
Polymer-based microfluidic chip for rapid and efficient immunomagnetic capture and release of Listeria monocytogenes†
L.
Malic
a,
X.
Zhang
a,
D.
Brassard
a,
L.
Clime
a,
J.
Daoud
a,
C.
Luebbert
b,
V.
Barrere
b,
A.
Boutin
a,
S.
Bidawid
b,
J.
Farber
b,
N.
Corneau
b and
T.
Veres
*acd
aLife Sciences Division, National Research Council of Canada, 75 de Mortagne Boulevard, Boucherville, QC J4B 6Y4, Canada. E-mail: teodor.veres@cnrc-nrc.gc.ca
bBureau of Microbial Hazards, Health Canada, 251 Sir Frederick Banting Driveway, Ottawa, ON K1A 0K9, Canada
cBiomedical Engineering & Bioengineering Departments, McGill University, 845 Rue Sherbrooke Ouest, Montreal, QC H3A 0G4, Canada
dInstitut National de la Recherche Scientifique (INRS), 1650 Boulevard Lionel-Boulet, Varennes, QC J3X 1S2, Canada
First published on 28th August 2015
Abstract
Infections caused by foodborne pathogens such as Listeria monocytogenes pose a threat to public health while timely detection is challenging due to pathogen low numbers. The development of robust and efficient sample preparation techniques is crucial to improve detection sensitivity and workflow. Immunomagnetic separation using magnetic nanoparticles (MNPs) is attractive, as it can efficiently capture target cells. For food safety applications, a platform is needed to rapidly process large sample volumes, allowing capture and release of target bacteria conjugated to immunomagnetic nanoparticles (IMNPs). Herein, we demonstrate a method for magnetic capture and release of bacteria-IMNPs complex based on a 3D magnetic trap integrated on a polymeric microfluidic device. The 3D magnetic capture region consist of a dense array of high-aspect ratio (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) cylindrical pillars embossed in thermoplastic polymer and coated with soft ferromagnetic nickel by an electroless deposition technique. This allows the generation of strong and switchable magnetic capture regions due to the very low remanence of the nickel shell. We propose and validate an optimized configuration of capture regions for efficient localized capture and rapid release of MNPs and IMNPs conjugated to L. monocytogenes. A maximum recovery rate for MNPs corresponded to 91% while a maximum capture efficiency of 30% was obtained for live bacteria, with a minimum detectable sample concentration of ~10 cfu ml−1 in 1 ml volume using plate-culture method. We believe that the flexible design and low-cost fabrication process of the proposed system will allow rapid sample preparation for applications beyond food and water safety, including point-of-care diagnosis.
:1) cylindrical pillars embossed in thermoplastic polymer and coated with soft ferromagnetic nickel by an electroless deposition technique. This allows the generation of strong and switchable magnetic capture regions due to the very low remanence of the nickel shell. We propose and validate an optimized configuration of capture regions for efficient localized capture and rapid release of MNPs and IMNPs conjugated to L. monocytogenes. A maximum recovery rate for MNPs corresponded to 91% while a maximum capture efficiency of 30% was obtained for live bacteria, with a minimum detectable sample concentration of ~10 cfu ml−1 in 1 ml volume using plate-culture method. We believe that the flexible design and low-cost fabrication process of the proposed system will allow rapid sample preparation for applications beyond food and water safety, including point-of-care diagnosis.
1. Introduction
Foodborne infections pose a significant burden on public health and the worldwide economy. Despite major efforts to develop rapid, accurate and ultrasensitive technologies to detect the presence of low cell numbers in food samples, the number of reported food-borne infections is continuously rising.1 In the United States alone, the total cost related to foodborne-infections amounts to US$78 billion annually,2–5 which represents a staggering 15-fold increase in a little over a decade.6 Herein, 48 million foodborne illnesses are estimated annually, that are responsible for 128000 hospitalizations, and 3000 deaths.2 Of these, 20 to 30 percent of clinical infections resulting in death are attributed to human listeriosis (Listeria monocytogenes infections).7 In response to several large outbreaks of listeriosis over the past three decades,8 U.S. regulatory agencies have issued testing mandates for various food products9 and established a “zero tolerance” policy for L. monocytogenes in ready-to-eat foods.10
Conventional assays based on classical microbiology techniques represent the gold standard for detection of L. monocytogenes in food samples. Multiple steps involving culture enrichment, selective plating, followed by biochemical identification make the process lengthy, taking up to a week for the results to be finalized. While the development of sensitive detection techniques that rely on polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA) or DNA/RNA hybridization has shortened the time to detection from 5 days to 2–3 days, the requirement for culture enrichment for all these assays is still the rate-limiting step.11 Consequently, these tests cannot quickly provide accurate data for many emergency events or comply with the objective of detection and identification in less than 8 hours.
Pre-analytical sample preparation techniques, where target pathogens are separated, concentrated, and purified from the initial sample matrix before detection, have the potential to further reduce the overall sample-to-answer time.12 Additional benefits of this approach include the removal of matrix-associated reaction inhibitors, possibility to detect multiple bacterial strains, and reduction of the dilution ratio allowing for the use of representative food sample sizes and/or smaller media volumes.13
Immunomagnetic separation (IMS) is one of the most widely used approaches for sample preparation,2,14,15 where the separation of target cells is achieved by using magnetic particles conjugated to antibodies specific to cell surface receptors. Conventional immunomagnetic separation is typically performed in tubes with several commercially available kits, including MACS® system (Miltenyi Biotec, USA), CellSearch System (Veridex, USA), and Dynabeads MPC separator series (Life technologies, USA). In the past decade, microfluidics approaches combined with magnetism have also emerged as viable high throughput and low cost alternatives.16,17
Most microfluidic methods rely on positioning a permanent magnet in the vicinity of a microfluidic channel. The generated magnetic field gradient acts on the magnetically labeled targets, and deflects and separates them out of the main stream.18–21 This method is suitable for trapping targets conjugated to micrometer sized magnetic beads. Unfortunately, for applications where the target size is in the micron range (as is the case of most bacteria) suspended in complex samples such as large volumes of food filtrates or blood, the use of micron-size commercial magnetic carriers may not provide enough surface to volume capture area to achieve the required trapping efficiency. The use of magnetic nanoparticles can be more advantageous due to much smaller size, higher surface area to volume ratio, and increased mobility, resulting in enhanced attachment on the surface of the micron-sized target. However, for small magnetic nanoparticles, the magnetic field gradient generated by external magnets is too low and typically requires amplification. Thus to fully exploit the benefits of nanoparticles, high magnetic gradients necessary for their capture need to be generated and operated in an easy and affordable way.
Among other approaches, a soft magnetic material can be patterned in microscopic elements to create local distortions of the externally applied magnetic field and thus generate stronger magnetic forces for very efficient target separation in microfluidic format.22–33 Unfortunately, the integration of soft magnetic materials in microfluidic devices often requires very complex and costly fabrication process,25,27–33 which would prevent the deployment of the technology for large-scale food safety or diagnostic applications. The self-assembled structures of magnetic microparticles on top of printed magnetic ink arrays34 could potentially lower the cost of fabrication of magnetic elements. However, the throughput of this method is limited by the low flow rates required to prevent the detachment of self-assembled structures from the printed arrays. Additionally, most of the reported microfluidic devices featured small channel sizes and low density of magnetic microstructures which typically result in low bead capturing capacity27,29–32,35 and further limit the applicability of these systems for high-throughput and large-volume applications where a large number of magnetic beads have to be processed.
In this manuscript, we discuss a method and the development of a microfluidic device for immuno-magnetic sample preparation fabricated using materials and techniques suitable for mass-production. The proposed high gradient magnetic separation (HGMS) device is tailored to immunomagnetic nanoparticles (IMNPs) and integrates a 3D magnetic trap into a polymeric microfluidic chip. The magnetic capture chip (M-chip) consists of a sandwich of soft and hard structured thermoplastic layers fabricated using hot-embossing. The 3D magnetic capture region is engraved in the hard material and the chip-to-world microfluidic interface is embossed in the softer one. The former consist of a dense array of flow-invasive high aspect-ratio soft-magnetic cylindrical shells that simultaneously provide strong capture force throughout a wide capture chamber and enhanced release capabilities. We demonstrate both numerically and experimentally that the topology of the capture regions is strongly influenced by the magnetic shell, the geometry of the pillar and the strength of the magnetic field. Specifically, the design and layout of the device is optimized for efficient capture and rapid release of target species magnetically-labeled with functionalized silica-shell-iron-oxide-core nanoparticles. The capability of the proposed system was demonstrated for the capture and release of L. moncytogenes in both buffer and beef filtrate, representing a relative food matrix. A maximum capture efficiency of 30% was obtained for live bacteria, while the 24 hour plate-culture method allowed the detection of initial sample concentration of ~10 cfu ml−1 in 1 ml volume. The device was also very efficient in concentrating the sample 2.8-fold from a 10 ml initial volume in only 17 minutes, demonstrating the suitability of the system for throughput-intensive applications such as food safety.
