DOI:
10.1039/C4MB00436A
(Method)
Mol. BioSyst., 2015,
11, 71-76
In vitro selection of DNA-based aptamers that exhibit RNA-like conformations using a chimeric oligonucleotide library that contains two different xeno-nucleic acids†
Received
24th July 2014
, Accepted 10th October 2014
First published on 10th October 2014
Abstract
We successfully generated chimeric DNA aptamers that contained six nucleoside analogs of 2′-O,4′-C-methylene bridged/locked nucleic acid (2′,4′-BNA/LNA) in the primer region and multiple guanosine analogs of 2′-deoxy-2′-fluoro-ribonucleic acid (FNA) in the non-primer region using capillary electrophoresis-systematic evolution of ligands by exponential enrichment (CE-SELEX). Active species enrichment became saturated only after five selection rounds, and we obtained DNA-based xeno-nucleic acid (XNA) aptamers that had high binding affinities for the target human thrombin, with dissociation constant (Kd) values of ≥10 nanomolar. Based on sequence and circular dichroism (CD) analyses, these XNA aptamers exhibited RNA-like conformations, which could cause DNA-based strands to adopt structurally diverse conformations.
Introduction
Xeno-nucleic acids (XNAs),1 such as 1,5-anhydrohexitol nucleic acid (HNA),2 cyclohexenyl nucleic acid (CeNA),3 α-L-threofuranosyl nucleic acid (TNA),4,5 FNA,6–8 and 2′,4′-BNA/LNA (hereafter referred to as 2′,4′-BNA),9–13 have recently been recognized for their role in heredity and evolution of engineered polymerases.14 The availability of XNA transcription and reverse transcription for XNA aptamer selection was demonstrated by classical SELEX15–17 selections using an HNA library. Some DNA polymerases and their mutants can accept sugar-modified nucleoside-5′-triphosphates as a substrate to provide the corresponding modified oligodeoxyribonucleotide (ODN) strands.18 For example, we previously demonstrated that KOD Dash DNA polymerase and KOD mutants could elongate modified DNA strands with 2′-O,4′-C bridged 5-methyluridines using natural DNA as a template.19 However, only few polymerases that can tolerate this type of bulky sugar modification have been reported thus far. In contrast, the 2′-fluoro substitution, which is one of the smallest sugar modifications, was acceptable for some RNA and DNA polymerases.12,20 In fact, 2′-deoxy-2′-fluoro-pyrimidine nucleotides (U, C) are used in a therapeutic RNA-based modified aptamer, i.e., MACUGEN® (pegaptanib sodium injection), to enhance structural stability and nuclease resistance.21 However, to date, no reports have described acquiring DNA-based FNA aptamers by SELEX methods.
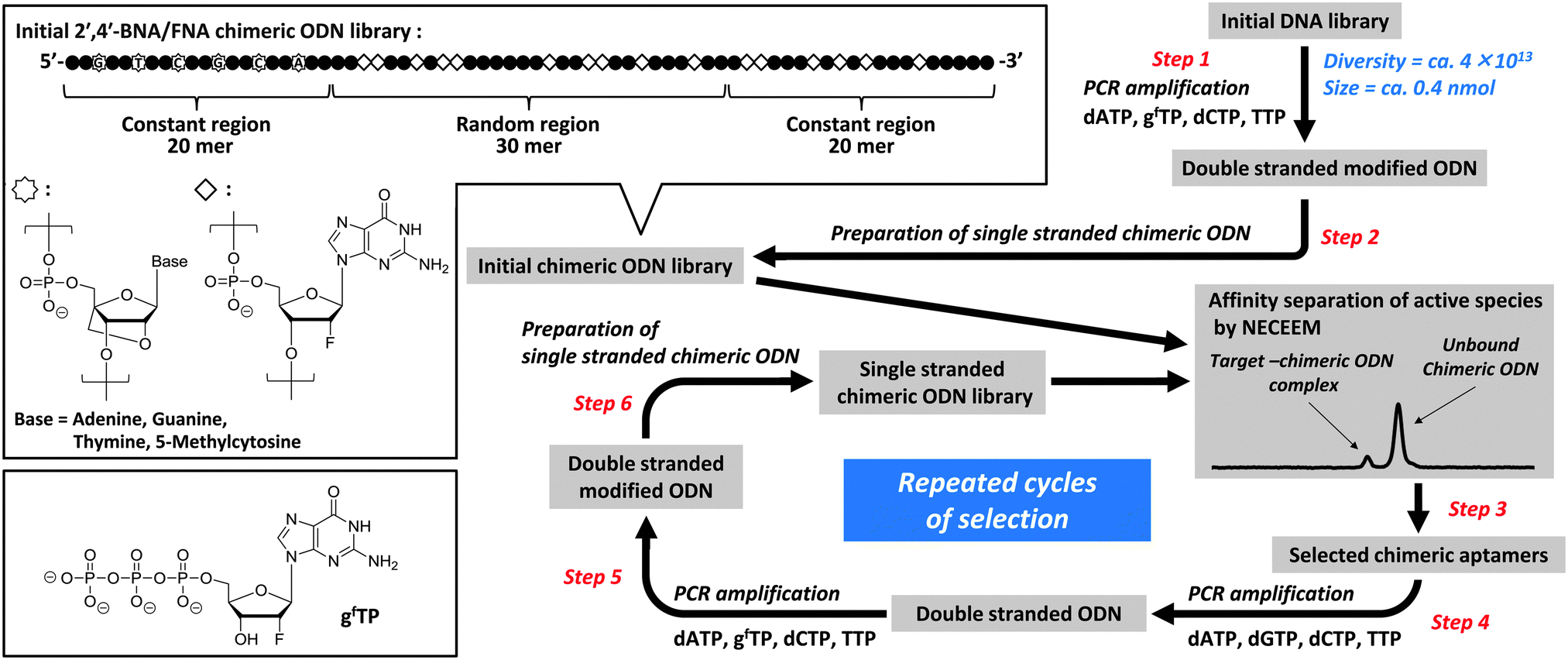
