
Microfluidic device for mechanical dissociation of cancer cell aggregates into single cells†
Xiaolong
Qiu‡§
a,
Janice
De Jesus‡§
a,
Marissa
Pennell§
a,
Marco
Troiani§
a and
Jered B.
Haun§
*abc
aDepartment of Biomedical Engineering, University of California Irvine, 3107 Natural Sciences II, Irvine, CA 92697, USA. E-mail: jered.haun@uci.edu; Tel: +1 949 824 1243
bDepartment of Chemical Engineering and Materials Science, University of California Irvine, Irvine, CA 92697, USA
cChao Family Comprehensive Cancer Center, University of California Irvine, Irvine, CA 92697, USA
First published on 29th October 2014
Abstract
Tumors tissues house a diverse array of cell types, requiring powerful cell-based analysis methods to characterize cellular heterogeneity and identify rare cells. Tumor tissue is dissociated into single cells by treatment with proteolytic enzymes, followed by mechanical disruption using vortexing or pipetting. These procedures can be incomplete and require significant time, and the latter mechanical treatments are poorly defined and controlled. Here, we present a novel microfluidic device to improve mechanical dissociation of digested tissue and cell aggregates into single cells. The device design includes a network of branching channels that range in size from millimeters down to hundreds of microns. The channels also contain flow constrictions that generate well-defined regions of high shear force, which we refer to as “hydrodynamic micro-scalpels”, to progressively disaggregate tissue fragments and clusters into single cells. We show using in vitro cancer cell models that the microfluidic device significantly enhances cell recovery in comparison to mechanical disruption by pipetting and vortexing after digestion with trypsin or incubation with EDTA. Notably, the device enabled superior results to be obtained after shorter proteolytic digestion times, resulting in fully viable cells in less than ten minutes. The device could also be operated under enzyme-free conditions that could better maintain expression of certain surface markers. The microfluidic format is advantageous because it enables application of well-defined mechanical forces and rapid processing times. Furthermore, it may be possible to directly integrate downstream processing and detection operations to create integrated cell-based analysis platforms. The enhanced capabilities enabled by our novel device may help promote applications of single cell detection and purification techniques to tumor tissue specimens, advancing the current understanding of cancer biology and enabling molecular diagnostics in clinical settings.
Introduction
Tumors are now viewed as an ecosystem of diverse cell types, and this cellular heterogeneity has been identified as a key factor underlying tumor progression, metastasis, and the development of drug resistance.1,2 This has led to an increase in studies that are focused on defined cellular subsets within tumors to address biological and therapeutic questions. Cell-based analysis platforms such as flow cytometry are ideally suited to this task because they offer high-throughput and multiplexed information at the single cell level, allowing the entire population to be analyzed. Other platforms include mass cytometry,3 microfabricated magnetic and optical detectors,4–6 cytology,7 single cell gene sequencing,8 as well as physical measurements such as density and deformability.9,10 Reducing tumor tissue into single cells is a critical step in providing material for identification and analysis of specific tumor cell subsets such as cancer stem cells, metastatic precursors, or drug resistant clones for more detailed study.11–14Tumor tissues and cancer cell aggregates are dissociated into single cells using proteolytic enzymes that digest cellular adhesion molecules and/or the underlying extracellular matrix. Large clinical specimens such as surgical resections and core biopsies are first minced with a scalpel into approximately 1–2 mm pieces to facilitate digestion. Samples are then subjected to fluid shear forces by vortexing and/or repeated pipetting to mechanically liberate individual cells. These methods generate poorly defined shear flow environments that do not allow control over sample exposure, potentially resulting in variations among samples and across different laboratories. The gentleMACS™ Dissociator is a commercial system that has been developed to standardize mechanical dissociation for large tumor tissues,15 but its use is not common and performance is not well documented. Thus, there is a critical need for develop new technologies to improve mechanical dissociation of digested tumor tissue and cancer cell aggregates into single cells, particularly for smaller samples. Improving mechanical dissociation would enhance cell recovery, and potentially shorten digestion times or enable use of milder enzymes or even non-enzymatic treatments such as the calcium chelator ethylenediaminetetraacetic acid (EDTA).
Microfabrication technologies have advanced the fields of biology and medicine by miniaturizing devices to the scale of cellular samples. In particular, microfluidic systems have enabled precise manipulation of cells and other reagents to achieve systems with high throughput, cost efficiency, minimal sample requirement, integration of multiple procedures on the same device, and point-of-care operation.16,17 Sample processing has been a major focus area, specifically for on-chip cell purification, sorting, and lysis.17–19 However, little attention has been given to processing tissues. One example is a microfluidic device designed to maintain and interrogate tissue samples, including on-chip digestion with collagenase.20 The Biogrid is another example, which employs a 100 μm mesh with sharp edges to cut large cell aggregates into smaller units that still contain numerous cells.21 More recently, an array of microstructures was used to dissociate small neurospheres into single cells under fluid flow.22 A microfluidic device for mechanically dissociating tumor tissue and cancer cell aggregates across a large range of sizes, from millimeter tissues down to tens of micron cells, has not been described to date. Ideally such a device would maintain a flow through format to integrate the resulting single cell suspension directly with downstream operations such as purification/sorting, physical analysis,9,10 and/or probe detection4–6 to achieve integrated cancer cell analysis platforms.
In this work, we present a novel microfluidic device for processing tumor tissue samples into single cells. The device employs channel features ranging in size from millimeters down to hundreds of microns. The channels also contain constriction and expansion regions that generate hydrodynamic fluid jets at varying size scales and shear force magnitudes to progressively break down tissue fragments and aggregates into single cells. Using in vitro cancer cell models of varying complexity, we show that the microfluidic dissociation device significantly augments enzymatic digestion by increasing the number of single cells liberated, while still maintaining viability. This is because the device is significantly more effective than standard mechanical dissociation procedures (pipetting and vortexing) at dissociating small clusters, while limiting cell destruction or holdup. We also demonstrate that efficient dissociation can be obtained under enzyme-free conditions, in which the device is used in combination with EDTA treatment or even alone for certain samples. These samples had lower viability, but could be utilized directly for diagnostic applications, and the lack of enzyme treatment could better preserve molecular expression levels. Furthermore, all these results were obtained in less than ten minutes total processing time. We envision our device operating as a standalone unit or as a component of an integrated processing and analysis platform. Sample types could include tumors or other tissues that are 1 mm in size or less, such as laboratory-scale tissue models, small volume specimens such as fine needle aspirate (FNA) biopsies, and larger surgical or core biopsy specimens that have been finely cut with a scalpel. The improved dissociation capabilities will help promote laboratory and clinical investigations that utilize cell-based detection and isolation platforms, thereby advancing our understanding of cancer biology and enabling molecular diagnostics.
