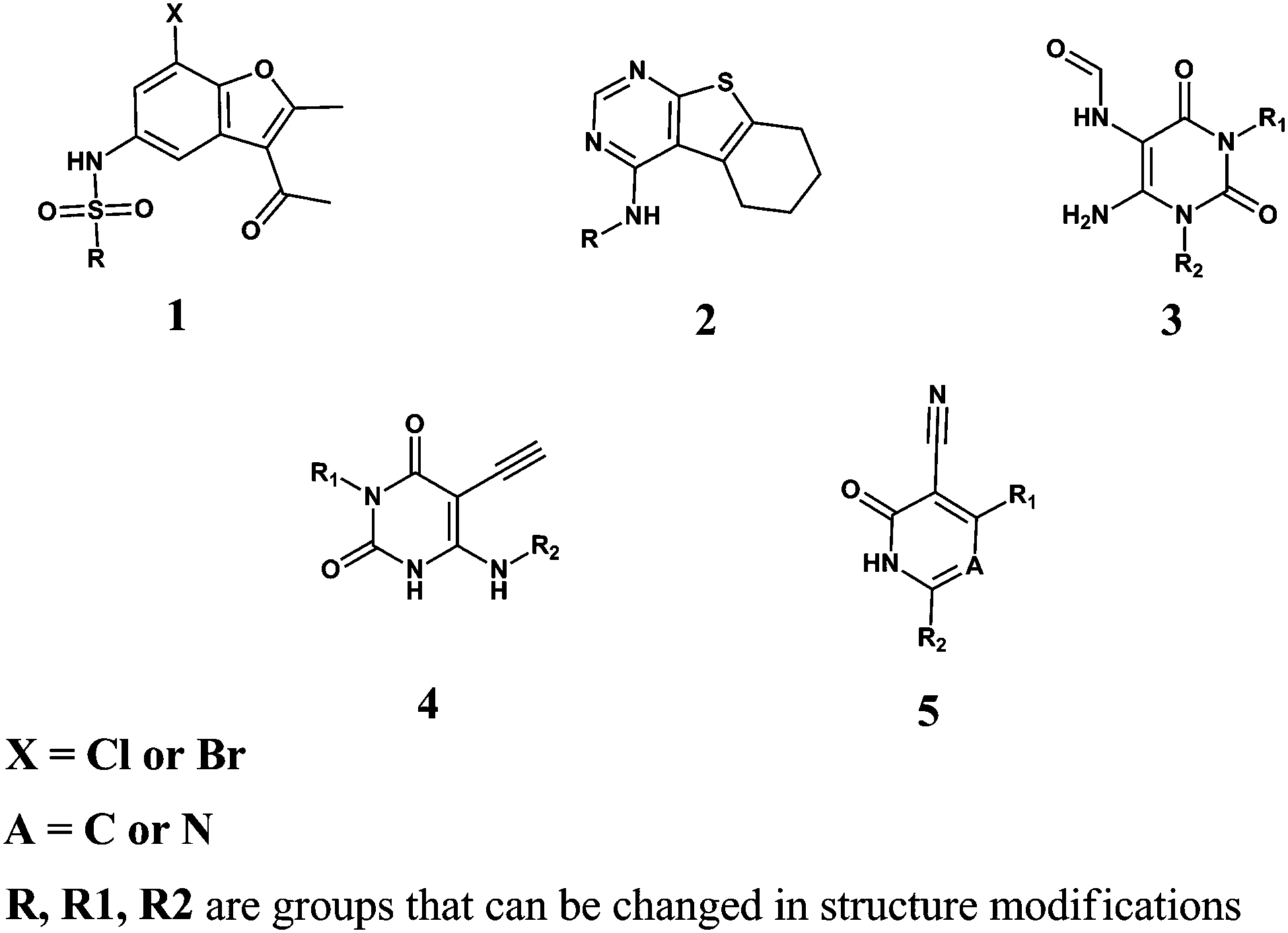 | ||
| Fig. 5 The five new scaffolds for PDE9A inhibitors. | ||
From the binding modes of the ligands, scaffolds 2, 3, 4, and 5 may have great potential for further structural modification. Among the four scaffolds, 2, 4, and 5 have very rigid nuclei, and due to a lack of rotatable bonds, rigid molecules are more favorable in the entropy when binding the receptor. For example, structural modification of compound AG-690/40135604 (IC50 = 8.0 μM) in Scheme 1 led to a new one, 16, with an improved inhibitory affinity of 2.1 μM as expected. Scaffold 3, although not as rigid as the other three scaffolds, forms stronger hydrogen bond interactions with the receptor. Scaffold 5 is not a totally novel one when A = N, because there is a report of this kind of molecule.50 But as far as we know, no reports of PDE9A inhibitors exist when A = C.