In this study, we first demonstrated the applicability of FNA to CE-SELEX selection22–28 using a chimeric ODN library with a 2′,4′-BNA primer (Fig. 1). Before the SELEX experiment, we assessed the substrate properties of 2′-deoxy-2′-fluoro-nucleoside-5′-triphosphates in a polymerase reaction. The data obtained showed that KOD Dash DNA polymerase can accept both pyrimidine (T, C) and purine (A, G) nucleotides (Fig. S1, ESI†), although T7 RNA polymerase, which was used for development of pegaptanib, hardly tolerates the latter (A, G).29 Furthermore, inclusion of 2′-deoxy-2′-fluoro-guanosines (gf's) in the ODN library could be considered to destabilize G-quadruplex formation6,30 and thus provide alternative motifs for target-binding, because FNA prefers the northern-biased sugar pucker (N-type sugar pucker, C3′-endo puckering) while DNA prefers the southern-biased sugar pucker (S-type sugar pucker, C2′-endo puckering). Therefore, we chose gf as a purine nucleotide for library construction. The binding affinities and specificities of the 2′,4′-BNA/FNA chimeric aptamers that were obtained were determined and collated with those of 2′,4′-BNA aptamers and 2′,4′-BNA primer-containing DNA aptamers with identical sequences, in which all g′s in the non-primer region were enzymatically replaced with 2′-O,4′-C-methylene-bridged/linked bicyclic guanosines (gb's), which are constrained to the N-type sugar pucker, and natural 2′-deoxyguanosines (G's), respectively. We also analyzed their structural conformations and stabilities in human serum.
 |
| | Fig. 1 The schematic illustration of TBA selection from the 2′,4′-BNA/FNA chimeric ODN library using CE-SELEX. | |
Results and discussion
Enrichment and sequence analyses
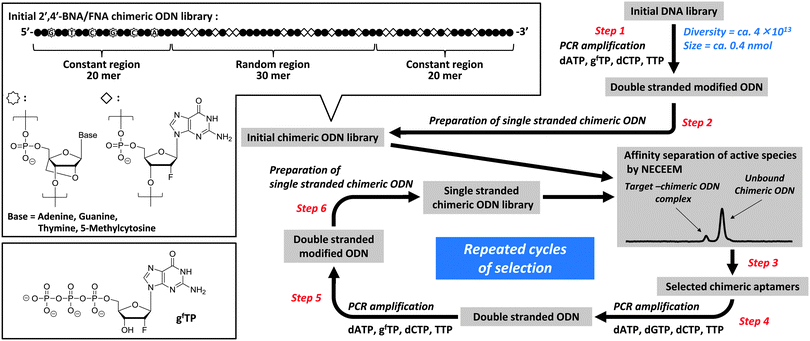
Application of a similar CE-SELEX protocol to the selection of thrombin-binding aptamers (TBAs) required four and 11 rounds to achieve the enrichment of natural DNA and DNA-based 2′,4′-BNA libraries, respectively.18,31 In the present study, enrichment of the 2′,4′-BNA/FNA chimeric ODN library was more rapid than with the DNA-based 2′,4′-BNA library and comparable to that with the natural DNA library (Fig. 2), possibly because of the sufficient efficiency and accuracy with replicating FNA strands with active sequences. Eventually, 42 sequences were isolated from the final enriched library and identified as shown in Table 1. The sequences were classified into seven groups, and multiple incorporations of gf's, but no G-quadruplex motifs32,33 were found in every sequence recovered except for group III, assuming that the length of the loop was less than 12 nucleotides.