Results and discussion
Device design
The concept for our dissociation device is to employ a series of branching channels that resemble a physiological microvascular capillary network or reported microfluidic droplet splitters.23 We believe this design will make it possible to process tissue fragments and cell aggregates of different length scales in an effective but gentle manner. The device has a total of 5 stages that are each approximately 1 cm in length, and with branch points placed at the end of each stage (Fig. 1A). The first stage is a single channel with a minimum channel width of 2 mm. We have chosen for channel dimensions to decrease by half after branching, thus the total width across each stage is maintained constant throughout the device. We have included an additional design feature to facilitate dissociation, continuous expansion and constriction of the channel width. This will modulate fluid velocity, actively mixing the sample and generating shear forces across cell aggregates. The expansion and constriction regions are connected by smooth curved lines to avoid turbulent mixing and the generation of microvortices that can trap cells in recirculating flows.24 The maximum width in the expansion region is 3-fold greater than the minimum width in the constriction, and this ratio is maintained throughout the device. Finally, constriction regions are separated by a distance equal to the expansion region width, which results in an increase in the number of constrictions per channel with each stage. Since channel width dimensions decrease by half as channel number doubles, and channel height is constant (300 μm, discussed below), average velocity (vavg) is constant in each channel throughout the device. Channel specifications for each stage are listed in Table 1. For reference, the orifice diameter for standard P-1000 pipette tips that are commonly used to shear tumor tissue samples is approximately 1 mm.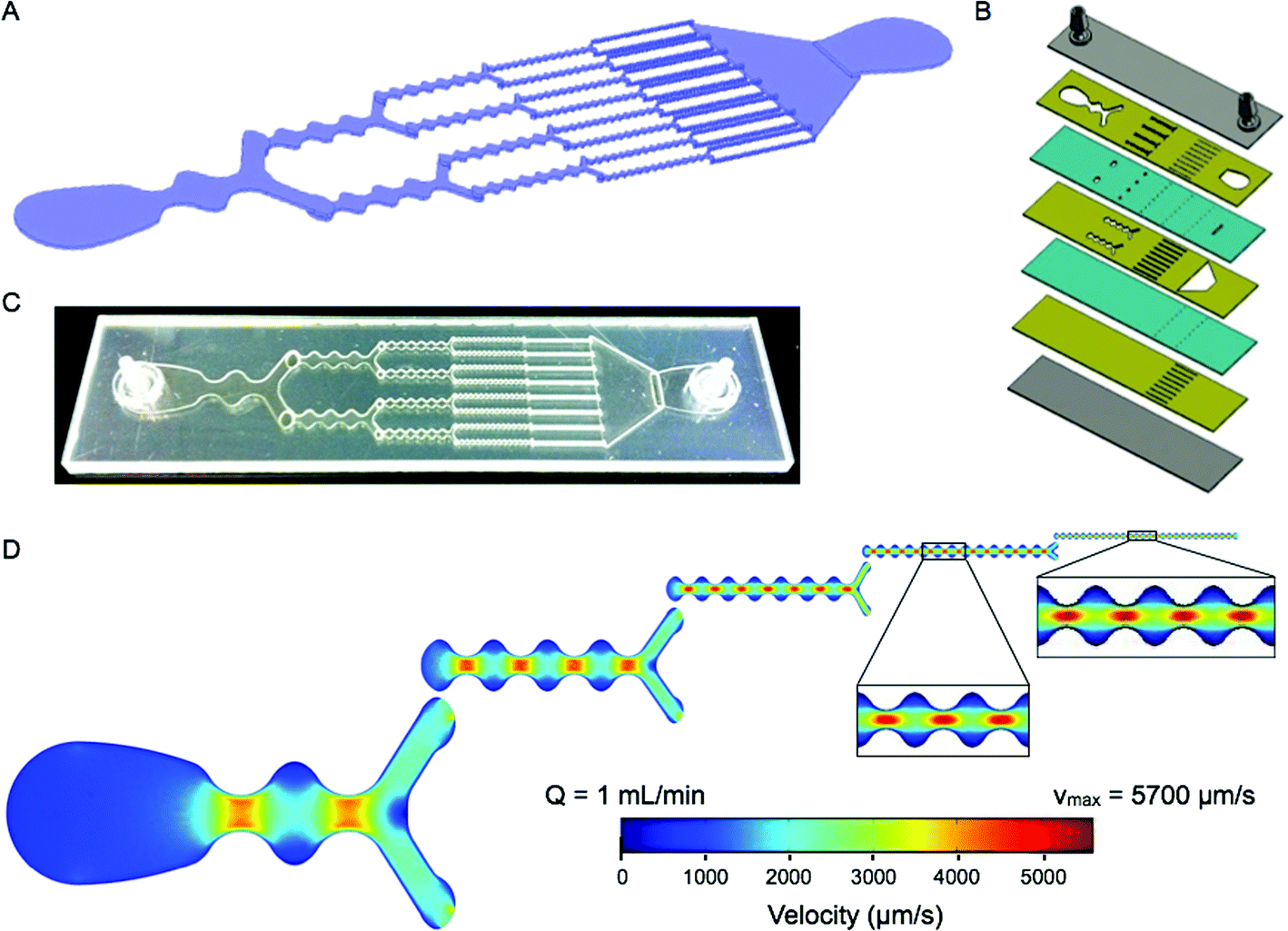 | ||
| Fig. 1 Microfluidic dissociation device. (A) Schematic showing the branching channel design containing alternating constriction and expansion regions. Channel dimensions are listed in Table 1. (B) Expanded view of the laminate format containing 7 plastic layers. Features are etched in plastic using a laser, with the channels placed on three layers (gold) that are connected by via layers (teal). Two layers are used to seal the device (gray), with hose barbs placed in the top layer to serve as the inlet and outlet. (C) Picture of a fabricated microfluidic device. (D) Finite-element fluid dynamics simulations showing velocity profiles in each stage of the device. Fluidic jets are generated in the constriction regions with high flow velocities and shear stresses. The fluidic jets decrease in size scale and increase in shear magnitude with each stage, leading to gradual dissociation of tissues and cell aggregates. Simulations were performed at 1 mL min−1, and flow velocities and shear stresses are listed in Table 1. | ||
| Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 | |
|---|---|---|---|---|---|
| a All flow properties are given for Q = 1 mL min−1. b v avg = 2800 μm s−1. c ΔPTotal = 100 Pa. | |||||
| Channels | 1 | 2 | 4 | 8 | 16 |
| Min. width (mm) | 2 | 1 | 0.5 | 0.25 | 0.125 |
| Constrictions | 2 | 4 | 7 | 14 | 27 |
| Remaxa,b | 160 | 80 | 40 | 20 | 10 |
| v max , (μm s−1) | 4300 | 4600 | 5200 | 5300 | 5700 |
| τ W,max (dyne cm−2) | 0.4 | 0.9 | 2 | 4 | 9 |
| τ H (dyne cm−2) | 2.9 | 3.1 | 3.4 | 3.6 | 3.8 |
| ΔPChannela (Pa) | 50 | 40 | 45 | 60 | 160 |
| ΔPStage (Pa)a,c | 50 | 20 | 12 | 8 | 10 |
We chose to fabricate the device using a laminate approach, with channel features etched in hard plastic (polyethylene terephthalate, PET) using a laser. This format should provide a more robust product than alternative options that utilize photolithography and polydimethyl siloxane (PDMS), and thus better support high flow rates and pressures that may be needed to effectively dissociate tissues. We employed multiple plastic layers, which were bonded with adhesive and pressure lamination. Consecutive device stages were placed on different layers to maximize device strength and integrity, as well as change the direction of fluid flow to mix and agitate the sample between stages. Seven layers were used in total, including three for the channel features, two for the vias that pass the sample between the channel layers, and two to seal the top and bottom (Fig. 1B). Channel height includes contributions from both the plastic layer (250 μm) and adhesive (~50 μm), and is approximately 300 μm.
While vavg is constant in each channel, the continuous variations in width will result in complex, size-dependent velocity profiles. Therefore we have performed computational fluid dynamics simulations using COMSOL Multiphysics. This will be important for understanding the mechanisms of tissue dissociation achieved through hydrodynamic forces. We separately analyzed single channels within each stage to simplify simulations and because the multi-layer laminate design is not continuous in the axial direction. However, we have confirmed that single channel outputs do closely match results obtained from consecutive, multistage simulations (see ESI†). Simulations were performed under laminar flow conditions, which are expected because changes in channel width are gradual and the Reynolds number remains less than 25 at all points in the device when operating at 1 mL min−1 flow rate. Maintaining laminar flow is important to achieve well-defined flow properties and shear forces. Velocity profiles across the channel widths are shown in Fig. 1D, viewed from the center of the height dimension. Flow velocity increases in the constriction regions to form discrete jets, with the maximum velocity (vmax) concentrated in the central region for all but the first stage. The magnitude of vmax is similar in each stage, but does increase slightly throughout the device. However, hydrodynamic shear force scales with the shear rate, which depends on the change in velocity (vmax) divided by the channel half-width. Thus, the fluidic jets increase in dissociation power as they become smaller in scale. We envision that these regions act as “hydrodynamic micro-scalpels” that become sharper and finer throughout the device, breaking tissue down with increasing precision until finally obtaining single cells. Since the “hydrodynamic micro-scalpels” operate on a smaller size scale than the channels, we did not feel that it was necessary for the dimensions of the final channel to approach the 10–20 μm diameter of typical epithelial cells. For this reason we left the minimum dimension of the final stage at 125 μm. The shear stress generated across the device width (τW) was calculated by multiplying the shear rate by the fluid viscosity, which was assumed to be the same as water. The shear stress across the height dimension (τH) can also be calculated using vmax and the half-height, which will remain constant in each stage. It should be noted that τH exceeds τW, even within the constrictions, for the first 3 stages. Velocity and shear stress values are listed for each stage in Table 1. Note that physiological values for wall shear stresses in human blood vessels are in the range of 1–10 dynes cm−2.
Dissociation of small cell clusters
We first introduced cell suspensions into the device to determine whether cells are lost under different operating conditions due to holdup or damage induced by shearing. We employed HCT 116 colon cancer cells that were grown in culture flasks, digested with trypsin-EDTA, and mechanically sheared (pipetting and vortexing) per routine procedures. Cell suspensions were applied to the microfluidic device using a syringe pump under different flow rate and cell concentration conditions. Cell recovery was then assessed using a commercial cell counter, and compared to the initial value measured before passing through the device. We found that recovered cell counts increased progressively with flow rate, ranging from approximately 40% of the initial count at 0.2 mL min−1 to approximately 100% at 12.5 mL min−1 (Fig. 2A). In fact, recovery was slightly greater than 100% and we observed a shift in the population to smaller sizes (Fig. 2B). We believe that the larger species correspond to small clusters of 2 or more cells that remained after trypsin digestion and vortexing/pipetting treatments. This is a common occurrence in routine cell culture, but additional treatment is not encouraged because it could decrease cell viability and the small clusters do not negatively affect sub-culturing or most downstream assays. The device was significantly more effective at dissociating these small cell clusters, resulting in a truer single cell suspension. Evidence supporting our conclusion can be found in the fact that the device yielded a cell population that was evenly distributed around an average diameter of 13–14 μm, which was consistent with microscopic analysis of HCT 116 cells (Fig. 2B). Control samples had a similar peak size, but also showed significant species at larger sizes. We defined single cells by gating the histograms equally around the 13.5 μm mean, from 9.5 μm to 17.5 μm. Using this size range to define single cells, we found that only 75% of the events were single cells prior to device treatment. Single cell percentage increased with flow rate up to 94% at 12.5 mL min−1 (Fig. 2C, see ESI†). We believe the clusters were dissociated rather than simply retained in the device, as the latter case would have lowered total recovery yield for the 12.5 mL min−1 sample. However, since recovery at this condition did not significantly exceed 100%, increases in cell number from cluster dissociation were likely offset by losses that could have been incurred from device holdup, including non-specific sticking or entrapment in low flow regions, or cell destruction. Recovery results did decrease slightly for lower input cell concentrations, but remained greater than 90% even when only 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were tested (Fig. 2D). Finally, we employed a different cell type, LS 174T colon cancer cells. Cell suspensions contained 79% single cells prior to device treatment, and this was enhanced to 92%, with a similar total cell count, after processing at 12.5 mL min−1 (Fig. 2E). Representative cell histograms and micrographs are shown in Fig. 2F. Note that it is possible that single cell percentage is over-represented in control samples because clusters larger than 30 μm, such as those pictured in the micrograph, were not assessed by the cell counter. Other methods, such as flow cytometry, may be able to capture these larger clusters. From these studies, it is clear that the microfluidic dissociation device can significantly improve single cell content in trypsin-treated cell cultures. Moreover, results were consistent and robust for low sample concentrations and for different cell lines.
