
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast size-determination of intact bacterial plasmids using nanofluidic channels†
K.
Frykholm
a,
L. K.
Nyberg
a,
E.
Lagerstedt
b,
C.
Noble‡
b,
J.
Fritzsche
c,
N.
Karami
d,
T.
Ambjörnsson
b,
L.
Sandegren
e and
F.
Westerlund
*a
aDepartment of Biology and Biological Engineering, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden. E-mail: fredrik.westerlund@chalmers.se
bDepartment of Astronomy and Theoretical Physics, Lund University, Box 43, SE-221 00 Lund, Sweden
cDepartment of Applied Physics, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden
dDepartment of Clinical Microbiology, Sahlgrenska University Hospital and Department of Infectious Diseases, Institute of Biomedicine, Sahlgrenska Academy of the University of Gothenburg, Gothenburg, Sweden
eDepartment of Medical Biochemistry and Microbiology, Uppsala University, Box 582, SE-751 23 Uppsala, Sweden
First published on 7th May 2015
Abstract
We demonstrate how nanofluidic channels can be used as a tool to rapidly determine the number and sizes of plasmids in bacterial isolates. Each step can be automated at low cost, opening up opportunities for general use in microbiology labs.
Antibiotic resistance in bacteria is a global and rapidly increasing threat to human health.1 Antibiotic resistance is acquired and spread among bacterial pathogens mainly through horizontal transfer of resistance genes located on plasmids which are extrachromosomal, in most cases circular, double-stranded DNA molecules capable of autonomous replication.2,3
Identification and characterization of plasmid content in bacterial samples is crucial for studies of the spread of antibiotic resistance. Two commonly used methods for characterization of the sizes of plasmids are S1 nuclease linearization of plasmid DNA in cells embedded in agarose followed by pulsed field gel electrophoresis (S1/PFGE), enabling plasmid detection and size determination,4 and whole-plasmid DNA sequencing using next generation sequencing techniques.5 The latter provides base-pair resolution and is the most accurate method for plasmid characterization, but is hindered by the non-trivial assembly of DNA reads from large plasmids, especially when more than one plasmid is present in the sample. Furthermore, the highly dynamic behavior of plasmids makes sequencing of all possible plasmid compositions more or less infeasible. Therefore, S1/PFGE is still the gold standard for size determination of large plasmids, but the protocols are typically lab intensive and take several days to complete and the results are often of varying quality (see discussion below).
We here present nanofluidic channels as an alternative method to characterize the plasmid content in bacterial isolates by size. Nanofluidic channels, in combination with fluorescence microscopy, have become increasingly utilized for studies of fluorescently labeled single DNA molecules, including investigations of fundamental DNA polymer physics,6,7 optical DNA mapping8–12 and the physical properties of protein–DNA complexes.13–17 When confined to channels of dimensions smaller than their radius of gyration, DNA molecules are forced to stretch out along the channel. The extension of DNA scales linearly with its contour length, enabling direct measurements of the sizes of individual DNA molecules.18 Since the DNA molecules confined to nanofluidic channels are manipulated and stretched without being attached to any handles, circular DNA, like plasmids, can be conveniently studied.19
In a recent study, we compared the physical properties of confined circular and linear DNA. We concluded that two main characteristics clearly distinguish circular DNA pieces from linear ones.19 First, since a circular DNA molecule is double folded in the nanochannel, its emission intensity is approximately two times higher compared to that of a linear DNA molecule. Second, since the local DNA concentration is higher for the double folded circular DNA, it fluctuates less in extension than that for the corresponding linear DNA and hence has a lower standard deviation of extension. We here use these two observables to automatically discriminate intact circular plasmids from linear DNA fragments in a nanofluidic device.
We investigate the plasmid content of five clinical bacterial isolates (denoted as 008, 033, 071, 163 and 299). First, we demonstrate how to discriminate circular DNA from linear ones by introducing cut-off values for the emission intensity and the standard deviation of fluctuation. Next, we show how to obtain the sizes of the selected molecules with the help of a reference plasmid of known size. The plasmids of the isolates in this study have previously been characterized by conventional molecular methods, such as S1/PFGE and high-throughput sequencing.20 All five isolates carry plasmids coding for extended-spectrum beta-lactamases (ESBLs); in addition, isolates 008, 033 and 299 contain additional beta-lactamases and genes encoding resistance to aminoglycosides, trimethoprim, sulphonamides and macrolides. Isolate 033 also contains genes for resistance to chloramphenicol and tetracycline.