2. Experimental
2.1. Magnetic capture platform
The magnetic capture (M-chip) device for sample concentration and purification consists of a hard thermoplastic bottom substrate that contains the capture chamber and a soft thermoplastic elastomer top substrate housing the microfluidic channel network (Fig. 1a and b). The inlet and outlet regions of the microfluidic channel network are designed to ensure uniform distribution of the flow throughout the wide capture chamber (Fig. 1c) which contains a dense array of cylindrical micro-pillars with an aspect ratio of approximately 3:1 (Fig. 1d). The micro-pillars (16 μm diameter) are coated with a conformal layer (2 μm thick) of soft magnetic material (nickel) (Fig. 1e and SI Fig. S1a and b†) which is subsequently passivated to prevent non-specific absorption. When a pair of permanent magnets is placed at the opposite edges of the device, a quasi-uniform magnetic field is generated within the capture chamber (Fig. 1f). This external magnetic field is strongly perturbed around each nickel-coated pillar, which leads to a magnetic field gradient and a related local attractive force that can be exploited to efficiently and rapidly trap magnetic species in pre-determined locations on each pillar (Fig. 1g). In order to improve magnetic trapping efficiency, the micropillar array is designed to ensure a distribution wherein each column (50 μm pitch) is slightly shifted relative to the next column (by ~4.4 μm). As such, the magnetic objects flowing through the microfluidic chamber are successively forced to come in close contact with a pillar along the chamber length, where the local magnetic field gradient is high. This configuration ensures that there is no streamline where the target species can escape preferentially, which improves trapping efficiency and minimizes the magnetic force required. The low remanence of the magnetic material (Ni) (see SI Fig. S1c†) renders the local magnetic field perturbations fully reversible such that the magnetic trapping force becomes negligible upon removal of the external magnetic field. This allows for efficient subsequent release of the captured magnetic species.
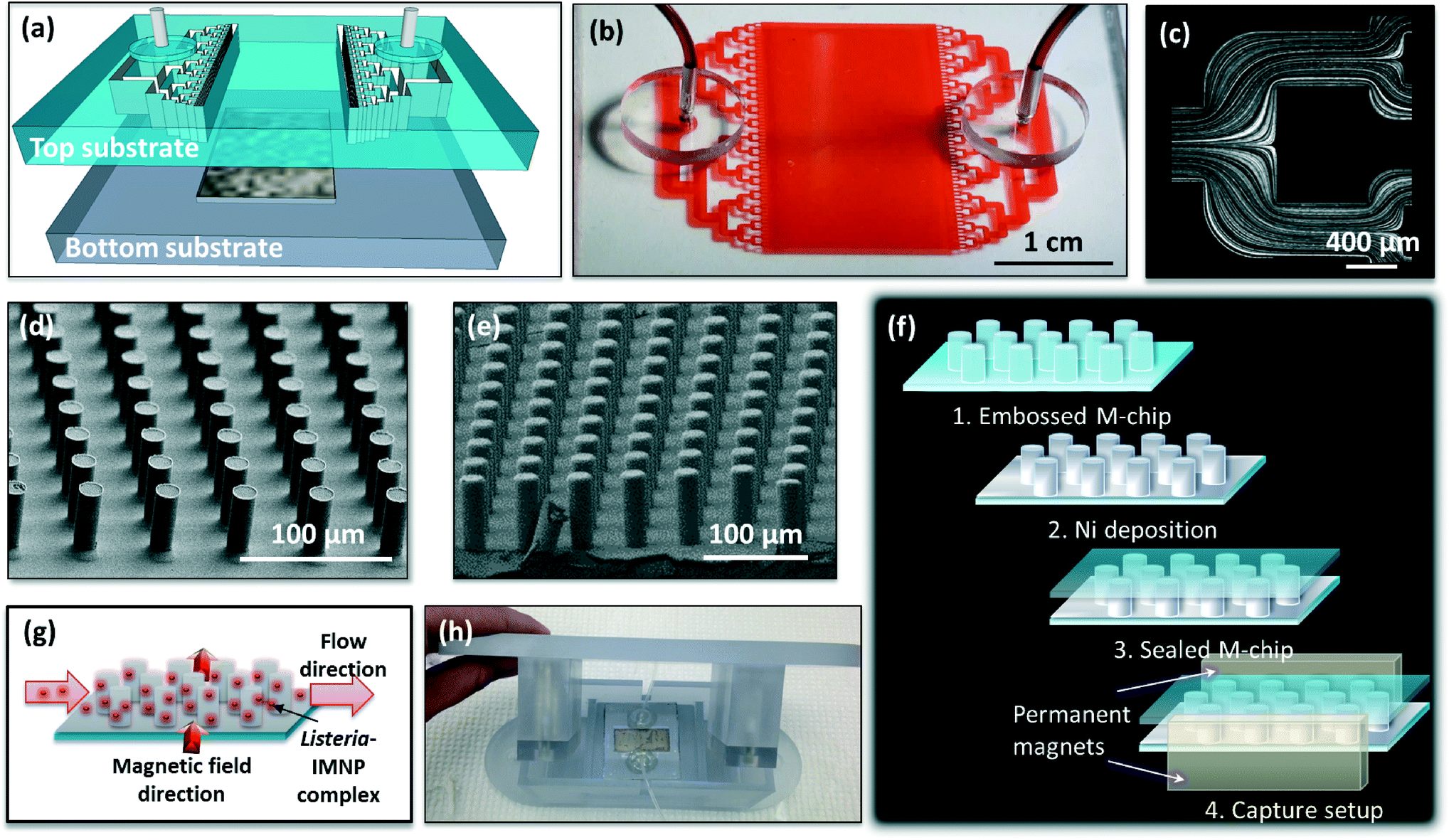 | ||
| Fig. 1 (a) Magnetic capture microfluidic device illustration showing a top substrate comprising a microfluidic inlet and outlet network and a bottom substrate with integrated capture chamber; (b) photograph of the assembled plastic magnetic capture microfluidic device (before Ni deposition) filled with a food dye for clarity; (c) micrograph showing fluorescence traces left by particles flowing in a selected inlet channel region of the M-chip revealing the flow lines and uniform flow distribution; (d and e) scanning electron microscopy images showing pillar array in the capture chamber of M-chip replicated in plastic before and after Ni deposition; (f) schematic showing different M-chip device layers and external magnet capture setup; (g) illustration of M-chip capture function with magnetic field direction perpendicular to the flow direction; (h) photograph of M-chip manifold comprising top holder for two permanent magnets and a bottom microfluidic device holder. | ||
Using antibody-functionalized magnetic nanoparticles, the combined capture and release capabilities of the M-chip can be used to concentrate and isolate bacteria from complex samples for subsequent culture and downstream processing, such as PCR detection. The complete process consists of three steps: (i) by applying an external magnetic field, a strong trapping force is first generated in the M-chip to efficiently trap bacteria conjugated to immunomagnetic nanoparticles (IMNPs). (ii) With the magnetic field still applied, a wash step is subsequently applied to remove sample impurities. (iii) Following the washing of the trapped species, by simply removing the external magnets, the bacteria-nanoparticle complex is released and eluted using a small and clean volume of buffer solution, yielding a concentrated sample for downstream processing.
In order to apply a uniform magnetic field at the chamber region, two permanent neodymium rare-earth magnets 5 cm × 1 cm in size (K&J Magnetics Inc, USA) were centered (2.5 cm from either edge) at both sides of the chip perpendicular to the sample flow. For this purpose, the magnets were mounted into a custom made holder with opposing poles facing each other and separated by a distance of 5.8 cm. The magnetic field at the center of the magnetic plate system was measured to be approximately 100 kA m−1 using a commercial gaussmeter (LakeShore Cryotronics, USA). For this purpose the M-chip was mounted onto a custom made M-chip holder and enclosure device (Fig. 1h) ensuring safe operation with pathogenic samples. The top enclosure served as a permanent magnet holder and featured curved edges for simple manual manipulation. Manipulation and control of fluid flow through the device was achieved using two programmable multirack syringe pumps (PHD Ultra Syringe Pump, Harvard Apparatus, USA). Due to the inherent weak bonding of the device at the top of the Ni coated pillars, the pumps were operated in negative-pressure withdrawal mode in order to avoid delamination of the chamber. Mechanical pinch valves placed at the outlet tubing were used to redirect the flow to the waste and collection syringes.
2.2. Device fabrication, assembly, and characterization
We have developed a process for low-cost fabrication of the thermoplastic microfluidic device containing an array of micron-size pillars coated with a conformal layer of low-remanence nickel film. The structuring of the plastic parts is performed using hot embossing36,37 as detailed in the ESI† section 1. Briefly, the bottom substrate containing the capture chamber was embossed in cyclo olefin polymer using a polymeric working stamp (Solvay MD700 with 1% Darocur 1173). The top substrate comprising the microfluidic channel network connecting the inlet and outlet to the capture chamber was fabricated in thermoplastic elastomer (TPE) using hot embossing and an epoxy mold. Fig. 1d shows images of the plastic devices and high-aspect ratio pillars (16 μm wide, 50 μm high) that were successfully replicated from a silicon master mould using hot embossing.Post-embossing, a conformal nickel coating was deposited in the chamber area via an electroless deposition process, as detailed in the ESI.† The electroless process was precisely controlled to grow an approximately 2 μm thick nickel coating (Fig. 1e and SI Fig. S1†) to allow both efficient capture and prompt release.
Following nickel deposition, the film was passivated using hydroxy-terminated tri (ethylene glycol) undecane thiol (thiolated PEG-OH, Nanoscience, USA). Surface modification was carried out using in situ chemical reduction,38 as described in the ESI.†
Finally, the devices were bonded by simply aligning and bringing in contact the top and bottom substrates at room temperature. The chip assembly was completed by inserting disposable fluidic connectors (Teflon PTFE Tubing, Scientific Commodities, USA) and tubing (Silastic, Dowcorning, USA) at the inlet and outlet holes. An epoxy adhesive (Lepage, Canada) was then applied over the connections to ensure leak-free device operation. The outlet tubing was split into waste and collection tubing using a T-barb tube fitting (Warner Instruments, USA).
The flow profile of fabricated devices was characterized using fluorescent nanoparticles, visualized with a fluorescence microscope (Nikon Eclipse Ti, Nikon, USA). Clogging tests were performed by flowing beef filtrate possessing a particulate size of <5 μm for the duration of 10 to 60 min at a flow rate of 100 μl min−1.