000 cells were tested (Fig. 2D). Finally, we employed a different cell type, LS 174T colon cancer cells. Cell suspensions contained 79% single cells prior to device treatment, and this was enhanced to 92%, with a similar total cell count, after processing at 12.5 mL min−1 (Fig. 2E). Representative cell histograms and micrographs are shown in Fig. 2F. Note that it is possible that single cell percentage is over-represented in control samples because clusters larger than 30 μm, such as those pictured in the micrograph, were not assessed by the cell counter. Other methods, such as flow cytometry, may be able to capture these larger clusters. From these studies, it is clear that the microfluidic dissociation device can significantly improve single cell content in trypsin-treated cell cultures. Moreover, results were consistent and robust for low sample concentrations and for different cell lines.
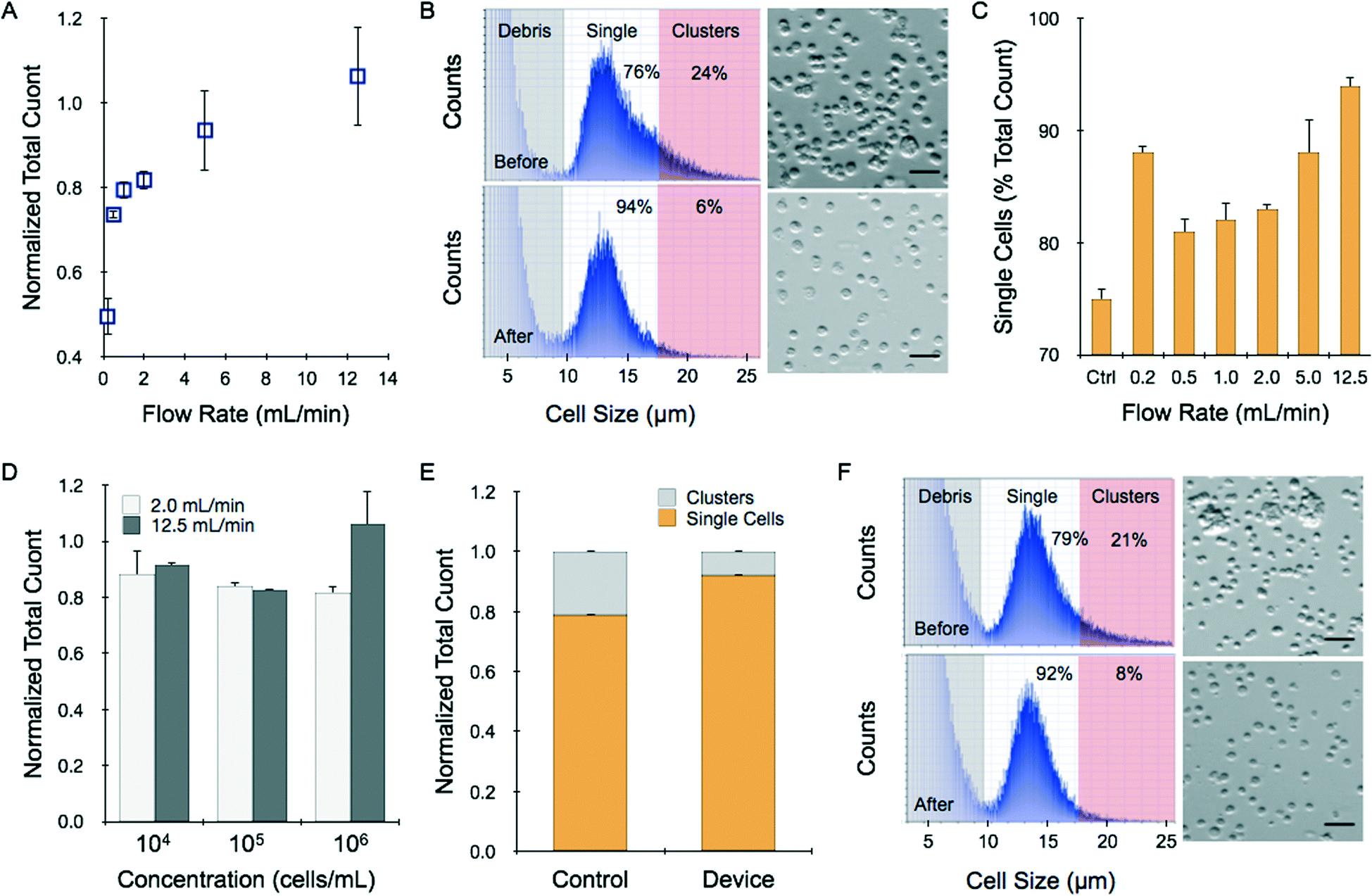 | ||
| Fig. 2 Dissociation of small clusters in cell suspensions. (A–D) Cultured HCT 116 colon cancer cells were treated with trypsin-EDTA and the cell suspensions were passed through the microfluidic device. (A) Increasing flow rate led to progressively higher cell counts in the recovered sample, reaching approximately the same value as before addition to the device. (B) While total count was similar, the size distribution before (top) and after treatment at 12.5 mL min−1 (bottom) changed significantly. The peak size was approximately 13.5 μm for both cases, however prior to device treatment there was also a population at larger sizes corresponding to cell clusters. The device-processed sample was evenly distributed around the mean, indicating that nearly all events were single cells. The accompanying micrographs confirm average cell size and single cell content. (C) The percentage of single cells was determined as the counts between 9.5 to 17.5 μm compared to the total count. In general, increasing flow rate resulted in more efficient dissociation of cell clusters. (D) Total recovery was similar at lower initial concentrations, down to only 10000 cells. (E–F) Single cell and total counts obtained for LS 174T colon cancer cell suspensions prior to and after passing through the microfluidic device at 12.5 mL min−1. (E) Total recovery was again similar, with a greater percentage of single cells due to dissociation of cell clusters. (F) Population size distribution shifted to lower values after device treatment, indicating higher single cell content. All total count results obtained after device treatment were normalized to the value before treatment. Scale bars represent 50 μm. Error represent the standard error from at least three independent experiments. | ||
Dissociation of intact cell monolayers
We next created a simple tissue model consisting of cell monolayers that were released as intact sheets. This was accomplished by growing HCT 116 cells to confluency in collagen-coated wells and then treating with collagenase. These monolayer tumor sheets contained approximately 1 million cells that indeed remained connected to each other after being suspended (ESI†). Dissociation experiments were conducted by passing a single tumor sheet through the device in buffer at 2 and 12.5 mL min−1 flow rates. To increase dissociation efficiency, some samples were repositioned for multiple device passes. After testing was complete, the sample was recovered and a cell count was obtained. Finally, we tested whether large aggregates passed through the device by treating the effluent with trypsin-EDTA, vortexing, and pipetting before performing a second cell count. Control sheets only received trypsin-EDTA, vortexing, and pipetting procedures, and less than 60% of the cell counter events were single cells. At 2 mL min−1 operating flow rate, total recovery including contributions from single cells, small clusters, and large aggregates was only 60% of the control count (Fig. 3A). Utilizing more device passes progressively increased single cell yield, but total recovery was similar suggesting that significant sample remained within the device or samples were damaged. Increasing flow rate to 12.5 mL min−1 improved results substantially (Fig. 3B). Total recoveries after 1 and 3 passes were both nearly the same as the control, with single cell yield increasing upon further treatment. However, after 10 passes the total count exceeded the control by 30%. This was possible because the sample now contained 95% single cells with no appreciable large aggregates (Fig. 3B and C). Note that these promising results were obtained exclusively using the hydrodynamic forces generated within the microfluidic device following release of the intact monolayers using collagenase.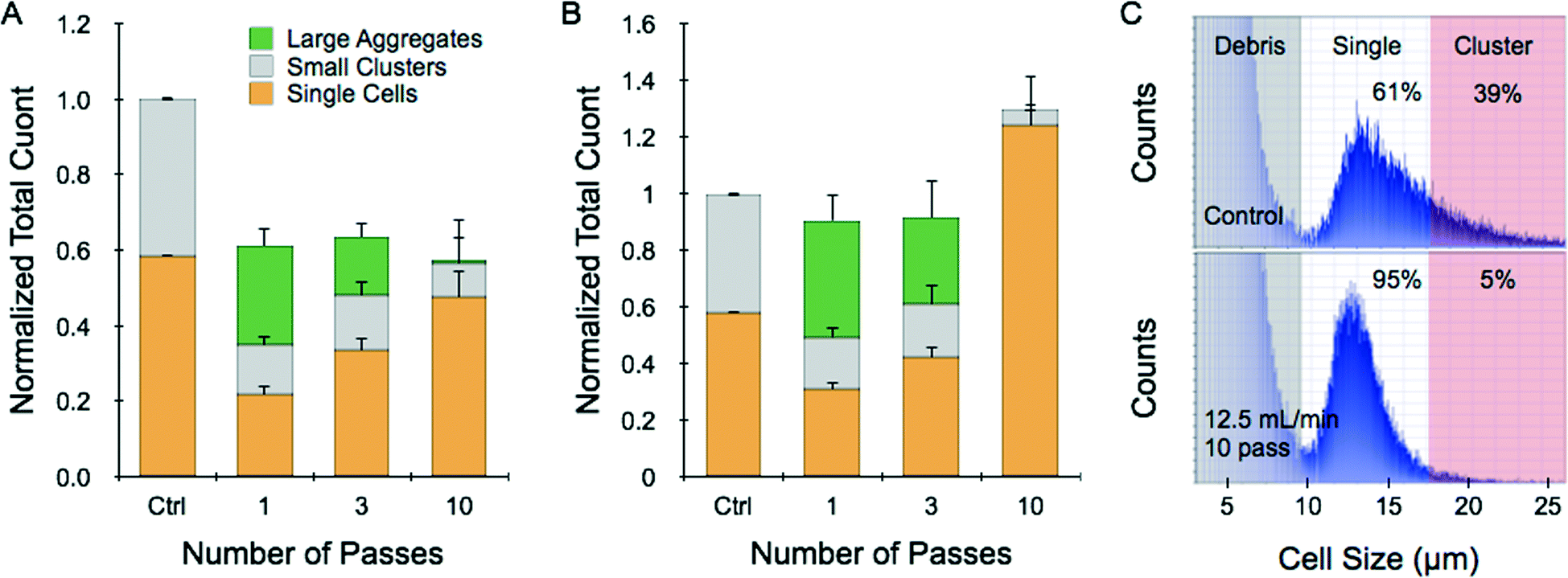 | ||
