
PLS/OPLS models in metabolomics: the impact of permutation of dataset rows on the K-fold cross-validation quality parameters†
Mohamed N.
Triba
*a,
Laurence
Le Moyec
b,
Roland
Amathieu
c,
Corentine
Goossens
a,
Nadia
Bouchemal
a,
Pierre
Nahon
d,
Douglas N.
Rutledge
e and
Philippe
Savarin
a
aUniversité Paris 13, Sorbonne Paris Cité, Laboratoire Chimie, Structures, Propriétés de Biomatériaux et d'Agents Thérapeutiques (CSPBAT), Unité Mixte de Recherche (UMR) 7244, Centre National de Recherche Scientifique (CNRS), Equipe Spectroscopie des Biomolécules et des Milieux Biologiques (SBMB), 74 rue Marcel Cachin, 93037, Bobigny, France. E-mail: mohamed.triba@univ-paris13.fr
bUniversité d'Evry Val d'Essonne, Unité de Biologie Intégrative des Adaptations à l'Exercice (UBIAE), U902, INSERM, Bd François Mitterrand, 91025 Evry Cedex, France
cService d'Anesthésie et des Réanimations Chirurgicales, Université Paris 12, Hôpital Henri Mondor, Assistance Publique des Hôpitaux de Paris (AP-HP), Créteil, France
dService d'Hépatologie et Université Paris 13, Hôpital Jean Verdier, Assistance Publique des Hôpitaux de Paris (AP-HP), 93143 Bondy Cedex, France
eLaboratoire de Chimie Analytique, AgroParisTech, 16 rue Claude Bernard, 75231 Paris, France
First published on 23rd October 2014
Abstract
Among all the software packages available for discriminant analyses based on projection to latent structures (PLS-DA) or orthogonal projection to latent structures (OPLS-DA), SIMCA (Umetrics, Umeå Sweden) is the more widely used in the metabolomics field. SIMCA proposes many parameters or tests to assess the quality of the computed model (the number of significant components, R2, Q2, pCV-ANOVA, and the permutation test). Significance thresholds for these parameters are strongly application-dependent. Concerning the Q2 parameter, a significance threshold of 0.5 is generally admitted. However, during the last few years, many PLS-DA/OPLS-DA models built using SIMCA have been published with Q2 values lower than 0.5. The purpose of this opinion note is to point out that, in some circumstances frequently encountered in metabolomics, the values of these parameters strongly depend on the individuals that constitute the validation subsets. As a result of the way in which the software selects members of the calibration and validation subsets, a simple permutation of dataset rows can, in several cases, lead to contradictory conclusions about the significance of the models when a K-fold cross-validation is used. We believe that, when Q2 values lower than 0.5 are obtained, SIMCA users should at least verify that the quality parameters are stable towards permutation of the rows in their dataset.
Introduction
Projection to latent structures (PLS) and orthogonal projection to latent structures (OPLS) are popular methods for multivariate statistical analysis in metabolomics.1 For classification or discrimination problems these methods are referred to as PLS-DA and OPLS-DA where DA stands for discriminant analysis. Many software packages are available for these types of analysis in commercial or academic statistical packages: R Project for Statistical Computing (http://www.r-project.org),2 MetaboAnalyst,3 MVAPACK,4 Multibase (Numerical Dynamics), IFRNOPLS-DA,5,6 STATISTICA (StatSoft), Unscrambler (CAMO Software), SAS (SAS Institute Inc, Cary NC), the PLS-toolbox for Matlab (Eigenvector Research Inc, Wenatchee WA) and SIMCA (Umetrics, Umeå Sweden). However, in comparison to the other software packages, SIMCA seems to be much more often used in the metabolomics field (Table 1). Consequently, an appropriate use of this statistical package is necessary to assure the quality of the results published by the metabolomics community.| Bibliographic database | No. of articles containing metabolomics and PLS | Using Metabo analyst | Using SIMCA | Using R | Using Statistica | Using Unscrambler |
|---|---|---|---|---|---|---|
| Science direct | 1117 | 51 | 464 (42%) | 49 | 37 | 37 |
| Royal Society of Chemistry | 135 | 9 | 54 (40%) | 5 | 3 | 2 |
| PLOS One | 245 | 13 | 108 (44%) | 28 | 16 | 3 |
| Springer link | 654 | 32 | 274 (42%) | 45 | 22 | 18 |
| ACS | 473 | 32 | 278 (59%) | 30 | 14 | 22 |
PLS/OPLS models try to find a linear relationship between a X predictor matrix (e.g. spectrometric data of biological samples) and a Y response matrix (e.g. clinical results, treatment…). In metabolomics, the X predictor matrix frequently has more columns (predictor variables) than rows (individuals). Because of this property of metabolomics data, PLS/OPLS models can easily be overfitted and their predictability overestimated.