2.3. Nanoparticle synthesis, surface modification, and isolation efficiency
The superparamagnetic Fe3O4 nanoparticles were synthesized using a well-known protocol, in which polyacrylic acid (PAA) was used as cross-linker for growth of 50–100 nm magnetic nanoparticles consisting of ultra-small iron oxide cores of 2–5 nm.39 The ratio between the magnetic component and nonmagnetic PAA was optimized to obtain desirable magnetic properties. Furthermore, these iron oxide nanoparticles were coated with a layer of hydroxyl-terminated silica shells to reduce the magnetic inter-particle interactions and enhance the aqueous dispersibility.The silica-coated iron oxide nanoparticles with hydroxyl (–OH) groups were purified by dispersing them in a de-ionized water solution (concentration: 500 μg ml−1) by a 1 hr ultrasonication treatment, and then slowly separated by a permanent magnet. This purification process was repeated three times. Subsequently, the nanoparticles were re-dispersed in PBS buffer (the pH value was adjusted to 8.5 with 0.1 M NaOH). In parallel, 1 g cyanogen bromide (CNBr) was dissolved in 0.5 mL acetonitrile. The cyanogen bromide solution was then slowly added in a dropwise manner to the nanoparticle suspension under rapid stirring. The activation reaction was allowed to continue for precisely 2 min at room temperature, immediately followed by an ice-cold water wash (30 ml) under ultrasonication for 1 min, and finally centrifugation at 7000 rpm for 5 min at room temperature. The separated nanoparticles were then washed three times with 5 ml of PBS pH 8.5 and centrifuged again at 7000 rpm for 5 min. Finally, 100 μg of BacTrace anti-Listeria antibody (KPL, USA) was added to the above 5 ml nanoparticle suspension in PBS, and mixed using vortex for 1 h at room temperature. Subsequently, Bovine Serum Albumin (BSA) was added to a final concentration of 0.1% (w/v) in order to block the unreacted functional groups. Finally, the antibody-modified nanoparticles were magnetically separated and dispersed in 5 ml PBS pH 7.4 buffer containing 30 mM glycine and 0.1% (w/v) BSA. The functionalized nanoparticles were used immediately or stored at −20 °C for up to a month.
Following the surface modification, we have investigated the trapping efficiency of the immunomagnetic nanoparticles (IMNPs) and compared their performance with commercially available Dynabeads. For this purpose, 1 ml volume of live L. monocytogenes at a concentration of 3.27 × 103 cfu ml−1 was incubated with: (i) IMNPs at a final concentration of 4 μg ml−1, (ii) Dynabeads anti-Listeria (Life Technologies, USA) or (iii) Dynabeads M-270 Amine beads (Life Technologies, USA) conjugated to BacTrace anti-Listeria antibodies according to the manufacturer's protocol. For bacteria isolation with anti-Listeria Dynabeads, manufacturer's recommendations for optimum capture conditions were followed using 20 μl of beads solution in 1 ml sample volume. The Amine beads were counted and added as per the Dynabeads anti-Listeria manufacturer suggested amounts. For all experiments, conjugations were performed in a 2 mL microcentrifuge tube from 15 to 120 min using gentle vortex mixing, after which a permanent magnet was employed for bacteria capture. Following a capture for 60 min, a wash step was performed using careful pipetting to remove non-captured species. A plate-culture method followed by colony counting was then employed to assess the relative isolation efficiencies of the three different methods. Isolation efficiencies were calculated as a ratio of isolated Listeria-particle complex and initial Listeria concentration.
2.4. Numerical modeling
Numerical and analytical modeling was used to characterize and optimize the magnetic capture in the microfluidic device and corroborate the experimental results. The theoretical work has been oriented on three directions: (i) simulation of the global magnetic field landscape generated by the pair of two permanent magnetic plates; (ii) topology of local perturbations and related magnetic gradients induced by the presence of the magnetized Ni pillars and (iii) magnetic capture and separation of colloidal suspensions of magnetic nanoparticles.2.5. M-chip recovery rate and sample concentration efficiency
The efficiency of M-chip for concentration of unconjugated magnetic nanoparticles only and dead and live Listeria conjugated to IMNPs from buffer and beef filtrate solutions was evaluated using optical absorbance measurements, fluorescence microscopy and plate-culture methods, respectively. Optimization of operating conditions for bacterial capture was also performed by varying the nanoparticle concentration and incubation times. For live bacteria experiments, L. monocytogenes strain HPB582 was kindly provided by the Listeriosis Reference Centre for Canada (Dr. Pagotto). The serotype was 4b, isolated from the spinal fluid of a child with meningitis from Germany (ATCC13932). Prior to experiments, bacteria was grown overnight in TSBYE broth (Tryptic Soy Broth, BD cat# 211825, Yeast Extract, BD cat# 212750) at 37 °C.Unless otherwise noted, the flow rates used for all experiments were optimized in order to allow maximum recovery rates while ensuring reasonable assay time (<20 min) and corresponded to 100 μl min−1 during capture and 250 μl min−1 during release functions. The experimental sequence consisted of (i) priming the chip with HBS-EP buffer (GE Healthcare Life Sciences, USA) for 2 min prior to sample injection. (ii) With the magnetic field applied, the sample was then allowed to flow through the chamber at 100 μl min−1 flow rate. (iii) A wash step was subsequently performed by flowing HBS-EP buffer for 2 min in order to remove any impurities. (iv) Finally, the magnetic field was removed, and the sample was released from the chamber at the flow speed of 250 μl min−1 for the duration of 2 min in a small and clean sample volume for further analysis.
3. Results and discussion
The main objective of this work is to propose and demonstrate a method for the fabrication and integration of 3D magnetic capture and release integrated into a microfluidic chip based on HGMS. The proposed chip geometry can be tailored to optimize the size, shape and aspect ratio of magnetic pillars in order to allow the implementation of both capture and release functions. This is in contrast to most HGMS devices designed to create magnetic field gradients as high as possible to maximize the capture forces. Consequently, solid magnetic wires42 or solid magnetic micropillars31,33 are often used since they provide maximum perturbation effects and non-uniformities in the applied field. However, there are several drawbacks in this approach when re-suspension of captured biological species is desired. First, the topology of the capture regions is not controlled accurately as the resulting magnetic field gradient leads to an attractive capture force everywhere on the surface of the pillar.35 Secondly, the magnetic release is challenging since large amounts of magnetic material usually possess strong remnant magnetization; in turn creating significant capture forces persisting after removal of the external magnetic field. Very high flow rates and associated drag forces may force the release of trapped particles from these regions. However the effectiveness is attenuated in low flow regions and stagnation points on the pillars, which are difficult to clear solely by a unidirectional liquid flush.3.1. M-chip device characterization
In the present work, we propose a device with completely reengineered magnetic pillar structures with the objectives (i) to improve the release of trapped magnetic species while conserving high gradients necessary for efficient magnetic trapping and (ii) to enable fabrication techniques compatible with mass production. To this extent, magnetic pillars are fabricated on non-magnetic thermoplastic substrates by hot embossing and then coated with a layer of soft nickel magnetic material by batch electroless deposition process. We demonstrate here that this approach provides the key prospect to limit the magnetic capture to two diametrically opposed regions that are aligned with the direction of the magnetic field (see section 3.4). In the presence of liquid flow parallel to the magnetizing field, these capture regions are coincident with the upstream and downstream stagnation points. Although the resulting magnetic capture could be enhanced by low fluid velocities near these points, this will drastically impede the release of the captured material. Conversely, with the magnetic field perpendicular to the flow, the capture regions and the stagnation points are at 90° with respect to each other. The trapped material is then localized far from the stagnation points, which facilitates the release when the magnetic field is removed.Additionally, the M-chip for sample concentration and cleaning was designed to accommodate processing of large volumes, typically required for testing of food samples (Fig. 1a). For this purpose, a wide capture chamber (Fig. 1b) having x–y dimensions of 30 × 17 mm was populated with high aspect ratio pillars (20 μm diameter and 67 μm height), allowing operation at flow rates of up to 800 μl min−1. Thus, a 10 mL sample can be concentrated into a volume of 500 μL or less in approximately 15 minutes. It is important to minimize variations in the sample distribution across the chamber and consequently prevent capture saturation or clogging of the chip. This is accomplished through implementation of a distributed microfluidic channel network at the inlet. This allowed homogeneous flow throughout the entire chamber and uniform capture across its width (Fig. 1c). Additionally, the chip was able to withstand the continuous flow of 6 ml volume of beef filtrate (particulate size <5 μm) at a flow rate of 100 μl min−1 for one hour without device failure. While larger particles accumulated at the entrance of the chip, no significant clogging was observed (SI Fig. S3†). Similarly, the microfluidic outlet and collection network ensured a parallel moving front for sample concentration, thus achieving fast and efficient release of the captured species into a small volume. This microfluidic design prevents dead flow zones within the chamber locations which would otherwise require order-of-magnitude higher wash and release times.
