
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast electrically assisted regeneration of on-chip SERS substrates†
T.-A.
Meier
a,
E.
Poehler
a,
F.
Kemper
b,
O.
Pabst
b,
H.-G.
Jahnke
c,
E.
Beckert
b,
A.
Robitzki
c and
D.
Belder
*a
aInstitut für Analytische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: belder@uni-leipzig.de; Fax: +49 341 97 36115; Tel: +49 341 97 36091
bFraunhofer-Institut für Angewandte Optik und Feinmechanik (IOF), Albert-Einstein-Straße 7, 07745 Jena, Germany
cInstitut für Molekularbiologisch-Biochemische Prozesstechnik, Universität Leipzig, Deutscher Platz 5, 04103 Leipzig, Germany
First published on 27th May 2015
Abstract
A microfluidic chip approach utilising integrated electrically connected stationary SERS targets based on inkjet-printed silver nanoparticles is presented. It enables multiple interference-free consecutive surface-enhanced Raman measurements inside chip channels by electrically assisted regeneration of the stationary SERS substrate. Thereby it circumvents common adsorption and memory effect problems associated with stationary SERS targets allowing multiple consecutive measurements in a continuous-flow system.
Introduction
Within the past ten years surface-enhanced Raman spectroscopy (SERS) has become increasingly important within the field of microfluidics due to its potential for label-free, non-destructive and sensitive on-chip detection.1 In addition, it provides structural information making it an attractive tool for usage in a wide application area. Fields of interest are chemical2–4 and biological5–7 analyses as well as detection in separation techniques8–10 and reaction monitoring.11Traditionally, suspended nanoparticles as SERS substrates have played a key role in microfluidic SERS measurements. They can be used straightforwardly in both continuous12 and segmented-flow13–16 systems.
The use of particle suspensions to promote SERS-detection in microfluidics, however, can be problematic as it can contaminate or even clog microfluidic channels13,17 and it can also interfere negatively with downstream processes. Stationary substrates such as roughed silver surfaces18 or immobilized nanoparticles19,20 provide interesting alternatives with the additional promise of being more suitable for quantitative analytics.21,22
The utilisation of stationary substrates in flow-through systems is, however, hampered by the strong adsorption of analyte molecules to the SERS substrate, causing a so-called memory effect.23 This often irreversible adsorption of analytes to the SERS-active surface causes sample carry-over and impedes the broad application of SERS as detection technique in advanced lab-on-a-chip applications such as flow synthesis24–27 or separation techniques.28 Especially in chip-based separation techniques there is a great demand for label-free detection methods.29–32 The corresponding application of SERS could thus be a break-through in chip electrophoresis, chip chromatography or flow chemistry. A prerequisite, however, is a fast desorption of molecules from such immobile SERS-active substrates present in integrated detection flow cells. This is necessary to avoid both, signal carry-over and a potential blockade of the SERS-active spots.4 To the best of our knowledge a necessary fast and straightforward regeneration of SERS surfaces immobilised in microfluidic devices has not yet been described. Literature on conventional macroscopic flow cells, however, indicates that this could be achieved by electrically assisted desorption.33–36
We present a microfluidic system using electrically assisted substrate regeneration for inline SERS detection in chip-based systems. A chip-integrated electrically regenerable SERS substrate is used giving access to sensitive interference-free Raman spectroscopic monitoring of different successively transported compounds in flow (see Fig. 1).
