
Research highlights: enhancing whole genome amplification using compartmentalization
Andy
Tay
a,
Rajan P.
Kulkarni
a,
Armin
Karimi
a and
Dino
Di Carlo
*abc
aDepartment of Bioengineering, University of California, Los Angeles, CA 90095, USA. E-mail: dicarlo@ucla.edu
bCalifornia NanoSystems Institute, Los Angeles, California, USA
cJonsson Comprehensive Cancer Center, Los Angeles, California, USA
First published on 21st October 2015
Abstract
The ability to break up a larger liquid volume into an array of smaller confined volumes that do not chemically communicate is a key enabling technology driving microfluidic innovations. We highlight recent work using drop-based confinement to improve on whole genome amplification, reducing amplification bias and contaminant amplification by bringing reactions to saturation within each confined drop. We also highlight a complementary technique to target whole genome amplification to a subset of nucleic acids within a sample by combining drop-based PCR with sorting and downstream sequencing. These new approaches have the potential to enhance our ability to categorize the diversity of microorganisms (especially difficult to culture species) that contribute to complex microbial communities, and in particular assemble the individual genomes of the species involved in biologically and environmentally important microbiomes.
Emulsion whole genome amplification
Single-cell characterization of genetic material is critical for analysis of rare samples and to probe heterogeneity in a population of cells, particularly in disease states such as cancer or to survey diversity in viral or bacterial populations.1 Some of the techniques for whole genome amplification (WGA) includes multiple displacement amplification (MDA), degenerate oligonucleotide-primed PCR (DOP-PCR), and multiple annealing and looping-based amplification cycles (MALBAC). MDA using phi29 DNA polymerase and random primers can generate sufficient DNA quantity in a high fidelity manner, making it one of the most popular techniques for single cell whole genome amplification (WGA).2 Nonetheless, this technique suffers from issues such as amplification bias, formation of genomic chimeras and amplification of contaminating DNA which are particularly detrimental to amplification of for example a single bacterial cell genome which is present in extremely low quantity.3To overcome the aforementioned limitations in MDA, Nishikawa and colleagues incorporated an additional step using pico-liter sized droplets to compartmentalize each amplification reaction on DNA fragments, running amplification reactions to completion in each droplet and then recombining amplified products for sequencing.4 Using this approach produced two distinct advantages: (i) the researchers observed a decreased contribution from contaminating DNA, presumably because contaminating DNA was confined to a drop where amplification could proceed only to the exhaustion of the reagents in the drop, rather than competing for the whole pool of reagents in a larger volume reaction. (ii) Less bias in the WGA was observed, which is also related to the fact that kinetic factors play less of a role in driving differential amplification between genome regions, given sufficient time for each DNA fragment to amplify in its own compartment.
Briefly, single bacteria were sorted using flow cytometry into well plates and lysed. The lysed suspension of 10 μL was then converted into ~1.5 × 105 droplets each 67 pL where compartmentalized reactions can occur followed by emulsion breaking. The authors showed that their proposed method could produce a time-dependent increase in fluorescence that saturated indicative of uniform amplification (Fig. 1a). When the authors compared their technique to conventional MDA i.e. in-tube, they also found that the amplicon yield using their technique was proportional to the starting DNA content while in-tube MDA generated unwanted amplification of contaminant DNA fragment (Fig. 1b). Further emphasizing the reduced effect of contaminant DNA in the drop reaction, the total length of contigs that were unaligned to the reference sequence were 0.77 Mbp in emulsion MDA compared to 3.4 Mbp for in-tube MDA (Fig. 1c). Overall, this work shows that emulsion MDA could reduce the number of unexpected contigs due to DNA contamination and presence of chimeric fragments as well as reduce amplification bias. Compartmentalized droplets for DNA amplification can potentially provide high quality genome assembly from a single bacterium.