 |
| | Fig. 2 Capillary electrograms for the 2′,4′-BNA/FNA chimeric ODN library of each round with (A) or without (B) human thrombin. All electrograms recorded fluorescent intensity of 5′-labelled 6-FAM versus tm (migration time). (C) Process of active species enrichment in selection rounds. | |
Table 1 Sequences and affinities of 2′,4′-BNA/FNA chimeric aptamers obtained using CE-SELEX
| Group |
2′,4′-BNA/FNA chimeric aptamera |
Sequence in the variable regionb |
K
d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (nM) c (nM) |
|
Each aptamer's name indicates the clone number.
Sequences are aligned in the 5′ to 3′ direction. 2′-Deoxy-2′-fluoro-guanosine is shown in bold letters (gf).
K
d values were determined by NECEEM. n.d., not determined.
|
| I |
A#1,5,6,8,9,13,14,19,22,24,26,35,38,39,41 |
gfTAgfAAgfgfTAgfgfgfAgfTgfgfATCCCTCTgfCA |
10 ± 0.4 |
| A#10 |
gfTAgfAAgfgfTTgfgfgfAgfTgfgfATCCCTCTgfCA |
n.d. |
| II |
A#7,16,27 |
gfgfTATgfTgfACgfTTTTTCACgfgfgfTCgfCACgf |
22 ± 0.2 |
| A#23 |
gfgfTATgfTgfATgfTTTTTCACgfgfgfTCgfCACgf |
n.d. |
| III |
A#11,12,33,36 |
gfgfTgfgfAgfgfTTgfgfATCAAgfTCATTTACTgfCT |
84 ± 8 |
| IV |
A#15,20,34 |
CCgfgfAAgfCgfgfTTTTAAgfTTgfgfCCgfATCgfTC |
12 ± 0.5 |
| A#18,29 |
CCgfgfAAgfCgfgfTTTTAAgfATgfgfCCgfATCgfTC |
n.d. |
| V |
A#4,17,28,31,32,40 |
gfTgfCCTACTTACgfgfgfTACTACCTTCATTgfgf |
27 ± 2 |
| VI |
A#3,21,30 |
AgfTTTACgfgfAAATgfCgfATCgfTCATTTCgfgfC |
47 ± 2 |
| A#2 |
AgfTTTACgfgfAAATgfCgfATCTTCATTTCgfgfC |
n.d. |
| VII |
A#25,37 |
TTTgfATCgfgfgfTgfACCTTgfAgfgfgfTCgfCTTCC |
45 ± 2 |
| A#42 |
TTTgfATCgfgfgfTgfACCTTgfATgfgfTCgfCTTCC |
n.d. |
Affinity and specificity analyses
The dissociation constants (Kd's) for the target were determined by affinity analyses using nonequilibrium capillary electrophoresis of equilibrium mixtures (NECEEM).34–36 All representative sequences of each group exhibited target-binding activities with Kd values in the range of 10–84 nM (Table 1 and Fig. S2, ESI†). Although rapid, significant enrichment was obtained during 2′,4′-BNA/FNA chimeric aptamer selection, the selected aptamers unexpectedly exhibited inferior thrombin-binding activities compared with the previously reported 2′,4′-BNA primer-containing DNA aptamers with low nanomolar affinities.31 Furthermore, the best Kd values of TBAs reported until date are in the several hundred picomolar range. This may be because the N-type sugar pucker of the 2′-fluoro analog gf and its preference for the anti glycosidic conformation would suppress the generation of two-tiered G-quadruplex motifs that fold into an antiparallel topology,37–41 which is commonly found in DNA aptamers of thrombin.42–44 However, a non-G-quadruplex TBA recovered from an RNA-based library that contained 2′-deoxy-2′-fluoro-pyrimidine nucleotides (U, C) was reported;45 it exhibited a very high binding affinity with a Kd value of 0.54 ± 0.1 nM. This suggests that higher affinity DNA-based aptamers with 2′-fluoro modifications can be obtained by further improvements in the current selection method.