| Fig. 3 Dissociation of tumor monolayer sheets. HCT 116 cells were grown to confluency in collagen-coated 24-well plates and released as intact monolayer sheets using collagenase. Sheets were then passed directly through the microfluidic dissociation device at different flow rates for a total of 1, 3, or 10 passes. Control sheets were treated with trypsin-EDTA, vortexed, and pipetted. (A) At 2 mL min−1 flow rate, total count was only 60% of the trypsin control after a single device pass. Additional passes did not change total recovery, but did increase single cells at the expense of large aggregates. (B) Increasing flow rate to 12.5 mL min−1 enhanced total recovery to values similar to the trypsin-control for 1 and 3 passes. After 10 passes, total recovery exceeded the trypsin control by 30%, and single cells accounted for 95% of the recovered sample. (C) Representative cell counter histograms for the (top) trypsin control and (bottom) 12.5 mL min−1, 10-pass cases. All total count results were normalized to controls that were digested with trypsin. Error represent the standard error from at least three independent experiments. | ||
Dissociation of tumor spheroids
Tumor spheroids are a more advanced in vitro model with three-dimensional structure that more closely resembles solid tumors. Spheroids were prepared using the hanging drop method and collected after reaching 250–300 μm diameter. We employed HCT 116 and LS 174T cells as already discussed, as well as NCI-H1650 lung cancer cells. In each case, the spheroids contained approximately 1000 cells. Dissociation experiments were conducted in a similar manner to the tumor sheet studies. Results obtained for 12 pooled HCT 116 spheroids that were processed at 12.5 mL min−1 flow rate for different number of device passes are presented in Fig. 4A. Overall, recovery results were strikingly similar to the HCT 116 tumor sheets processed at 2 mL min−1 flow rate (Fig. 3A). Total count was approximately 60% of the control after 1 pass, but actually decreased with additional treatment, possibly indicating cell damage. Single cell content did increase progressively, nearly approaching the value present within the control. We also performed 10-pass experiments with different numbers of HCT 116 spheroids, and found recovery results that scaled directly with sample size (Fig. 4B). Remarkably, this includes using only a single spheroid that initially contained only 1000 cells. Finally, we performed dissociation tests with NCI-H1650 (Fig. 4C) and LS 174T (Fig. 4D) spheroids, and observed recovery results that were similar to the HCT 116 case but with even lower yields. This is likely due to the fact that these spheroids are more cohesive, as they do require longer digestion times with trypsin (see ESI†). These data suggest that the device was not able to generate high enough shear forces to effectively dissociate tumor spheroids without inducing cell damage. Thus, low recovery was due either to sample remaining entrapped within the device in the form of large aggregates or cell destruction incurred as shear forces ripped cells away from multiple, strongly adherent neighbors. Improving recovery results through additional device passes did not appear to offer much potential, and we were not able to investigate this further by increasing flow rate because we were already operating at the maximum for our syringe pump.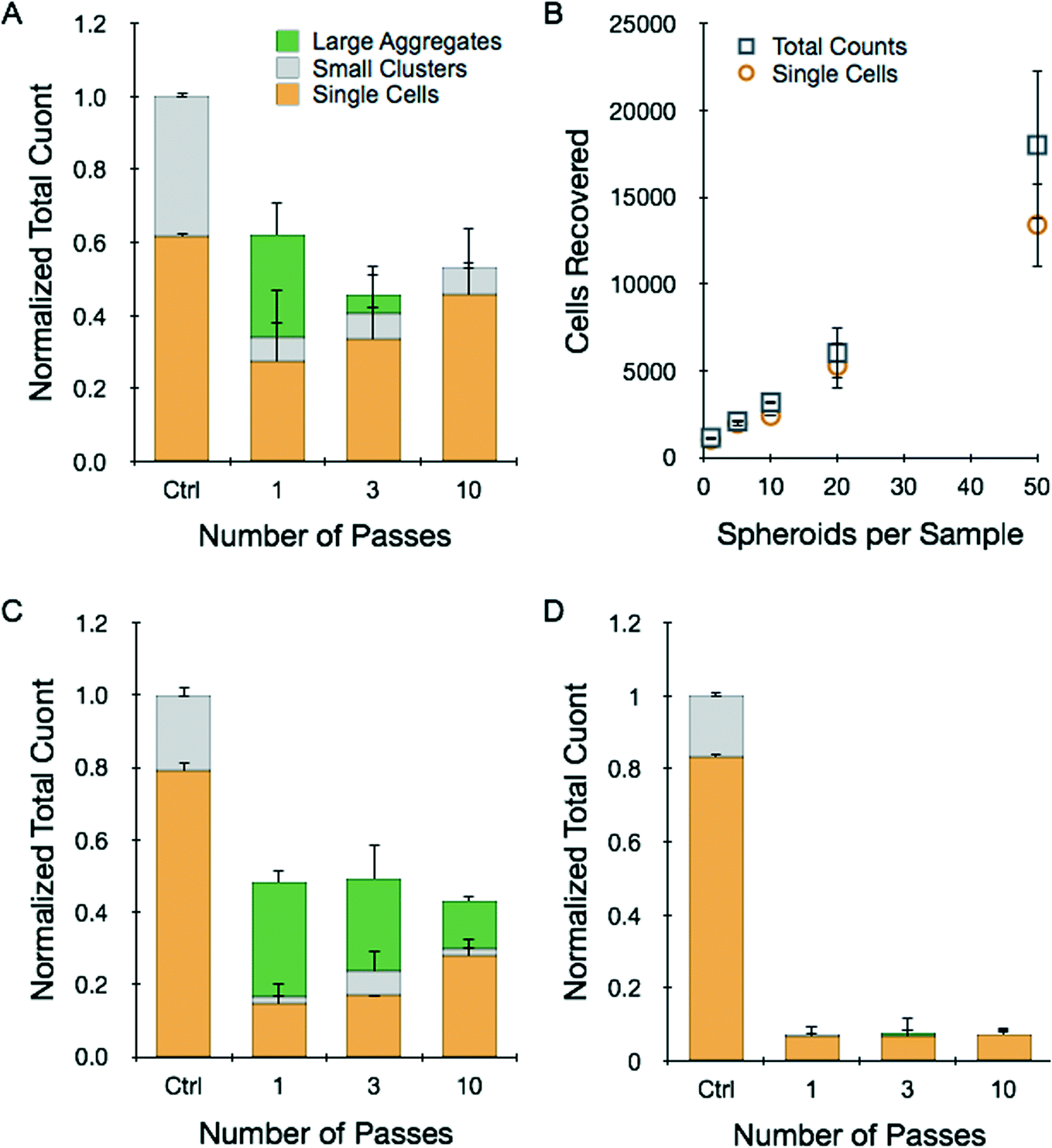 | ||
| Fig. 4 Dissociation of tumor spheroids. Tumor spheroids were grown with different cell lines using the hanging drop method and passed directly through the microfluidic dissociation device at 12.5 mL min−1 for a total of 1, 3, or 10 passes. Control spheroids were treated with trypsin-EDTA, vortexed, and pipetted. (A) For HCT 116 spheroids, total recovery was only 60% of the control after a single pass. Additional device passes decreased total recovery, but did shift the population to higher single cell percentages that were similar to the control. (B) Total and single cell recoveries scaled with initial spheroid concentration, even down to a single spheroid. Results for (C) NCI-H1650 lung cancer and (D) LS 174T spheroids were similar, but with lower total recoveries even after 10 passes. All total count results were normalized to the maximum count determined by fully digesting each spheroid type with trypsin. Error represent the standard error from at least three independent experiments. | ||
Although device dissociation of tumor spheroids was inefficient, this would be an unnecessarily stringent goal for practical purposes, as tumor tissues are typically treated with proteolytic enzymes prior to mechanical treatments. Therefore we tested device performance after brief exposure of spheroids to trypsin-EDTA. We also tested brief EDTA treatment to explore an enzyme-free alternative. After a 5 min trypsin digestion, total recovery of HCT 116 spheroids improved 2.75-fold after a single pass, with negative effects observed for additional processing (Fig. 5A). Single cell content only increased from 75 to 85% (Fig. 5D), and thus the dramatic increase in total count is likely to have arisen from enhanced dissociation of larger aggregates that were not measured by the cell counter. EDTA treatment