
A quantitative microfluidic angiogenesis screen for studying anti-angiogenic therapeutic drugs†
Choong
Kim
ad,
Junichi
Kasuya
a,
Jessie
Jeon
b,
Seok
Chung
e and
Roger D.
Kamm
*abc
aDepartments of Biological Engineering and Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: rdkamm@mit.edu
bDepartments of Biological Engineering and Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cBiosystems and Micromechanics IRG, Singapore-MIT Alliance for Research and Technology, Singapore 117543
dSchool of Mechanical and Automotive Engineering, Kyungil University, Daegu, 712-701, South Korea
eSchool of Mechanical Engineering, Korea University, Seoul 136-701, South Korea
First published on 5th November 2014
Abstract
Anti-angiogenic therapy, which suppresses tumor growth by disrupting oxygen and nutrient supply from blood to the tumor, is now widely accepted as a treatment for cancer. To investigate the mechanisms of action of these anti-angiogenesis drugs, new three dimensional (3D) cell culture-based drug screening models are increasingly employed. However, there is no in vitro high-throughput screening (HTS) angiogenesis assay that can provide uniform culture conditions for the quantitative assessment of physiological responses to chemoattractant reagents under various concentrations of anti-angiogenesis drugs. Here we describe a method for screening and quantifying the vascular endothelial growth factor (VEGF)-induced chemotactic response on human umbilical vein endothelial cells (HUVECs) cultured with different concentrations of bortezomib, a selective 26S proteasome inhibitor. With this quantitative microfluidic angiogenesis screen (QMAS), we demonstrate that bortezomib-induced endothelial cell death is preceded by a series of morphological changes that develop over several days. We also explore the mechanisms by which bortezomib can inhibit angiogenesis.
Introduction
Drug screening technologies in two dimensions (2D) (i.e. as a flat layer of cells) have been widely adopted in the pharmaceutical industry for drug discovery. These approaches are still in use, however considerable challenges remain in identifying new drugs for cancer treatment due to in particular the critical role of multiple cell types in disease progression and the inability to accurately mimic the cellular environment in vivo.1–5 Consequentially, a wide gap exists between 2D cell culture assays and animal testing.6 Moreover, pressure is mounting to reduce animal testing due to its expense, the long time required to obtain results, ethical considerations, and the limitations of an animal model in predicting human responses.7,8 In order to overcome the drawbacks of 2D cell culture assays and potentially reduce the need for animal testing, new analytical screening assays in three dimensions (3D) employing human cells are needed. Cells in vivo are subject to multiple cues that vary in time and space, including gradients of cytokines and secreted proteins from neighboring cells of similar or different types, and their behavior is strongly influenced by mechanical and biochemical interactions with the extracellular matrix (ECM). To meet these challenges, microfluidic lab-on-a-chip (LOC) technologies offer promising strategies for addressing the inherent complexity of cellular systems with spatio-temporal multiple cues.9,10 In addition, microfluidic approaches have been adopted to create 3D tissue scaffolds with realistic physical, mechanical, and biological properties.3D cell culture-based microfluidic devices have been previously reported, which enable the formation of cell spheroids in 3D to mimic the complexity of heterogeneous tumor tissue, for cytotoxicity tests of anticancer drugs.11–15 These systems could be used as an assay for screening anticancer drugs in cancer treatment, but, currently, anti-angiogenic therapies (vascular-targeted therapies), which suppress tumor growth by cutting off the supply of nutrients and oxygen from blood to tumor cells, have become an important advancement in cancer treatment with a significant reduction in the adverse effects caused by other anti-cancer drugs.16–19 The potential now exists to develop new approaches to anti-angiogenesis drug screening which would enable monitoring and quantification of cell responses to anti-angiogenesis drugs in addition to screening for cell viability. Recent efforts have produced in vitro microfluidic cell culture models that apply physical and biochemical stimuli within a 3D hydrogel scaffold integrated in the channels and offer a viable solution for monitoring cellular behaviors in response to drugs.17–31 For example, these models have been employed to investigate heterotypic cell–cell interactions in 3D,22 to study neurite responses to growth factor gradients,23 to evaluate and quantify capillary sprouting and angiogenesis from an intact cell monolayer,24,26 and to examine the effects of interstitial flow on cancer cell morphology and migration.26
However, the previously developed microfluidic based-platforms are ill-suited for the purpose of screening the effects of various anti-angiogenesis drugs over a range of concentrations. For example, preparing and handling many previously developed microfluidic based-platforms can be time-consuming and tedious. Moreover, higher variability may occur between different chips due to slight variations in the protocol caused by testing a large number of chips manually.
Here, we propose a new microfluidic platform (termed the quantitative microfluidic angiogenesis screen (QMAS)) that can monitor and quantify cellular behaviors, such as morphological changes, endothelial cell viability, and formation of angiogenic sprouts, depending on the various concentrations of drugs applied (i.e., bortezomib, a selective 26S proteasome inhibitor). This model incorporates a perfusion culture-based system to produce stable concentration gradients in multiple 3D collagen matrices by generating a uniform pressure-driven flow rate through each of the microfluidic channels. We demonstrate these capabilities by studying a drug, bortezomib, which is known to inhibit angiogenesis in vivo. To gain insight into the anti-angiogenic effects of bortezomib on human umbilical vein endothelial cells (HUVECs), we monitored cell behaviors daily and investigated the changes in the morphological shape and viability of HUVECs treated with various concentrations of bortezomib. Capillary growth and the formation of circular lumen-like structures from an intact endothelial cell monolayer in response to the vascular endothelial growth factor (VEGF) were quantified, which showed that the size and the number of lumen-like structures are directly affected by the concentrations of bortezomib. We demonstrate that bortezomib inhibits angiogenesis in a dose-dependent manner and also that it causes the cell death of HUVECs at sufficiently high concentrations.
Results and discussion
Characterization of the QMAS for screening anti-angiogenic therapeutic drugs
Vascular sprouts grow from existing blood vessels into the interstitial matrix guided by a concentration gradient of factors (e.g., vascular endothelial growth factor (VEGF)) secreted from the tumor during tumor angiogenesis. When an anti-angiogenesis drug is employed in vivo for the treatment of cancer, it inhibits the formation of tumor-associated blood vessels, thereby disrupting the supporting perivascular niche for cancer stem cells16–18 (Fig. 1A). This in vivo anti-angiogenic therapeutic process was modeled in the QMAS (Fig. 1B & C) incorporating the following significant features: (1) 3D hydrogel scaffolds in gel cages (fourteen) that mimic the extracellular matrix (ECM) in vivo, (2) confluent endothelial cell (ECs) monolayers in the cell culture channels (eight) that mimic pre-existing blood vessels in vivo, (3) VEGF-supplemented media in conditioned media channels (seven) that mimic the presence of growth factors (VEGF) secreted by tumor cells in vivo, and (4) media supplemented with various concentrations of bortezomib in the cell culture channels (eight) that mimic the introduction of anti-angiogenesis drugs (i.e. bortezomib, a selective 26S proteasome inhibitor, marketed by Millennium Pharmaceuticals as Velcade) in vivo via circulation (Fig. 1B & C). The QMAS makes it possible for us to monitor, screen, and quantify the angiogenic responses in the 3D hydrogel scaffolds due to various concentrations of bortezomib. | ||
| Fig. 1 In vivo anti-angiogenesis application and microfluidic assay. (A) Potential anti-angiogenic therapeutic application in cancer treatment in vivo. (B) Schematic view of the QMAS for screening anti-angiogenic therapeutic drugs. (C) The significant parts of the QMAS are the: (1) 3D hydrogel scaffolds in gel cages (fourteen) that mimic the extracellular matrix (ECM) in vivo, (2) confluent endothelial cell (EC) monolayers in the cell culture channels (eight) that mimic pre-existing blood vessels in vivo, (3) VEGF-supplemented media in the conditioned media channels (seven) that mimic the presence of growth factors secreted by tumor cells in vivo, and (4) media supplemented with various concentrations of bortezomib in the cell culture channels (eight) that mimic the introduction of anti-angiogenesis drugs in vivo via circulation. | ||
The QMAS, fabricated using soft lithography and PDMS replica molding (Fig. S1,† see Materials and methods), consists of: (1) reservoirs used for providing fresh conditioned media (VEGF-supplemented or bortezomib-supplemented media) into all of the channels made by cutting the ends of commercial pipette tips (i.e. MBP (Molecular BioProducts)) (the media in the reservoirs were replaced daily during cell culture using a multi-tip pipette); (2) a 1st PDMS layer including microchannels for media supplemented with various concentrations of bortezomib, endothelial cell monolayers and collagen gel cages; (3) a 2nd PDMS layer as a manifold layer used only for filling collagen gels; and (4) a large cover slip at the bottom (Ted Pella, Inc., 3 × 3 1/4", thickness: 0.19–0.25 mm) (Fig. S1A & S1B†).