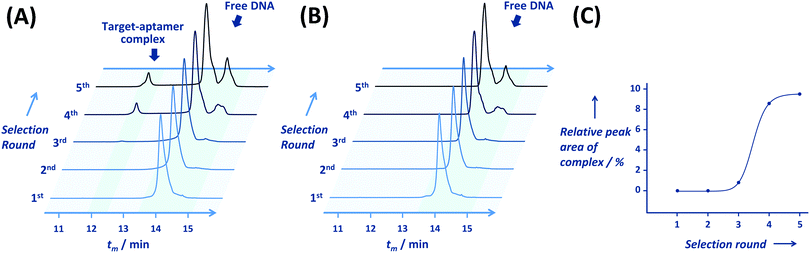
Additional analyses of binding properties were done using the two best aptamers A#1 and A#15. A significant increase in the human thrombin–A#1 complex peak with an ascending target concentration was observed, as shown in Fig. 3. The critical effect of the 2′-fluorine group on target binding was established using A#1N and A#15N, which contain only natural nucleotides in the non-primer region; both A#1N and A#15N exhibited no binding affinity for the target (Fig. S2, ESI†).
 |
| | Fig. 3 Capillary electrograms of the assays using A#1 (10 nM) in addition to 0–4-fold equivalents of human thrombin (0, 10, and 40 nM). The asterisk indicates the peak of the thrombin–aptamer complex. As the target concentration was raised, the peak strengths of the complex and free aptamers increased and decreased, respectively. tm = migration time, If = fluorescence intensity at 520 nm. | |
The binding specificities of A#1 and A#15 were also assessed using different proteins as targets, including rat thrombin, lysozyme, soybean lectin, and streptavidin (Fig. S3, ESI†). A#1 showed no binding affinity to lysozyme, soybean lectin, and streptavidin, while it exhibited high binding affinity to rat thrombin, with a Kd value of 1.3 nM, which is about 7.7-fold lower than that observed for human thrombin. Similar experimental data were also obtained using A#15 and in our recent study on DNA-based 2′,4′-BNA aptamer selection.19 This may be because of the higher net charge of rat thrombin (+11) than that of human thrombin (+6), in addition to sequence and structural similarities between the two types of thrombin,46,47 implying that electrostatic interactions between the aptamer and protein may largely contribute to the enhancement of target-binding affinities.
Analyses of structure and activity
Typically, the CD spectra of B-form oligonucleotides display a positive band at approximately 280 nm and a negative band at approximately 250 nm, the base absorption region. Subsequently, CD spectra of the A-form are characterized by a dominant positive band at approximately 260 nm and an absent or weak negative band at approximately 250 nm. RNA is known to prefer the A-form, whereas DNA comprising 2′-deoxynucleotides can adopt either the A- or B-form. B-form DNAs are observed in solutions under various conditions. As shown in Fig. 4, A#1, A#1B, A#15, and A#15B adopt A-type helical structures, indicating that the presence of N-puckered sugars (gf or gb) can induce an A-form in a oligonucleotide predominantly (>66%) composed of natural 2′-deoxynucleosides. Here, A#1B and A#15B contain gb's in place of gf's in A#1 and A#15, respectively. By contrast, A#1N and A#15N adopt the B-form, indicating that despite the inclusion of the 2′,4′-BNA primer region these oligonucleotides do not retain their original conformation; this observation is consistent with the observed loss in binding activity (Fig. S2, ESI†). Little change in the CD spectra of the oligonucleotides except for A#15N was observed when the concentration of salt (NaCl) increased to 100 mM (Fig. S4, ESI†), indicating these structures adopt a particular conformation in the range of ionic strength examined. Intriguingly, CD spectral patterns and band intensities of A#1 and A#15 were similar to those of A#1B and A#15B. These results indicated that substitution of 2′-hydroxyl with 2′-fluorine, a minor structural modification, induced the A-form in a manner comparable to that induced by the presence of 2′,4′-bridged modification, which ensures N-puckering in a sugar. Although A#1B and A#15B retained their original conformations, replacement of gf with gb led to loss of binding activity. This was attributed to the loss of direct interactions between 2′-fluorine and protein residues and/or steric hindrance imposed by the bulky 2′,4′-bridged ring during target binding.