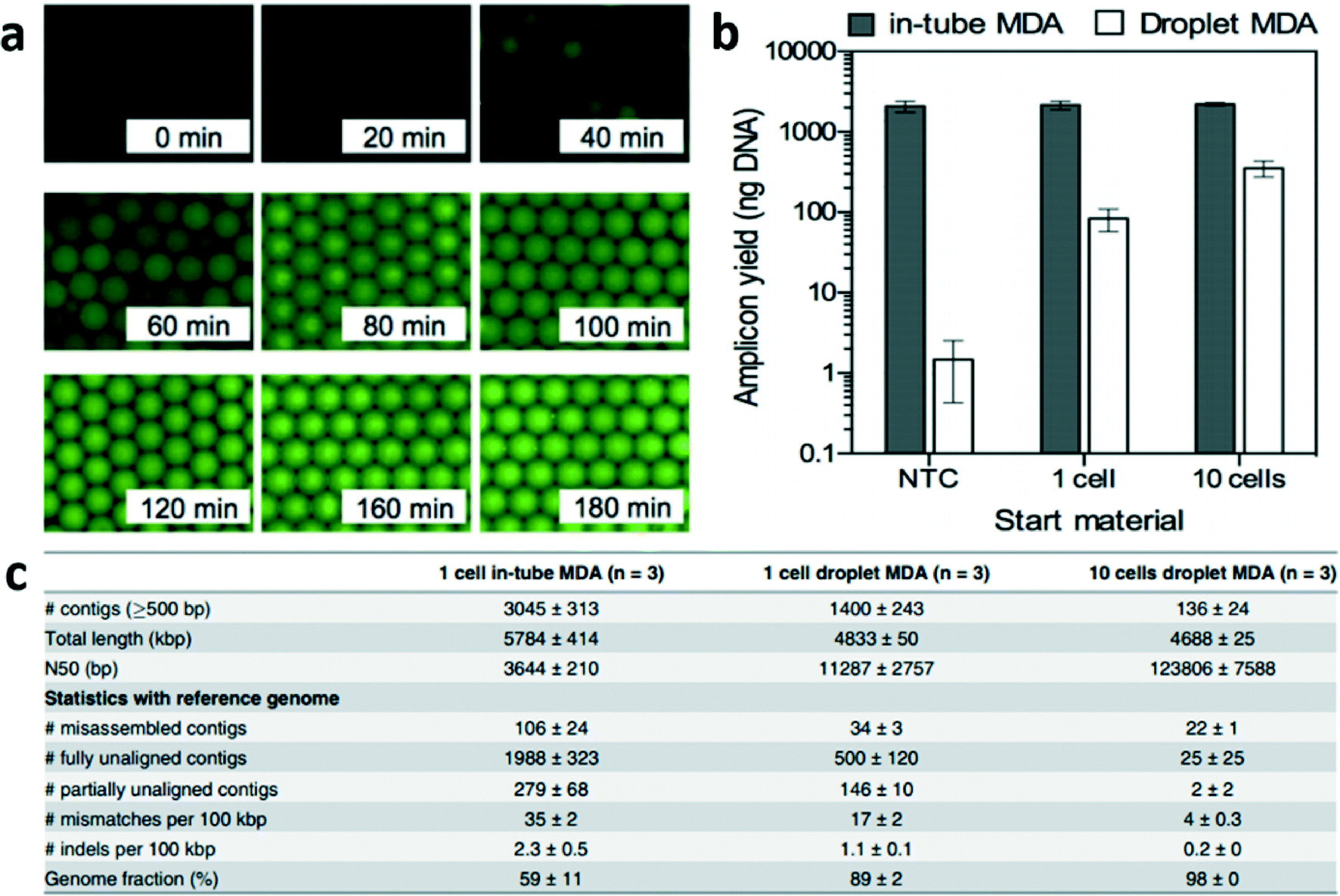 | ||
| Fig. 1 Droplet multiple displacement reaction (MDA) can increase the quality of genome assembly from single E. coli. (a) Time-dependent increase in fluorescence intensity showing DNA amplification saturating for each drop. (b) Droplet MDA produced amplicon yield proportional to starting DNA content while in-tube MDA was affected by contaminant DNA fragments. (c) Droplet MDA generated less unexpected (misassembled, fully unaligned, partially unaligned and mismatches) contigs. Reproduced with permission from PLoS One under the Creative Commons Attribution License, ref. 4. | ||
Using a similar approach, Fu et al. divided the genomic DNA of a single cell into many aqueous droplets prior to performing WGA, a method they termed emulsion WGA (eWGA).5 The DNA of each cell was fragmented into 105 fragments and the resulting fragments were encapsulated into small droplets using a microfluidic device (Fig. 2). On average, one fragment was present per droplet.