The only way to reliably estimate the ability of the model to predict Y values of new individuals is to predict individuals from an independent dataset (i.e. that were not used to build this model). This can be achieved by splitting the dataset into a training set and a test set. The training set is used to build the model and the test set is used to estimate the predictability. However, the cost of this splitting is that the model is built with only a fraction of the information that is present in the whole dataset. This may reduce the ability of this model to correctly predict a new dataset. Thus, splitting the dataset into a training set and a test set can be done only if enough individuals are available to build a reliable model. As in univariate statistics, the significance of the results of multivariate models depends on the sample size. However, the minimum number of individuals needed to attain a given significance threshold for the PLS models is very application-dependent and no easily applicable rules have been proposed to estimate this number.7
When no test set is available, the cross-validation method is the main strategy proposed by commercial or academic statistical packages to assess the quality of a model. Different cross-validation procedures exist. The default SIMCA cross-validation is the so-called K-fold cross-validation. Results of the cross-validation procedure are summarized by the value of different quality parameters. The most frequently mentioned in the metabolomics literature are R2 and Q2 parameters (also called cross-validated R2). R2 measures the goodness of fit while Q2 measures the predictive ability of the model. R2 = 1 indicates perfect description of the data by the model, whereas Q2 = 1 indicates perfect predictability. R2 increases monotonically with the number of components (NC) and will automatically approach 1 if NC approaches the rank of the X matrix. Q2 will not necessarily approach 1. At a certain value of NC, Q2 reaches a plateau and usually will finally decrease with addition of more components. This indicates that at a certain degree of complexity the predictive ability of the model decreases.8 At this stage, it is very likely that the model is trying to fit dataset characteristics that are no longer representative of the studied population. A large discrepancy between R2 and Q2 indicates an overfitting of the model through the use of too many components. According to the SIMCA users' guide, Q2 > 0.5 is admitted for good predictability (SIMCA P12 users' guide, p. 514).9 It has been shown that in practice it is difficult to give a general limit that corresponds to a good predictability since this strongly depends on the properties of the dataset.8,10 For example, an acceptable Q2 threshold will strongly depend on the number of observations included. During the last few years, a large number of SIMCA PLS-DA/OPLS-DA models have been published with Q2 below 0.4 or even below 0.3 (for example, see ref. 11 and 12). These models with poor predictability are frequently validated by a permutation test that consists in comparing the Q2 obtained for the original dataset with the distribution of Q2 values calculated when original Y values are randomly assigned to the individuals.10 The cross-validation procedure also provides the possibility to calculate a p-value to estimate the significance of PLS/OPLS models (pCV-ANOVA).13
As recently published in this journal,14 metabolomic results based on PLS/OPLS models should always give the values of the quality parameters of the multivariate models. The number of components used in the final model, Q2 and pCV-ANOVA values should be presented to allow the reader to assess the quality of the model calculated by SIMCA. However, in this Opinion piece, we want to point out that in some cases, because of the way in which the default SIMCA cross-validation procedure selects members of the calibration and validation subsets, permutation of the rows of a dataset can result in variations in the values of the quality parameters. As a consequence, in these circumstances, different conclusions on the quality of the PLS/OPLS models may be drawn from the same dataset. In a first part, we will show that under some conditions a random permutation of rows in the dataset strongly affects the quality parameter values obtained when default SIMCA cross-validation settings are used. In a second part, we will discuss three different types of situations frequently encountered in metabolomics studies where the K-fold cross-validation procedure fails to calculate a Q2 that is not strongly dependent on the arbitrary order of the rows in a dataset.
Default SIMCA cross-validation procedure
We give here a very basic description of the default SIMCA cross-validation procedure. Only the way the validation sets are built will be discussed in detail. For an exhaustive description of the procedure, the reader should refer to the Umetrics documentation.8Cross-validation allows to estimate the ability of a model to correctly predict the Y response matrix of new individuals. In the SIMCA software, cross-validation is also used to avoid overfitting by estimating the number of significant components (NSCs) to use in the model. Many cross-validation procedures are used in the metabolomic community (K-fold, Leave One Out, Monte-Carlo, 2CV, etc.). The default SIMCA cross-validation procedure is a 7-fold cross-validation8 where the dataset is split into 7 different subsets. For a fixed number of components (NC), the Y values of all individuals of each subset are predicted using a submodel built with the 6 other subsets (calibration subset). The differences between the predicted Y values and the observed Y values are used to calculate the QNC2 parameter for this number of components. The procedure starts at NC = 1 and is repeated by incrementing NC as long as the increase of QNC2 is larger than a limit value fixed by various rules.9
Each subset is constituted by selecting one row every seven rows in the dataset. The first subset is built with the individuals corresponding to rows 7, 14, 21 and so on. The second subset is constituted with the individuals corresponding to rows 1, 8, 15,…. The other subsets are built in the same way (Scheme 1a).