3.2. Microfluidic device fabrication and surface functionalization
M-chip fabrication materials and processes were adapted to methods amenable to mass fabrication. In particular, low-cost thermoplastics and thermoplastic elastomers were employed that can be easily formed thermally and bonded for final device assembly. Additionally, electroless deposition was used to grow uniform and conformal Ni coating (Fig. 1e and SI Fig. S1a and b†). This method allowed high metal film performances such as high adhesion and filling capability, as well as low magnetic remanence. Finally, the surface of the Ni film was passivated to prevent Ni oxidation and non-specific biological sample adsorption (see ESI† section 1 and SI Fig. S4a for details). SI Fig. S4b and c† demonstrate capture and release of fluorescently labelled heat-killed L. monocytogenes, conjugated to IMNPs, using M-chip without and with surface functionalization, respectively. From SI Fig. S4b,† the non-specific adsorption of Listeria-IMNP complex to the Ni-coated pillars, prior to the application of the magnetic field, is higher for non-passivated surfaces. In addition, following a 10 minute capture, once the magnetic field is removed, the release is significantly more efficient for the passivated chip. This can also be observed from the normalized fluorescence intensity plot shown in SI Fig. S4d.†3.3. Nanoparticle characterization and immunomagnetic separation
The fabricated superparamagnetic nanoparticles are shown in Fig. 2a. The MNPs consist of multiple 2–5 nm diameter iron-oxide cores embedded in PAA and coated with a 50 nm thick layer of hydroxyl-terminated silica shell (Fig. 2a).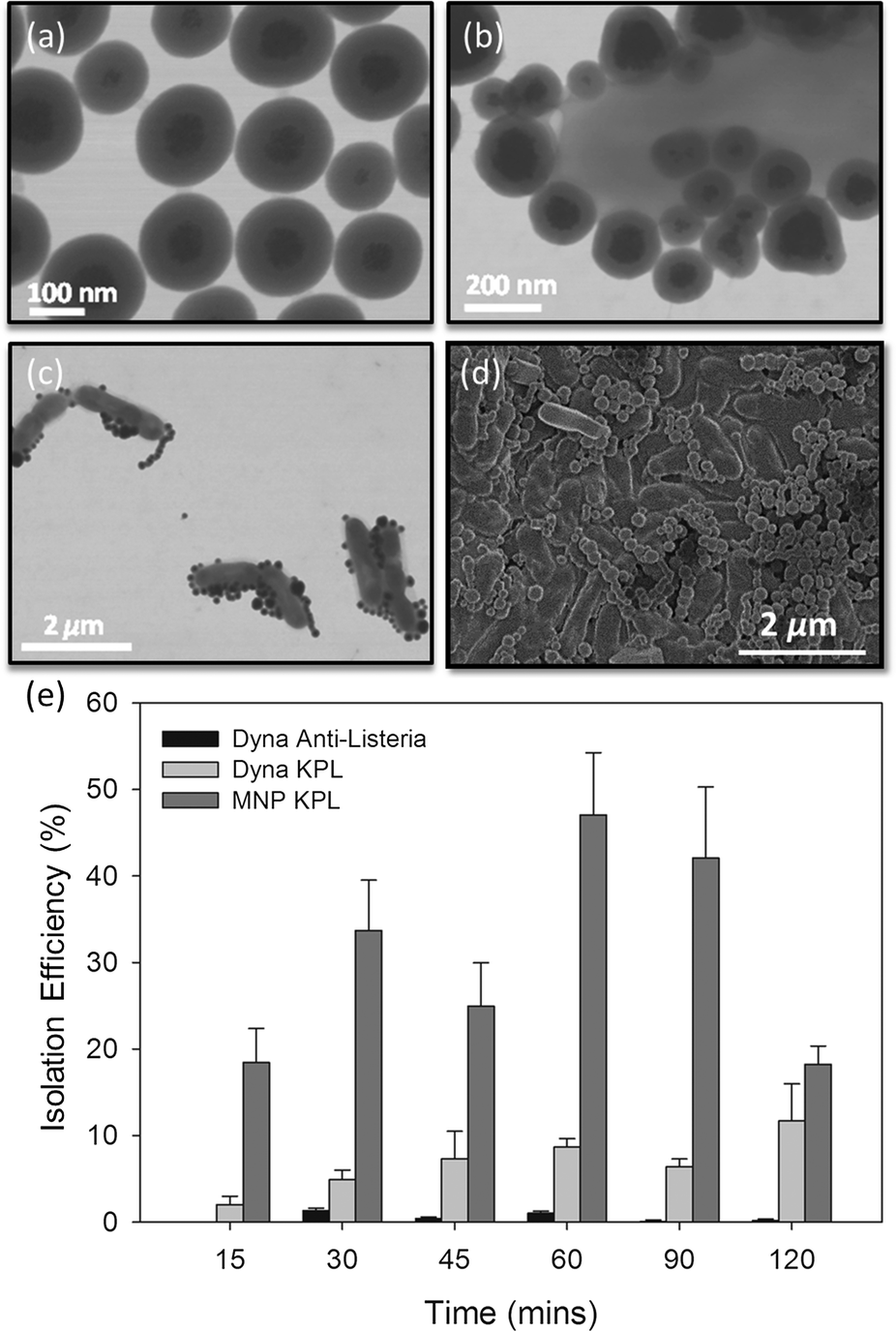 | ||
| Fig. 2 (a) TEM image showing the synthesized Fe3O4/Silica nanoparticles; (b and c) TEM image showing IMNPs-listeria conjugates; (d) SEM image showing IMNPs-Listeria conjugates; (e) comparison of L. monocytogenes immunomagnetic separation using commercially available Dynabeads (Dynabeads – anti-Listeria and Dynabeads conjugated to KPL BacTrace anti-Listeria antibody) and synthesized IMNPs via tube capture as a function of bacteria-particle incubation time. Constant nanoparticle concentration of 4 μg ml−1 and constant initial Listeria concentration of approximately 3.27 × 103 cfu ml−1 were used. | ||
The functionality of the silica shell is three-fold: i) it reduces magnetic interaction between particles; ii) enhances aqueous dispersibility; and iii) allows simple functionalization with specific antibodies. MNP surface chemistry was optimized to reduce non-specific adsorption and allow the immobilization of anti-Listeria antibodies. The antibody-nanoparticle conjugates did not show aggregation and demonstrated full functionality while stored at −20 ° C for a period of up to one month with virtually no degradation. The IMNPs were then conjugated to L. monocytogenes for 15 minutes, as shown in Fig. 2b–d, which allowed for subsequent magnetic trapping of the pathogen.
The performance of IMNPs was compared to commercially available Dynabeads in terms of isolation efficiency of live L. monocytogenes, according to the procedure described in section 2.3. From Fig. 2e, the commercially available antibody-coated Dynabeads anti-Listeria enabled poor recovery rates ranging from 0.2 to 1.32%, while the recovery rate of Dynabeads functionalized with BacTrace anti-Listeria antibodies was found to be higher, in the range of 2.1 to 11.7%. These isolation efficiencies correspond roughly to other previous reports,43–48 with the difference between the two being attributed to different antibodies used for bead conjugation. Indeed, it is well known that the sensitivity of any capture assay can be directly linked to the capture efficiency of the binding ligand employed, whereas BacTrace anti-Listeria antibodies seem to have higher affinity for L. monocytogenes strain HPB582. In comparison to Dynabeads, IMNPs conjugated to BacTrace anti-Listeria antibodies were significantly more efficient in capturing bacteria for all incubation times, with isolation efficiencies in the range of 18.2 to 47.1%. The small nanoparticles have faster kinetics in solution as compared to their micrometer-sized counterparts. Since the diameter of IMNPs is an order of magnitude smaller than the diameter of Dynabeads, the inherent diffusion time is lower, resulting in faster and more effective attachment on the bacteria surface.49,50 Additionally, the use of nanoparticles is more advantageous since high surface/volume ratio offers larger contact surface area for attaching antibodies and for capturing pathogens.51,52 In contrast to Dynabeads, IMNP size is an order of magnitude smaller than the size of Listeria, which allows the attachment of several tens to hundreds of IMNPs onto a bacterial cell. This should in turn yield stronger coupling between Listeria and IMNPs in comparison to a single or a few antibody bonds provided by equally sized Dynabeads. It is interesting to note that for long incubation times (120 min), the measured isolation efficiency of IMNPs decreased. This is most likely attributed to aggregation effects, which reduces artificially the number of colonies visible in the plate-culture method. This aggregation results from a time-dependent increase in the magnetic interaction in competition with repulsive forces of the electric double layer that is further increased in electrolyte salt containing media.53,54 In addition, prolonged incubation times may lead to increased nanoparticle cytotoxicity, resulting in lower bacterial counts.55
3.4. Modeling of the M-chip magnetic capture-release properties
Numerical magnetostatic simulations coupled to magnetic measurements of the magnetic field generated by the permanent magnetic plate system (see sections 2.4.1 and 2.4.2) indicate a value of about H0 = 100 kA m−1 at the center of the system and field gradients of 2.48 × 106 A m−2 and 1.21 × 106 A m−2 at the edges of the magnetic pillar array (Fig. 3a and SI Fig. S2a†). To evaluate the efficiency of the magnetic capture, we also investigated the topology of the magnetic field generated by interacting magnetic cylindrical shells (SI Fig. S2c†). As shown in Fig. 3b and SI Fig. S2b,† the presence of the pillars distorts the external magnetic field and leads to an increase of the magnetic field by approximately 100 kA m−1 at the surface of the pillar (point A) and 35 kA m−1 at the middle point (B) between neighboring pillars.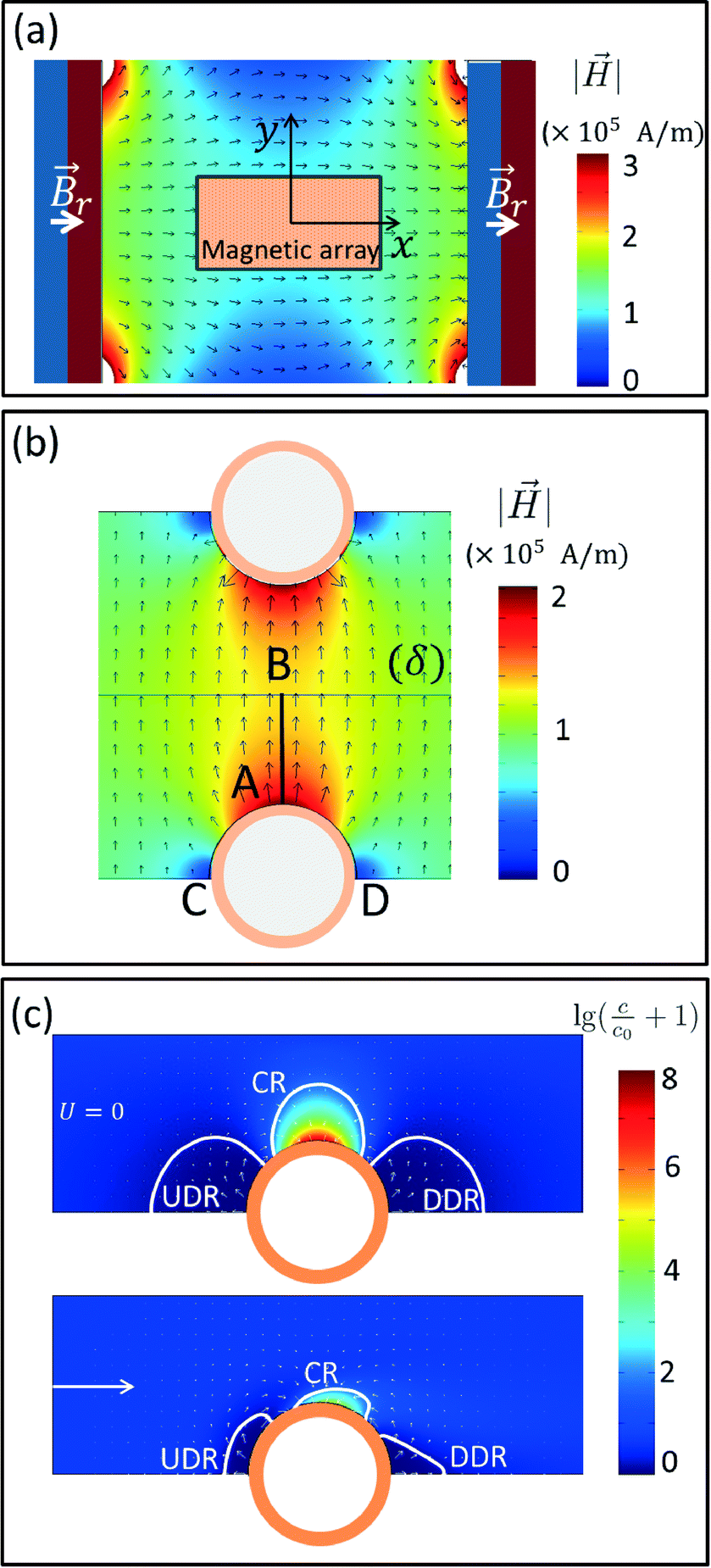 | ||