The novel scaffolds only occupy the Q-pocket of PDE9, which is similar to the classical pyrazolopyrimidinone-like inhibitors. So the strategies used in the structural modifications of the purine-like structures may also be suitable for the inhibitors with the five new scaffolds. As shown in our previous work, the modification based on the Tyr424 and H-pocket can increase the inhibitory activity of the pyrazolopyrimidinone-like or purine-like structures greatly,28 so we have a good reason to believe that the modification of inhibitors with new novel scaffolds is feasible in a similar way to the pyrazolopyrimidinone-like or purine-like inhibitors.
4 Conclusions
To find new scaffolds for PDE9A inhibitors rather than pyrazolopyrimidinones, a combinatorial method containing pharmacophores, molecular docking, MD simulations, binding free energy calculations, and bioassay was adopted in this study. 29 non-pyrazolopyrimidinone compounds were selected by our MD-based virtual screening, and 15 of them with affinities of no more than 50 μM were proved to be active compounds via bioassay. The high hit ratio of 52% for this virtual screening method indicated that the combinatorial method used in this study is a good compromise between computational cost and accuracy. Based on the 15 novel PDE9A inhibitors, 5 kinds of new scaffolds were summarized. The new scaffolds occupy the Q-pocket of PDE9A, and have great potential in further structural modifications. Modification of compound AG-690/40135604 (IC50 = 8.0 μM) led to a new one, 16, with an improved affinity of 2.1 μM as expected. Our further modifications based on this scaffold gave inhibitory affinities of no more than 500 nM (not shown), which is beyond the scope of this study.This study has provided five new scaffolds different from those of the classical PDE9A inhibitors and also provided a clue to the development of PDE9A inhibitors in the future.
Acknowledgements
We cordially thank Prof. H. Ke of the Department of Biochemistry and Biophysics at the University of North Carolina, Chapel Hill, for his help with molecular cloning, expression, purification, and bioassay of PDE9A2, and the High Performance Supercomputer Center at Sun Yat-Sen University. This work was supported by the Natural Science Foundation of China (21103234 and 81373258), the Guangdong Natural Science Foundation (S2011030003190 and S2013010014867), the Guangdong Science Foundation (2012A080201007), Science Foundation of Guangzhou City (2014J4100165), and the Research Fund for the Doctoral Program of Higher Education of China (20130171110096).Notes and references
- M. D. Houslay, Semin. Cell Dev. Biol., 1998, 9, 161–167 CrossRef CAS PubMed.
- M. Zaccolo and M. A. Movsesian, Circ. Res., 2007, 100, 1569–1578 CrossRef CAS PubMed.
- J. S. O'Neill, E. S. Maywood, J. E. Chesham, J. S. Takahashi and M. H. Hastings, Science, 2008, 320, 949–953 CrossRef PubMed.
- F. A. Antoni, Front. Neuroendocrinol., 2000, 21, 103–132 CrossRef CAS PubMed.
- S. S. Hannila and M. T. Filbin, Exp. Neurol., 2008, 209, 321–332 CrossRef CAS PubMed.
- M. Piper, F. van Horck and C. Holt, Adv. Exp. Med. Biol., 2007, 621, 134–143 CrossRef.
- A. Horvath and C. A. Stratakis, Curr. Opin. Endocrinol., Diabetes Obes., 2008, 15, 227–233 CrossRef CAS PubMed.
- C. Lugnier, Pharmacol. Ther., 2006, 109, 366–398 CrossRef CAS PubMed.
- S. H. Francis, M. A. Blount and J. D. Corbin, Physiol. Rev., 2011, 91, 651–690 CrossRef CAS PubMed.
- S. H. Soderling, S. J. Bayuga and J. A. Beavo, J. Biol. Chem., 1998, 273, 15553–15558 CrossRef CAS PubMed.