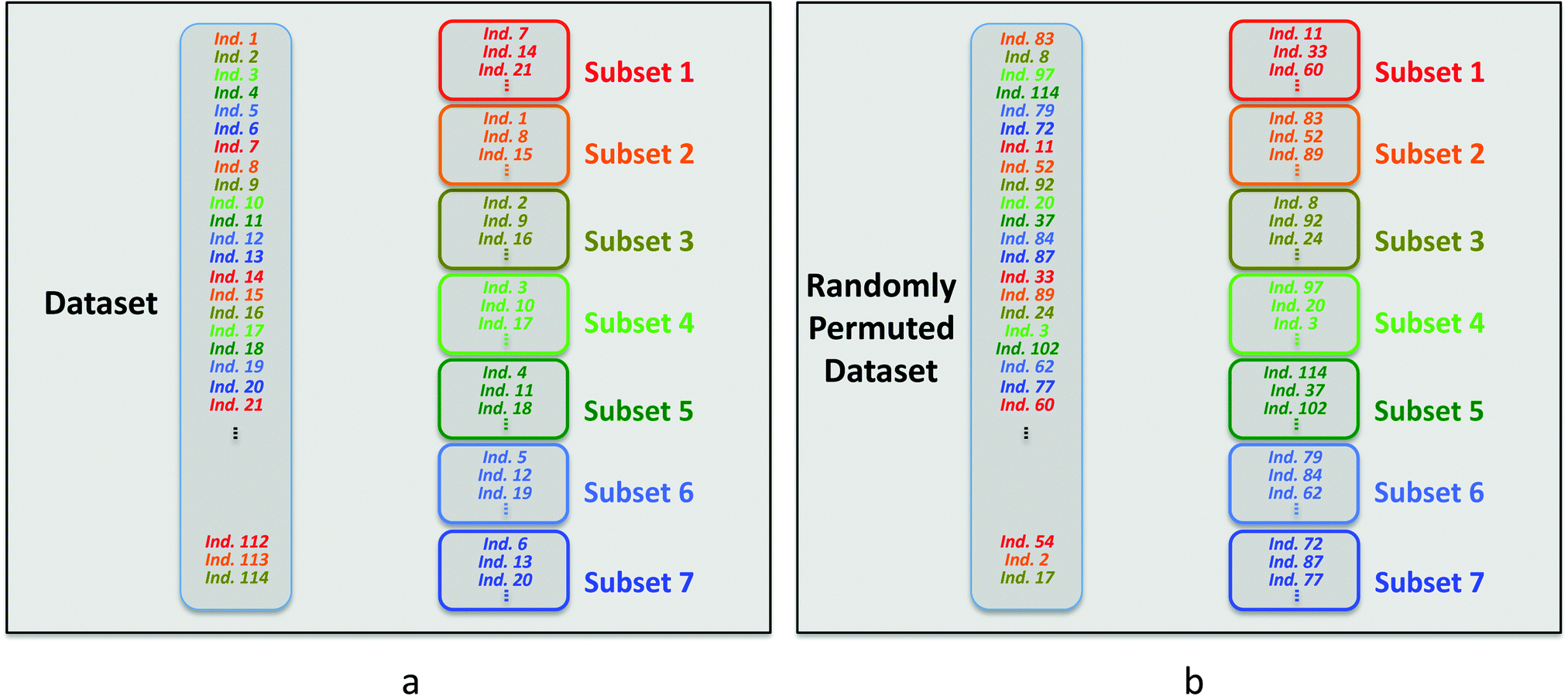 | ||
| Scheme 1 Selection of the individuals used to build the cross-validation subsets in SIMCA for the original dataset (a) and when the rows of this dataset are randomly permuted (b). | ||
Considering the way the subsets are built, it is clear that a permutation in row order of the X and Y dataset changes the individual positions and modifies the composition of these subsets (Scheme 1b). Thus, submodels and predicted Y values calculated during the cross-validation procedure are also affected by a permutation of rows.
The major consequences of this are:
– Row permutations can potentially change the number of components considered as significant (NSC) by SIMCA.
– For the same number of significant components, row permutations will change the value of the QNSC2 parameter.
– The CV-ANOVA p-value, which depends on the cross-validation procedures, is also affected by row permutations in the dataset.
– The conclusion of the permutation test can be different when the order of rows is changed.