Fig. 3 Numerical simulations of the magnetic field layout driving the M-chip magnetic capture: (a) contour fill plot and normalized vector cut plane for the magnetic field generated by a pair of two strong plates magnetized through thickness at a remnant magnetic induction Br = 1.35T as indicated in the figure. The position of the magnetic chip is highlighted by the small rectangle centered at the origin of the coordinate frame xOy. (b) Zoomed view of the magnetic field at the gap between two magnetized pillars: contour fill plot represents the norm of the magnetic field while arrows indicate its orientation. (c) Numerical simulations for the magnetic capture of particles with magnetic moment μP = 4 × 10−19 A M−2 at stop flow (U = 0) and at a flow rate of 10 μL min−1 (U = 10 μm s−1) indicating the formation of a capture region (CR), an upstream-depleted region (UDR) and a downstream-depleted region (DDR). Contour fill plots represents the decimal logarithm of the quantity  (c0 – initial concentration, c – actual concentration) while small proportional arrows indicate magnetic forces acting upon individual particles. Force values in the capture region CR have a maximum value of 12.1 × 10−15 N on the surface of the pillar. (c0 – initial concentration, c – actual concentration) while small proportional arrows indicate magnetic forces acting upon individual particles. Force values in the capture region CR have a maximum value of 12.1 × 10−15 N on the surface of the pillar. | ||
For superparamagnetic particles, the intensity of the applied magnetic field is responsible for the magnetic moment 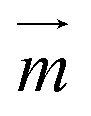 of the particles, while the magnetic field gradient induced by pillars gives the total magnetic force
of the particles, while the magnetic field gradient induced by pillars gives the total magnetic force 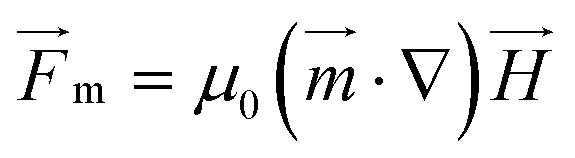 .The actual magnetic moment
.The actual magnetic moment 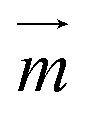 for the particles will sum up the individual magnetic moments of all magnetite nanoparticles embedded in the core (SI Fig. S2c†). The overall superparamagnetic behavior was confirmed by the major hysteresis loop measurements (SI Fig. S2c†). These results were then used together with magnetic fields from magnetostatic simulations (Fig. 3a and b and SI Fig. S2a and b†) to evaluate the magnetic moments and related magnetic forces experienced by particles. For the setup presented in this study, we estimated a maximum magnetic actuation force of FA = 12.1 fN and a maximum related magnetic velocity of UA = 12.8 = μm s−1, which represents the maximum terminal velocity of the particle in water induced only by the magnetic force FA.
for the particles will sum up the individual magnetic moments of all magnetite nanoparticles embedded in the core (SI Fig. S2c†). The overall superparamagnetic behavior was confirmed by the major hysteresis loop measurements (SI Fig. S2c†). These results were then used together with magnetic fields from magnetostatic simulations (Fig. 3a and b and SI Fig. S2a and b†) to evaluate the magnetic moments and related magnetic forces experienced by particles. For the setup presented in this study, we estimated a maximum magnetic actuation force of FA = 12.1 fN and a maximum related magnetic velocity of UA = 12.8 = μm s−1, which represents the maximum terminal velocity of the particle in water induced only by the magnetic force FA.
Finite element numerical simulations of the magnetic transport of particles in liquid flows obtained by coupling the above magnetic field landscapes and the particle–pillar interaction model (see section 2.4.3) were used to study the particle concentration field around the magnetized pillars. At stop-flow conditions (U = 0), the particle transport is given by the magnetic drift only and particles accumulate near the pillar surface. Herein, higher densities of magnetic field lines are present thus forming capture regions (CR) with highly non-uniform distributions of magnetic material (top panel of Fig. 3c). Near tangential (lateral) points we indeed observe that the particle concentration field is depleted and two regions are formed where the concentration (c) decreases below the initial concentration c0 of the solution: the upstream depleted region (UDR) and the downstream depleted region (DDR). Under these conditions of stop flow, the two regions are symmetrical with respect to the magnetic field direction and expand over distances of the same order of magnitude as the diameter of the pillars. Conversely, under continuous flow conditions (U ≠ 0), we observe an important reduction in size of all three regions, with the capture region slightly sliding clockwise on the surface of the pillar while the two depleted regions become asymmetric and deformed by the presence of the flow (bottom panel of Fig. 3c). Since the solutions of the convection-diffusion equation presented in Fig. 3c are actually steady state (after a sufficiently large interval of time), we deduce that the actual capture process evolves in two stages: (i) in a first instance, the particles flowing in the channel are continuously drifted by the magnetic field gradients toward the capture regions CR and away from depleted regions (UDR and DDR); (ii) a steady state distribution of the concentration field is achieved and the capture process actually stops. This means that the pillars will have an intrinsic capture capacity that will depend not only on the properties of the individual particles and the topology of the magnetic field landscape but also on the overall flow rate of the magnetic solution. Consequently, after pillar saturation, the concentration of the magnetic solution at the outlet is expected to reach the same initial value of the inlet. Therefore, large arrays (with large numbers of pillars) are necessary for processing of large sample volumes. Additionally, the results in Fig. 3c indicate that the two depleted regions UDR and DDR will keep particles away from the fluid stagnation points, such that the release will simply consist of flowing a resuspension liquid through the array of pillars at a sufficiently high flow rate, such that the capacity of the capture region CR is reduced to a negligible value. The huge advantage in this approach is that both sample and resuspension buffers are flown through the same inlet and outlet connection as the sample magnetic solute itself with no need of reconfiguring the chip ports and the manipulation protocol.
In the results presented in the bottom panel of Fig. 3c, an average velocity of the microfluidic flow of about 10 μm s−1 has been considered. This corresponds to a flow rate of 10 μL min−1 for the M-chip configuration used in this study. While the concentration in the capture region was lower at this flow rate value in comparison to the stop flow conditions, it still increased to at least a few thousand times to the value of the initial concentration. In practice, this increase is limited by (i) by the finite size of the beads as well as their respective packing and by (ii) the flow rate that limits the capture region to a size at which advection and diffusion drift forces equilibrate, resulting in finite capacity for magnetic capture of each pillar. Consequently, in addition to geometry and physical properties of both pillar array and magnetic particles, initial concentration of magnetic species and the total volume of processed sample should also be considered when designing and optimizing the performance of the M-chip.