- D. A. Fisher, J. F. Smith, J. S. Pillar, D. S. St and J. B. Cheng, Biochem. Biophys. Res. Commun., 1998, 246, 570–577 CrossRef CAS PubMed.
- P. Wang, P. Wu, R. W. Egan and M. M. Billah, Gene, 2003, 314, 15–27 CrossRef CAS.
- H. Wang, X. Luo, M. Ye, J. Hou, H. Robinson and H. Ke, J. Med. Chem., 2010, 53, 1726–1731 CrossRef CAS PubMed.
- K. U. Domek-Lopacinska and J. B. Strosznajder, Mol. Neurobiol., 2010, 41, 129–137 CrossRef CAS PubMed.
- M. P. Deninno, M. Andrews, A. S. Bell, Y. Chen, C. Eller-Zarbo, N. Eshelby, J. B. Etienne, D. E. Moore, M. J. Palmer, M. S. Visser, L. J. Yu, W. J. Zavadoski and G. E. Michael, Bioorg. Med. Chem. Lett., 2009, 19, 2537–2541 CrossRef CAS PubMed.
- D. A. Fryburg and E. M. Gibbs, US Pat., 6 967 204, 2005 Search PubMed.
- A. S. Bell, M. P. DeNinno, M. J. Palmer and M. S. Visser, PDE9 inhibitors for treating type 2 diabetes, metabolic syndrome, and cardiovascular disease, US Pat., 20 040 220 186, 2004 Search PubMed.
- M. P. Deninno, M. Andrews, A. S. Bell, Y. Chen, C. Eller-Zarbo, N. Eshelby, J. B. Etienne, D. E. Moore, M. J. Palmer, M. S. Visser, L. J. Yu, W. J. Zavadoski and G. E. Michael, Bioorg. Med. Chem. Lett., 2009, 19, 2537–2541 CrossRef CAS PubMed.
- F. Wunder, A. Tersteegen, A. Rebmann, C. Erb, T. Fahrig and M. Hendrix, Mol. Pharmacol., 2005, 68, 1775–1781 CAS.
- A. Garcia-Osta, M. Cuadrado-Tejedor, C. Garcia-Barroso, J. Oyarzabal and R. Franco, ACS Chem. Neurosci., 2012, 3, 832–844 CrossRef CAS PubMed.
- D. P. Rotella, Nat. Rev. Drug Discovery, 2002, 1, 674–682 CrossRef CAS PubMed.
- U. Gresser and C. H. Gleiter, Eur. J. Med. Res., 2002, 7, 435–446 CAS.
- A. Daugan, P. Grondin, C. Ruault, D. G. A. Le Monnier, H. Coste, J. M. Linget, J. Kirilovsky, F. Hyafil and R. Labaudiniere, J. Med. Chem., 2003, 46, 4533–4542 CrossRef CAS PubMed.
- P. R. Verhoest, C. Proulx-Lafrance, M. Corman, L. Chenard, C. J. Helal, X. Hou, R. Kleiman, S. Liu, E. Marr, F. S. Menniti, C. J. Schmidt, M. Vanase-Frawley, A. W. Schmidt, R. D. Williams, F. R. Nelson, K. R. Fonseca and S. Liras, J. Med. Chem., 2009, 52, 7946–7949 CrossRef CAS PubMed.
- J. Hou, J. Xu, M. Liu, R. Z. Zhao, H. B. Luo and H. M. Ke, PLoS One, 2011, 6, e1809 Search PubMed.
- P. R. Verhoest, K. R. Fonseca, X. Hou, C. Proulx-Lafrance, M. Corman, C. J. Helal, M. M. Claffey, J. B. Tuttle, K. J. Coffman, S. Liu, F. Nelson, R. J. Kleiman, F. S. Menniti, C. J. Schmidt, M. Vanase-Frawley and S. Liras, J. Med. Chem., 2012, 55, 9045–9054 CrossRef CAS PubMed.
- M. M. Claffey, C. J. Helal, P. R. Verhoest, Z. Kang, K. S. Fors, S. Jung, J. Zhong, M. W. Bundesmann, X. Hou, S. Lui, R. J. Kleiman, M. Vanase-Frawley, A. W. Schmidt, F. Menniti, C. J. Schmidt, W. E. Hoffman, M. Hajos, L. McDowell, R. E. O'Connor, M. Macdougall-Murphy, K. R. Fonseca, S. L. Becker, F. R. Nelson and S. Liras, J. Med. Chem., 2012, 55, 9055–9068 CrossRef CAS PubMed.