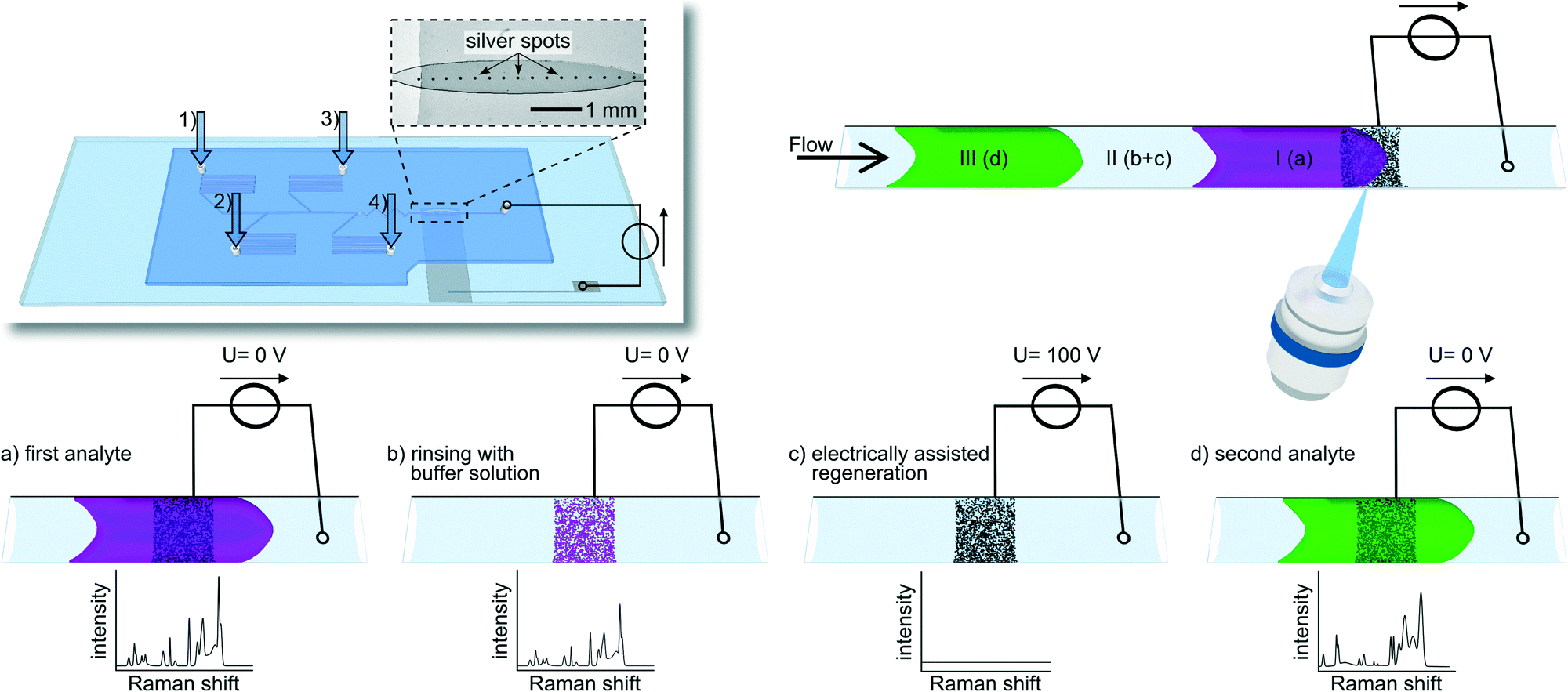 | ||
| Fig. 1 Schematic drawing of the experimental set-up and the process for electrochemically assisted regeneration of integrated SERS substrates in a microfluidic chip. The insert in the upper left depicts the microfluidic chip with four inlets (1 – flushing buffer, 2 – malachite green, 3 – brilliant green and 4 – crystal violet) and an outlet channel connected to a power supply which defines the potential at the ITO/silver electrode. A light microscopic photograph of the SERS-active silver dots printed on an ITO strip substrates is shown in the enlarged area. In the upper right part a microfluidic channel, containing two different analyte plugs (I + III) and the buffer (II), as well as the detection area with a silver spot on an ITO strip is depicted. The actual regeneration process is schematically sketched below. If an analyte passes the detection zone a strong SERS spectrum is obtained (a). After the analyte has passed the detection area, traces of the analyte still reside at the silver surfaces with a remaining strong Raman signal withstanding subsequent rinsing steps (b). The application of a high electrical potential induces a fast removal of the analyte (c) allowing to detect a following compound zone (d). | ||
Experimental section
Chemicals
MicroChem SU-8 2050 polymer and developer were purchased from Micro Resist Technology (Germany) and Sylgard 184 Polydimethylsiloxane (PDMS) from Dow Corning (Germany). 2-Propanol was purchased from Roth (Germany). Crystal violet (CV), brilliant green (BG), malachite green (MG) and phosphoric acid were obtained from Sigma Aldrich (Germany). Suntronic EMD5603 silver ink was purchased from Sun Chemical (USA). Purified water was used as solvent.ITO/silver electrode
The bottom layer of the microfluidic chip, containing an indium-tin-oxide (ITO)/silver electrode was fabricated using a commercial microscope glass slide (Carl Roth, Germany). On top, a 5 mm broad and 500 nm thick strip of ITO was sputtered (CREAMET® 500, Creavac, Germany) approximately 2 cm parallel shifted from the short edge.Orthogonally, a row of 100 μm broad silver spots was inkjet-printed in the middle of the ITO strip (see Fig. 1). Printing was performed using a commercial inkjet deposition system (Omnijet 100, Unijet, South Korea) equipped with printing cartridges that exhibit a nominal droplet volume of 10 pL (DMC-11610, Dimatix Fujifilm, USA). Silver structures were printed using commercial silver nanoparticle dispersion in an ethanol/ethanediol mixture with a solid content of 20 wt% and particle diameters of 30 to 50 nm. After printing, the structures were partially sintered in a convection oven and used for microfluidic chip fabrication (see ESI S1†).
Microfluidic setup
Microfluidic streams were applied using neMESYS high precision syringe pumps (Cetoni, Germany) equipped with gastight syringes (Hamilton, Switzerland) linked to the chip inlets with Teflon tubes (Supelco, USA). The microfluidic structure itself consists of four feeding channels with pressure-stabilising serpentines. Downstream the fluids are lead through a zigzag-shaped mixing structure before flowing over the printed silver ink dots. All channels were approximately 150 μm wide and 100 μm high. As an exception, the area containing the dots was oval-formed with a maximum width of 650 μm. A schematic sketch of the microfluidic system can be seen in Fig. 1. The first of the four channels is used for flushing with phosphate buffer solution (pH = 3, 22.89 mM), while the other three provide the model analyte solution streams from crystal violet (1 × 10−5 mol L−1) brilliant green (1 × 10−4 mol L−1) and malachite green (5 × 10−5 mol L−1). All solutions were pumped with 5 μL min−1.Electronic circuit