Row permutation can strongly affect the K-fold cross-validation procedure
In this example we used serum NMR spectra of a metabolomics study on the influence of hepatitis viruses on patients with Hepatocellular Carcinoma (HCC).15 OPLS-DA was used to discriminate 57 HCC patients with hepatitis infection from 57 HCC patients without viral infection. Spectra were normalized with the probabilistic quotient normalization method16 to eliminate any dilution effect. They were divided into 230 domains of 0.05 ppm and the water signal region was suppressed. The resulting X predictor matrix was composed of 114 rows and 196 non-null columns. X and Y matrices are available as ESI† (Dataset1.xlsx). The X matrix was centered and scaled (UV scaling) prior to multivariate analysis. OPLS-DA analyses were performed with an in-house Matlab 2012b (The Mathworks Inc., Natick, Massachusetts, USA) code based on the Trygg and Wold method.17 The number of significance components is determined according to SIMCA rules (SIMCA P12 users' guide, p. 529).9X and Y dataset rows were randomly permuted in the same way and for each permutation various quality parameters (i.e. NSC, QNSC2, pCV-ANOVA) were calculated. In Fig. 1a, b and c are represented, respectively, the distributions of the NCS, QNSC2 and pCV-ANOVA calculated for 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 random permutations. For each permutation, an area under ROC curve is calculated with Y values predicted during the cross-validation process (CV-AUROC).10 The distribution of this parameter is shown in Fig. 1d. In Fig. 1b–d, the black lines represent the distributions of the quality parameters calculated for the 50000 random permutations. In order to estimate the variability of the quality parameters for a given number of components, these distributions were decomposed according the NSC value calculated for each permutation (colored lines). It clearly appears that an important part of the variability of these parameters is a consequence of NSC variability with row re-arrangements.
000 random permutations. For each permutation, an area under ROC curve is calculated with Y values predicted during the cross-validation process (CV-AUROC).10 The distribution of this parameter is shown in Fig. 1d. In Fig. 1b–d, the black lines represent the distributions of the quality parameters calculated for the 50000 random permutations. In order to estimate the variability of the quality parameters for a given number of components, these distributions were decomposed according the NSC value calculated for each permutation (colored lines). It clearly appears that an important part of the variability of these parameters is a consequence of NSC variability with row re-arrangements.
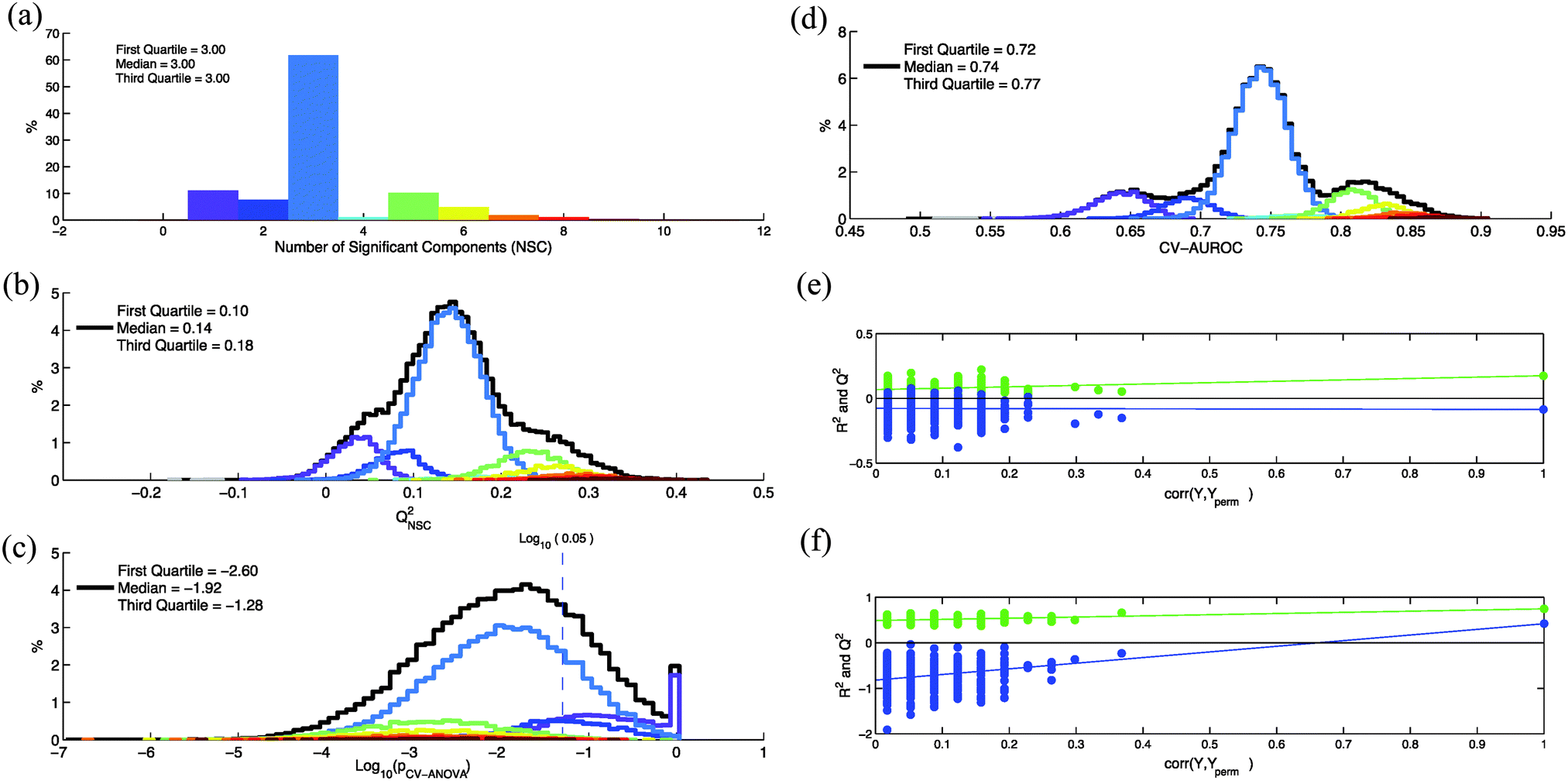 | ||
| Fig. 1 Variation of the OPLS quality parameters when 50000 random permutations of the dataset lines were performed. For each permutation, values of NSC, QNSC2, pCV-ANOVA and CV-AUROC were calculated (a): distribution of the number of significant components (NSC). Each color is associated to a specific value of NSC. (b)–(d): distributions of QNSC2, pCV-ANOVA and CV-AUROC parameters calculated for the 50000 random permutations (black lines). These distributions were decomposed according the NSC value calculated for each permutation (colored lines). (e) and (f): 500 random permutation tests (permutation of group membership) performed on the datasets that corresponded to the lowest and highest QNSC2 values among the 50000 models. The vertical axis corresponds to R2 (green points) and Q2 (blue points) values of each model. The horizontal axis corresponds to the correlation coefficient between the original Y and the permuted Y. | ||
Thus, a better estimation of the number of components could help to reduce the variability of the quality parameters. According to Wheelock and Wheelock,14 “the default automatic fitting in SIMCA extracts the maximal number of significant components, which in most cases results in an overfitted model”. These authors suggested that the optimal number of components (ONC) can be estimated by using the pCV-ANOVA parameter: when this ONC is reached then the addition of another component would increase pCV-ANOVA. As shown in Fig. 1c, values of pCV-ANOVA can strongly depend on the arbitrary order of the rows in the dataset. This dependence is also observed for a given number of components (colored lines). As a consequence, an ONC based on pCV-ANOVA may also strongly vary when the dataset lines are permuted. In order to estimate this variability, we performed 1000 permutations of the rows and, for each permutation, the value of ONC was determined by looking for the first local minimum of pCV-ANOVA when NC is incremented. We found a large variability of the ONC with rows rearrangement (Fig. S1, Supplementary Data A, ESI†). Thus, an ONC determined by using pCV-ANOVA can also strongly depend on the arbitrary order of the lines in the dataset if the K-fold cross-validation procedure is used. More generally, the number of components estimated by using parameters that depend on row order (such as Q2, pCV-ANOVA,…) can potentially exhibit a large variability with row permutations.