For experiments, extracted plasmids were labeled with the fluorescent dye YOYO-1 (YOYO) and inserted into nanofluidic channels with dimensions of 150 × 150 nm2 using pressure-driven flow (Fig. 1A). Subsequently, 200 images were taken from each field of view (for details on the experimental procedure, refer to the ESI†). Fig. 1B shows a snapshot image of one of the plasmid samples (isolate 163) in the nanochannels where many individual DNA molecules are observed in the same field of view. This particular sample contains plasmids of two different sizes; examples of both are seen in the image. Furthermore, a long DNA fragment (white arrow) with a weaker emission intensity is observed, suggesting that this is a linear piece of DNA.
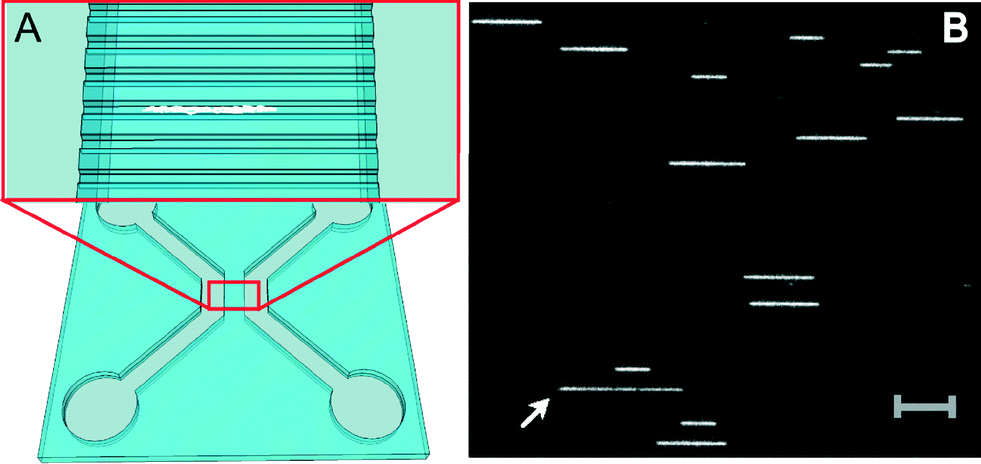 | ||
| Fig. 1 (A) Schematic illustration of the design of the nanofluidic chip with four inlets connected in twos by microchannels and spanned by nanochannels. The close-up illustrates the nanochannels with an enclosed DNA molecule in white. (B) A snapshot microscopy image of one field of view of nanoconfined plasmids in 150 × 150 nm2 nanochannels. This sample (isolate 163) contains plasmids of two different sizes. Note the longer DNA fragment, most likely a linearized plasmid, with lower emission intensity (indicated by white arrow). Scale bar corresponds to 10 μm. | ||
The presence of linearized plasmids is expected since YOYO in its excited state gives rise to reactive oxygen species that cause single-strand breaks in DNA.21 The occurrence of two such breaks on opposite strands and in close proximity to each other destabilizes the duplex between the breaks and results in a double-strand break and linearization of the circular DNA.19 This linearization can occur before introduction of the plasmids into the nanochannels but, importantly, also while the movie is recorded. Also, there is a possibility of finding residual linear DNA fragments from the bacterial chromosome from the preparation of the sample.
To analyze the data, a MatLab-based, in-house developed and automated program was used (described in detail in the ESI†). In short, the program receives the microscopy image stacks as input, and in each frame detects the edges of the DNA molecules and extracts extensions and intensities, with standard deviations, for each piece of DNA. Plasmids that break and unfold to the linear configuration while the movie is recorded are easily discarded from data analysis, since they show a large decrease in emission intensity and simultaneous increase in extension as the movie progresses. We construct colormaps, where the average intensity of the plasmid is plotted against its average extension. The color of the symbol represents a cut-off value of 10% for the standard deviation normalized with the mean extension (Fig. 2). The colormaps in Fig. 2A–C show groups of molecules with common features such as uniform size, high emission intensity and a small standard deviation of the extension. Our previous study suggests that these molecules are circular.19 We can thus set thresholds, both in intensity and fluctuations, to automatically discriminate circular DNA molecules from linear fragments in the sample. The threshold values used here can be justified by plotting the normalized standard deviation as a function of the mean extension of the plasmid, with intensities indicated by color (ESI,† Fig. S2).