To mimic the presence of pro-angiogenic factors secreted from the tumor, VEGF (40 ng ml−1) was introduced into the QMAS, which then diffused into the 3D hydrogel scaffold regions (fourteen) simultaneously. However, establishing stable concentration profiles within the QMAS with integrated porous matrices requires that we eliminate any interstitial flow across the matrix resulting from pressure imbalances; however this proved challenging. To achieve stability of the concentration profiles, the QMAS was designed as a perfusion-based system. The stable concentration profiles could be created in the fourteen 3D hydrogel scaffold matrices by minimizing interstitial flow across them; this was accomplished by a combination of introducing identical volumetric flow rates into the fifteen independent flow channels and connecting the ends of all flow channels to a single syringe pump at the common outlet junction (Fig. S2A†). The distribution of the volumetric flow rates was controlled via design of the hydrodynamic resistance of the microchannels (Fig. S2B†). We confirmed that uniform flow rates were generated in each of the fifteen microchannels during perfusion using a commercial finite element solver (COMSOL, Burlington, MA) (Fig. S2C†).
Numerical simulations based on a transient solution of the Brinkman equation for porous medium flow and the convection–diffusion equation for a growth factor having the diffusivity of VEGF (D = 5 × 10−11 m2 s−1) demonstrated the generation of a nearly linear concentration gradient of the growth factor; details of the numerical simulations are provided in the ESI,† Materials and methods (Fig. S3). In order to maintain this gradient over the entire length of the gel regions, the media should be introduced at a flow rate corresponding to a Péclet number (Pe) >50. To satisfy this condition, the withdrawal flow rate in the syringe pump (Infuse/Withdraw syringe pump, Harvard) was set to 1.33 μl min−1; we used this flow rate for all experiments.
In the previously developed systems as well as the QMAS, the collagen hydrogel was confined to the gel regions by a series of equally spaced posts (110 μm, Fig. 1C) that prevent liquid from leaking into adjacent channels. Generally, collagen gel filling by a pipette was performed manually; this is a simple method, but leakage between adjacent channels is sometimes difficult to avoid. In order to improve the consistency of filling, we modified the collagen gel filling method using the manifold layer that can insert the gel to the fourteen gel regions simultaneously (Fig. S4†). When the collagen solution is injected into an inlet of the manifold layer by the syringe pump, the injected solution is distributed among the fourteen microchannels in the manifold layer (2nd PDMS layer), and then simultaneously enters the fourteen collagen gel regions (1st PDMS layer) (Fig. S4A†). The volumetric flow rates in the microchannel network of the manifold layer were controlled by specifying the dimensions, and thereby the hydrodynamic resistance, of the microchannels (Fig. S4B†). We again demonstrated that the same volumetric flow rates were generated in the 14 collagen gel regions using hydrodynamic simulation (COMSOL Inc., MA, USA) (Fig. S4C†). Experimentally, we observed that the consistency of collagen gel filling in the gel cages was influenced by the surface properties of the bottom layer; for example, a glass surface and a PDMS surface behaved differently. When a PDMS surface was used as the bottom layer, the collagen filling was more uniform than when glass was used due to its low wettability (data not shown). All reported experiments, however, were conducted with glass as the bottom layer to provide optimal imaging quality. Chip preparation details are provided in Materials & methods and in the ESI,† Movie 1.
High throughput angiogenesis assay
To demonstrate the capabilities of the QMAS as an angiogenesis assay in response to chemoattractant gradients, VEGF-supplemented media (40 ng ml−1) were introduced into the VEGF injection channels (#1, #3, #5, & #7) (Fig. S5† & 2A) and the control media without VEGF were injected into the VEGF injection channels (#2, #4, #6, & #7) as reference conditions (Fig. S5† & 2A). The response of HUVECs was monitored daily for 5 days by phase-contrast microscopy (CKK 41, Olympus, Tokyo, Japan). On day 5, the HUVECs were fixed and stained to examine the cell morphologies. Cells rapidly and actively migrated from the endothelial channel into the 3D hydrogel scaffolds (condition-side) toward the VEGF-supplemented media channel (Fig. 2B); on the other hand, significantly less migration was observed into the opposite scaffold (control-side) toward the control channel without VEGF (Fig. 2D). Confocal microscopy revealed the growth of circular lumen-like structures from the HUVEC monolayer into the condition-side hydrogel scaffolds in response to VEGF (Fig. 2C). No morphological change was evident in the control-side hydrogel scaffolds in the immunostained and confocal microscopy images except that several cells migrated. These cells were not connected to the endothelial monolayer by vascular stalks presumably due to the absence of a VEGF concentration gradient across the collagen gel (Fig. 2E). These qualitative observations were quantified by counting the number of cells that migrated from each opening (total openings: 6, Fig. 2A) into the collagen gel scaffolds using ImageJ (http://rsbweb.nih.gov/ij/). We found that 79 ± 9.8 cells migrated and proliferated toward the condition-side hydrogel scaffolds; in contrast, 8 ± 2.3 cells migrated toward the control-side hydrogel scaffolds (Fig. 2F). Using this migration assay, we could assess the response of an endothelial monolayer to various biochemical stimuli simultaneously via achieving a stable concentration gradient toward the fourteen 3D hydrogel scaffolds. The QMAS therefore allows us to monitor and quantify angiogenesis in in vivo-like situations under uniform experimental conditions (i.e. uniform 3D hydrogel structure conditions & hydrostatic conditions between multiple microchannels) simultaneously and with little variability due to the operator's technique.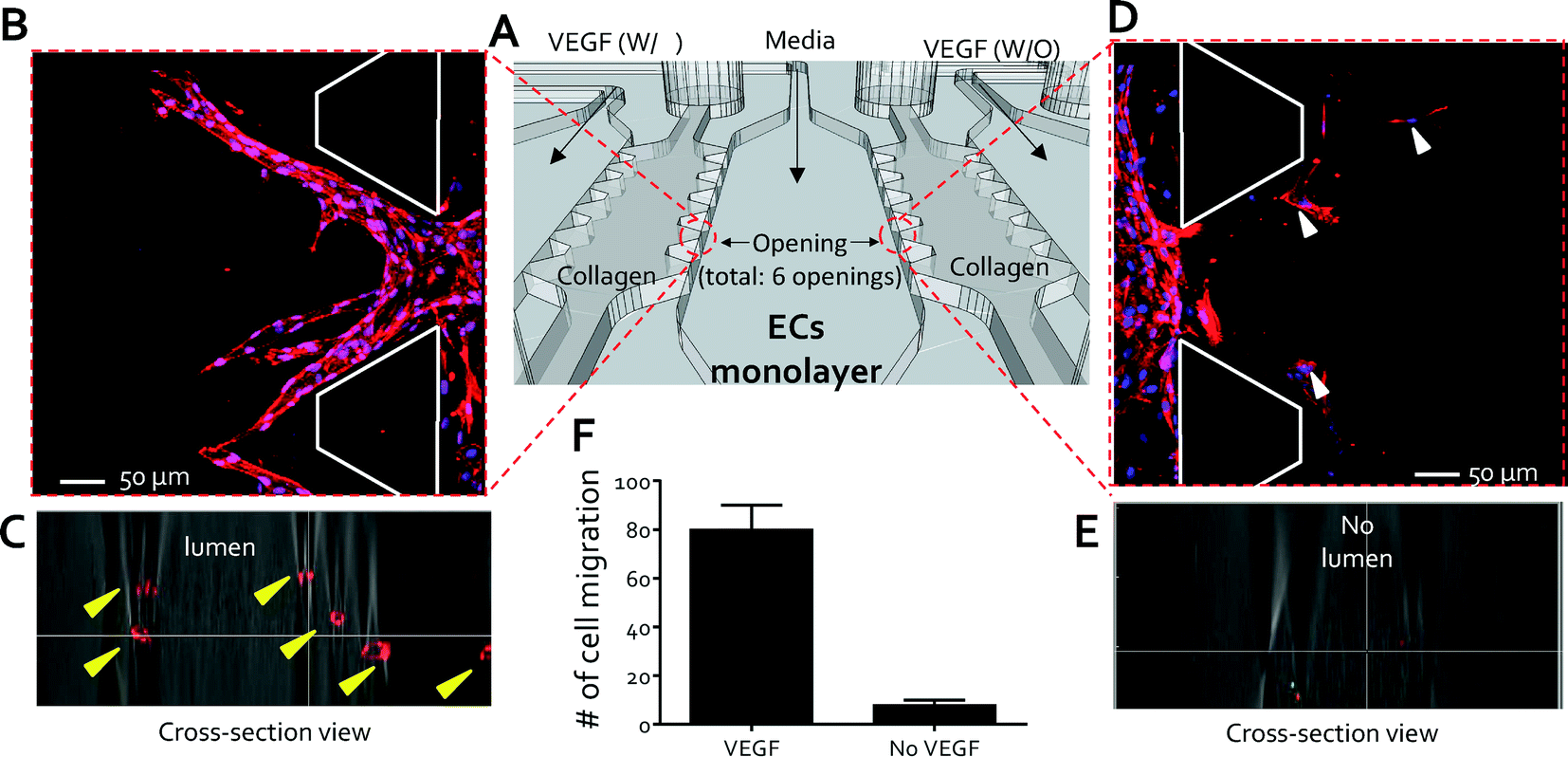 | ||