The datasets with the row arrangement corresponding to the lowest and the highest calculated values of QNSC2 (i.e. −0.09 and 0.42) were compared by calculating the quality parameters of the OPLS models. These permuted datasets are available as ESI† (Dataset2.xlsx and Dataset3.xlsx). We observed that, for the same experimental result, quality parameters of the two models (Table 2) lead to contradictory conclusions on the significance of the metabolic differences between the two classes. Contradictory conclusions are also obtained when permutation tests (random permutation of group affiliation) were performed on these two models (Fig. 1e and f). This particular dataset proves that, in some situations, quality parameter values calculated with the default SIMCA cross-validation procedure are strongly determined by chance. This result also suggests that performing row permutations allows an estimation of confidence intervals for the various quality parameters.
| Permuted dataset 1 | Permuted dataset 2 | |
|---|---|---|
| NSC | 1 | 10 |
| R 2 | 0.18 | 0.75 |
| Q 2 | −0.09 | 0.42 |
| p CV-ANOVA | 1 | 0.00004 |
| CV-AUROC | 0.57 | 0.91 |
In which situations?
Considering this result, an important question is now: in which situations do row permutations strongly affect the K-fold cross-validation procedure? We will not try to give an exhaustive or theoretical answer to this question but considering our practical experience in the PLS/OPLS analysis of metabolomic datasets, we will discuss some situations where row permutations can have a strong effect. According to Eriksson et al. “a necessary condition for PLS-DA to work reliably is that each class is tight and occupies a small and separate volume in X-space.”8 This condition is fulfilled when interclass variability is large enough relative to the intraclass variability and is observed for example when classes can be discriminated by a simple principal component analysis (PCA). When this situation was observed in our experimental results, we noticed that row permutation in the dataset did not significantly affect the K-fold cross-validation procedure. In particular, under these conditions no contradictory conclusions can be drawn from the same experimental dataset. However, Eriksson et al. also noticed that “… when some of the classes are not homogeneous and spread significantly in X-space, the discriminant analysis does not work”.8 According to our experience, when this situation is encountered, row permutations can have serious consequences on the conclusions of the K-fold cross-validation procedure. This is related to the fact that we can no longer neglect the probability to build by chance calibration and validation subsets that are not representative of the whole dataset in terms of difference between classes. These non-representative subsets can lead to an underestimation or an overestimation of the capability of the model to predict new data.To illustrate this point, we modified the experimental results of a second metabolomics study where we evaluated the influence of HCC on the metabolism of cirrhotic patients.18 OPLS-DA was used to discriminate 33 patients without HCC from 33 patients with large HCC. Spectra were normalized with the probabilistic quotient normalization method. They were divided into 230 domains of 0.05 ppm and the water signal region was suppressed. The resulting X and Y matrices are available as ESI† (Dataset4.xlsx). The properties of this dataset correspond to the first situation mentioned by Eriksson et al.8 (i.e. interclass variability is large enough relative to the intraclass variability) and no strong effect of row permutations on the Q2 parameter was observed for this dataset (Fig. 2a). We modified this dataset until we reached the second condition mentioned by Eriksson and coworkers8 (i.e. non-homogeneous classes and large intraclass variability). The modifications introduced in the original dataset were chosen to simulate three types of circumstances frequently observed in metabolomic studies.