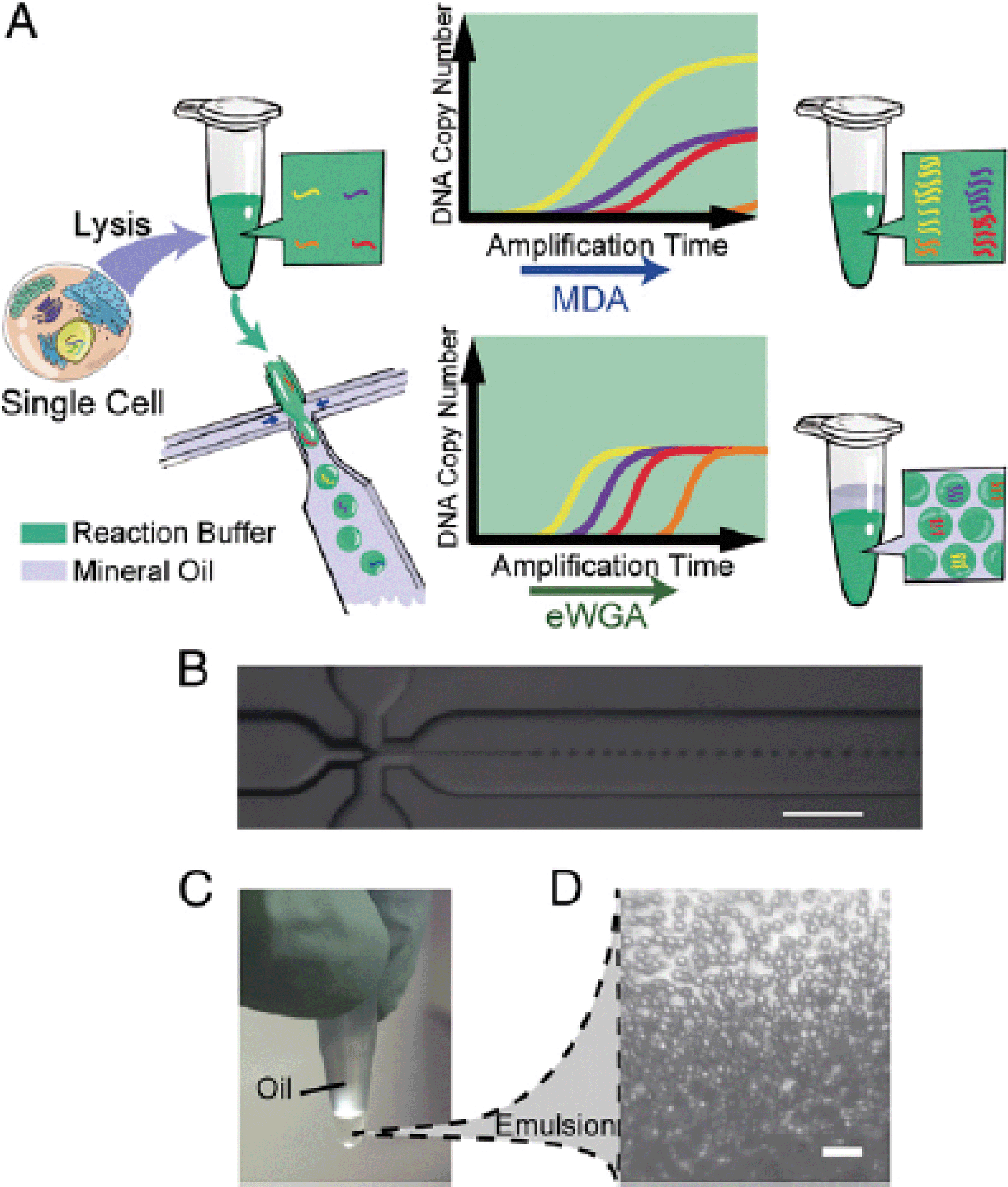 | ||
| Fig. 2 The experimental process of eWGA-seq and emulsion generation. (A) A single cell is lysed and then mixed with MDA reaction buffer in a tube. The solution was either directly used for conventional MDA, generating unevenly amplified DNA fragments, or used for emulsion generation in a microfluidics cross-junction device, resulting in uniformly distributed aqueous reaction droplets and more evenly amplified DNA fragments. (B) The microfluidics cross-junction. (C) All droplets are collected into a microcentrifuge tube and incubated at 30 °C to perform eWGA. (D) The emulsion is stable during the reaction (scale bar: 100 μm). Reproduced with permission from Proceedings of the National Academy of Sciences of the United States of America, ref. 5. | ||
Using emulsion MDA (eMDA), the authors claimed to detect copy number variations (CNV) with increased resolution and higher accuracy compared to regular MDA. They also attained increased coverage compared to non-microfluidic amplification methods and had the highest fidelity of single nucleotide variant (SNV) calls. While the accuracy of eMDA was less than that of DOP-PCR and the allelic dropout greater than MALBAC, the overall performance characteristics of eMDA were comparable or improved compared to the other three conventional methods.