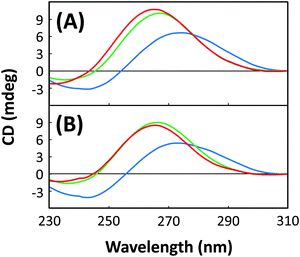 |
| | Fig. 4 CD spectra for (A) A#1, A#1N, and A#1B and (B) A#15, A#15N, and A#15B in 20 mM Tris-HCl buffer containing 1 mM MgCl2 and 10 mM NaCl, pH 7.4: A#1 and A#15 (green), A#1N and A#15N (blue), and A#1B and A#15B (red). | |
Biostability
Half-lives of A#1 and A#15 in the human serum were approximately 4.5 and 3.7 h, respectively (Fig. 5). These values were 2.1- and 1.8-fold higher than those of A#1N and A#15N, respectively, and demonstrated that 2′-fluoro modification significantly enhances biostability of DNA-based aptamers. Similar enhancements in the lifetime of RNA aptamers have been reported previously. Further improvements in half-lives were observed for A#1B and A#15B. The half-lives of A#1B and A#15B were 4.8- and 4.4-fold higher than that of A#1N and A#15N, respectively. Different sequences containing the same modified nucleotide exhibited different half-lives; the half-lives of A#1 and A#1B were 1.2- and 1.1-fold higher than that of A#15 and A#15B, respectively. This could be attributed to the number of modified guanosines inserted in the non-primer region (17 residues for A#1/A#1B and 16 residues for A#15/A#15B), in a hairpin loop (3 residues for A#1/A#1B and 1 residue for A#15/A#15B), and/or in a bulge of the predicted secondary structures (Fig. S5, ESI†).48
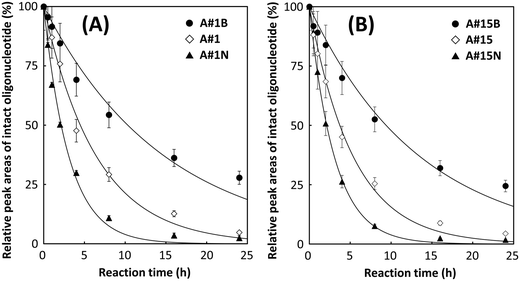 |
| | Fig. 5 Time-dependent degradation of (A) A#1, A#1N, and A#1B and (B) A#15, A#15N, and A#15B in 80% human serum at 37 °C. The half-lives of A#1N, A#15N, A#1, A#15, A#1B, and A#15B were approximately 2.1, 2.1, 4.5, 3.7, 10.3, and 9.4 h, respectively. | |
Conclusion
The significance of this work is that DNA-based 2′,4′-BNA/FNA chimeric aptamers were rapidly and conveniently obtained using CE-SELEX selection because of the type of sugar modification used, 2′-fluorination, which resulted in minimum obstruction allowing for efficient and accurate polymerase reactions. Despite this minimum obstruction, 2′-fluorination could clearly enhance biostability and induce a DNA-based strand to form an A-type conformation that is generally found in RNA, as does 2′-O,4′-C-methylene linkage. Thus, our study results provide a means for the further development of XNA aptamers,49 which are broader in terms of structural diversity to fit targets and are stable under those physiological conditions used in practical applications.