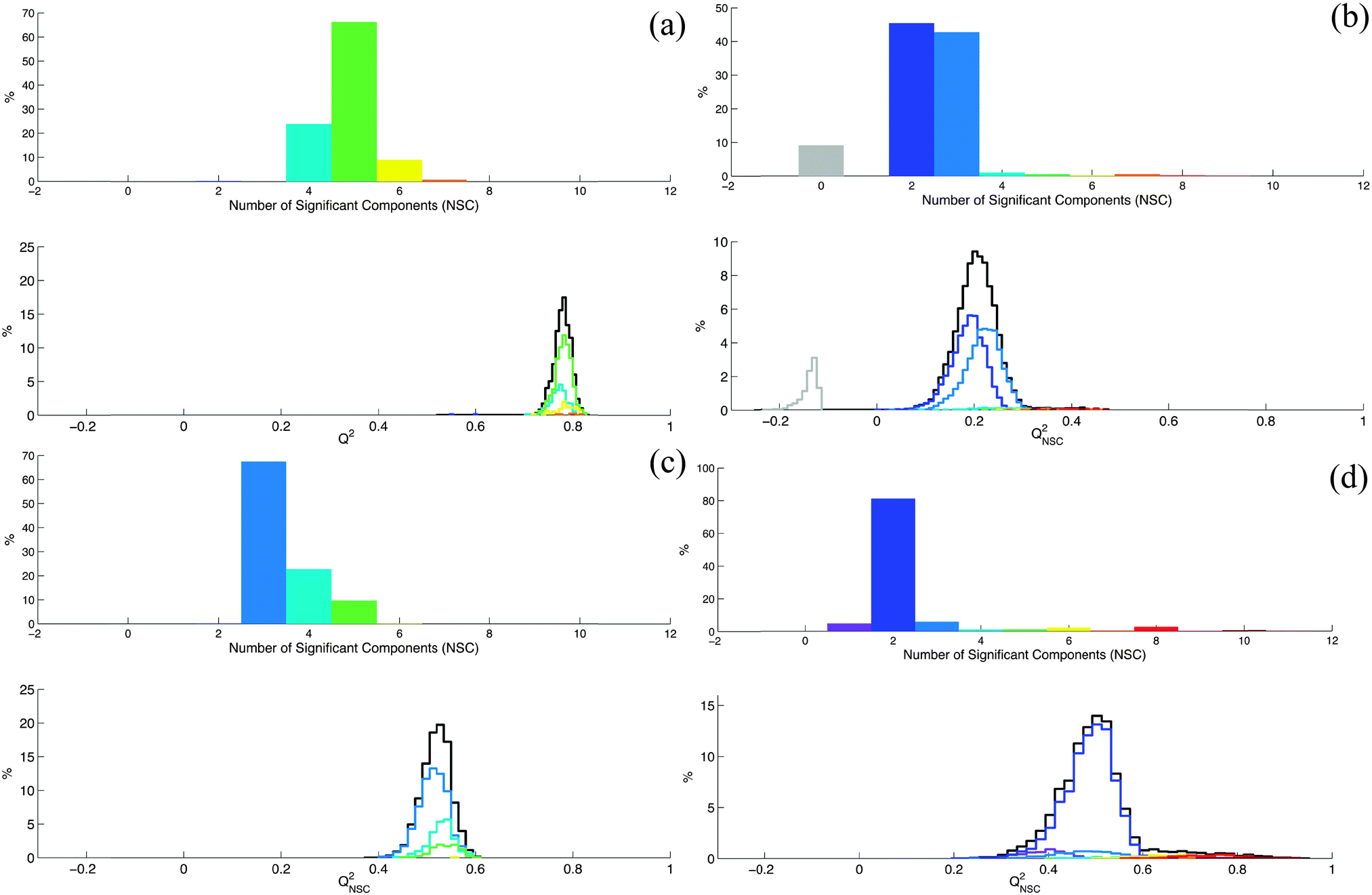 | ||
| Fig. 2 The first figures correspond to the original dataset (a) while the others correspond to the original dataset modified to simulate the effects of dilution (b), class noise (c) and small dataset size (d). Each figure illustrates the variation of the NSC and QNSC2 parameters when 10000 random permutations of the dataset lines are performed. Each color is associated to a specific value of NSC. The QNSC2 distributions (black lines) correspond to the 10000 random permutations. These distributions were decomposed according to the NSC value calculated for each permutation (colored lines). | ||
– The first situation is when the main source of variability in the dataset is uncorrelated with the Y response variable. This can be observed for example when incorrect sample normalization has been applied to correct for dilution effects. To simulate this situation we multiplied each line of the original dataset by a dilution factor randomly chosen between 1 and 50 (ESI,† Dataset5.xlsx). We randomly permuted the rows of the resulting dataset and calculated the NSC and Q2 values for each permutation (Fig. 2b). We observed a larger distribution of Q2 compared to Fig. 2a. For some permutations, no significant component was obtained.