| Fig. 2 High throughput angiogenesis assay. (A) Schematic view of a set of experiments to demonstrate the capabilities of the QMAS as an angiogenesis assay in response to chemoattractant gradients: VEGF-supplemented media (40 ng ml−1) were introduced into the VEGF injection channels (#1, #3, #5, & #7, (Fig. S5†)) and control media without VEGF were injected into the VEGF injection channels (#2, #4, #6, & #7, (Fig. S5†)) as reference conditions. (B) Migration of the ECs from the endothelial channel into the 3D hydrogel scaffolds toward the VEGF-supplemented media channel (condition-side). (C) Growth of circular lumen-like structures into the condition-side hydrogel scaffolds in response to VEGF (yellow arrowheads, cross-section view). (D) Minimal migration of the ECs (white arrowheads) into the opposite scaffold toward the control channel without VEGF (control-side). (E) Confocal microscopy confirming the absence of circular lumen-like structures toward the control channel without VEGF (cross-section view). (F) Qualitative analysis showing the number of cells that migrated from each opening into the collagen gel scaffolds. | ||
Quantitative analysis of HUVEC morphology by bortezomib
Bortezomib (Millennium Pharmaceuticals, Inc., Cambridge, MA) was chosen to demonstrate the utility of the QMAS for drug screening. Bortezomib, one of the 14 anti-angiogenic therapeutic drugs which have been approved by the US Food and Drug Administration (FDA), exerts its anti-angiogenic effect by decreasing the secretion of the vascular endothelial growth factor (VEGF) from myeloma cells.32,33 We wanted to explore the possibility that bortezomib could act directly on the endothelium, independent of the myeloma cells. In order to model the in vivo situation (Fig. 1A), we first established endothelial cell monolayers on the walls of the channels to mimic in vivo blood vessels through the injection of endothelial cells (HUVECs) using a multiple pipette (drug injection channels: #1–#8). Second, VEGF (40 ng ml−1) supplemented media mimicking the growth factors (i.e. VEGF) secreted from the tumor were introduced in the VEGF injection channels, and finally, we added media containing a range of concentrations of bortezomib to each reservoir using the multiple pipette (drug injection channels: #1–#8, over a logarithmic range (Fig. S6) & a linear range (Fig. S7)).† The response of the HUVECs was monitored daily by phase-contrast microscopy for 5 days. Subsequent steps are shown in the ESI,† Movie 1.We first assessed whether the morphological changes were observed in bortezomib treated HUVECs (logarithmic concentrations: 0, 10−5, 10−4, 10−3, 10−2, 10−1, and 1 μM (Fig. S6†)). The typical cobblestone-like morphology of HUVECs was observed at low concentrations of bortezomib (0, 10−5, and 10−4 μM) by day 5, whereas the HUVECs showed some elongated morphological shapes without spindle-like shapes when conditioned with a dose of 10−3 μM (Fig. S8A†). Almost all cells died when exposed to bortezomib at higher concentrations (10−2, 10−1, and 1 μM). We investigated whether or not bortezomib-induced endothelial cell death was preceded by observable morphological changes34 by monitoring cell behaviors daily with a dose of 10−2 μM (Fig. S9†). The morphological transition to a spindle-like shape was discernible from day 1, and dead cells, which were detached from the cell surface, were observed from days 2 to 5. Consequently, we determined that bortezomib-induced endothelial cell death was preceded by a series of morphological changes that develop over an extended period. We then screened for the morphological changes in finer detail over a reduced range of bortezomib concentrations between 10−3 μM (1 nM) and 10−2 μM (10 nM) (Fig. S7†). Using a linear variation in concentrations we were able to identify that the morphological changes in the HUVECs occurred in the range of 4 nM to 6 nM bortezomib (Fig. S8B†). Within this narrow range, the HUVECs neither died nor detached from the surface and only showed slight changes in their structures. Almost all cells died at doses of 8 nM and 10 nM, became detached from the surface and were washed away during media changes.
We next studied the morphological changes of HUVECs quantitatively but limited either the time period of study or the concentration of bortezomib since HUVECs were unable to survive at higher concentrations (10−2, 10−1, and 1 μM) after 3 days. To analyze the morphological changes of HUVECs quantitatively, circularity was calculated from the area and perimeter of the traced ECs (see the ESI,† Materials and methods). The former equals 4π (area/perimeter2); a circularity of 1.0 indicates a perfect circle, and as the circularity approaches 0.0, the shape becomes an increasingly elongated ellipse. At low concentrations of bortezomib (0, 10−5, 10−4, and 10−3 μM), the circularities were >0.6, whereas circularities were <0.4 when treated with a dose of 10−2 μM (Fig. 3A). From these results, we observed that the HUVEC morphology changed significantly in the concentration range between 10−3 μM and 10−2 μM, so we performed a second set of experiments over a linear concentration range (0, 1 (10−3 μM), 2, 4, 6, 8, and 10 nM (10−2 μM)). We found the circularity of the HUVECs to be a sensitive precursor to cell death, which decreased as the concentration and treatment time increased (Fig. 3B). These show that we can demonstrate the effects of bortezomib on the morphogenesis of HUVECs in a dose and time-dependent manner by using the QMAS.