Methods
CE-SELEX selection of 2′,4′-BNA/FNA chimeric aptamers
A library was prepared enzymatically using 2′-deoxy-2′-fluoro-guanosine-5′-triphosphate (gfTP) as a modified substrate, KOD Dash DNA polymerase as a catalyst, and a 20-mer 2′,4′-BNA strand, as a primer that exhibited improved biostability (steps 1 and 2; Fig. 1). The single-stranded chimeric ODN library was mixed with the target human thrombin, followed by rapid separation of active species from unbound chimeric ODNs with high resolution using NECEEM (step 3). The active species were exponentially amplified by a polymerase chain reaction (PCR) using four natural 2′-deoxyribonucleoside-5′-triphosphates (dNTPs) to yield the corresponding double stranded ODNs (step 4), which were further amplified by PCR using gfTP instead of dGTP (step 5). This was followed by single-strand preparation to provide an increasingly enriched pool, which was used for the next-round library (step 6). The selection round involving separation and amplification of active species was repeated five times, resulting in substantial saturation of the active species (Fig. 2 and 3). The detailed protocols for the selection procedure and subsequent aptamer isolation and affinity analyses using CE are provided in the ESI.†
Enzymatic syntheses of 2′,4′-BNA aptamers and 2′,4′-BNA primer-containing DNA aptamers
Although FNA exhibits superior nuclease resistance compared with natural RNA/DNA, the effects of 2′,4′-BNA on biostability are known to be greater than that of FNA. To investigate the effects of the 2′-fluorine group as a sugar modification on binding properties, biostability, and structure, two 2′,4′-BNA/FNA chimeric aptamers with the highest target-binding affinities, A#1 and A#15, were converted to 2′,4′-BNA aptamers (A#1B and A#15B) and 2′,4′-BNA primer-containing DNA aptamers (A#1N and A#15N), which had sequences identical to those of A#1 or A#15, but in which all g's in the non-primer region were replaced with gb's and with natural G's, respectively. These oligonucleotides were obtained by PCR amplification (16 PCR cycles, each cycle composed of the following steps: 94 °C for 30 s, 54 °C for 30 s, and 74 °C for 60 s) of the appropriate plasmid template (for A#1 or A#15) in a TC-312 thermal cycler (Techne, Staffordshire, UK) with the forward primer P1p7A (5′-/phosphate/AAA AAA ATT CGC CTT GCC GGA TCG CAG A-3′; 0.4 μM), the reverse primer P2 (5′-GGT GTC AGG CTC ACG GAC CA-3′; 0.4 μM) in the presence of four natural dNTPs (0.2 mM each), and 1× polymerase reaction buffer. The crude amplified dsDNA was obtained after the removal of excess dNTPs by ethanol precipitation. The P1p7A-elongated oligonucleotide product was selectively degraded by incubating the dsDNA with λ exonuclease (0.025 U μL−1) in 1× nuclease buffer (800 μL) at 37 °C for 30 min. Subsequently, PCR was performed using the P2-elongated strand as the template, with the use of only one primer, P1fb (5′-/6-FAM/TCG CCT TGC CGG ATC GCA GA-3′), which contained 2′,4′-BNA monomer nucleotides indicated by italic letters in the sequence. The reaction mixture (800 μL) contained the P2-elongated template (80 nM; containing the sequence corresponding to either A#1 or A#15), primer P1fb (0.4 μM), mix of substrate triphosphates (dATP, TTP, dCTP, gfTP; 0.2 mM each), KOD Dash DNA polymerase (0.01 U μL−1), and 1× polymerase reaction buffer. The reaction mixture was subjected to 16 thermal cycles under conditions described above. Excess dNTPs were removed by ethanol precipitation, and the purification of the resulting crude product by denaturing polyacrylamide gel electrophoresis (PAGE) yielded single-stranded oligonucleotides (A#1 or A#15, as appropriate). Single-stranded oligonucleotides A#1N and A#15N were prepared similarly, except that dGTP was used in place of gfTP in the PCR with a P2-elongated product and the concentration of the DNA polymerase was 0.005 U μL−1. Simultaneously, for the preparation of A#1B and A#15B, 2′-O,4′-C-methylene-bridged/linked bicyclic guanosine-5′-triphosphate (gbTP) was used instead of gfTP, a 19:1 mix of KOD Dash and KOD mutant DNA polymerases (0.25 U μL−1)19 was used instead of only KOD Dash DNA polymerase, and 80 thermal cycles for one-primer PCR were used instead of 16.
CD measurements
Sample solutions that contained 1.0 μM (final concentration) of different oligonucleotides (A#1, A#1B, A#1N, A#15, A#15B, or A#15N) in 20 mM Tris-HCl buffer (pH 7.4) containing 1 mM MgCl2 with different NaCl concentrations (10 and 100 mM) were analyzed. Prior to CD measurements, each of these oligonucleotides was dissolved in the Tris-HCl buffer, followed by refolding by denaturing at 94 °C for 0.5 min and cooling to 25 °C at a rate of 0.5 °C min−1 using a thermal cycler. CD spectroscopic data were acquired using a JASCO J-820 spectrometer. CD spectra were measured over a wavelength range of 200–340 nm using a quartz cuvette with a 1.0 cm optical path length (Fig. 4). The scanning speed was set at 100 nm min−1 and the response time was 1 s. Each spectrum was an average of five measurements made at 25 °C.