– A second situation corresponds to the inaccurate labeling of group membership of individuals. The situation is known as class noise.19 It is frequently encountered in metabolomic studies applied to clinical problems especially when a reliable diagnostic tool is unavailable. To simulate this situation, 10% of the individuals of each group were incorrectly labeled in the original dataset (ESI,† Dataset6.xlsx). NSC and Q2 distributions after random permutations of rows were calculated (Fig. 2c). Here again, we observed a larger distribution of Q2 compared to Fig. 2a.
– Finally, a third situation is when the number of individuals used to build the model is too small. In this case, the probability to build by chance a sample with at least one non-homogeneous or non-representative class is not negligible even if classes are homogeneous in the population. We selected 8 individuals of each class from the original dataset and we randomly permuted the rows of the resulting 16 rows dataset (ESI,† Dataset7.xlsx). For each permutation we calculated the NSC and Q2 parameters (Fig. 2d). In this case, Q2 values spread from 0.23 to 0.92.
These results showed that when the situations mentioned above are encountered, quality parameters could be strongly affected by row permutations in the dataset if the K-fold cross-validation procedure is used. Moreover, many combinations of these three situations can be encountered in metabolomics studies.
Suggestions
We agree with Wheelock et al.14 that the number of components used in the final model, Q2 and pCV-ANOVA values should be presented to allow the reader to assess the quality of the multivariate model published on metabolomics studies. Our opinion is that the cross-validation method used to build the model should also be described since it determines the value of the parameters mentioned above. This is particularly necessary if the K-fold procedure is used and the Q2 value found is smaller than 0.5. Indeed, we have shown in the first example (Fig. 1) that Q2 values of 0.4 and −0.1 can be obtained for the same dataset. We also strongly recommend SIMCA users to permute the lines of their dataset to control that the Q2 value calculated is stable regarding this permutation. In the supplementary material, a simple procedure is proposed to estimate Q2 variability with row rearrangements (Supplementary Data B, ESI†).The SIMCA software allows users to modify the cross-validation procedures by changing the number of cross-validation sets and/or selecting the individuals of each set. We believe that this possibility can also help users to estimate a confidence interval of the calculated quality parameters. The Leave One Out (LOO) procedure can also be tested on SIMCA by setting the number of subsets to the number of samples. This method does not depend on the order of the rows in the dataset, however, as pointed out by several authors,20,21 the LOO procedure can lead to over-fitting and over estimation of Q2.
Other cross-validation methods that (to our knowledge) are not yet implemented in SIMCA should be tested. We particularly recommend the double cross-validation (2CV) method.22,23 As the K-fold method, the 2CV uses all the available individuals to build the models and to estimate their predictability. However, in 2CV, the estimation of NSC and Q2 are decoupled. This is a very important issue since, as illustrated by the Fig. 1b, an overestimation/underestimation of NSC frequently leads to overestimation/underestimation of Q2. Thus, even if the double validation loop process is time consuming compared to the simple validation loop performed in the K-fold procedure, the risk of overestimation or underestimation of the predictability is reduced with the 2CV procedure.
Another interesting method is the Monte Carlo Cross-validation (MCCV) procedure.24 By randomly building many subsets with many combinations of individuals, this procedure averages the opposite effects of too optimistic and too pessimistic cross-validation submodels.