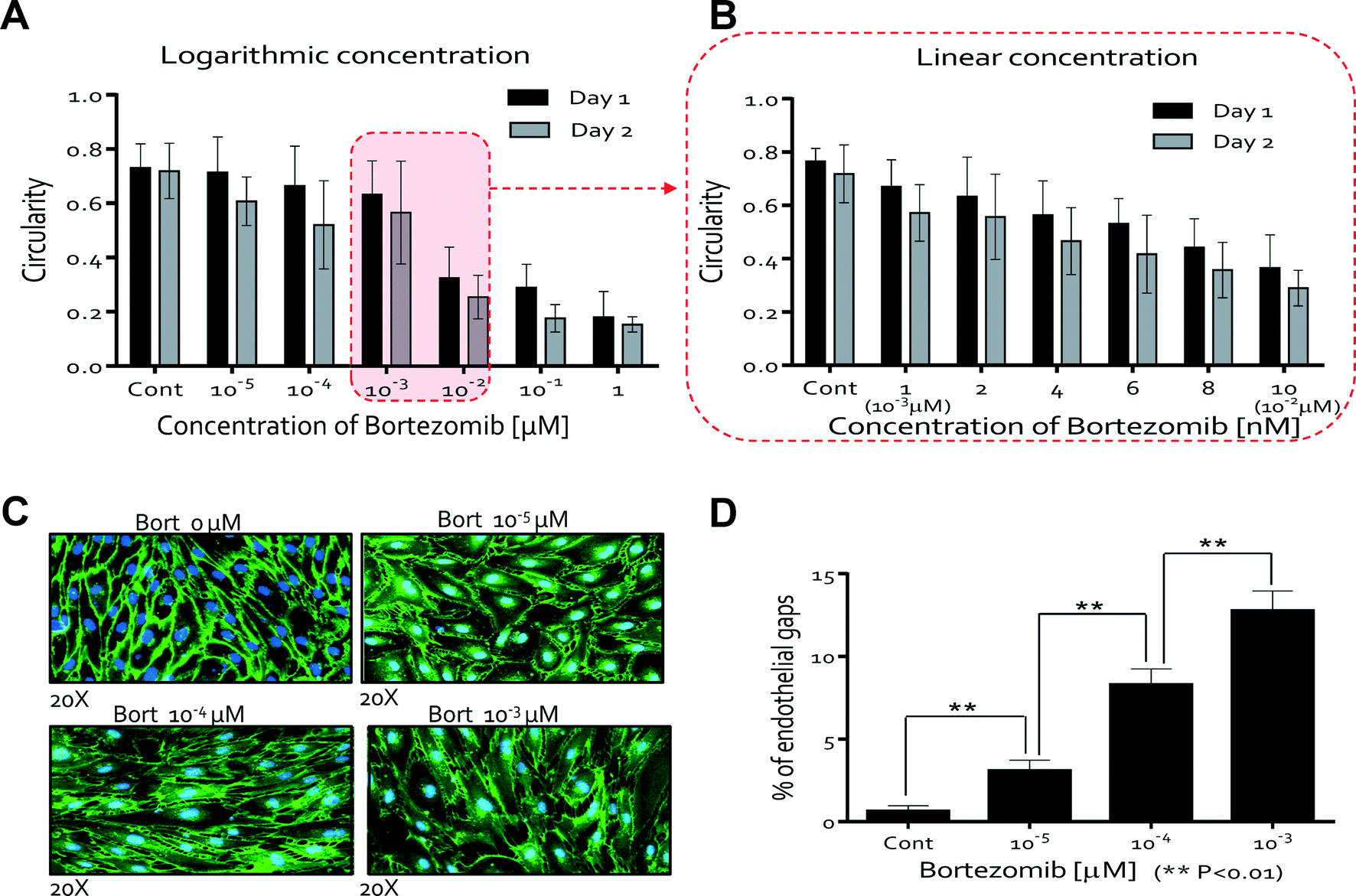 | ||
| Fig. 3 Quantitative analysis of the influence of bortezomib on HUVEC morphology. (A) Circularities of ECs as a function of bortezomib concentration (0, 10−5, 10−4, 10−3, 10−2, 10−1, and 1 μM). Circularities were >0.6 at <10−3 μM and <0.4 at 10−2 μM. (B) Circularities of ECs as a function of bortezomib concentration (0, 1 (10−3 μM), 2, 4, 6, 8, and 10 nM (10−2 μM)). The circularity of the ECs, a sensitive measure of cell health, decreased as the concentration and treatment time increased. (C) The HUVECs were stained with anti-VE-cadherin followed by Alexa 488. Breakdown of VE-cadherin at 10−5, 10−4, & 10−3 μM bortezomib doses was evident through the discontinuity of the green fluorescent line labeling the VE-cadherin junctions compared with the intact VE-cadherin junctions in the case of HUVECs cultured in the control medium (0 μM). (D) Qualitative analysis of the cell junction integrity by measuring the number of gaps observed in the VE-cadherin staining patterns; the endothelial gap number increased with increasing concentration of bortezomib after 5 days (0, 10−5, 10−4, and 10−3 μM). | ||
Fluorescence imaging of HUVECs stained for VE-cadherin allowed us to quantitatively assess the disruption of VE-cadherin junctions when cells were cultured for 5 days. The breakdown of VE-cadherin was evident through the discontinuity of the green fluorescent line labeling of the VE-cadherin junctions compared with the intact VE-cadherin junctions in the case of HUVECs cultured in the control medium with no bortezomib (Fig. 3C). We quantified the cell junction integrity by measuring the number of gaps observed in the VE-cadherin staining patterns (see Materials and methods). Endothelial gap formation increased as the concentration of bortezomib increased for 5 days (0, 10−5, 10−4, and 10−3 μM) (Fig. 3D), demonstrating significant but sub-lethal effects at these lower concentrations. No data are presented for the concentrations of 10−2, 10−1, and 1 μM due to cell death and detachment from the surface. These results show that the disruption of adherent junctions contributes to the increase in vasculature permeability.