The outlet tube of the microfluidic chip was equipped with an in-house developed flow-through platinum electrode.37 This electrode was connected to the cathode of a power supply (HCV 35–6500, F.u.G. Elektronik, Germany). The anode was connected to the ITO/silver electrode imprinted on the bottom glass slide (Fig. 1). A potential of 100 V, resulting in a current of 44 μA, was applied short-timed for substrate cleaning purpose.Raman measurements
For SERS measurements the microfluidic chip was placed onto an inverted IX71 epifluorescence microscope (Olympus, Japan) which was part of a modular confocal Raman system. The system was equipped with a 473 nm laser (Cobolt, Sweden) as excitation light source, with the laser beam passing a double neutral density filter wheel for intensity tuning. Excitation intensity was kept at 0.5 mW for all experiments. The laser beam was focussed by a 40-fold objective (Olympus, Japan) onto the individual silver spots to obtain SERS signals. The scattered light passes an edge filter (473 nm), which is eliminating Rayleigh and anti-Stokes scattering, and a 150 μm wide entrance slit. Raman measurements were taken with an Acton SP2750 monochromator (Princeton instruments, USA) using a 600 lines mm−1 grating and a peltier-cooled EM-CCD camera (ProEM 1600, Princeton instruments, USA) as detector. Acquisition time in all experiments was 1000 ms.Results and discussion
The setup described above was tested by acquiring SERS spectra from model compounds. We investigated if SERS substrates which are contaminated with adsorbed analytes can be regenerated by applying electrical potentials via an integrated ITO electrode. For that purpose crystal violet was first flushed through the chip including the silver dots. When the analyte solution approaches the detection area with the immobilised SERS substrates this was monitored by a strong increase in the respective Raman signals. These signals, however, did not disappear if the analyte solution was removed from the channel. Even extensive rinsing with buffer solution caused only little decrease in signal intensities of only approximately −0.16% per s (see ESI S2†). While rinsing steps were not effective for analyte desorption from the SERS substrates this could be promoted by applying an electric potential. When a voltage of 100 V was applied between the SERS substrate and the counter electrode, a fast signal intensity decrease occurs, denoting a half-life period of 4.6 s. This phenomenon is visualised in Fig. 2. | ||
| Fig. 2 Decreasing intensity of the SERS spectra of a 10 μM crystal violet solution on a silver spot while potential (100 V, 44 μA) and buffer flow (phosphate buffer, pH = 3, 22.89 mM, 5 μL min−1) is applied. The dashed box depicts the intensity of the indicator Raman band at 1585 cm−1. | ||