Finally, we want to remind readers that a truly reliable estimation of the predictability of a model is obtained with individuals that are independent of those used to build this model.25
Conclusion
PLS-DA/OPLS-DA is a powerful method for multivariate problems and SIMCA is a very robust and well-adapted software for this type of analysis. However, as mentioned by Erikson et al., the PLS method fails to build reliable discriminant models under some conditions (i.e. non-homogeneous classes and large intraclass variability). We have shown that when these conditions are encountered, the K-fold cross-validation procedure results can be strongly determined by the composition of the different subsets. As a consequence, a simple permutation of the row order in the dataset can strongly modify the value of the quality parameters calculated by the K-fold cross-validation procedures. In some cases, contradictory conclusions concerning the significance of the multivariate model can be drawn from the same dataset.Abbreviations
| PLS | Projection to latent structures |
| OPLS | Orthogonal projection to latent structures |
| DA | Discriminant analysis |
| PCA | Principal components analysis |
| NC | Number of components |
| NSC | Number of significant components |
| ONC | Optimal number of components |
| LOO | Leave one out |
| MCCV | Monte Carlo cross-validation |
References
- B. Worley and R. Powers, Curr. Metabolomics, 2013, 1, 92–107 CAS.
- B.-H. Mevik and R. Wehrens, Journal of Statistical Software, 2007, 18, 1–24 CrossRef.
- J. Xia, R. Mandal, I. V. Sinelnikov, D. Broadhurst and D. S. Wishart, Nucleic Acids Res., 2012, 40, 127–133 CrossRef PubMed.
- B. Worley and R. Powers, ACS Chem. Biol., 2014, 9, 1138–1144 CrossRef CAS PubMed.
- H. S. Tapp and E. K. Kemsley, TrAC, Trends Anal. Chem., 2009, 28, 1322–1327 CrossRef CAS PubMed.
- E. K. Kemsley and H. S. Tapp, J. Chemom., 2009, 23, 263–264 CrossRef CAS.
- G. A. Marcoulides and C. Saunders, Management Information Systems Quarterly, 2006, 30, iii–ix Search PubMed.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold, J. Trygg, C. Wikström and S. Wold, Multi- and Megavariate Data Analysis, Part I: Basic Principles and Applications, Umetrics AB, Umeå, Sweden, 2006 Search PubMed.
- Umetrics, User Guide SIMCA-+ 12, Umetrics, 2008 Search PubMed.
- J. Westerhuis, H. J. Hoefsloot, S. Smit, D. Vis, A. Smilde, E. J. van Velzen, J. M. van Duijnhoven and F. van Dorsten, Metabolomics, 2008, 4, 81–89 CrossRef CAS.
- H. L. Cai, H. D. Li, X. Z. Yan, B. Sun, Q. Zhang, M. Yan, W. Y. Zhang, P. Jiang, R. H. Zhu, Y. P. Liu, P. F. Fang, P. Xu, H. Y. Yuan, X. H. Zhang, L. Hu, W. Yang and H. S. Ye, J. Proteome Res., 2012, 11, 4338–4350 CrossRef CAS PubMed.
- B. Dong, J. Jia, W. Hu, Q. Chen, C. Jiang, J. Pan, Y. Huang, W. Xue and H. Gao, Clin. Biochem., 2013, 46, 346–353 CrossRef CAS PubMed.
- L. Eriksson, J. Trygg and S. Wold, J. Chemom., 2008, 22, 594–600 CrossRef CAS.
- A. M. Wheelock and C. E. Wheelock, Mol. BioSyst., 2013, 9, 2589–2596 RSC.
- C. Goossens, M. N. Triba, L. Le Moyec, O. Seror, P. Nahon and P. Savarin, 2014, in preparation.
- F. Dieterle, A. Ross, G. Schlotterbeck and H. Senn, Anal. Chem., 2006, 78, 4281–4290 CrossRef CAS PubMed.
- J. Trygg and S. Wold, J. Chemom., 2002, 16, 119–128 CrossRef CAS.
- P. Nahon, R. Amathieu, M. N. Triba, N. Bouchemal, J. C. Nault, M. Ziol, O. Seror, G. Dhonneur, J. C. Trinchet, M. Beaugrand and L. Le Moyec, Clin. Cancer Res., 2012, 18, 6714–6722 CrossRef CAS PubMed.
- D. B. Kell and R. D. King, Trends Biotechnol., 2000, 18, 93–98 CrossRef CAS.
- A. Golbraikh and A. Tropsha, J. Mol. Graphics Modell., 2002, 20, 269–276 CrossRef CAS.
- H. A. Martens and P. Dardenne, Chemom. Intell. Lab. Syst., 1998, 44, 99–121 CrossRef CAS.
- S. Smit, M. l. J. van Breemen, H. C. J. Hoefsloot, A. K. Smilde, J. M. F. G. Aerts and C. G. de Koster, Anal. Chim. Acta, 2007, 592, 210–217 CrossRef CAS PubMed.
- M. Stone, Journal of the Royal Statistical Society: Series B (Statistical Methodology), 1974, 36, 111–147 Search PubMed.
- Q.-S. Xu, Y.-Z. Liang and Y.-P. Du, J. Chemom., 2004, 18, 112–120 CrossRef CAS.
- D. Broadhurst and D. Kell, Metabolomics, 2006, 2, 171–196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00414k |
| This journal is © The Royal Society of Chemistry 2015 |