 | ||
| Fig. 2 Colormaps of four different plasmid samples from bacterial isolates: 033 (A), 163 (B), 008 (C) and 071 (D), showing measured fluorescence intensity versus extension. Each square represents a single molecule and the red color indicates a normalized standard deviation of the extension lower than 10%. The dashed lines in (A), (B) and (C) represent a threshold in intensity, above which groups of molecules with a small standard deviation of extension (red squares) are observed. The isolates in (A), (B) and (C) are shown to contain one, two and three plasmids of different sizes, respectively. In (D), there is no clear intensity threshold, but molecules with a low standard deviation of extension are still clustered in a narrow extension range (indicated by the dashed ellipse). | ||
It is important to point out that the cut-offs in the intensity and standard deviation of extension can be more or less strict. By using a cut-off value of 10% in extension fluctuation, we discard some molecules having a size and an emission intensity that suggest that they are circular. However, we prefer this fairly strict cut-off to avoid the incorrect classification of linear fragments as plasmids. The fact that we can separate circular DNA from linear fragments with two parameters furthermore enables the identification of intact plasmids in lower quality data, such as in Fig. 2D. Finally, we note a spread in emission intensity of the plasmids, which stems from uneven staining by YOYO, a well-established fact for this DNA dye.22 Interestingly, the emission intensity does not seem to affect the extension of the plasmids, suggesting that circular plasmids potentially are less sensitive to the amount of dye bound than linear DNA.
The extensions of the plasmids presented in Fig. 2 are given in pixels. To convert the observed molecular extensions to actual plasmid sizes in base-pairs, a circular reference DNA of known size is needed. Here, we have used the fully sequenced plasmid R100 (ref. 23) and retrieved its size, 94.3 kbp, from the RefSeq database (http://www.ncbi.nlm.nih.gov/refseq/). Under the experimental conditions used here, the average extension of R100 was determined to be 49.9 ± 3.1 pixels based on three individual experiments (192 single plasmids in total). Using this size standard, the sizes of the plasmids in the bacterial isolates were calculated and found to be reproducible (ESI,† Table S1). For all isolates, the sizes obtained using nanofluidics are in good agreement with what is presented in the previous study using S1/PFGE.20 This is shown in Fig. 3 where a linear regression of the data for the two techniques (from Table S1†) yields an R2 value of 0.9986 and a slope of 1.056. It is, however, important to remember that we, at this moment, cannot judge whether the largest errors are in the S1/PFGE method or in the nanochannel measurements.
 | ||
| Fig. 3 A comparison of the results obtained from nanofluidic and S1/PFGE measurements. Each isolate was measured twice, indicated by circles and squares. The isolates are indicated by color with 008 in black, 033 in red, 071 in blue, 163 in magenta and 299 in green. The black line is a linear regression with R2 = 0.9986 and a slope of 1.056. The data can also be found in Table S1 (ESI†). | ||
There are several advantages of using nanofluidics compared to the gold standard for plasmid sizing, S1/PFGE. First, in nanofluidic channels, the extension of DNA in the channels varies linearly with the contour length for all relevant sizes.18 This is a clear advantage for large plasmids. S1/PFGE has a non-linear separation that requires a reference ladder of DNAs with different sizes and even with the ladder, it is sometimes hard to accurately interpolate the size of the plasmid. Also, the gel resolution interval requires separate conditions to be used for small and large plasmids for accurate size estimation. Plasmids can be detected up to the limit of S1/PFGE separation but with decreasing accuracy of size determination with increasing size. With the experimental setup used here, we detected plasmids within a size range of 8–180 kbp (Fig. 3) from the same field of view, where the extension of the largest plasmid corresponds to about 100 pixels out of 512 available. Hence, the upper limit of what fits into one field of view is over 900 kbp, well beyond the expected size of plasmids of interest.24
Second, nanofluidic measurements are carried out at the single plasmid level and hence it is possible to detect “rare” plasmids that would be difficult to detect by S1/PFGE. For example, plasmids from less abundant bacteria would be rare in plasmid extracts from a mix of bacteria but could still be efficiently detected in our device.
Third, since we measure an independent extension of each plasmid, we improve the statistics of our measurements with each additional molecule investigated. In Fig. S3 (ESI†), we show that with the cut-offs used here, the standard error of the mean extension is in general as low as about 1%. Furthermore, as expected for independent measurements, the standard error of the mean extension decreases as approximately 1/√N for N > 20 (with N being the number of molecules). This is in sharp contrast to S1/PFGE measurements where all plasmids of a certain size in a sample jointly form one band in the gel.