Proliferation rate and cell viability in bortezomib treated HUVECs
We next studied whether or not HUVEC proliferation was affected by the treatment of bortezomib using Ki67 expression, a marker of proliferation also used for drug discovery.35–37 Studies were conducted on HUVECs cultured for 2 days with logarithmic (Fig. S6†) and linear concentrations (Fig. S7†) of bortezomib. We found that Ki67 was expressed at higher levels in the absence of bortezomib (0 μM, average = 18.2%), and its expression ratio gradually decreased with increasing bortezomib concentration at day 1 (from 18.2% at 0 μM to 7.8% at 1 μM), but declined more by day 2 (from 18.2% at 0 μM to 3.6% at 1 μM, Fig. 4A). The Ki67 expression ranged from 18.5% at 0 nM to 6.70% at 10 nM on day 1 and 18.34% at 0 nM to 4.24% at 10 nM on day 2 (Fig. 4B). When conditioned with 6 nM of bortezomib, which indicates that the proliferation rate varies in a time and dose dependent manner, Ki67 was expressed in less than 5% of the HUVECs.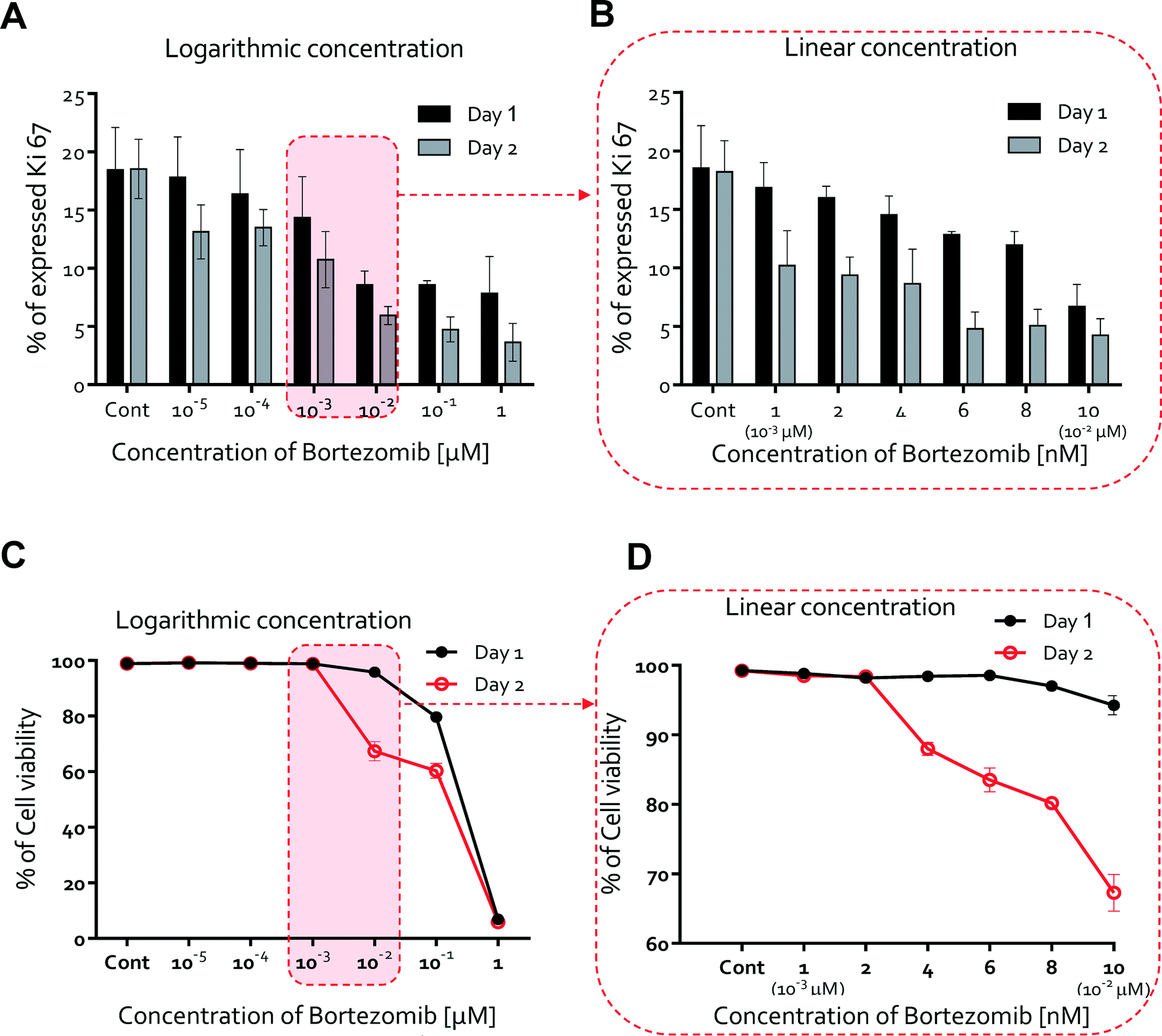 | ||
| Fig. 4 Proliferation rate and cell viability in bortezomib treated HUVECs. (A) Percentages of the proliferation rate over a logarithmic range of bortezomib concentrations (0, 10−5, 10−4, 10−3, 10−2, 10−1, and 1 μM). Ki67 was expressed at higher levels in the absence of bortezomib (0 μM, average = 18.2%), and its expression ratio gradually decreased with increasing bortezomib concentration at day 1 (from 18.2% at 0 μM to 7.8% at 1 μM), but declined more by day 2 (from 18.2% at 0 μM to 3.6% at 1 μM). (B) Percentages of the proliferation rate over a linear range of bortezomib concentrations (0, 1 (10−3 μM), 2, 4, 6, 8, and 10 nM (10−2 μM)). Ki67 expression ranged from 18.5% at 0 nM to 6.70% at 10 nM on day 1 and 18.34% at 0 nM to 4.24% at 10 nM on day 2. (C) Cell viability percentages over a logarithmic range of bortezomib concentrations. Cell viability did not decrease with the addition of <10−2 μM bortezomib on day 1 but significantly decreased at 10−2 μM (cell viability < 67 %, day 2). (D) Percentages of cell viability over a linear concentration range of bortezomib. Cell viability was still mostly maintained for doses <4 to 6 nM (cell viability: >80%, day 2). | ||
We also hypothesized that the potent anti-angiogenic capacity of bortezomib is in part a consequence of the regression of blood vessels, so we next evaluated the HUVEC viability upon exposure to bortezomib. Cell viability did not decrease with the addition of bortezomib on day 1 (Fig. 4C & D). On day 2, cell viability was still mostly maintained for doses <4 to 6 nM (cell viability > 80%, Fig. 4D), but was significantly decreased at a concentration of 10−2 μM (cell viability < 67%, Fig. 4D). According to these results, we have also shown that the cell viability varies in a time and dose-dependent manner and that bortezomib is capable of inducing the death of HUVECs.
Quantitative analysis of anti-angiogenesis in bortezomib-treated endothelial cells
Vascular endothelial growth factor (VEGF) induced endothelial cell migration is an important component of tumor angiogenesis. VEGF regulates key steps in the angiogenic process, including endothelial cell migration and tube formation.38 To find treatment conditions that would not result in significant toxicity to HUVECs and would inhibit the growth of new blood vessels from pre-existing ones, we studied the effects of bortezomib over a logarithmic concentration range of bortezomib on the VEGF induced migration of HUVECs using the QMAS.We found that bortezomib treatment significantly suppressed VEGF-induced migration at higher doses of bortezomib (10−2, 10−1, & 1 μM). Confocal microscopy revealed the growth of circular lumen-like structures from the HUVEC monolayer into the collagen scaffold in response to VEGF at lower bortezomib concentrations (0, 10−5, 10−4, & 10−3 μM), but no morphogenesis at doses >10−2 μM was evident in the immunostained and confocal microscopy images (Fig. S10A†). The formation of circular lumen-like structures was preceded by a period of VEGF-induced migration and was also reduced with increasing dose of bortezomib. We also monitored daily the cellular behaviors at a dose of 10−2 μM to assess defects in cell migration due to bortezomib. HUVECs migrated toward the 3D hydrogel scaffolds in response to VEGF for 2 days, but those migrated cells regressed after 3 days (Fig. S10B†).
The number of migrated cells generally decreased at doses <10−3 μM, but the number drastically declined when 10−2 μM was applied (Fig. 5A). The percentage of the image area occupied by the migrated cells fell dramatically at ~10−2 μM, in agreement with our results on the number of migrated cells (Fig. 5C). For finer discrimination, we screened for the cellular behaviors in the linear concentration range of bortezomib (1, 2, 4, 6, 8, & 10 nM). The number of migrated cells and % of the area occupied by the migrated cells exhibited a nearly linear dependence along the linear dose range region (Fig. 5B & D). As expected, cells actively migrated at all heights inside the collagen gel scaffolds at concentrations of 0 nM, 1 nM, and 2 nM (Fig. 6A) with lumen-like structures, but at concentrations of 4 nM and 6 nM, although endothelial cells started out in the gel, they wound up at the bottom with little or no lumen-like structures (Fig. 6B). Neither cell migration nor lumen-like structures formed at doses of 8 nM and 10 nM (Fig. 6C). From these results, we determined that bortezomib by itself inhibited VEGF-induced angiogenesis, and we were able to identify the appropriate dosage of bortezomib (4 nM & 6 nM) that did not prove to be fatally toxic to HUVECs while being able to inhibit the growth of new blood vessels from a pre-existing monolayer.