- F. Meng, J. Hou, Y. X. Shao, P. Y. Wu, M. Huang, X. Zhu, Y. Cai, Z. Li, J. Xu, P. Liu, H. B. Luo, Y. Wan and H. Ke, J. Med. Chem., 2012, 55, 8549–8558 CrossRef CAS PubMed.
- N. Huang, B. K. Shoichet and J. J. Irwin, J. Med. Chem., 2006, 49, 6789–6801 CrossRef CAS PubMed.
- S. Vadivelan, B. N. Sinha, S. J. Irudayam and S. A. Jagarlapudi, J. Chem. Inf. Model., 2007, 47, 1526–1535 CrossRef CAS PubMed.
- K. Boppana, P. K. Dubey, S. A. Jagarlapudi, S. Vadivelan and G. Rambabu, Eur. J. Med. Chem., 2009, 44, 3584–3590 CrossRef CAS PubMed.
- K. C. Shih, C. Y. Lin, J. Zhou, H. C. Chi, T. S. Chen, C. C. Wang, H. W. Tseng and C. Y. Tang, J. Chem. Inf. Model., 2011, 51, 398–407 CrossRef CAS PubMed.
- A. N. Jain, J. Med. Chem., 2003, 46, 499–511 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. I. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. P. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 10, University of California, San Francisco, 2008 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, O. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 (Revision E.01), Gaussian, Inc., Pittsburgh PA, 2004 Search PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- R. H. Stote and M. Karplus, Proteins, 1995, 23, 12–31 CrossRef CAS PubMed.
- Y. Xiong, H. T. Lu, Y. Li, G. F. Yang and C. G. Zhan, Biophys. J., 2006, 91, 1858–1867 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, D. York and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- M. Shuichi and P. A. Kollman, J. Comput. Chem., 1992, 8, 952–962 Search PubMed.
- J. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- I. Massova and P. A. Kollman, Perspect. Drug Discovery Des., 2000, 1, 113–135 CrossRef.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
- Z. Li, Y. H. Cai, Y. K. Cheng, X. Lu, Y. X. Shao, X. Li, M. Liu, P. Liu and H. B. Luo, J. Chem. Inf. Model., 2013, 53, 972–981 CrossRef CAS PubMed.
- M. Liu, M. Yuan, M. Luo, X. Bu, H. B. Luo and X. Hu, Biophys. Chem., 2010, 147, 28–34 CrossRef CAS PubMed.
- S. K. Chen, P. Zhao, Y. X. Shao, Z. Li, C. Zhang, P. Liu, X. He, H. B. Luo and X. Hu, Bioorg. Med. Chem. Lett., 2012, 22, 3261–3264 CrossRef PubMed.
- J. C. Aponte, A. J. Vaisberg, D. Castillo, G. Gonzalez, Y. Estevez, J. Arevalo, M. Quiliano, M. Zimic, M. Verastegui, E. Malaga, R. H. Gilman, J. M. Bustamante, R. L. Tarleton, Y. Wang, S. G. Franzblau, G. F. Pauli, M. Sauvain and G. B. Hammond, Bioorg. Med. Chem., 2010, 18, 2880–2886 CrossRef CAS PubMed.
- B. E. Sleebs, G. Nikolakopoulos, I. P. Street, H. Falk and J. B. Baell, Bioorg. Med. Chem. Lett., 2011, 21, 5992–5994 CrossRef CAS PubMed.
- M. Hendrix, L. Bärfacker, B. Beyreuther, U. Ebert, C. Erb, et al., 6-Arylamino-5-cyano-4-pyrimidinones as PDE9A inhibitors, US Pat., 7 488 733, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Chemical structures of classical PDE9 inhibitors, compounds with “appropriate” binding modes with PDE9, the RMSD curve of the PDE9A crystal structure (PDB ID: 4GH6) during the molecular dynamics simulations, inhibitory curves of the four most potent compounds towards PDE9A, the pharmacophore model generated automatically by MOE 2008.10., and rmsd curves of all the 29 selected compounds during molecular dynamics simulations. See DOI: 10.1039/c4mb00389f |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2015 |