The authors determined that eMDA could reduce amplification bias, ensuring greater and more even coverage of the entire cell's genome while still maintaining high replication accuracy. eMDA is the first technique that can enable simultaneous identification of both small CNVs and SNVs with a low error rate. The emulsion method is generalizable and could potentially incorporate other enzymes and chemistries in the future for even greater improvements in amplification efficiency. Future theoretical and experimental work will be important to determine what droplet size leads to the highest level of uniformity in amplified DNA while minimizing contributions from contamination and maximizing amplification.
Whole-genome sequencing of a single viral species from a heterogeneous sample
A lack of technologies to efficiently enrich various types of viruses and characterize unknown viral or other microbial genomes have been a roadblock to identify potential infectious diseases.6 Less than 0.01% of existing viruses have been sequenced7 and improvement in the understanding of virus diversity, ecology, adaptation, and evolution is highly needed.In this study Han et al. developed a platform utilizing droplet digital PCR (ddPCR) and microfluidics sorting techniques to selectively enrich specific types of viruses from a complex mixture for downstream pure whole genome amplification and sequencing (Fig. 3a–e).8 An aqueous mixture containing a virus sample, PCR buffer, primers, dUTP/dATP/dCTP/dGTP, and SYBR Green I, is encapsulated to compartmentalize the sample such that most drops contain zero or one virion.