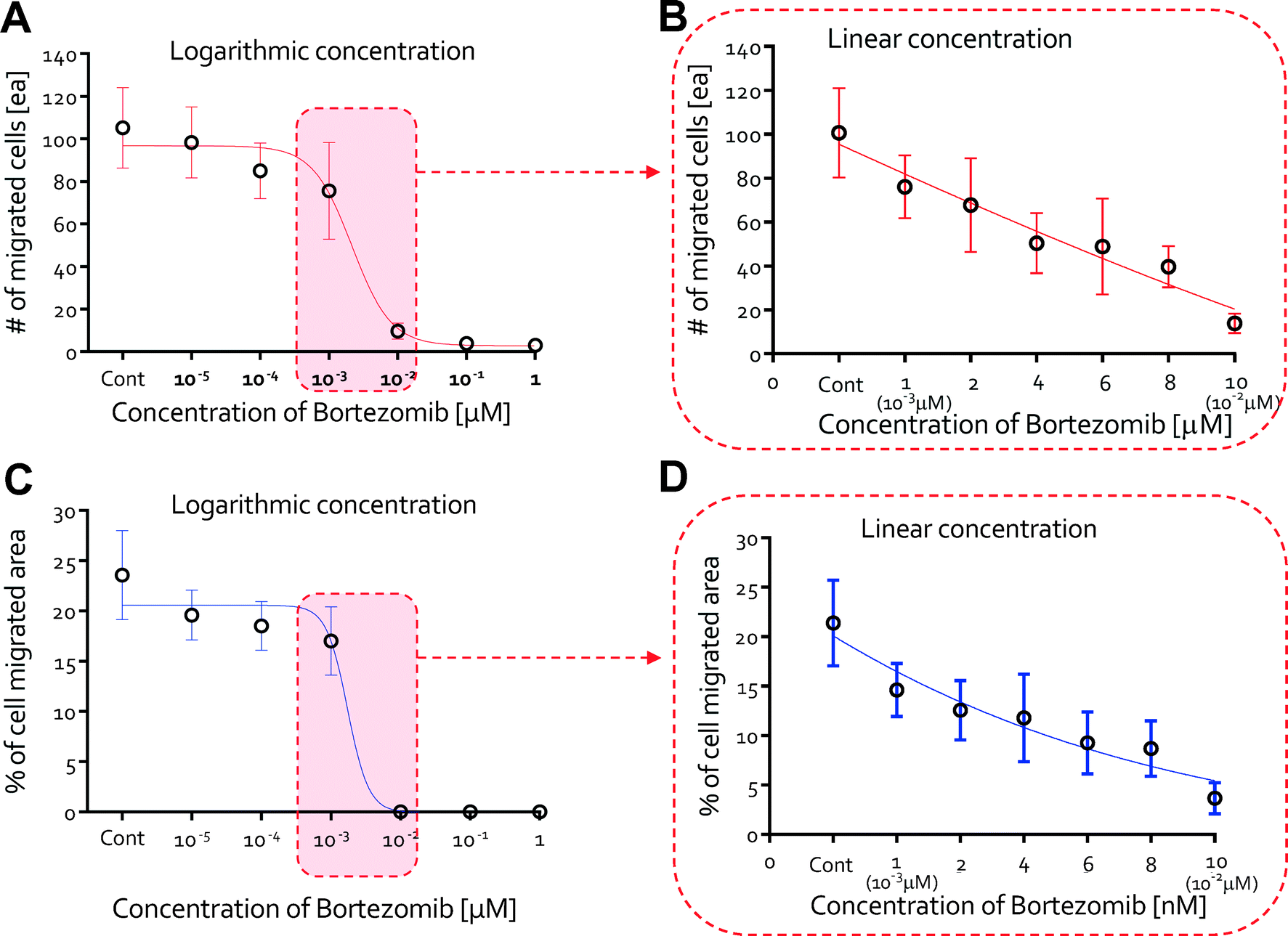 | ||
| Fig. 5 Quantitative analysis of anti-angiogenesis in bortezomib-treated endothelial cells. (A) The number of migrated cells as a function of dose with logarithmic variations in the concentration of bortezomib (0, 10−5, 10−4, 10−3, 10−2, 10−1, and 1 μM). The # of migrated cells generally decreased at doses <10−3 μM, but the number drastically declined when 10−2 μM was applied. (B) The number of migrated cells as a function of dose with linear variations in the concentration of bortezomib (0, 1 (10−3 μM), 2, 4, 6, 8, and 10 nM (10−2 μM)). The # of migrated cells exhibited a nearly linear dependency along the linear dose range region. (C) Percentages of the cell migrated area as a function of dose of bortezomib (0, 10−5, 10−4, 10−3, 10−2, 10−1, and 1 μM). The % of the area occupied by the migrated cells fell dramatically at a dose of 10−2 μM, in agreement with our results on the number of migrated cells. (D) Percentages of the cell migrated area as a function of dose with linear variations in the concentration of bortezomib. The % of the area occupied by the migrated cells exhibited a nearly linear dependency along the linear dose range region. | ||
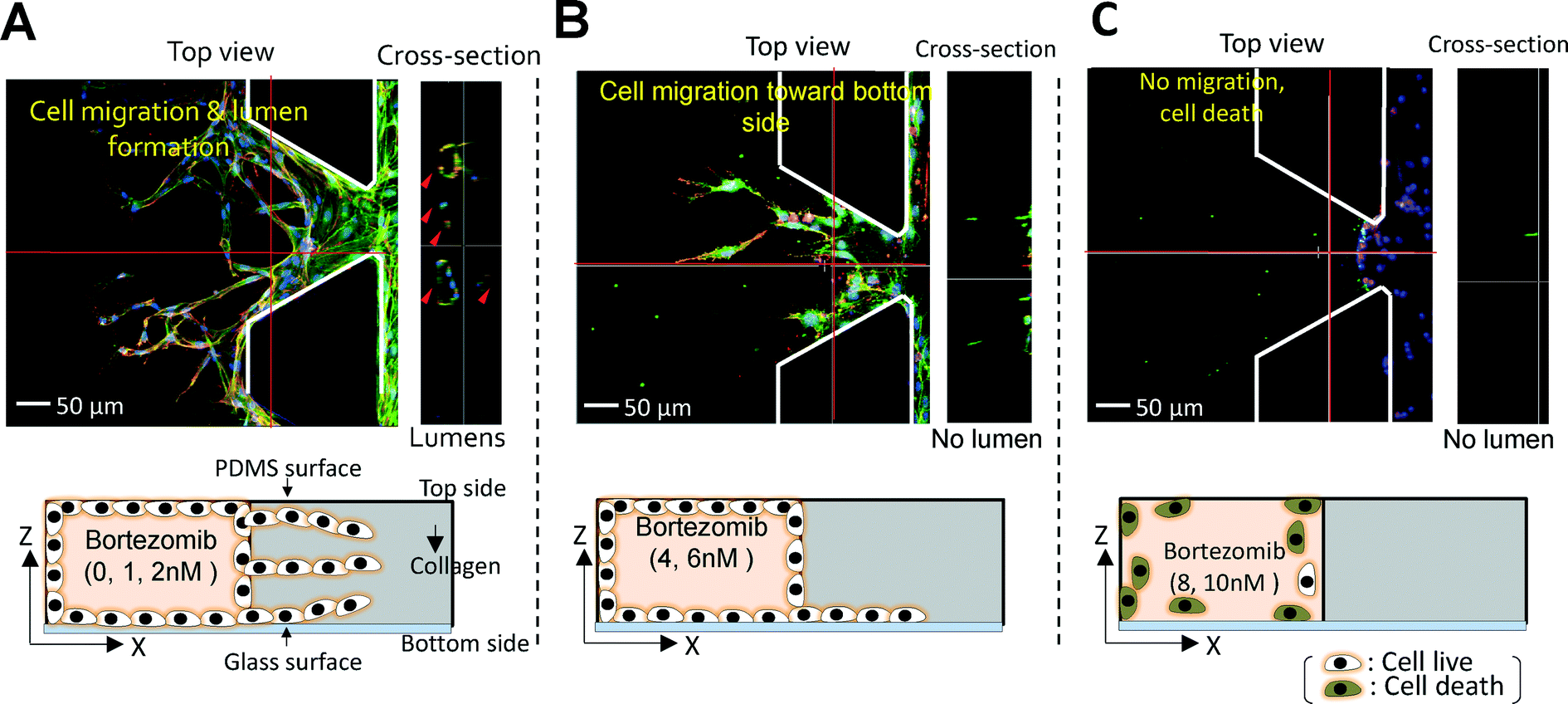 | ||
| Fig. 6 Projected images showing the effect of bortezomib dose. (A) Cells actively migrated towards the upper, middle, and bottom regions of the collagen gel scaffolds and formed lumen-like structures at concentrations of 0 nM, 1 nM, and 2 nM. (B) At concentrations of 4 nM and 6 nM, although endothelial cells started out throughout the gel, they wound up migrating to the bottom and failed to create lumen-like structures. (C) Neither cell migration nor lumen-like structures formed at doses of 8 nM and 10 nM. | ||
Conclusions
In the current study, we have proposed and described the QMAS as a quantitative anti-angiogenesis screening assay. Its utility was demonstrated using a known anti-angiogenic agent, bortezomib. An important attribute of the QMAS is its ability to screen multiple concentrations simultaneously, allowing a side-by-side comparison of the dose- and time-dependent effects of drugs in in vivo-like situations. Minimizing the need for repeated experiments on multiple devices will likely reduce experiment-to-experiment variability and lead to assays that are less sensitive to user experience. With this quantitative microfluidic angiogenesis screen (QMAS), we explored the possibility that bortezomib, which has been reported to act by decreasing the VEGF secretion from myeloma cells.32,33 Our results suggest an additional mechanism, that bortezomib can inhibit angiogenesis by influencing the endothelium directly. In addition, we successfully identified a dosage of bortezomib that did not result in fatal toxicity for HUVECs while inhibiting the growth of lumen like structures from the intact HUVEC monolayer. Our results show a similar behavior to the previously reported results obtained from the 3D cell culture39 and in vivo model,40 in that the drug was tolerated up to concentrations of approximately 0.5 mg kg−1 (corresponding to ~1 μM). A more detailed comparison including measures of effectiveness would be needed to make a more direct assessment. The proposed QMAS allows experiments in a more biologically relevant microenvironment that represents a compromise between mimicking all the complexity of in vivo conditions and current in vitro 2D-based screening systems. Thus, the QMAS can provide quantitative information on the intricacies of cellular behaviors using a less labor intensive, time-saving method as compared to previously developed in vitro angiogenesis assays. The QMAS can also be a useful screening tool for studying resistance to chemotherapy via embedding cancer cell spheroids in the collagen scaffold regions. Recently, much effort is being directed towards the design of cancer treatment combining anti-angiogenesis drugs and conventional anti-cancer drugs. The present QMAS lends itself to assays for combination therapies to search for synergistic effects; currently such studies are under way in our group.We acknowledge that there could be several outstanding issues which must be overcome in subsequent designs. These include (1) establishing a longer-term culture in the model; (2) conducting a detailed study comparing the QMAS and animal experimental results; (3) increasing the number of screening locations; and (4) testing various cell types. Nevertheless, this assay can provide representative tissue specific responses; it can be used throughout early drug discovery, from target identification and validation to primary screening, lead identification and optimization, and safety and toxicology screening.