After electrical regeneration the substrates provided SERS-properties equal to fresh substrates which was demonstrated by repeated flushing with crystal violet (see Fig. 3). The deviations of the absolute signal intensities are quite high (RSE = 33%) as shown in Fig. 3. As similar results are achieved if several fresh substrates are compared (RSE = 40%, n = 5, ESI S3†) the strong deviations in absolute intensities can be attributed to the inhomogeneity of the sintered silver particle layer. This could in principle be overcome using better defined targets such as engineered nanostructures38,39 allowing for a more quantitative analysis.
 | ||
| Fig. 3 Consecutively acquired SERS spectra of clean silver substrates (grey lines) and substrates rinsed with crystal violet solution (black lines). In the dashed box the mean intensity of the crystal violet indicator band (1585 cm−1) of fresh/cleaned and used substrates are depicted (n = 3). | ||
In order to investigate the electrical regeneration process a bit closer we studied the influence of the applied potentials and electrical currents. For this purpose the voltages were varied between 0 and 120 V in a corresponding series of experiments. It was found that an increase in voltage correlates with an accelerated regeneration speed (see ESI S2†). We also observed a degradation of the ITO surface above 100 V could, which could be circumvented by using alternative conductive sub-layers such as sputtered platinum. As the effective applied voltages of around 100 V are far beyond any relevant electrochemical potential including that for the decomposition of the solvent water, it appears that analyte specific electrochemical33–36,40 processes are less relevant. Although the actual desorbing mechanism is still unclear initial indications suggest that, electrolytic decomposition of the solvent and the corresponding generation of reactive and convection promoting gas at the surface supports the desorption. This assumption is supported by the finding that an increase in current at constant voltages, using buffer solutions of different conductivity, accelerates substrate regeneration (see ESI S4†). The regeneration of the SERS substrate, or more specifically the removal of the analytes from the surface, could also be monitored via fluorescence microscopy. This was shown for crystal violet which showed strong decrease of emission if applying an electric potential as above (see ESI S5†).
The presented system reveals great potential for the detection of different analytes in flow applications such as separations or segmented flow. In order to study if this can, in principle, be achieved with the presented approach, three different analytes, crystal violet (1 × 10−5 mol L−1) brilliant green (1 × 10−4 mol L−1) and malachite green (5 × 10−5 mol L−1), were sequentially flushed over the target. In the aftermath of every injection, 5 minutes flushing with phosphate buffer solution was performed to monitor the memory effect in the absence of an applied potential. For monitoring an indicator wavenumber at 1585 cm−1 was chosen, which was present in spectra of all three substances. As depicted in Fig. 4 the rates of decrease in intensity were rather low (BG: −0.11% per s; MG: −0.25% per s; CV: −0.16% per s) as expected. If we apply an electrical potential of 100 V to the SERS substrate we find an immediate drop of intensity (t1/2(BG) = 23.2 s, t1/2(MG) = 13.4 s, t1/2(CV) = 4.6 s), for every analyte. This demonstrates the feasibility to detect different compounds in a microflow process with the same regenerable SERS target.