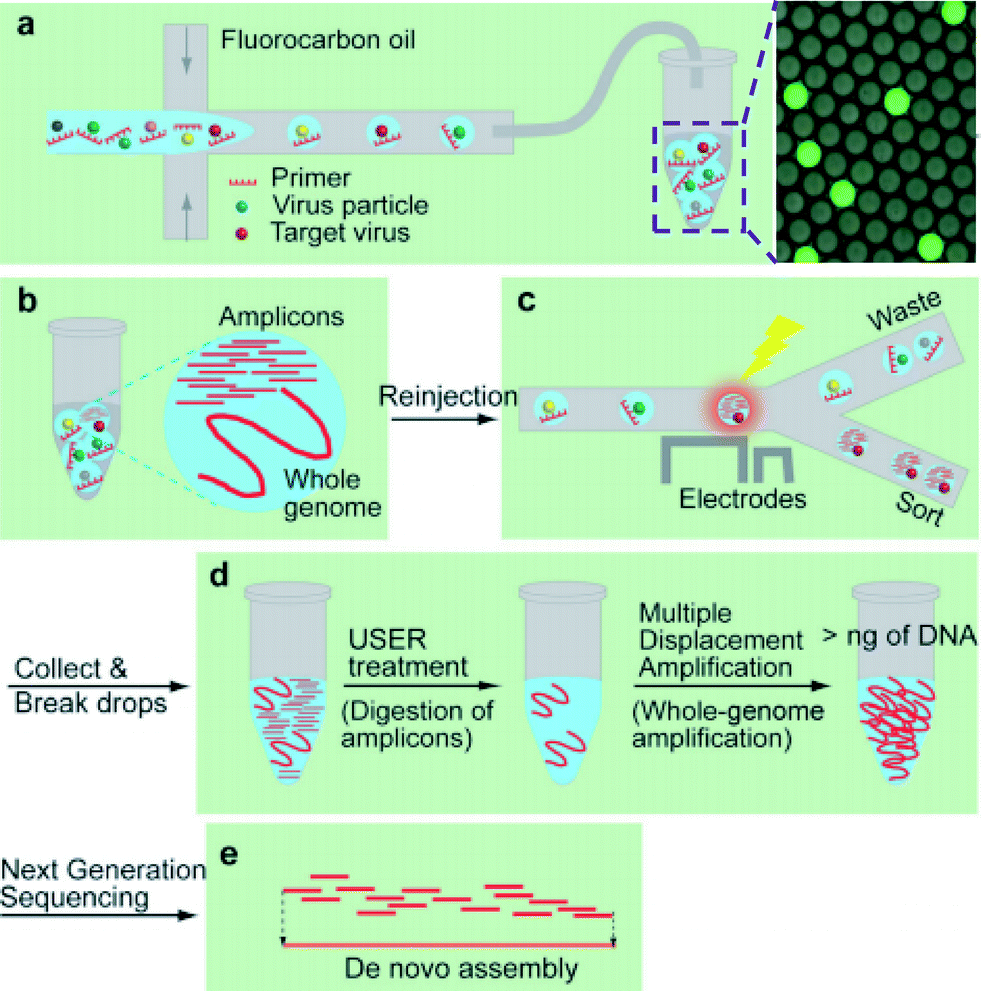 | ||
| Fig. 3 Using compartmentalization to sequence viral genomes from an environmental sample. (a) Viruses are encapsulated into drops. (b) After thermocycling with dUTP replacing dTTP, amplicons are generated in the drops containing the target virus yielding higher fluorescence intensity. (c) Drops are sorted based on their fluorescence intensities. (d) The enriched virus solution is treated with USER to digest amplicons containing uracil, followed by whole-genome amplification. (e) The amplified products are sequenced and assembled. Reproduced with permission from Angewandte Chemie, ref. 8. | ||
The method presented herein is shown to be more efficient, less labor-intensive, and more importantly applicable to various subsets of viruses, unlike traditional enrichment methods such as cell culture, hybridization, or immunoscreening followed by sequence-independent PCR, and the most recently developed flow cytometry method which does not employ a selection strategy to enable an efficient usage of sequencing power.
In their study, SV40, a well characterized virus, is spiked into wastewater and the mixture is encapsulated into 8 pL drops. After thermocycling, the presence of SV40 virions, in a small fraction of drops, is confirmed by detecting a high level of fluorescence under excitation at 470 nm (Fig. 3a and b). Using a microfluidic sorter, these drops are selected (Fig. 3c), and the PCR amplicons in the collected solution are digested using Uracil-Specific Excision Reagent (USER). Importantly, during the initial amplification dTTP is replaced with dUTP, such that amplicons can be selectively digested during this step, preventing later bias during whole genome amplification. Whole-genome amplification is then conducted with Φ29DNA polymerase (Fig. 3d).
