
Design of a 2D no-flow chamber to monitor hematopoietic stem cells†
Théo
Cambier
a,
Thibault
Honegger
b,
Valérie
Vanneaux
c,
Jean
Berthier
d,
David
Peyrade
b,
Laurent
Blanchoin
a,
Jerome
Larghero
c and
Manuel
Théry
*ac
aLaboratoire de Physiologie Cellulaire et Végétale, Institut de Recherche en Technologie et Science pour le Vivant, UMR5168, CEA, INRA, CNRS, Université Grenoble-Alpes, Grenoble, France. E-mail: manuel.thery@cea.fr
bLaboratoire des Technologies de la Microélectronique, CNRS, CEA, Université Grenoble-Alpes, Grenoble, France
cUnité de Thérapie Cellulaire et Centre d'Investigation Clinique en Biothérapies, Hôpital Saint Louis, Institut Universitaire d'Hematologie, UMRS1160, INSERM, AP-HP, Université Paris Diderot, Paris, France
dLaboratoire d'Electronique et de Technologie de l'Information, CEA, Université Grenoble-Alpes, Grenoble, France
First published on 23rd October 2014
Abstract
Hematopoietic stem cells (HSCs) are the most commonly used cell type in cell-based therapy. However, the investigation of their behavior in vitro has been limited by the difficulty of monitoring these non-adherent cells under classical culture conditions. Indeed, fluid flow moves cells away from the video-recording position and prevents single cell tracking over long periods of time. Here we describe a large array of 2D no-flow chambers allowing the monitoring of single HSCs for several days. The chamber design has been optimized to facilitate manufacturing and routine use. The chip contains a single inlet and 800 chambers. The chamber medium can be renewed by diffusion within a few minutes. This allowed us to stain live human HSCs with fluorescent primary antibodies in order to reveal their stage in the hematopoiesis differentiation pathway. Thus we were able to correlate human HSCs' growth rate, polarization and migration to their differentiation stage.
Introduction
Blood, milk, lymph, urine, sweat and mucus are some of the body fluids that run through our organs and tissues. Fluid flow is intimately coupled with cell function and organ physiology. Cells produce, sense and respond to fluid flow in various ways. Fluid flow generates shear stress on adherent cells, such as epithelial or endothelial cells, which tend to align their shape and polarity with respect to the flow.1 Shear stress also regulates cell size,2 cell proliferation and differentiation3 and thus tissue patterning4 and morphogenesis.5 In contrast, most cells of the lymphoid and myeloid lineages are less adhesive and thus much less resistant to high fluid-flow-induced shear stress. They are transported along the blood flow and home in to flow-protected regions in adjacent tissues. Shear stress modulates leukocyte adhesion, architecture and migration properties.6 This sensitivity to fluid flow is a matter of concern under classical cell culture conditions where temperature-induced convection permanently displaces cells. These movements prevent long-term monitoring of their behavior by video microscopy. In addition, they are also likely to have consequences on hematopoietic stem cell (HSC) differentiation since shear stress and mechanical forces in general are major regulators of the stem cells' fate.7–9 Analysis and control of the HSC's fate is of paramount importance since HSCs are currently the most commonly used cell type for stem cell-based therapy.10,11 HSCs have been used clinically since 1959 and their use is now established as a standard therapeutic modality for a variety of malignant and non-malignant diseases.12 Our incapacity to protect HSCs from fluid flow and monitor single cell behavior as they are submitted to biochemical signals is a serious matter of concern. While numerous microfluidic devices have been designed to control the flow speed and shear stress applied on adherent cells,13 much less attempts have been made to completely stop the flow in order to monitor and analyze HSCs in their unbiased state.Multilayered chips with fluidic valves have been used to start/stop the flow and isolate reaction chambers.14 HSCs have also been protected from fluid flow in the bottom of deep microwells. Multilayered chips were used to start/stop the flow above the wells, load the HSCs and test the influence of several combinations of soluble factors in parallel.15 Multilayered chips have also been used to supply cells with soluble factors through a porous membrane protecting the cells from convection flow.16 The same concept was applied to single-layer chips in which the supply channels were connected to a reaction chamber via several thin channels in which the flow was very low.17 Simpler devices made of a single layer of channels of equal height were also tested. As an alternative to thin channels, alignments of pillars were used to separate the supply channels from the reaction channel and limit the flow between them.18,19 In some other devices, the flow was prevented by using dead-end channels as reaction chambers.20,21 All of these devices have pros and cons. To be usable on a routine basis in biology, the device has to be easy and fast to manufacture. It also has to be robust and should allow the visualization of numerous chambers in parallel. Here we describe a 2D chip with a single inlet and 800 no-flow chambers per square centimeter.