Materials and methods
Cell seeding & perfusion culture
Human umbilical vein endothelial cells (HUVECs) (Lonza, NJ, USA) or RFP-expressing HUVECs (Angio-Proteomie; Boston, MA, USA ) were seeded and cultured in the cell culture channels in direct contact with collagen hydrogel scaffolds prepared by diluting a 4.0 mg ml−1 solution of type I collagen (BD Biosciences, MA, USA) to 2.5 mg ml−1 and polymerizing at an initial pH of 7.4. The cell culture channels were filled with HUVEC suspensions prepared in medium at a density of 2 × 106 cells ml−1. After forming an endothelial cell monolayer, the reservoirs were added and each conditioned medium was injected using a multi-tip pipette. Flow was then generated through the microfluidic channels by an external pump (withdrawal flow rate = 1.33 μl min−1, microsyringe pump, Harvard, USA) and continued throughout the experiment in order to maintain a stable concentration profile across the 3D hydrogel scaffolds. The device was then placed in a 37 °C incubator for 5 days. The subsequent steps are shown in the ESI,† Movie 1.Preparation of the microfluidic device
Chips were fabricated using an SU-8 fabrication method and replica molding. An SU-8 (Microchem, Newton, MA, USA) photoresist was used for the development of SU-8 microstructures. Both the fabricated PDMS device and the glass cover slip were autoclaved and dried at 80 °C overnight. They were then plasma treated (PDC-001, Harrick, CA, USA) in air and bonded together to form a closed microfluidic channel. After recovering the hydrophobic surface characteristics of the gel filling region, the scaffold material was introduced into the fourteen gel regions to form a gel scaffold using the manifold layer. In these experiments, type I collagen (BD Biosciences, MA, USA) was used as the scaffold material and was gelled by incubating for 30 minutes at 37 °C. Gel stiffness was varied by adjusting the solution pH; we used pre-polymerized collagen solutions (2.5 mg ml−1) of pH 7.4. Following gelation, fibronectin (dilution ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, Sigma-Aldrich) was introduced into all microfluidic channels to improve the cell attachment, and after treatment for 1 day, fresh cell culture medium was injected and the device was placed in an incubator (37 °C) for cell seeding.
:10, Sigma-Aldrich) was introduced into all microfluidic channels to improve the cell attachment, and after treatment for 1 day, fresh cell culture medium was injected and the device was placed in an incubator (37 °C) for cell seeding.
Fixation, imaging, and quantification of cell migration
The ImageJ software (http://rsbweb.nih.gov/ij/) was used to measure the % of projected areas and the number (#) of migrated cells. The F-actin distribution and the number of cells involved in the “capillary-like” networks or tube structures were assessed after 5 days of culture in the device. Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min at room temperature. During fixation, the pressure head and flow were maintained in the experimental devices to preserve protein expression and activation. After fixation with 4.0% PFA, actin filaments and nuclei were then stained with rhodamine-phalloidin (Sigma-Aldrich) and 4′6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich), respectively. Fluorescent images were obtained using a confocal microscope (Olympus FLUOVIEW FV1000, Tokyo, Japan).Analysis of gaps and disruption of VE-cadherin
The disruption of VE-cadherin was identified from the analysis of discontinuity of green fluorescence at VE-cadherin junctions between HUVECs. The gap area within the disrupted VE-cadherin junctions was determined from six images. The gap area was quantified as the ratio of pixels within all the gaps and the total number of pixels in one image. The average percentage of endothelial gaps was calculated using the ImageJ software.HUVEC proliferation and cell viability assays
HUVECs were treated with various concentrations of bortezomib in the model for the indicated periods of time. Ki-67, a common proliferation marker, was labeled with rabbit polyclonal antibody (Abcam) at 1:500 dilutions to analyze the HUVEC proliferation ratio. Cell viability was tested using the LIVE/DEAD reduced-biohazard viability/cytotoxicity test (Molecular Probes, Invitrogen, Cergy-Pontoise, France). Staining and analysis were performed as recommended by the manufacturer. The cell number was determined using ImageJ. All experiments were repeated at least twice at different times.