In the present study, we present an application where the magnetic solute consists of bacteria covered with magnetic particles that form a composite aggregate of an approximate radius R = 1 μm. This size, with high magnetic load that can be hundred fold higher than an individual magnetic particle, renders the convection-diffusion type numerical simulation very difficult. This is due to unavoidable low diffusion constants and high Peclet numbers associated with the particle size and the magnetic load, respectively. However, despite these difficulties some interesting qualitative assessment of the efficiency of capture of these special solutes can still be made. Since the overall magnetic moment of a bacteria complex is just the sum of the individual magnetic moments of all n particles attached to its surface, the force responsible for the magnetic drift can be written simply as n × FA, with FA as defined above. It is however more difficult to trap these bacteria aggregates, since the drag forces that drift particles along the liquid flow streamlines are now about ten times larger, reflecting the difference in size between the bacteria and magnetic nanoparticle (bacteria are approximately ten-fold larger than the magnetic particles). Under these conditions in the present magnetic setup, we evaluate that the highest magnetic velocity a bacteria-particle aggregate can achieve corresponds to n × 1.2 μm s−1 where n is the number of magnetic particles conjugated to the bacteria surface. Therefore, in order to achieve efficient capture at a flow rate of about 100 μL min−1 (that is 1 mm s−1 average velocity of the liquid flow), about few hundreds of magnetic particles have to be attached to one bacterial cell. A simple condition that the average drag in the fluid flow and the magnetic terminal velocity of the particle be identical has been used in the above reasoning. However, despite its simplicity, this actually gives rather satisfactory results at least qualitatively as demonstrated in the subsequent experimental sections.
3.5. Experimental evaluation of M-chip performance
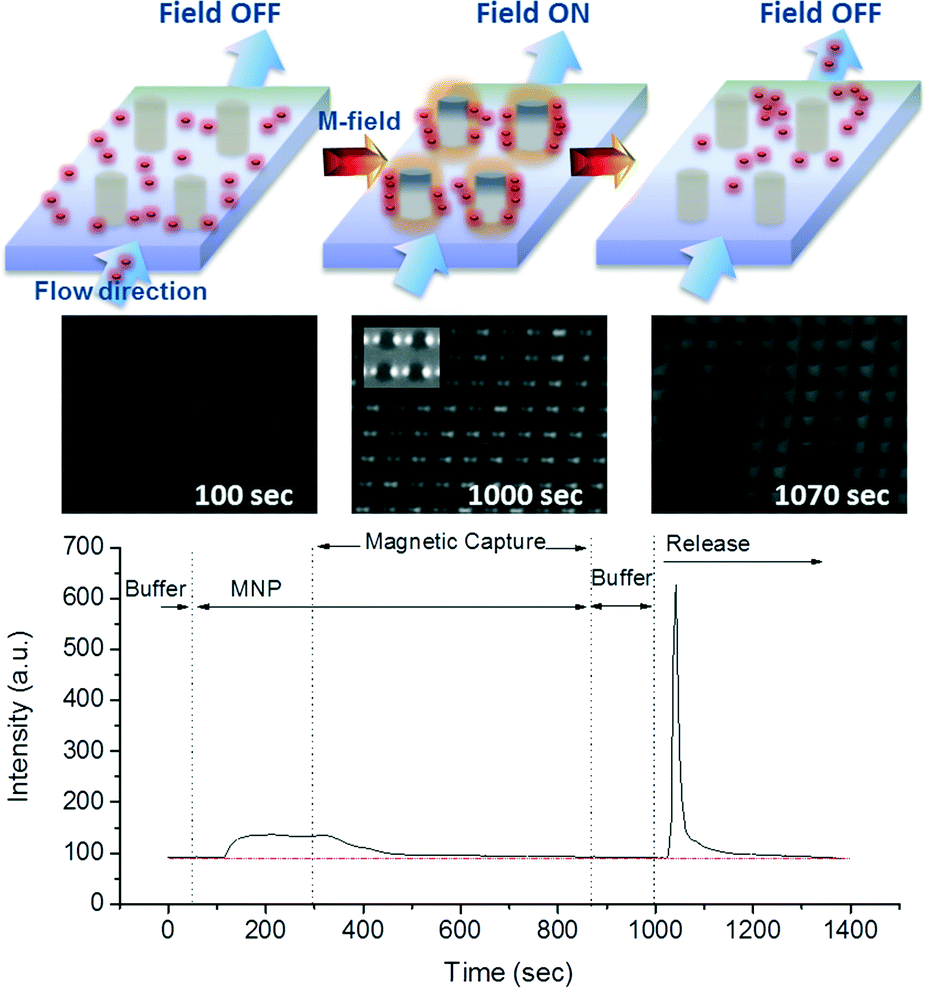 | ||
| Fig. 4 Kinetic capture and release profile of the magnetic nanoparticles using the M-chip obtained from absorption intensity measurements and the corresponding illustrations and fluorescence images of the chip showing each step in the process. | ||
At time 0, a buffer sample is allowed to flow through the device and the baseline absorption spectrum is measured at the outlet. Following the recording of a baseline buffer signal, the solution containing MNPs is allowed to flow through the chip at 100 μl min−1. For visualisation purposes, the MNP concentration of 50 μg ml−1 was used. The corresponding absorption spectrum is shown to increase until it reaches saturation. It is important to note that the delay between the injection of the solution and the change in the recorded signal is attributed to the length of the tubing and the flow rate, while the gradual signal change is due to Taylor dispersion inside the tubes. Subsequently, as the magnetic field is applied and MNPs become captured in the chamber of the chip, the concentration of the particles in the outlet tubing decreases, and the signal rapidly reaches the baseline level. During the 10 min of magnetic capture time (i.e. 1 ml total volume), capture efficiency remained greater than 95%, indicating that the chip does not saturate. Following the MNP capture, a wash step is performed, during which the signal remains stable, indicating no loss of captured particles occurs. Finally, the flow rate is increased to 250 μl min−1 and particles are released by removing the magnetic field, as shown by the abrupt increase in the signal. The release is found to be highly efficient, and the signal returns to the baseline position after about 5 min. By computing the integral of the area under the curve, we obtain recovery rate of 82% when releasing in a volume of 250 μl, which translates to a MNP concentration factor of 3.3. A slight increase in recovery rate efficiency is obtained by increasing the released volume to 500 μl, corresponding to 91%, but at the expense of a MNP concentration factor which decreases to 1.8. These results demonstrate that the developed M-chip can both capture and release very efficiently the MNPs.
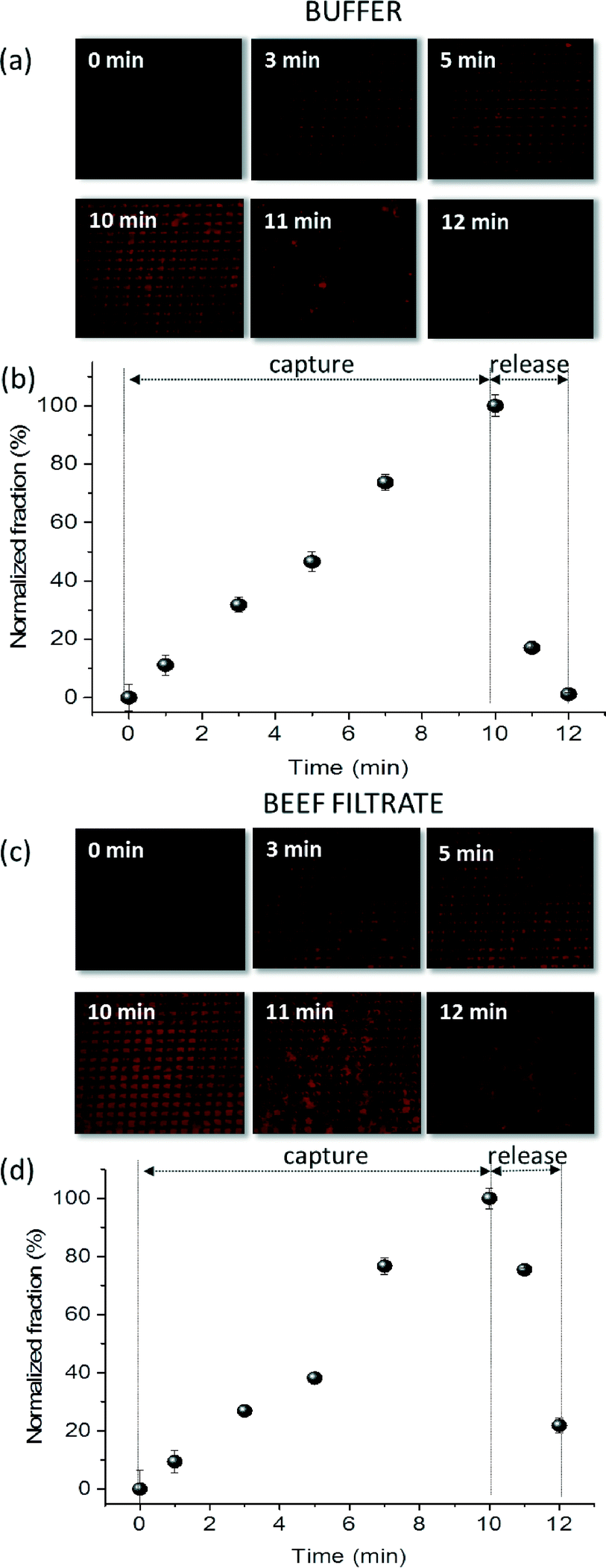 | ||
| Fig. 5 Fluorescence images (a and b) and normalized fluorescence intensity fraction plots (c and d) showing capture and release of stained dead Listeria-IMNP complex on a passivated M-chip in (a and c) buffer and (b and d) beef filtrate for the 104 cfu mL−1 concentration. At time 0, no magnetic field is applied and florescence intensity is at 0. Following the application of the magnetic field, the intensity increases for the entire duration of the capture experiment. At time t = 10 min, the intensity is at its maximum. Finally, as the magnetic field is removed, during the release-step, the intensity decreases all the way down to its initial value for buffer medium. Release of species captured in beef medium is less efficient, the fluorescent intensity decreasing to the 22nd percentile after two minutes. | ||
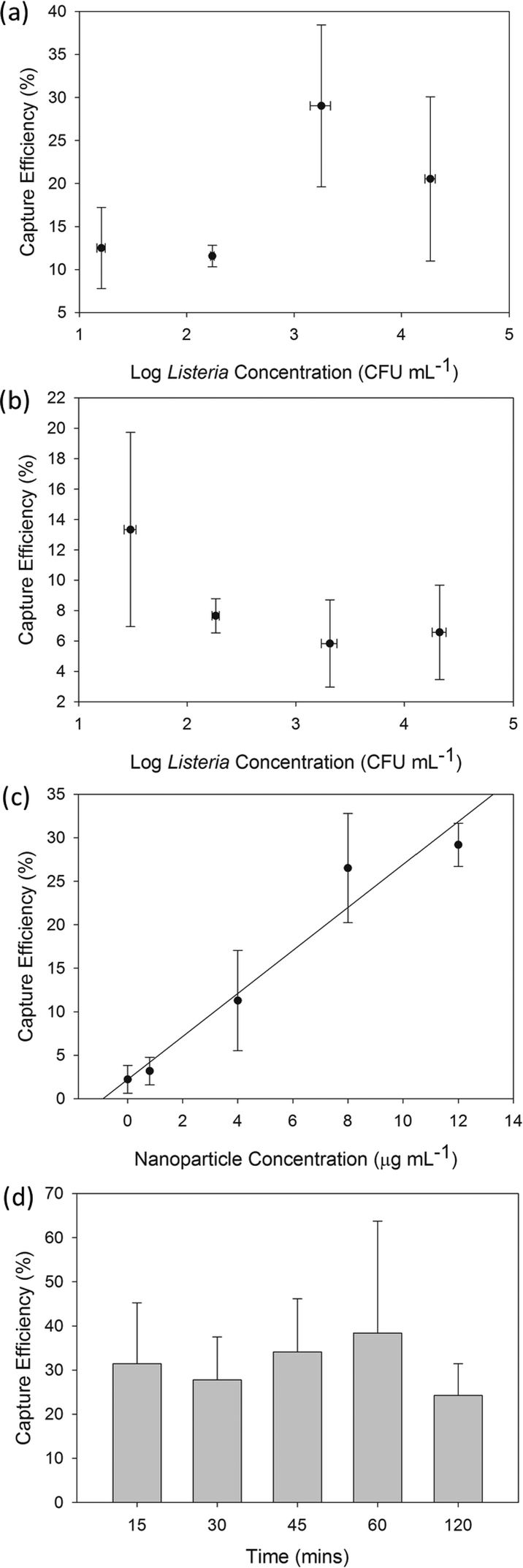 | ||
| Fig. 6 Capture efficiency of live L. monocytogenes-IMNP complex using M-chip platform in: (a) buffer and (b) beef filtrate as a function of Listeria concentration for constant nanoparticle concentration of 4 μg ml−1 and 15 min bacteria-particle pre-capture mixing/incubation time; (c) buffer as a function of varying nanoparticle concentration for constant Listeria-IMNP incubation time of 15 min and constant initial listeria concentration of approximately 1400 cfu ml−1. (d) Buffer as a function of Listeria-IMNP incubation time for constant initial listeria concentration of approximately 2300 cfu ml−1 and constant nanoparticle concentration of 4 μg ml−1. Error bars represent standard deviation including taylor expansion of error propagation. Statistical analysis was performed using student t-test; n = 3 samples for all experimental data points. | ||
From previous sections, it is observed that obtained capture efficiency values using live L. monocytogenes are lower than the M-chip's capacity to capture magnetic species (see section 3.5.1). The actual capture efficiency of the complete system depends on the ability of the nanoparticles to coat the bacterial surface, which in turn depends on the nanoparticle concentration, incubation time, as well as avidity and specificity of antibodies. In our experiments, we used a 4 μg ml−1 nanoparticle concentration and limited the incubation time to 15 minutes. Furthermore, as antibodies have varying degrees of target specificity and avidity, it is expected that not all Listeria in the sample will be conjugated which will consequently impact the overall capture efficiency.11 Nevertheless, it is noteworthy that even for the lowest initial Listeria sample concentration of approximately 10 cfu ml−1, we have been able to capture, release, culture and provide a positive identification of the target cells from 1 ml sample volumes.