Stability in human serum
The effects of sugar modification on their stability in human serum (from human male AB plasma) were examined using A#1, A#1B, A#1N, A#15, A#15B, and A#15N (Fig. 5). These oligonucleotides were degraded in a 4-μL reaction volume that contained 0.4 μL of A#1, A#1B, A#1N, A#15, A#15B, or A#15N (400 nM), 3.2 μL of human serum, and 0.4 μL of 10× reaction buffer (500 mM Tris-HCl and 100 mM MgCl2, pH 8.0). These reaction mixtures were incubated at 37 °C for 0.5, 1, 2, 4, 8, 16, and 24 h and then separated by capillary gel electrophoresis using an ssDNA 100-R kit. The fluorescence emissions (520 nm) of the 6-FAM labeled 5′-ends of these oligonucleotides were monitored with excitation at 488 nm. Data were recorded at 4 Hz and analyzed using 32 Karat software ver. 8.0. The relative amounts of intact oligonucleotides were calculated from peak areas, and the decay curves of intact oligonucleotides were fitted by the least squares method using OriginPro ver.8 to determine the half-lives of intact forms.
Author contributions
KH and HF performed the analytical experiments, prepared figures and wrote the manuscript. YK and YI performed the SELEX experiments. SO contributed to the study design and discussed results. MK organized the research group, designed the study, analyzed the data, prepared figures and wrote the manuscript. All the authors commented on the manuscript.
Acknowledgements
This study was partly supported by a Grant for Adaptable and Seamless Technology Transfer Program through Target-driven R & D, No. AS2525029M, from Japan Science and Technology Agency (JST), and by a Grant-in-Aid for Scientific Research (C), No. 25350962, from the Japan Society for the Promotion of Science (JSPS).
References
- V. B. Pinheiro and P. Holliger, Curr. Opin. Chem. Biol., 2012, 16, 245 CrossRef CAS PubMed.
- P. Herdewijn, Chem. Biodiversity, 2010, 7, 1 CAS.
- K. Robeyns, P. Herdewijn and L. Van Meervelt, Nucleic Acids Res., 2008, 36, 1407 CrossRef CAS PubMed.
- J. K. Ichida, K. Zou, A. Horhota, B. Yu, L. W. McLaughlin and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 2802 CrossRef CAS PubMed.
- K. Schoning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347 CrossRef CAS.
- C. G. Peng and M. J. Damha, Nucleic Acids Res., 2007, 35, 4977 CrossRef CAS PubMed.
- J. Ruckman, L. S. Green, J. Beeson, S. Waugh, W. L. Gillette, D. D. Henninger, L. Claesson-Welsh and N. Janjic, J. Biol. Chem., 1998, 273, 20556 CrossRef CAS PubMed.
- T. Ono, M. Scalf and L. M. Smith, Nucleic Acids Res., 1997, 25, 4581 CrossRef CAS PubMed.
- R. N. Veedu, B. Vester and J. Wengel, J. Am. Chem. Soc., 2008, 130, 8124 CrossRef CAS PubMed.
- M. Kuwahara, S. Obika, J. Nagashima, Y. Ohta, Y. Suto, H. Ozaki, H. Sawai and T. Imanishi, Nucleic Acids Res., 2008, 36, 4257 CrossRef CAS PubMed.
- S. Obika, D. Nanbu, Y. Hari, J. Andoh, K. Morio, T. Doi and T. Imanishi, Tetrahedron Lett., 1998, 39, 5401 CrossRef CAS.
- S. K. Singh, A. A. Koshkin, J. Wengel and P. Nielsen, Chem. Commun., 1998, 455 RSC.
- S. Obika, D. Nanbu, Y. Hari, K. Morio, Y. In, T. Ishida and T. Imanishi, Tetrahedron Lett., 1997, 38, 8735 CrossRef CAS.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S. Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818 CrossRef CAS PubMed.
- M. Kuwahara, Y. Takano, Y. Kasahara, H. Nara, H. Ozaki, H. Sawai, A. Sugiyama and S. Obika, Molecules, 2010, 15, 8229 CrossRef CAS PubMed.