was extremely inefficient, but the microfluidic device increased recovery dramatically. Total yield was similar to the trypsin control following a single pass, and increased almost 2.5-fold with further processing. Increasing EDTA exposure time enhanced recovery for all but the 10-pass case. NCI-H1650 and LS 174T spheroids required at least 20 min for complete trypsin dissociation (ESI†), and after 5 min total recoveries were only 30% for NCI-H1650 and 8% for LS 174T cases. The microfluidic device improved recovery by 2- and 10-fold, respectively (Fig. 5B and C). Combined EDTA-device treatments had lower total recoveries, but still exceeded the short trypsin treatment. In all cases, the microfluidic device improved single cell content (Fig. 5D–F). Representative cell counter histograms for all conditions are shown in the ESI.† Based on these findings, microfluidic dissociation significantly augments enzymatic digestion, resulting in enhanced single and total cell yields. Notably, comparable recoveries were obtained using the enzyme-free combination of EDTA and device treatments, which could be beneficial for preserving key surface proteins. As a final note, EDTA may also serve as an anticoagulant for clinical specimens, which can be contaminated with blood from procurement procedures.4
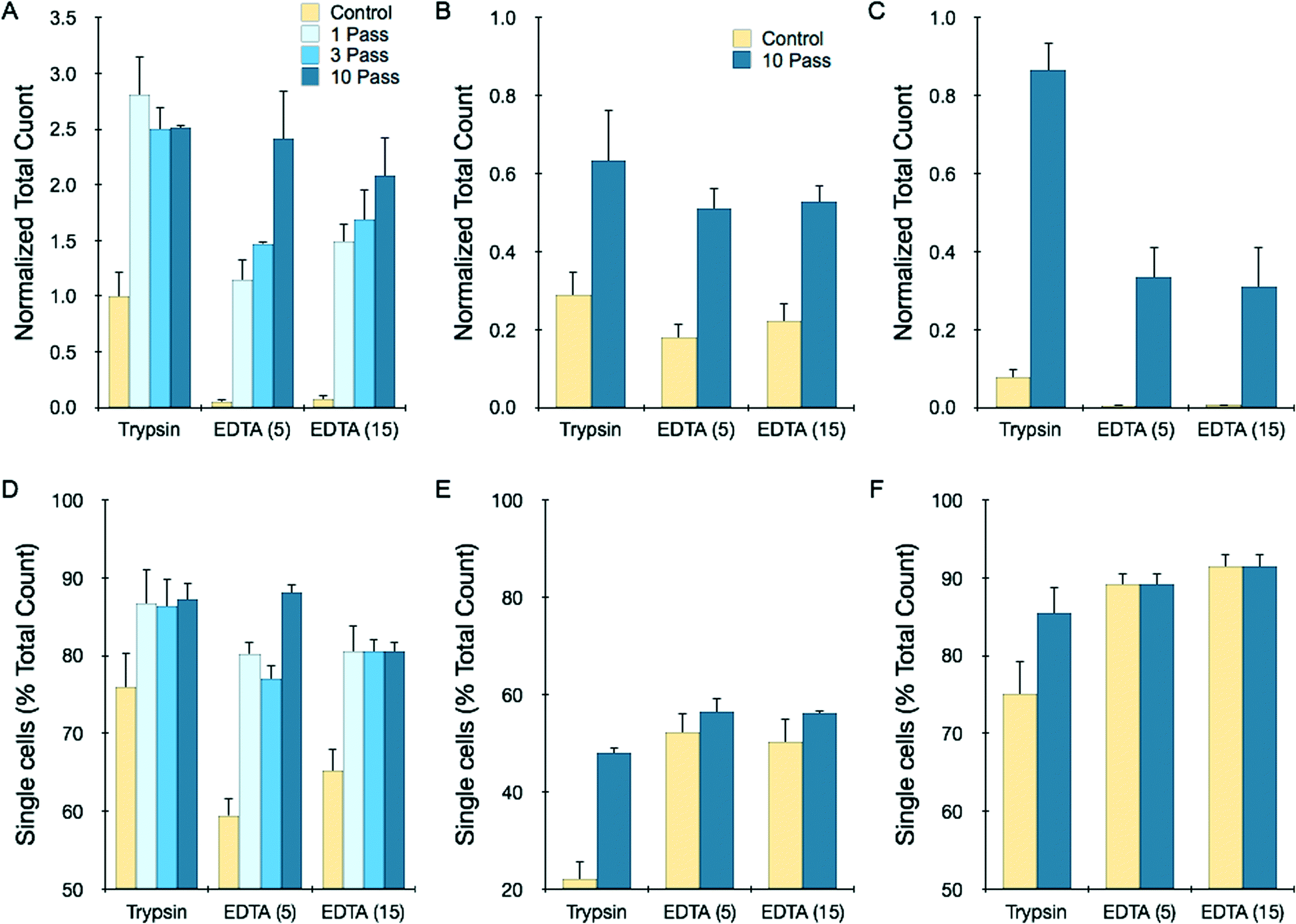 | ||
| Fig. 5 Microfluidic dissociation augments trypsin and EDTA treatments. Total recovery (A–C) and single cell percentage (D–F) for different spheroid types following treatment with trypsin for 5 min, EDTA for 5 min, or EDTA for 15 min, with or without microfluidic device processing at 12.5 mL min−1. Controls received mechanical treatment by pipetting and vortexing. (A,D) For HCT 116 spheroids, the device increased total recovery for both treatments by 2- to 3-fold, with improved single cell content as well. Maximal results were obtained after 1 pass for the trypsin case, while additional passes enhanced EDTA results. (B,E) For NCI-H1650 spheroids, samples were only partially dissociated after brief trypsin and EDTA treatments, but 10 passes through the microfluidic device improved total recovery by more than 2-fold. (C,F) For LS 174T spheroids, brief trypsin and EDTA treatments were extremely inefficient, but were enhanced by more than 10-fold following microfluidic device processing (10 passes). All total count results were normalized to the maximum count determined by fully digesting each spheroid type with trypsin (see ESI†). Error represent the standard error from at least three independent experiments. | ||
Analysis of cell suspensions by flow cytometry
We further characterized the cell suspensions achieved from the different dissociation procedures using flow cytometry. First, we utilized light scattering information to confirm our conclusions regarding single cell content. This was done by plotting the forward-scatter width (FSC-W) versus the forward-scatter area (FSC-A). These values are directly related for single cells, uniformly aligning data points along a constant axis. Representative results for HCT 116 spheroids treated with trypsin, trypsin followed by device processing (12.5 mL min−1, 10 passes), and EDTA followed by device processing are shown in Fig. 6A. Note the data points located within the gated rectangle region are the expected single cells. Non-symmetric components such as doublets and larger order clusters have a relatively larger FSC-A, shifting data points to the right of the plot. Single cell percentages were calculated based on the number of events inside the gated region relative to total events, and are superimposed on the flow cytometry plots. Results were similar for NCI-H1650 and LS 174T spheroids (ESI†), and for all cases closely matched cell counter results shown in Fig. 5D–F.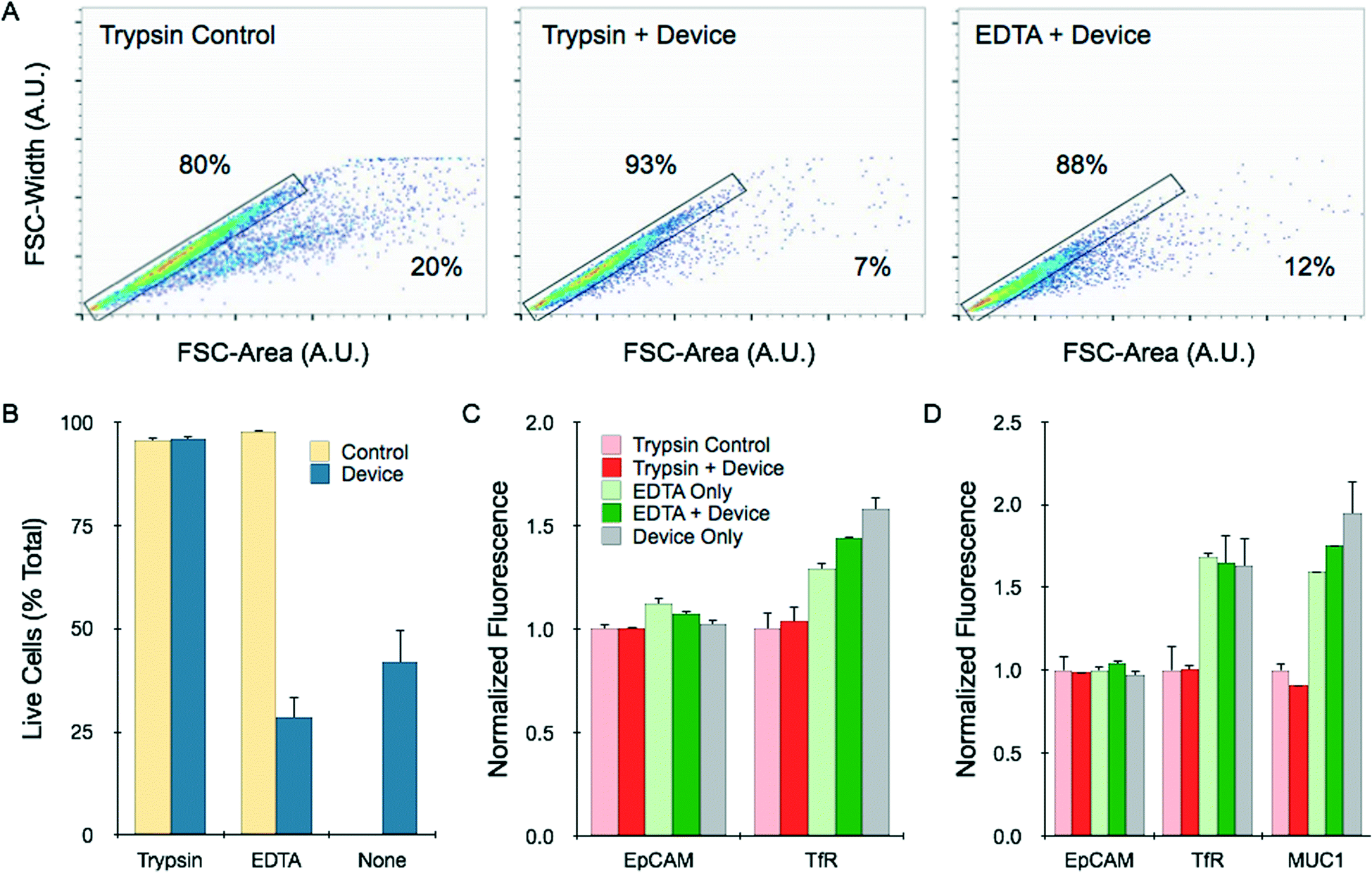 | ||
| Fig. 6 Flow cytometry analysis of single cell content, viability, and molecular expression. (A) Single cells and aggregates were identified by plotting the forward scatter (FSC)-width versus FSC-area for HCT 116 spheroids under different dissociation conditions. Controls received mechanical treatment by pipetting and vortexing. Single cells fall within the gated region, while aggregates are shifted to the right, and percentages are shown. (B) Cell viability was assessed by PI exclusion assay. Live cells were gated based on PI fluorescence signal, using unstained control cells as a baseline (see ESI†). Viability was not affected by device processing if the sample was treated first with trypsin, but did decrease significantly if treated first with EDTA or not treated at all. (C) HCT 116 spheroids were stained for the surface biomarkers EpCAM and TfR. EpCAM is a homotypic cell adhesion molecules, and expression was similar under all dissociation conditions. TfR is sensitive to trypsin cleavage, resulting in lower expression. Device treatment did not alter expression in either case. (D) Similar results were observed for NCI-H1650 spheroids. MUC1 expression was also measured, and showed trypsin sensitivity. All fluorescent signals were normalized to controls that were digested with trypsin. Error represent the standard error from at least three independent experiments. | ||