Statistical analysis
Numerical values were expressed as means ± SDs of more than three independent experiments. Statistical significance was determined via one-way analysis of variance (ANOVA). Paired Student's t-tests were conducted to compare the means when the ANOVA indicated a significant difference. Statements of significance were based on P-values < 0.01.Acknowledgements
This research was supported in part by the US National Institutes of Health (R33 CA174550) and the National Science Foundation (CBET-0939511). S. Chung was supported by the National Research Foundation of Korea (Pioneer Research Center Program 2012-0009565 and 2014M3A7B4052193) and the Ministry of Trade, Industry & Energy of Korea (No. 20124010203250). This work was also supported by a 2013 Kyungil University research grant.References
- J. Takei, AATEX, 2006, 11, 170–176 Search PubMed.
- C. Fang, I. Avis, D. Salomon and F. Cuttitta, J Cancer., 2013, 4, 402–415 CrossRef CAS PubMed.
- J. L. Horning, S. K. Sahoo, S. Vijayaraghavalu, S. Dimitrijevic, J. K. Vasir and T. K. Jain, Mol. Pharmaceutics, 2008, 5, 849–862 CrossRef CAS PubMed.
- K. Takara, T. Sakaeda, T. Yagami, H. Kobayashi, N. Ohmoto, M. Horinouchi, K. Nishiguchi and K. Okumura, Biol. Pharm. Bull., 2002, 25, 771–778 CAS.
- T. Yamori, A. Matsunaga, S. Sato, K. Yamazaki, A. Komi, K. Ishizu, I. Mita, H. Edatsugi, Y. Matsuba, K. Takezawa, O. Nakanishi, H. Kohno, Y. Nakajima, H. Komatsu, T. Andoh and T. Tsuruo, Cancer Res., 1999, 59, 4042–4049 CAS.
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- K. Kobuke, F. Piccolo, K. W. Garringer, S. A. Moore, E. Sweezer, B. Yang and K. P. Campbell, Hum. Mol. Genet., 2008, 17, 1201–1213 CrossRef CAS PubMed.
- D. G. Hackam and D. A. Redelmeier, JAMA, J. Am. Med. Assoc., 2006, 296, 1731–1732 CAS.
- D. Wlodkowic and Z. Darzynkiewicz, World J. Clin. Oncol., 2010, 1, 18–23 CrossRef PubMed.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- M. C. Chen, M. Gupta and K. C. Cheung, Biomed. Microdevices, 2010, 12, 647–654 CrossRef CAS PubMed.
- R. Zang, D. Li, I.-C. Tang, J. Wang and S.-T. Yang, Int. J. Biotechnol. Wellness Ind., 2012, 1, 31–51 CAS.
- L. Zhao, J. T. Caot, Z. Q. Wu, J. X. Li and J. J. Zhu, J. Biomed. Nanotechnol., 2013, 9(3), 348–356 CrossRef CAS PubMed.
- L. Y. Wu, D. Di Carlo and L. P. Lee, Biomed. Microdevices, 2008, 10, 197–202 CrossRef CAS PubMed.
- C. Kim, J. H. Bang, Y. E. Kim, S. H. Lee and J. Y. Kang, Lab Chip, 2012, 12, 4135–4142 RSC.
- Y. Kubota, Keio J. Med., 2012, 61, 47–56 CrossRef CAS.
- H.-C. Wu, C.-T. Huang and D.-K. Chang, J. Cancer Mol., 2008, 4, 37–45 CAS.
- R. S. Samant and L. A. Shevde, Oncotarget, 2011, 2, 122–134 Search PubMed.
- A. Tandle, D. G. Blazer and S. K. Libutti, J. Transl. Med., 2004, 2, 1–20 CrossRef PubMed.
- N. Jeon, H. Baskaran, S. Dertinger, G. Whitesides, L. Van De Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–830 CrossRef CAS PubMed.
- N. W. Choi, M. Cabodi, B. Held, J. P. Gleghorn, L. J. Bonassar and A. D. Stroock, Nat. Mater., 2007, 6, 908–915 CrossRef CAS PubMed.
- R. Sudo, S. Chung, I. K. Zervantonakis, V. Vicherman, Y. Toshimitsu, L. G. Griffith and R. D. Kamm, FASEB J., 2009, 23, 2155–2164 CrossRef CAS PubMed.
- C. R. Kothapalli, E. van Veen, S. de Valence, S. Chung, I. K. Zervantonakis, F. B. Gertler and R. D. Kamm, Lab Chip, 2011, 11, 497–507 RSC.
- V. Vickerman, J. Blundo, S. Chung and R. D. Kamm, Lab Chip, 2008, 8, 1468–1477 RSC.
- S. Chung, R. Sudo, P. J. Mack, C. R. Wan, V. Vickerman and R. D. Kamm, Lab Chip, 2009, 9, 269–275 RSC.
- W. J. Polacheck, J. L. Charest and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11115–11120 CrossRef CAS PubMed.
- S. Chung, R. Sudo, I. K. Zervantonakis, T. Rimchala and R. D. Kamm, Adv. Mater., 2009, 21, 4863–4867 CrossRef CAS PubMed.
- C. Kim, S. Chung, L. Yuchun, M. C. Kim, J. K. Chan, H. H. Asada and R. D. Kamm, Lab Chip, 2012, 12, 2942–2950 RSC.
- D. H. Nguyen, S. C. Stapleton, M. T. Yang, S. S. Cha, C. K. Choi, P. A. Galie and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6712–6717 CrossRef CAS PubMed.
- Y. Zheng, J. Chen, M. Craven, N. W. Choi, S. Totorica, A. Diaz-Santana, P. Kermani, B. Hempstead, C. Fischbach-Teschl, J. A. López and A. D. Stroock, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9342–9347 CrossRef CAS PubMed.
- M. B. Chen, S. Srigunapalan, A. R. Wheeler and C. A. Simmons, Lab Chip, 2013, 13, 2591–2598 RSC.
- T. Hayashi, T. Hideshima and K. C. Anderson, Br. J. Haematol., 2003, 120, 10–17 CrossRef CAS.
- T. Hideshima, D. Chauhan, K. Podar, R. L. Schlossman, P. Richardson and K. C. Anderson, Semin. Oncol., 2001, 28, 607–612 CrossRef CAS.
- D. Belloni, L. Veschini, C. Foglieni, G. Dell'Antonio, F. Caligaris-Cappio, M. Ferrarini and E. Ferrero, Exp. Cell Res., 2010, 316, 1010–1018 CrossRef CAS PubMed.
- T. Scholzen and J. Gerdes, J. Cell. Physiol., 2000, 182, 311–322 CrossRef CAS.
- A. Urruticoechea, I. E. Smith and M. Dowsett, J. Clin. Oncol., 2005, 23, 7212–7220 CrossRef CAS PubMed.
- A. L. Johannessen and S. H. Torp, Pathol. Oncol. Res., 2006, 12, 143–147 CrossRef.
- L. Lamalice, F. L. Boeuf and H. Jacques, Circ. Res., 2007, 100, 782–794 CrossRef CAS PubMed.
- M. Lioni, K. Noma, A. Snyder, A. Klein-Szanto, J. A. Diehl, A. K. Rustgi, M. Herlyn and K. S. Smalley, Mol. Cancer Ther., 2008, 7, 2866–2875 CrossRef CAS PubMed.
- M. Boccadoro, G. Morgan and J. Cavenagh, Cancer Cell Int., 2005, 5, 18 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00866a |
| This journal is © The Royal Society of Chemistry 2015 |