- Y. Kasahara, Y. Irisawa, H. Ozaki, S. Obika and M. Kuwahara, Bioorg. Med. Chem. Lett., 2013, 23, 1288 CrossRef CAS PubMed.
- H. Aurup, D. M. Williams and F. Eckstein, Biochemistry, 1992, 31, 9636 CrossRef CAS.
- E. W. Ng, D. T. Shima, P. Calias, E. T. Cunningham, Jr., D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123 CrossRef CAS PubMed.
- J. Yang and M. T. Bowser, Anal. Chem., 2013, 85, 1525 CrossRef CAS PubMed.
- M. Jing and M. T. Bowser, Anal. Chem., 2013, 85, 10761 CrossRef CAS PubMed.
- L. N. Cella, P. Sanchez, W. Zhong, N. V. Myung, W. Chen and A. Mulchandani, Anal. Chem., 2010, 82, 2042 CrossRef CAS PubMed.
- E. Peyrin, J. Sep. Sci., 2009, 32, 1531 CrossRef CAS PubMed.
- R. K. Mosing, S. D. Mendonsa and M. T. Bowser, Anal. Chem., 2005, 77, 6107 CrossRef CAS PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2005, 127, 9382 CrossRef CAS PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2004, 126, 20 CrossRef CAS PubMed.
- J. F. Milligan, D. R. Groebe, G. W. Witherell and O. C. Uhlenbeck, Nucleic Acids Res., 1987, 15, 8783 CrossRef CAS PubMed.
- Z. Li, C. J. Lech and A. T. Phan, Nucleic Acids Res., 2014, 42, 4068 CrossRef CAS PubMed.
- Y. Kasahara, Y. Irisawa, H. Fujita, A. Yahara, H. Ozaki, S. Obika and M. Kuwahara, Anal. Chem., 2013, 85, 4961 CrossRef CAS PubMed.
- C. Sissi, B. Gatto and M. Palumbo, Biochimie, 2011, 93, 1219 CrossRef CAS PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402 CrossRef CAS PubMed.
- Y. Li, L. Guo, F. Zhang, Z. Zhang, J. Tang and J. Xie, Electrophoresis, 2008, 29, 2570 CrossRef CAS PubMed.
- A. P. Drabovich, M. Berezovski, V. Okhonin and S. N. Krylov, Anal. Chem., 2006, 78, 3171 CrossRef CAS PubMed.
- M. Berezovski and S. N. Krylov, Anal. Chem., 2005, 77, 1526 CrossRef CAS PubMed.
- D. Pradhan, L. H. Hansen, B. Vester and M. Petersen, Chemistry, 2011, 17, 2405 CrossRef CAS PubMed.
- J. T. Nielsen, K. Arar and M. Petersen, Angew. Chem., Int. Ed., 2009, 48, 3099 CrossRef CAS PubMed.
- P. K. Dominick and M. B. Jarstfer, J. Am. Chem. Soc., 2004, 126, 5050 CrossRef CAS PubMed.
- V. Dapic, V. Abdomerovic, R. Marrington, J. Peberdy, A. Rodger, J. O. Trent and P. J. Bates, Nucleic Acids Res., 2003, 31, 2097 CrossRef CAS PubMed.
- M. A. Keniry, Biopolymers, 2000, 56, 123 CrossRef CAS.
- O. Tatarinova, V. Tsvetkov, D. Basmanov, N. Barinov, I. Smirnov, E. Timofeev, D. Kaluzhny, A. Chuvilin, D. Klinov, A. Varizhuk and G. Pozmogova, PLoS One, 2014, 9, e89383 Search PubMed.
- D. M. Tasset, M. F. Kubik and W. Steiner, J. Mol. Biol., 1997, 272, 688 CrossRef CAS PubMed.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564 CrossRef CAS PubMed.
- R. White, C. Rusconi, E. Scardino, A. Wolberg, J. Lawson, M. Hoffman and B. Sullenger, Mol. Ther., 2001, 4, 567 CrossRef CAS PubMed.
- Protein Data Bank identification code: 2C8Y.
- Protein Data Bank identification code: 3HK3.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406 CrossRef CAS PubMed.
- J. Barciszewski, M. Medgaard, T. Koch, J. Kurreck and V. A. Erdmann, Methods Mol. Biol., 2009, 535, 165 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Five figures and additional text showing detailed experimental procedures and additional experimental results. See DOI: 10.1039/c4mb00436a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.