 | ||
| Fig. 4 Intensity of the indicator Raman band (1585 cm−1) of three consecutively injected different dyes during the cleaning process. Only slow decrease can be observed while flushing with buffer solution, which is dramatically increased by applying high voltage. This causes substrate regeneration in only a few seconds. | ||
Conclusions
We introduce an approach to perform multiple SERS measurements inside a single microfluidic device. A PDMS/glass hybrid-chip was built using moulding techniques embedding contactable silver spots as stationary SERS substrates. With the aid of integrated electrodes contacting the silver substrate electrical potentials was applied to promote analyte desorption. This led to fast and effective electrical substrate regeneration. As a proof-of-concept this approach allowed to investigate three different analytes flowed in a sequential manner over the same SERS substrate avoiding common memory effects.Acknowledgements
Support by the Deutsche Forschungsgesellschaft (DFG grant number: INST 268/231-1 FUGG) is gratefully acknowledged.References
- A. F. Chrimes, K. Khoshmanesh, P. R. Stoddart, A. Mitchell and K. Kalantar-zadeh, Chem. Soc. Rev., 2013, 42, 5880–5906 RSC.
- D. Cialla, A. März, R. Böhme, F. Theil, K. Weber, M. Schmitt and J. Popp, Anal. Bioanal. Chem., 2012, 403, 27–54 CrossRef CAS PubMed.
- J. M. Köhler, A. März, J. Popp, A. Knauer, I. Kraus, J. Faerber and C. Serra, Anal. Chem., 2012, 85, 313–318 CrossRef PubMed.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- J. Kneipp, H. Kneipp and K. Kneipp, Chem. Soc. Rev., 2008, 37, 1052–1060 RSC.
- P. Donfack, M. Rehders, K. Brix, P. Boukamp and A. Materny, J. Raman Spectrosc., 2010, 41, 16–26 CrossRef CAS PubMed.
- M. Srisa-Art, D.-K. Kang, J. Hong, H. Park, R. J. Leatherbarrow, J. B. Edel, S.-I. Chang and A. J. deMello, ChemBioChem, 2009, 10, 1605–1611 CrossRef CAS PubMed.
- L. C. Taylor, T. B. Kirchner, N. V. Lavrik and M. J. Sepaniak, Analyst, 2012, 137, 1005–1012 RSC.
- I. Talian and J. Huebner, J. Raman Spectrosc., 2013, 44, 536–539 CrossRef CAS PubMed.
- R. M. Connatser, R. Maggie, M. Cochran, R. J. Harrison and M. J. Sepaniak, Electrophoresis, 2008, 29, 1441–1450 CrossRef CAS PubMed.
- T.-A. Meier, R. J. Beulig, E. Klinge, M. Fuss, S. Ohla and D. Belder, Chem. Commun., 2015, 51, 8588–8591 RSC.
- C. Delhaye, J.-L. Bruneel, D. Talaga, M. Guirardel, S. Lecomte and L. Servant, J. Phys. Chem. C, 2012, 116, 5327–5332 CAS.
- K. R. Strehle, D. Cialla, P. Rösch, T. Henkel, M. Köhler and J. Popp, Anal. Chem., 2007, 79, 1542–1547 CrossRef CAS PubMed.
- A. März, K. R. Ackermann, D. Malsch, T. Bocklitz, T. Henkel and J. Popp, J. Biophotonics, 2009, 2, 232–242 CrossRef PubMed.
- G. Wang, C. Lim, L. Chen, H. Chon, J. Choo, J. Hong and A. deMello, Anal. Bioanal. Chem., 2009, 394, 1827–1832 CrossRef CAS PubMed.