The recovery rate for L. monocytogenes was subsequently investigated in a more complex sample matrix. Fig. 6b plots the recovery rate as a function of initial Listeria sample concentration suspended in beef filtrate. In contrast to buffer-based experiments, the capture efficiency is lower in these complex samples and appears to be similar for all Listeria concentrations with values around 10% ± 3%. This decrease in the recovery rate can be attributed to increased non-specific absorption of particulates, proteins and other molecules present in the beef filtrate. The abundance of these species in the sample can also cause steric hindrance and inhibit the Listeria-IMNP complexation.57 Additionally, the presence of particulates can possibly result in increased agglomeration of Listeria, resulting in lower count numbers of colony forming units. Finally, it is expected that some bacteria can attach to debris present in the solution, and can then escape without being captured by the magnetic field. However, even with a capture efficiency of 10%, we have been able to capture and identify the target cells using plate-culture method with a very low initial sample concentration of approximately 10 cfu ml−1.
Additionally, the effects of the incubation time on the recovery rate were explored in order to determine the optimum operating conditions for our system. Fig. 6d shows the plot of the capture efficiency as a function of incubation time. We observed that the increase in the incubation time beyond 15 min did not have a significant effect on the capture efficiency. This demonstrates that the developed IMNPs can attach very rapidly (less than 15 min) to the bacteria. Also, it is noteworthy that, at long incubation times (120 min), nanoparticle cytotoxicity can increase and agglomeration of Listeria-IMNP complexes could become more significant resulting in underestimated count numbers. Indeed, for long incubation times, we have observed the presence of agglomerates of dead Listeria-IMNP complex (see SI Fig. S5† for an example) which may be due to inter-particle attraction, as well as the crosslinking that would occur between bacteria, antibodies, and IMNPs. Therefore, for the optimum system performance a short incubation of 15 to 30 min is preferred in order to decrease the overall sample preparation time while allowing efficient pathogen concentration.
| V (ml) | C i (cfu ml−1) | SD | RR (%) | CF | ||
|---|---|---|---|---|---|---|
| 1 | Sample in | 1 | 173 | 55 | 0.27 | |
| Sample out | 0.275 | 47 | 11 | 7.54 | ||
| 2 | Sample in | 10 | 143 | 45 | 2.80 | |
| Sample out | 0.275 | 400 | 80 | 7.67 | ||
4. Conclusions
In this manuscript, we have demonstrated a flexible, customizable and low-cost 3D magnetic capture and release platform applicable to various targets in micron and sub-micron sizes for which traditional capture methodologies have limited efficiency. The magnetic capture chip can be easily fabricated and integrated in low-cost thermoplastics suitable for mass-production. As a proof of principle, we have demonstrated rapid and efficient capture and release of L. monocytogenes conjugated to IMNPs using an M-chip device enabling switchable high magnetic field gradient generation. The fabricated nanoparticles were optimized to achieve the desired magnetic moment, while the surface functionalization was tailored to allow efficient capture antibody immobilization. The integration, validation and further optimization of the capture and release protocol was demonstrated using both, dead and live L. monocytogenes through fluorescence microscopy and plate-culture methods. Numerical simulations and related theoretical analysis presented here were used to predict the distribution of magnetic species flowing around magnetized pillars and extract relevant parameters for efficient capture and prompt release. Magnetic pillars composed of a non-magnetic cylindrical core and a magnetically soft shell allowed precise localization and control of magnetic capture regions with relatively low external magnetizing fields. Moreover, we have demonstrated that, by adjusting the orientation of the external magnetizing field relative to the flow such that the depleted regions are coincident with upstream and downstream stagnant points, the release of captured magnetic species is greatly improved. The capture efficiency of the chip was found to vary as a function of bacterial cell to nanoparticle concentration ratio. The maximum capture efficiency was found to be approximately 30% and the 24-hour plate-culture method allowed detection of an initial sample concentration of ~10 cfu ml−1 in 1 ml sample volume. The device was also very efficient in concentrating the sample from a 10 ml initial volume. Specifically, a three-fold increase in concentration efficiency was achieved in only 17 minutes, demonstrating the suitability of the system for food safety applications. In addition, flexible design and low-cost fabrication processes allow for rapid sample preparation for applications beyond food and water safety, including point-of-care diagnostics. Ongoing work is focusing on the enhancement of capture efficiency by investigating aptamer-based nanoparticle surface functionalization and the integration of the on-chip mixing and incubation chamber coupled with real-time PCR.Acknowledgements
This work was supported by Health Canada and National Research Council of Canada through the GRDI-funded program “Strengthening Food and Water Safety in Canada through an Integrated Federal Genomics Initiative”. We would like to thank the members of Dr. Franco Pagotto's lab for providing strain of L. monocytogenes. We thank our colleague Gaétan Veilleux for technical assistance and useful discussion. We would also like to thank Sandeep Tamber and Jennifer Ronholm for critical review.References
- K. Ma, Y. Deng, Y. Bai, D. Xu, E. Chen, H. Wu, B. Li and L. Gao, Food Control, 2014, 42, 87–93 CrossRef CAS.
- A. K. Bhunia, Future Microbiol., 2014, 9, 935–946 CrossRef CAS PubMed.
- R. L. Scharff, J. Food Prot., 2012, 75, 123–131 CrossRef PubMed.
- T. McLinden, J. Sargeant, M. Thomas, A. Papadopoulos and A. Fazil, BMC Public Health, 2014, 14, 509 CrossRef PubMed.
- E. Scallan, R. M. Hoekstra, F. J. Angulo, R. V. Tauxe, M. A. Widdowson, S. L. Roy, J. L. Jones and P. M. Griffin, Emerging Infect. Dis., 2011, 17, 7–15 CrossRef PubMed.
- N. Balaban and A. Rasooly, Int. J. Food Microbiol., 2000, 61, 1–10 CrossRef CAS PubMed.