Fourth, with nanofluidics, there is no need for linearization of the plasmids. In S1/PFGE, plasmids are linearized by S1 nuclease. However, if the plasmid is cut in two places, it will appear as two bands and information regarding the number of plasmids in the sample and their sizes is misinterpreted. Furthermore, uncleaved and/or supercoiled plasmids might give false bands. In our nanofluidic experiments, on the other hand, circles are easily distinguished from linear fragments and supercoils, ensuring that we only measure the sizes of intact relaxed plasmids.
Fifth, the nanofluidic assay is significantly faster and less lab intensive than S1/PFGE. After extraction of the plasmids from the bacteria using standard microbiology kits, the only remaining step in sample preparation before loading the sample into the chip is to mix the DNA with the commercially available fluorescent dye YOYO. In the current study, the time from mixing the DNA and dye up to the final steps of the experiment, including data analysis, is ~2–3 hours, which should be compared to several days for completing an S1/PFGE experiment. Furthermore, there is room for improvement. Here, insertion of the plasmids into the channels and recording of the movies were performed manually, while a recent study demonstrates that nanofluidic experiments can be automated for high throughput measurements.25 Importantly, the assay presented here is directly applicable to the instrument used in the study in ref. 25, where the only technical difference is that we record several (200) images, whereas Cao et al. take a single snapshot of each field of view.
Sixth, our nanofluidic approach uses a significantly lower amount of sample compared to traditional techniques. In the experiments presented here, we add 20 μl of a solution with a typical DNA concentration of 0.25 ng μl−1. Since we measure the lengths of single DNA molecules, we foresee that the DNA concentration can be decreased by several orders of magnitude, the only difference will be that the measurements take longer time to complete. As an outlook, the small amount of sample required implies that it will be possible to use our technique to investigate the plasmid composition of a clinical sample directly after collection, without the time-consuming procedure of culturing bacteria. This could for example be beneficial during resistance outbreaks where it is important to quickly identify if a patient is carrying a specific plasmid. The small amounts of sample needed also means that the assay is directly compatible with fully integrated lab-on-a-chip devices, where the ultimate goal would be to have a clinical sample loaded into one end and the plasmids sized in the other.
We foresee that an automated version of size determination of bacterial plasmids using nanofluidics presented here has general applicability in microbiology labs around the world. The method is based on the use of fluorescence microscopy and the chemicals used are commercially available at low prices and employed in extremely small amounts. Furthermore, the possibility of making disposable plastic chips26 will significantly reduce the cost, in comparison to the nanofabricated devices used in the current version of the assay.
Conclusions
We demonstrate how nanofluidic channels can be used as a method for rapid and reliable characterization of the plasmid content in bacterial isolates. The method gives a direct indication of the diversity in plasmid content and plasmid sizes can be determined with high reproducibility in a way that opens up opportunities for future automation and general use. Our results are in good agreement with results obtained by traditionally used methods for plasmid characterization.Acknowledgements
This project was funded by the Chalmers Area of Advance in Nanoscience and Nanotechnology, the EuroNanoMedII-project NanoDiaBac and Torsten Söderbergs Stiftelse. LS acknowledges funding from the Swedish Research Council in Health and Medicine.Notes and references
- World Health Organization, http://www.who.int.
- L. S. Frost, R. Leplae, A. O. Summers and A. Toussaint, Nat. Rev. Microbiol., 2005, 3, 722–732 CrossRef CAS PubMed.
- A. Carattoli, Int. J. Med. Microbiol., 2013, 303, 298–304 CrossRef CAS PubMed.
- B. M. Barton, G. P. Harding and A. J. Zuccarelli, Anal. Biochem., 1995, 226, 235–240 CrossRef CAS PubMed.
- J. Shendure and H. Ji, Nat. Biotechnol., 2008, 26, 1135–1145 CrossRef CAS PubMed.
- S. L. Levy and H. G. Craighead, Chem. Soc. Rev., 2010, 39, 1133–1152 RSC.
- F. Persson and J. O. Tegenfeldt, Chem. Soc. Rev., 2010, 39, 985 RSC.
- L. K. Nyberg, F. Persson, J. Berg, J. Bergström, E. Fransson, L. Olsson, M. Persson, A. Stålnacke, J. Wigenius, J. O. Tegenfeldt and F. Westerlund, Biochem. Biophys. Res. Commun., 2012, 417, 404–408 CrossRef CAS PubMed.