The authors showed that without sorting, the amplified sequences contain 0.004% of SV40 reads, whereas, after their purification and amplification method, the reads increase to 94.0% and 98.6% for samples of 100 and 1000 selected drops, respectively. As was described above, during MDA, the larger DNA is preferentially amplified over shorter DNA, leading to bias and thus the sequences of high-molecular-weight contaminants are over-represented in the amplified product. This partly explains the low percentage of SV40 reads, and in particular it becomes a problematic issue in viral genome sequencing because viral genomes are generally much smaller than other abundant DNA in the sample. The results for the SV40 sample indicated a complete sequence read coverage. It also showed that without the digestion step (USER treatment), only amplicon sequences dominate the signal and are recovered after sequencing.
The final step of the process is to assemble sequence reads into contigs by using a computational tool (Fig. 3e). The BLAST search is used to analyze the contigs to identify their origin and sequence homology to the reference genomes and the result showed that the contig with the highest relative abundance aligned with the SV40 genomes, i.e. 97% of the genome sequence is covered. They indicated that their sequencing was performed with 50 bp single-end read and thus distinguishing the repeating sequences larger than that was challenging and could further improve. The authors used Sanger sequencing on the sorted drops and manually inserted that sequence based on the sequence overlap in order to correct the absence of the amplicon sequence in the contigs. For de novo assembly of unknown viral genomes, raw sequencing data can be computationally cleaned by removing the common contaminant sequences, however, the high-purity sequence reads ensure accurate de novo assembly with unknown contaminants.
In summary, the group showed a retrieval of more than 97% of the target genome sequence by using their proposed platform. They claim that this platform can be used for enrichment and sequencing of unknown viruses by designing primers specific to new contigs found in metagenomic (pooled) analysis of samples. Future work, could explore sequential droplet analysis, combining the described approach for viral particle selection with downstream drop-based eWGA to prevent amplification bias and reduce contaminant amplification.
References
- X. Ni, M. Zhuo, Z. Su, J. Duan, Y. Gao, Z. Wang, C. Zong, H. Bai, A. R. Chapman, J. Zhao, L. Xu, T. An, Q. Ma, Y. Wang, M. Wu, Y. Sun, S. Wang, Z. Li, X. Yang, J. Yong, X.-D. Su, Y. Lu, F. Bai, X. S. Xie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 21083–21088 CrossRef CAS PubMed.
- R. S. Lasken, Curr. Opin. Microbiol., 2007, 10, 510–516 CrossRef CAS PubMed.
- H. Yokouchi, Y. Fukuoka, D. Mukoyama, R. Calugay, H. Takeyama and T. Matsunaga, Environ. Microbiol., 2006, 8, 1155–1163 CrossRef CAS PubMed.
- Y. Nishikawa, M. Hosokawa, T. Maruyama, K. Yamagishi, T. Mori and H. Takeyama, PLoS One, 2015, 10, e0138733 Search PubMed.
- Y. Fu, C. Li, S. Lu, W. Zhou, F. Tang, X. S. Xie and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11923–11928 CrossRef CAS PubMed.
- P. G. Cantalupo, B. Calgua, G. Zhao, A. Hundesa, A. D. Wier, J. P. Katz, M. Grabe, R. W. Hendrix, R. Girones, D. Wang and J. M. Pipas, mBio, 2011, 2, e00180-11 CrossRef PubMed.
- S. J. Anthony, J. H. Epstein, K. A. Murray, I. Navarrete-Macias, C. M. Zambrana-Torrelio, A. Solovyov, R. Ojeda-Flores, N. C. Arrigo, A. Islam, S. Ali Khan, P. Hosseini, T. L. Bogich, K. J. Olival, M. D. Sanchez-Leon, W. B. Karesh, T. Goldstein, S. P. Luby, S. S. Morse, J. A. K. Mazet, P. Daszak and W. I. Lipkin, mBio, 2013, 4, e00598 CrossRef PubMed.
- H. S. Han, P. G. Cantalupo, A. Rotem, S. K. Cockrell, M. Carbonnaux, J. M. Pipas and D. A. Weitz, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201507047.
| This journal is © The Royal Society of Chemistry 2015 |