- V. Ramaswamy, V. M. Cresence, J. S. Rejitha, M. U. Lekshmi, K. Dharsana, S. P. Prasad and H. M. Vijila, J. Microbiol., Immunol. Infect., 2007, 40, 4 CAS.
- S. H. Suh and L. A. Jaykus, J. Biotechnol., 2013, 167, 454–461 CrossRef CAS PubMed.
- S. L. Gottlieb, E. C. Newbern, P. M. Griffin, L. M. Graves, R. M. Hoekstra, N. L. Baker, S. B. Hunter, K. G. Holt, F. Ramsey, M. Head, P. Levine, G. Johnson, D. Schoonmaker-Bopp, V. Reddy, L. Kornstein, M. Gerwel, J. Nsubuga, L. Edwards, S. Stonecipher, S. Hurd, D. Austin, M. A. Jefferson, S. D. Young, K. Hise, E. D. Chernak and J. Sobel, Clin. Infect. Dis., 2006, 42, 29–36 CrossRef PubMed.
- Y. Chen, W. H. Ross, V. N. Scott and D. E. Gombas, J. Food Prot., 2003, 66, 570–577 CrossRef PubMed.
- S. H. Suh and L.-A. Jaykus, J. Biotechnol., 2013, 167, 454–461 CrossRef CAS PubMed.
- S. W. Ruban, R. Sharada and T. G. Banday, J. Food Technol., 2011, 9, 106–111 CrossRef.
- Y. H. Hui, S. A. Sattar and W.-K. Nip, Foodborne Disease Handbook: Volume 2: Viruses: Parasites: Pathogens, and HACCP, CRC Press, 2000 Search PubMed.
- H. P. Dwivedi and L.-A. Jaykus, Crit. Rev. Microbiol., 2011, 37, 40–63 CrossRef CAS PubMed.
- B. Brehm-Stecher, C. Young, L.-A. Jaykus and M. L. Tortorello, J. Food Prot., 2009, 72, 1774–1789 CrossRef CAS PubMed.
- M. A. M. Gijs, F. Lacharme and U. Lehmann, Chem. Rev., 2010, 110, 1518–1563 CrossRef CAS PubMed.
- Q. Ramadan and M. M. Gijs, Microfluid. Nanofluid., 2012, 13, 529–542 CrossRef CAS.
- D. W. Inglis, R. Riehn, J. C. Sturm and R. H. Austin, J. Appl. Phys., 2006, 99, 08K101 CrossRef CAS.
- N. Pamme and C. Wilhelm, Lab Chip, 2006, 6, 974–980 RSC.
- D. Robert, N. Pamme, H. Conjeaud, F. Gazeau, A. Iles and C. Wilhelm, Lab Chip, 2011, 11, 1902–1910 RSC.
- V. I. Furdui and D. J. Harrison, Lab Chip, 2004, 4, 614–618 RSC.
- K. Smistrup, M. Bu, A. Wolff, H. Bruus and M. F. Hansen, Microfluid. Nanofluid., 2008, 4, 565–573 CrossRef CAS.
- D. W. Inglis, R. Riehn, R. H. Austin and J. C. Sturm, Appl. Phys. Lett., 2004, 85, 5093–5095 CrossRef CAS.
- J. D. Adams, U. Kim and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18165–18170 CrossRef CAS PubMed.
- S. Kim, S.-I. Han, M.-J. Park, C.-W. Jeon, Y.-D. Joo, I.-H. Choi and K.-H. Han, Anal. Chem., 2013, 85, 2779–2786 CrossRef CAS PubMed.
- H. Chen, M. D. Kaminski, A. D. Ebner, J. A. Ritter and A. J. Rosengart, J. Magn. Magn. Mater., 2007, 313, 127–134 CrossRef CAS.
- E. Mirowski, J. Moreland, S. E. Russek and M. J. Donahue, Appl. Phys. Lett., 2004, 84, 1786–1788 CrossRef CAS.
- K. Smistrup, B. Kjeldsen, J. Reimers, M. Dufva, J. Petersen and M. F. Hansen, Lab Chip, 2005, 5, 1315–1319 RSC.
- Y.-J. Liu, S.-S. Guo, Z.-L. Zhang, W.-H. Huang, D. Baigl, M. Xie, Y. Chen and D.-W. Pang, Electrophoresis, 2007, 28, 4713–4722 CrossRef CAS PubMed.
- T. Deng, M. Prentiss and G. M. Whitesides, Appl. Phys. Lett., 2002, 80, 461–463 CrossRef CAS.
- X. Yu, R. He, S. Li, B. Cai, L. Zhao, L. Liao, W. Liu, Q. Zeng, H. Wang, S.-S. Guo and X.-Z. Zhao, Small, 2013, 9, 3895–3901 CrossRef CAS PubMed.
- K. Smistrup, T. Lund-Olesen, M. F. Hansen and P. T. Tang, J. Appl. Phys., 2006, 99, 08P102 CrossRef.
- Q. Ramadan, V. Samper, D. P. Poenar and C. Yu, Biosens. Bioelectron., 2006, 21, 1693–1702 CrossRef CAS PubMed.
- A.-E. Saliba, L. Saias, E. Psychari, N. Minc, D. Simon, F.-C. Bidard, C. Mathiot, J.-Y. Pierga, V. Fraisier, J. Salamero, V. Saada, F. Farace, P. Vielh, L. Malaquin and J.-L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14524–14529 CrossRef CAS PubMed.
- S. Khashan, A. Alazzam and E. Furlani, Sci. Rep., 2014, 4 Search PubMed.
- L. Malic, K. Morton, L. Clime and T. Veres, Lab Chip, 2013, 13, 798–810 RSC.
- E. Roy, G. Stewart, M. Mounier, L. Malic, R. Peytavi, L. Clime, M. Madou, M. Bossinot, M. G. Bergeron and T. Veres, Lab Chip, 2015, 15, 406–416 RSC.
- J. E. Sadler, D. S. Szumski, A. Kierzkowska, S. R. Catarelli, K. Stella, R. J. Nichols, M. H. Fonticelli, G. Benitez, B. Blum, R. C. Salvarezza and W. Schwarzacher, Phys. Chem. Chem. Phys., 2011, 13, 17987–17993 RSC.
- H. Kim, J. Ge, J. Kim, S.-E. Choi, H. Lee, H. Lee, W. Park, Y. Yin and S. Kwon, Nat. Photonics, 2009, 3, 534–540 CrossRef CAS.
- H. Danan, A. Herr and A. J. P. Meyer, J. Appl. Phys., 1968, 39, 669–670 CrossRef CAS.
- L. Clime, X. Hoa, N. Corneau, K. Morton, C. Luebbert, M. Mounier, D. Brassard, M. Geissler, S. Bidawid, J. Farber and T. Veres, Biomed. Microdevices, 2015, 17, 1–14 CrossRef CAS PubMed.
- S. A. Khashan and E. P. Furlani, J. Phys. D: Appl. Phys., 2013, 46, 125002 CrossRef.
- A. C. Fluit, R. Torensma, M. J. Visser, C. J. Aarsman, M. J. Poppelier, B. H. Keller, P. Klapwijk and J. Verhoef, Appl. Environ. Microbiol., 1993, 59, 1289–1293 CAS.
- J. A. Hudson, R. J. Lake, M. G. Savill, P. Scholes and R. E. McCormick, J. Appl. Microbiol., 2001, 90, 614–621 CrossRef CAS PubMed.
- Y. S. Jung, J. F. Frank, R. E. Brackett and J. Chen, J. Food Prot., 2003, 66, 237–241 CrossRef CAS PubMed.
- Y. S. Jung, J. F. Frank and R. E. Brackett, J. Food Prot., 2003, 66, 1283–1287 CrossRef CAS PubMed.
- M. Mendonça, N. L. Conrad, F. R. Conceição, Â. N. Moreira, W. P. da Silva, J. A. G. Aleixo and A. K. Bhunia, BMC Microbiol., 2012, 12, 275–275 CrossRef PubMed.
- O. K. Koo, A. Aroonnual and A. K. Bhunia, J. Appl. Microbiol., 2011, 111, 93–104 CrossRef CAS PubMed.
- M. Varshney, L. Yang, X.-L. Su and Y. Li, J. Food Prot., 2005, 68, 1804–1811 CrossRef CAS PubMed.
- X. Zhao, L. R. Hilliard, S. J. Mechery, Y. Wang, R. P. Bagwe, S. Jin and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15027–15032 CrossRef CAS PubMed.
- K. El-Boubbou, C. Gruden and X. Huang, J. Am. Chem. Soc., 2007, 129, 13392–13393 CrossRef CAS PubMed.
- H. Gu, K. Xu, C. Xu and B. Xu, Chem. Commun., 2006, 941–949, 10.1039/B514130C.
- D. Rosická and J. Šembera, Nanoscale Res. Lett., 2013, 8, 1–9 CrossRef PubMed.
- P. Bradford, Ph.D. Thesis, University of Birmingham, 2012 Search PubMed.
- S. Naqvi, M. Samim, M. Z. Abdin, F. J. Ahmed, A. N. Maitra, C. K. Prashant and A. K. Dinda, Int. J. Nanomed., 2010, 5, 983–989 CrossRef CAS PubMed.
- E. Kaclíková, T. Kuchta, H. Kay and D. Gray, J. Microbiol. Methods, 2001, 46, 63–67 CrossRef.
- N. Xia, T. P. Hunt, B. T. Mayers, E. Alsberg, G. M. Whitesides, R. M. Westervelt and D. E. Ingber, Biomed. Microdevices, 2006, 8, 299–308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Addendum to experimental and results sections. See DOI: 10.1039/c5lc00852b |
| This journal is © The Royal Society of Chemistry 2015 |