To examine the impact of microfluidic dissociation on cell viability, we next performed a propodium iodide (PI) exclusion assay to identify whether recovered cells were likely to be alive or dead. Live cells were gated based on unstained cells that were dissociated using only trypsin (see ESI†). Dead cell percentages were in the range of 5% for HCT 116 cells treated with trypsin or EDTA (Fig. 6B). Results were similar for the combination of trypsin and 10-pass device treatments, and we further confirmed that device processing did not affect cell growth (ESI†). However, the combination of EDTA and device processing, as well as device treatment alone, decreased cell viability to less than 40%. Cell distress is further indicated for these samples by the population shift to lower forward scatter values in Fig. 6A. Therefore, we conclude that the device can rapidly produce single, intact cells in a purely non-enzymatic manner, but this is associated with significant damage.
Finally, we measured the expression of specific surface protein biomarkers to assess cell quality for diagnostic and cell sorting applications. We selected classic cell surface tumor biomarkers for this study, including epithelial cell adhesion molecule (EpCAM), transferrin receptor (TfR), and mucin 1 (MUC1). We have previously observed that TfR and MUC1 are cleaved by trypsin, leading to lower expression levels. EpCAM is not sensitive to trypsin, but is a homotypic cell–cell junction protein that could be affected by mechanical separation. We found that EpCAM expression on cells obtained from HCT 116 and NCI-H1650 spheroids was similar for all dissociation treatments, including when the device was used exclusively for dissociation (Fig. 6C and D). Representative histograms for all conditions are shown in ESI.† Brief exposure to trypsin did lower expression of TfR and MUC1 expression relative to EDTA treatment, by approximately 25 to 50%. Differences were more pronounced after longer trypsin digestion times (ESI†). Device treatment for 10 passes did not significantly alter trypsin or EDTA results. Slightly elevated expression of TfR and MUC1 was observed when the device was used alone. This could be related to the loss of membrane integrity noted in the PI assay (Fig. 6B), and potentially labeling of intracellular stores of the targets. Note that HCT 116 cells do not express appreciable levels of MUC1. These findings confirmed that non-enzymatic dissociation using the dissociation device can still be useful for direct diagnostic applications, and may help preserve surface biomarker expression.
Conclusions
Here, we present a novel microfluidic device for mechanically dissociating digested tissues and cell aggregates into single cells. This device utilizes a branching channel network and repeated constrictions to generate well-defined and multi-scaled fluid shear force challenges, acting as “hydrodynamic micro-scalpels”, to disaggregate samples. We demonstrated using in vitro cancer cell models that the microfluidic device produces significantly higher total cell numbers, with a greater percentage of single cells, when compared to vortexing and pipetting treatments. Microfluidic dissociation produced superior results for simple models such as small clusters and intact monolayer sheets without requiring digestion or chemical treatment. For more complex tumor spheroids, the device dramatically augmented trypsin and EDTA treatments. The combination of brief EDTA exposure followed by device processing is particularly interesting as a non-enzymatic method, resulting in higher cell recovery without affecting surface protein expression. This could significantly improve results for applications that rely on protease sensitive surface markers.We are currently pursuing ways to improve this initial prototype, investigating different channel dimensions and features to increase mechanical dissociation efficiency. This would improve overall capabilities and enable comparable results to be achieved at lower flow rates that may be more compatible with specific downstream applications. We are also evaluating new designs to minimize sample holdup and clogging. Further investigations into preserving cell viability using milder enzymes, different chemical treatments, and new microfluidic processing elements are currently being pursued. For processing speed, the high flow rates that were required to achieve efficient dissociation required only five seconds per pass for the 1 mL sample volumes. We should note that this is a reasonable processing rate to allow for manual flow actuation with a standard 1 mL volume laboratory pipetter. Multiple pass experiments required less than two minutes, and the brief trypsin or EDTA incubations employed led to processing times of less than ten minutes.