- C. D. Syme, C. Martino, R. Yusvana, N. M. S. Sirimuthu and J. M. Cooper, Anal. Chem., 2011, 84, 1491–1495 CrossRef PubMed.
- S. L. Poe, M. A. Cummings, M. P. Haaf, D. T. McQuade and D. Tyler, Angew. Chem., Int. Ed., 2006, 45, 1544–1548 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- J. Liu, I. White and D. L. DeVoe, Anal. Chem., 2011, 83, 2119–2124 CrossRef CAS PubMed.
- R. G. Freeman, K. C. Grabar, K. J. Allison, R. M. Bright, J. A. Davis, A. P. Guthrie, M. B. Hommer, M. A. Jackson, P. C. Smith, D. G. Walter and M. J. Natan, Science, 1995, 267, 1629–1632 CrossRef CAS PubMed.
- V. Joseph, C. Engelbrekt, J. Zhang, U. Gernert, J. Ulstrup and J. Kneipp, Angew. Chem., Int. Ed., 2012, 51, 7592–7596 CrossRef CAS PubMed.
- V. Joseph, M. Gensler, S. Seifert, U. Gernert, J. P. Rabe and J. Kneipp, J. Phys. Chem. C, 2012, 116, 6859–6865 CAS.
- D. Cialla, U. Hübner, H. Schneidewind, R. Möller and J. Popp, ChemPhysChem, 2008, 9, 758–762 CrossRef CAS PubMed.
- J. S. Moore and K. F. Jensen, Angew. Chem., Int. Ed., 2014, 53, 470–473 CrossRef CAS PubMed.
- L. Gitlin, C. Hoera, R. J. Meier, S. Nagl and D. Belder, Lab Chip, 2013, 13, 4134–4141 RSC.
- J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42 CrossRef CAS PubMed.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- S. Ehlert and U. Tallarek, Anal. Bioanal. Chem., 2007, 388, 517–520 CrossRef CAS PubMed.
- Y. Zhang, Y. Tang, Y.-H. Hsieh, C.-Y. Hsu, J. Xi, K.-J. Lin and X. Jiang, Lab Chip, 2012, 12, 3012–3015 RSC.
- K. B. Mogensen and J. P. Kutter, Electrophoresis, 2009, 30, S92 CrossRef PubMed.
- R. Beyreiss, S. Ohla, S. Nagl and D. Belder, Electrophoresis, 2011, 32, 3108–3114 CrossRef CAS PubMed.
- Q. Li and S. Seeger, Appl. Spectrosc. Rev., 2010, 45, 12–43 CrossRef CAS PubMed.
- R. K. Forcé and R. Ken, Anal. Chem., 1988, 60, 1987–1989 CrossRef.
- N. J. Pothier and R. K. Forcé, Anal. Chem., 1990, 62, 678–680 CrossRef CAS.
- J. P. V. Gouveia, J. P. Vitor, I. G. Gutz and J. C. Rubim, J. Electroanal. Chem., 1994, 371, 37–42 CrossRef.
- N. J. Pothier and R. K. Forcé, Appl. Spectrosc., 1992, 46, 147–151 CrossRef CAS.
- S. Köhler, C. Benz, H. Becker, E. Beckert, V. Beushausen and D. Belder, RSC Adv., 2011, 2, 520 RSC.
- A. J. Chung, Y. S. Huh and D. Erickson, Nanoscale, 2011, 3, 2903–2908 RSC.
- Y. Zhao, Y.-L. Zhang, J.-A. Huang, Z. Zhang, X. Chen and W. Zhang, J. Mater. Chem. A, 2015, 3, 6408–6413 CAS.
- D. P. Manica, Y. Mitsumori and A. G. Ewing, Anal. Chem., 2003, 75, 4572–4577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting measurements. See DOI: 10.1039/c5lc00397k |
| This journal is © The Royal Society of Chemistry 2015 |