- A. N. Nilsson, G. Emilsson, L. K. Nyberg, C. Noble, L. S. Stadler, J. Fritzsche, E. R. B. Moore, J. O. Tegenfeldt, T. Ambjornsson and F. Westerlund, Nucleic Acids Res., 2014, 42, e118 CrossRef PubMed.
- W. Reisner, N. B. Larsen, A. Silahtaroglu, A. Kristensen, N. Tommerup, J. O. Tegenfeldt and H. Flyvbjerg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13294–13299 CrossRef CAS PubMed.
- K. Jo, D. M. Dhingra, T. Odijk, J. J. de Pablo, M. D. Graham, R. Runnheim, D. Forrest and D. C. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2673–2678 CrossRef CAS PubMed.
- E. T. Lam, A. Hastie, C. Lin, D. Ehrlich, S. K. Das, M. D. Austin, P. Deshpande, H. Cao, N. Nagarajan, M. Xiao and P.-Y. Kwok, Nat. Biotechnol., 2012, 30, 771–776 CrossRef CAS PubMed.
- C. Zhang, D. Guttula, F. Liu, P. P. Malar, S. Y. Ng, L. Dai, P. S. Doyle, J. A. van Kan and J. R. C. van der Maarel, Soft Matter, 2013, 9, 9593–9601 RSC.
- C. Zhang, K. Jiang, F. Liu, P. S. Doyle, J. A. van Kan and J. R. C. van der Maarel, Lab Chip, 2013, 13, 2821–2826 RSC.
- K. Jiang, C. Zhang, D. Guttula, F. Liu, J. A. Van Kan, C. Lavelle, K. Kubiak, A. Malabirade, A. Lapp, V. Arluison and J. R. C. Van Der Maarel, Nucleic Acids Res., 2015 DOI:10.1093/nar/gkv268.
- K. Frykholm, M. Alizadehheidari, J. Fritzsche, J. Wigenius, M. Modesti, F. Persson and F. Westerlund, Small, 2014, 10, 884–887 CrossRef CAS PubMed.
- D. E. Streng, S. F. Lim, J. Pan, A. Karpusenka and R. Riehn, Lab Chip, 2009, 9, 2772–2774 RSC.
- J. O. Tegenfeldt, C. Prinz, H. Cao, S. Chou, W. W. Reisner, R. Riehn, Y. M. Wang, E. C. Cox, J. C. Sturm, P. Silberzan and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10979–10983 CrossRef CAS PubMed.
- M. Alizadehheidari, E. Werner, C. Noble, M. Reiter-Schad, L. K. Nyberg, J. Fritzsche, B. Mehlig, J. O. Tegenfeldt, T. Ambjörnsson, F. Persson and F. Westerlund, Macromolecules, 2015, 48, 871–878 CrossRef CAS.
- A. Brolund, O. Franzén, Ö. Melefors, K. Tegmark-Wisell and L. Sandegren, PLoS One, 2013, 8, e65793 CAS.
- M. A. Tycon, C. F. Dial, K. Faison, W. Melvin and C. J. Fecko, Anal. Biochem., 2012, 426, 13–21 CrossRef CAS PubMed.
- L. Nyberg, F. Persson, B. Åkerman and F. Westerlund, Nucleic Acids Res., 2013, 41, e184 CrossRef CAS PubMed.
- L. Silver, M. Chandler, E. B. de la Tour and L. Caro, J. Bacteriol., 1977, 131, 929–942 CAS.
- C. Smillie, M. P. Garcillán-Barcia, M. V. Francia, E. P. Rocha and F. de la Cruz, Microbiol. Mol. Biol. Rev., 2010, 74, 434–452 CrossRef CAS PubMed.
- H. Cao, A. R. Hastie, D. Cao, E. T. Lam, Y. Sun, H. Huang, X. Liu, L. Lin, W. Andrews, S. Chan, S. Huang, X. Tong, M. Requa, T. Anantharaman, A. Krogh, H. Yang, H. Cao and X. Xu, GigaScience, 2014, 3, 34 Search PubMed.
- P. Utko, F. Persson, A. Kristensen and N. B. Larsen, Lab Chip, 2011, 11, 303–308 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, nanofabrication, data analysis and additional figures. See DOI: 10.1039/c5lc00378d |
| ‡ Current address: Department of Systems Biology, Harvard Medical School, Boston, MA 02115, USA |
| This journal is © The Royal Society of Chemistry 2015 |