Considering future application to human clinical tumor specimens, these tissues may be substantially more complex than the cell culture models that we have employed in this work. Our model tissues only contained a single cancer cell type, did not have physiological structures such as blood vessels, and most likely had lower stromal content. Furthermore, sample dimensions were either centimeter scale monolayers or spheroids that were hundreds of microns in diameter. Human tumor tissue specimens obtained by resection, surgical biopsy, or core needle biopsy will be composed of heterogeneous cell populations held together by significant stroma. Future studies will seek to evaluate device performance using these types of samples, either obtained from xenograft tumor models or human specimens. Finally, we believe our device approach is particularly well-suited for small volume clinical specimens such as FNA biopsies that we have worked with previously.4 The FNA technique uses smaller 22 gauge needles (~400 μm orifice) to withdraw small tissue fragments and individual cells. This makes FNAs less invasive, minimizing patient morbidity while also increasing potential for repeated sampling in both spatial and temporal (i.e. before and after drug treatment) contexts.25,26 However, FNA samples are difficult to work with for diagnostic applications because of their low yields, potentially on the order of only 10000 cells.4,27–29 It is possible that the simple tissue models we have used in this work may have realistically reflected the small tissue fragments and cell aggregates that are characteristic of FNA specimens. We have already shown that the microfluidic device can operate with small sample sizes (10000 cells or single spheroids).
Materials and methods
Device fabrication
The dissociation device was produced using a commercial microfabrication process offered by ALine, Inc. (Rancho Dominguez, CA). This is a laminate technology that uses a laser to cut the desired pattern in hard plastic sheets. Multiple layers are then aligned and fused using adhesive and pressure lamination. The channel features were designed in AutoCAD and arranged over three polyethylene terephthalate (PET) layers that were each 250 μm thick (Fig. 1B). The channel layers were separated by two 125 μm thick PET layers that contained vias, which had diameters equal to the minimum cross-section of the subsequent stage. The device was sealed using two additional layers, for a total of seven. The top was a 1.5 mm thick layer of acrylic and had two holes drilled for the placement of hose barbs that served to interface the inlet and outlet tubing. The bottom layer was a 125 μm thick solid slab of PET.Fluid dynamics simulations
To characterize flow profiles and shear stresses within the device, particularly the channel constrictions where dissociation is primarily expected to occur, we performed finite-element fluid dynamics simulations using COMSOL Multiphysics software. Simulations were conducted by coupling the Navier–Stokes equations and continuity equation, assuming laminar flow and applying the no-slip condition at the channel walls. Fluid density and viscosity were assumed to be that of water. Simulations were performed for a single channel within each stage because full device calculations were complex and the laminate format causes the fluid to change directions as it passes between stages. Channel specifications were imported from the AutoCAD files used for device fabrication, and included the inlet, all expansions and constrictions, and the branch point (see Fig. 1B). Channel dimensions are given in Table 1. For the inlet condition, we used the average velocity (vavg), which is the same for all channels throughout the device because both channel height and total width across all channels within each stage are constants. Outlet pressure was assumed to be zero to determine the pressure drop (ΔP). Flow profiles are shown in Fig. 1C for a flow rate of 1 mL min−1, with maximum velocity (vmax) and pressure drop values given in Table 1. Total pressure drop (ΔPTotal) for the device was calculated by summing the channel pressure drops in parallel to determine the total value for the stage, and summing the results for each stage in series. For a 1 mL min−1 operating flow rate, ΔPTotal is only 60 Pa, or 0.009 PSI. Results for different flow rate conditions scale proportionally. Simulation outputs that included the first and second stages in sequence were similar to that of the individual channels (ESI†).Cell culture and tissue models
Human colon cancer cell lines HCT 116 and LS 174T and lung cancer cell line NCI-H1650 were obtained from ATCC (Manassas, VA). Cells were cultured in standard tissue culture flasks using DMEM media containing 10% FBS and 1% penicillin-streptomycin at 37 °C in 5% CO2. Confluent monolayers were passaged using trypsin-EDTA (Corning, Corning, NY) digestion for 5 to 15 min, depending on sensitivity of the cell line, at 37 °C. HCT 116 and LS 174T cell suspensions used for device testing were obtained in similar manner to the passaging procedures. Monolayer tumor sheets were prepared by growing HCT 116 cells in collagen-coated 24-well plates (Life Technologies, Carlsbad, CA) to confluency, washing with Hank’s Balanced Salt Solution (Corning, Corning, NY), incubating with 1 mg mL−1 Type II collagenase (Life Technologies, Carlsbad, CA) for 5 min, and releasing the intact monolayer by agitation. Tumor spheroids were grown in MicroWell MiniTrays (Thermo Fisher Scientific, Waltham, MA) using the hanging drop method.30 Briefly, 20 μL of media containing approximately 500 cells was added to each well, the plate was inverted, and the cells were cultured in the meniscus formed at the air-liquid interface until cohesive spheroids formed and reached approximately 300 μm diameter. Individual spheroids were recovered using a P-200 pipette, and pooled for experiments. For spheroids that received pre-treatment with trypsin-EDTA or EDTA (Cellstripper, Corning, Corning, NY), the sample was centrifuged, resuspended in the treatment, and incubated for the indicated time period. All samples (cell suspensions, tumor monolayer sheets, and tumor spheroids with or without pre-treatment) were prepared for experiments by centrifugation and resuspension in 1 mL PBS containing 1% BSA (PBS+). Control samples were only treated with trypsin-EDTA or EDTA, followed by vortexing and repeated pipetting.Dissociation studies
The dissociation device was prepared by affixing 3′′ PVC 1/32ID tubing (Nalgene, Rochester, NY) to the hose barbs at both the inlet and the outlet. A 3-way valve was added to the inlet tubing, with connections to a syringe pump (Harvard Apparatus, Holliston, MA) and a buffer reservoir. Prior to use, devices were filled with Superblock blocking buffer (Thermo Fisher Scientific, Waltham, MA) and incubated for 15 minutes to prevent non-specific adhesion of cells to the channel walls, and then flushed with PBS+. Cell or tumor tissue samples were loaded into a syringe, and administered to the device using a syringe pump with flow rate set between 0.2 to 12.5 mL min−1. The latter flow rate was the maximum we could generate with our syringe pump, and required only 5 s to run the 1 mL sample. For single pass studies, the sample was collected and the device was flushed at the same flow rate with 1 mL PBS+ that was obtained via the buffer reservoir at the inlet. Sample and wash effluents were combined prior to analysis. For multiple pass experiments, flow was reversed using the syringe pump operating at the same flow rate in withdraw mode to replace the sample back into the syringe in preparation for the next run. This process was repeated for the indicated number of passes, and afterward the sample was collected and device flushed as described above. Cell concentration was measured using a Moxi Z cell counter with type S cassettes (Orflo, Hailey, ID), which utilizes the Coulter principle. The cassettes contain a pre-filter to remove large cell aggregates and the device only measures cell sizes up to 26 μm diameter, thus only single cells and small aggregates are counted. To determine if large aggregates were present in a sample, the entire volume was centrifuged, resuspended in trypsin-EDTA, incubated for 5 min, vortexed, pipetted repeatedly, and a second cell count was obtained. The difference in the two cell counts was attributed to large aggregates. Single cells were defined by first identifying the peak size of the population from the cell counter histograms, and then applying an equal distribution to both smaller and larger sizes. Mean cell diameters were approximately 13 μm for cell suspensions and monolayer sheets and 17 μm for spheroids. Sizes were corroborated by visual inspection of samples using a microscope. Total recoveries for all experimental conditions were normalized to the control samples that were treated with trypsin-EDTA, vortexing, and repeated pipetting. Single cells were represented as the percentage of counts that fell within the single cell size range relative to the total count. Data are represented as the mean ± standard error determined from at least three independent experiments.Flow cytometry
Single cells were discriminated from aggregates in dissociated spheroid samples by plotting forward-scattering width (FSC-W) versus forward-scattering area (FSC-A). Single cells fall on a line of constant slope, and were gated. Aggregates have higher FSC-A, shifting data points away from the single cell region. Single cells were represented as the percentage of data points that fell within the gated region relative to the total population. Cell scattering was acquired using an LSR II flow cytometer (Becton Dickenson, Franklin Lakes, NJ), and analyzed using FlowJo software (Tree Star, Ashland, OR).Cell viability was assessed for HCT 116 cells by incubating with 2.7 μg mL−1 propidium iodide (Millipore, Billerica, MA) in 300 μL PBS+. PI cannot penetrate live cells with fully intact plasma membrane, but enters dead cells resulting in strong fluorescence signal. Cell scattering and fluorescence signal intensities were acquired using an Accuri C6 flow cytometer (Becton Dickenson, Franklin Lakes, NJ), and analyzed using FlowJo software (Tree Star, Ashland, OR). Dot plots were prepared by plotting side scatter (SSC) versus fluorescence intensity, and live cells were identified by creating a gate based on unstained control cells from spheroids that were treated only with trypsin. The percentage of live cells was determined based on the number of counts within the live gate versus total counts. Data are represented as the mean ± standard error determined from at least three independent experiments.
Biomarker labeling studies were performed by incubating samples with monoclonal antibodies specific to human EpCAM (mouse IgG2B, clone 158206, R&D Systems, Minneapolis, MN), transferrin receptor (TfR, mouse IgG1, clone 29806, R&D Systems, Minneapolis, MN), or mucin 1 (MUC1, mouse IgG1, clone M01102909, Fitzgerald Industries International, Acton, MA). A non-binding monoclonal antibody (rat anti-mouse IgG1, clone A85-1, BD Biosciences, San Jose, CA) was used as a control. Cell suspensions were centrifuged, resuspended in PBS+ containing 5 μg mL−1 primary antibody, and incubated for 30 min at room temperature. Samples were then washed twice with ice-cold PBS+ by centrifugation, resuspended in ice-cold PBS+ containing 2 μg mL−1 fluorescein-conjugated secondary antibody (rat anti-mouse IgG1, clone A85-1, or anti-mouse IgG2a/2b, clone R2-40, BD Biosciences, San Jose, CA), incubated for 30 min on ice, washed twice with PBS+, and resuspended in 0.25 mL PBS+. Cell scattering and fluorescein signal intensities were acquired using an LSR II flow cytometer (Becton Dickenson, Franklin Lakes, NJ), and analyzed using FlowJo software (Tree Star, Ashland, OR). Geometric mean fluorescence intensity values were measured for single cells, after subtracting background signals obtained with the control antibody. All signals were normalized to the control samples that were treated with trypsin-EDTA, vortexing, and repeated pipetting. Data are represented as the mean ± standard error determined from at least three independent experiments.
Acknowledgements
We thank Dr. Elliot Hui, Dr. Marian Waterman, and Dr. Robert Edwards for many helpful discussions. We would also like to thank Vanessa Scarfone and Trisha Westerhof for assistance with flow cytometry studies and Maha Rahim for critical review of the manuscript. This work was supported in part by the Hellman Foundation, the National Cancer Institute of the National Institutes of Health under Award Number P30CA062203, and the Defense Advanced Research Projects Agency (DARPA) N/MEMS S&T Fundamentals Program under grant number N66001-1-4003 issued by the Space and Naval Warfare Systems Center Pacific (SPAWAR) to the Micro/nano Fluidics Fundamentals Focus (MF3) Center.References
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- P. L. Bedard, A. R. Hansen, M. J. Ratain and L. L. Siu, Nature, 2013, 501, 355–364 CrossRef CAS PubMed.
- S. C. Bendall, E. F. Simonds, P. Qiu, E.-A. D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687–696 CrossRef CAS PubMed.
- J. B. Haun, C. M. Castro, R. Wang, V. M. Peterson, B. S. Marinelli, H. Lee and R. Weissleder, Sci. Transl. Med., 2011, 3, 71ra16 Search PubMed.
- D. Issadore, J. Chung, H. Shao, M. Liong, A. A. Ghazani, C. M. Castro, R. Weissleder and H. Lee, Sci. Transl. Med., 2012, 4, 141ra92 Search PubMed.
- D. A. Ateya, J. S. Erickson, P. B. J. Howell, L. R. Hilliard, J. P. Golden and F. S. Ligler, Anal. Bioanal. Chem., 2008, 391, 1485–1498 CrossRef CAS PubMed.
- J. Sun, M. D. Masterman-Smith, N. A. Graham, J. Jiao, J. Mottahedeh, D. R. Laks, M. Ohashi, J. DeJesus, K. Kamei, K. B. Lee, H. Wang, Z. T. Yu, Y. T. Lu, S. Hou, K. Li, M. Liu, N. Zhang, S. Wang, B. Angenieux, E. Panosyan, E. R. Samuels, J. Park, D. Williams, V. Konkankit, D. Nathanson, R. M. van Dam, M. E. Phelps, H. Wu, L. M. Liau, P. S. Mischel, J. A. Lazareff, H. I. Kornblum, W. H. Yong, T. G. Graeber and H. R. Tseng, Cancer Res., 2010, 70, 6128–6138 CrossRef CAS PubMed.
- A. K. White, M. VanInsberghe, O. I. Petriv, M. Hamidi, D. Sikorski, M. A. Marra, J. Piret, S. Aparicio and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13999–14004 CrossRef CAS PubMed.
- W. H. Grover, A. K. Bryan, M. Diez-Silva, S. Suresh, J. M. Higgins and S. R. Manalis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10992–10996 CrossRef CAS PubMed.
- D. R. Gossett, H. T. Tse, S. A. Lee, Y. Ying, A. G. Lindgren, O. O. Yang, J. Rao, A. T. Clark and D. Di Carlo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7630–7635 CrossRef CAS PubMed.
- A. D. Boiko, O. V. Razorenova, M. van de Rijn, S. M. Swetter, D. L. Johnson, D. P. Ly, P. D. Butler, G. P. Yang, B. Joshua, M. J. Kaplan, M. T. Longaker and I. L. Weissman, Nature, 2010, 466, 133–137 CrossRef CAS PubMed.
- P. Dalerba, S. J. Dylla, I. K. Park, R. Liu, X. Wang, R. W. Cho, T. Hoey, A. Gurney, E. H. Huang, D. M. Simeone, A. A. Shelton, G. Parmiani, C. Castelli and M. F. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10158–10163 CrossRef CAS PubMed.
- K. S. Chan, I. Espinosa, M. Chao, D. Wong, L. Ailles, M. Diehn, H. Gill, J. J. Presti, H. Y. Chang, M. van de Rijn, L. Shortliffe and I. L. Weissman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14016–14021 CrossRef CAS PubMed.
- M. E. Prince, R. Sivanandan, A. Kaczorowski, G. T. Wolf, M. J. Kaplan, P. Dalerba, I. L. Weissman, M. F. Clarke and L. E. Ailles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 973–978 CrossRef CAS PubMed.
- R. Strauss, Z. Y. Li, Y. Liu, I. Beyer, J. Persson, P. Sova, T. Moller, S. Pesonen, A. Hemminki, P. Hamerlik, C. Drescher, N. Urban, J. Bartek and A. Lieber, PLoS One, 2011, 6, e16186 CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- L. Y. Yeo, H. C. Chang, P. P. Chan and J. R. Friend, Small, 2011, 7, 12–48 CrossRef CAS PubMed.
- A. Lenshof and T. Laurell, Chem. Soc. Rev., 2010, 39, 1203–1217 RSC.
- D. R. Gossett, W. M. Weaver, A. J. Mach, S. C. Hur, H. T. Tse, W. Lee, H. Amini and D. Di Carlo, Anal. Bioanal. Chem., 2010, 397, 3249–3267 CrossRef CAS PubMed.
- S. M. Hattersley, C. E. Dyer, J. Greenman and S. J. Haswell, Lab Chip, 2008, 8, 1842–1846 RSC.
- L. Wallman, E. Akesson, D. Ceric, P. H. Andersson, K. Day, O. Hovatta, S. Falci, T. Laurell and E. Sundstrom, Lab Chip, 2011, 11, 3241–3248 RSC.
- C. H. Lin, D. C. Lee, H. C. Chang, I. M. Chiu and C. H. Hsu, Anal. Chem., 2013, 85, 11920–11928 CrossRef CAS PubMed.
- A. C. Hatch, J. S. Fisher, S. L. Pentoney, D. L. Yang and A. P. Lee, Lab Chip, 2011, 11, 2509–2517 RSC.
- A. J. Mach, J. H. Kim, A. Arshi, S. C. Hur and D. Di Carlo, Lab Chip, 2011, 11, 2827–2834 RSC.
- S. Krishnamurthy, Cancer, 2007, 111, 106–122 CrossRef PubMed.
- D. P. Clark, Cancer, 2009, 117, 289–297 Search PubMed.
- T. Crnogorac-Jurcevic, E. Efthimiou, P. Capelli, E. Blaveri, A. Baron, B. Terris, M. Jones, K. Tyson, C. Bassi, A. Scarpa and N. R. Lemoine, Oncogene, 2001, 20, 7437–7446 CrossRef CAS PubMed.
- W. F. Symmans, M. Ayers, E. A. Clark, J. Stec, K. R. Hess, N. Sneige, T. A. Buchholz, S. Krishnamurthy, N. K. Ibrahim, A. U. Buzdar, R. L. Theriault, M. F. Rosales, E. S. Thomas, K. M. Gwyn, M. C. Green, A. R. Syed, G. N. Hortobagyi and L. Pusztai, Cancer, 2003, 97, 2960–2971 CrossRef CAS PubMed.
- B. A. Centeno, S. A. Enkemann, D. Coppola, S. Huntsman, G. Bloom and T. J. Yeatman, Cancer, 2005, 105, 101–109 CrossRef PubMed.
- J. M. Kelm, N. E. Timmins, C. J. Brown, M. Fussenegger and L. K. Nielsen, Biotechnol. Bioeng., 2003, 83, 173–180 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc01126k |
| ‡ These authors contributed equally to this work. |
| § J.D.J. and J.B.H. proposed the concept of the work. X.Q., J.D.J., and M.P. carried out the experimental work. X.Q., J.D.J., and J.B.H. carried out the experimental analysis. X.Q., J.D.J, and M.T. carried out theoretical analysis. X.Q. and J.B.H. wrote the paper. |
| This journal is © The Royal Society of Chemistry 2015 |