Materials and methods
No-flow chamber manufacturing
The thick positive resist AZ9260 (AZ Electronic Materials) was spin-coated on a 4 inch silicon wafer at 750 rpm for 60 s to obtain a 20 micron thick layer. It was pre-baked on a hot plate for 1 min at 60 °C and then for 4 min at 110 °C. It was exposed to UV through a photomask (Toppan) bearing the features of interest for 50 s (35 mW mm−2 at 405 nm and 16 mW mm−2 at 365 nm) on a mask aligner (MJB4, SUSS MicroTec), developed in AZ400K (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 dilution in water) (AZ Electronic Materials) for 60 s and washed with deionized water. The developed resist was finally dried in air.
:4 dilution in water) (AZ Electronic Materials) for 60 s and washed with deionized water. The developed resist was finally dried in air.
Polydimethylsiloxane (PDMS) (Sylgard 184 kit, Dow Corning) was mixed with the curing agent (10:1 ratio), degassed, poured onto the mold and cured for 10 minutes at 100 °C on a hot plate. The PDMS layer was then peeled off and stored away from the dust. The PDMS chip and a standard glass slide (76 mm × 26 mm borosilicate) were oxidized in an oxygen plasma cleaner for 10 s at 100 W (Femto, Diener Electronic) and brought into contact to ensure bonding and prevent fluidic leakage. Prior to bonding, the PDMS chip was punched using a hole puncher (Ted Pella) with an outer diameter smaller than 1/16'.
Medium was injected into the single device inlet with a pipette. Then, without appling additional external pressure, medium filled in the whole microfluidic circuitry.
HSC isolation and culture
Human umbilical cord blood samples were collected from normal full-term deliveries after maternal informed consent according to approved institutional guidelines (Assistance Publique – Hôpitaux de Paris, Paris, France). CD34+ cells were isolated using the direct CD34 Progenitor Cell Isolation Kit (Miltenyi Biotec, Paris, France) and cryopreserved in SVF (Hyclone)/10% DMSO (B Braun). After thawing, cells were resuspended in the culture medium HPO1 (Macopharma, France) supplemented with growth factors 100 ng μL−1 SCF, 10 ng μL−1 G-CSF, 20 ng μL−1 TPO, and 100 ng μL−1 FLT3I (PeproTech) just before being injected into the microfluidic device at a density of 106 cells mL−1.CD34+ cells were stained by addition of PE-CD33, APC-CD34 and FITC-CD38 monoclonal antibodies (Becton Dickinson).
Image acquisition
Images were taken using an ECLIPSE Ti (Nikon) equipped with a confocal scanner unit (CSU-X1, Yokogawa), a 4-wavelength LED excitation device (CoolLED pE-2 collimator, Life Sciences and Analytical), and a CoolSNAP HQ2 camera (Photometrics). The microscope was controlled using MetaMorph software (Molecular Devices).When it contained cells, the chip was maintained on the microscope on a heating stage and humidified chamber at 37 °C supplied with 5% CO2 for pH buffering (Live Cell Instrument).
COMSOL simulations
Simulations of the fluid flow were performed by the finite element method using COMSOL Multiphysics (COMSOL). Each design was modeled in a 2D plane. A planar laminar flow was setup under the same input and output boundary flow conditions. A time-lapse diffusion model was monitored until full replacement of media in the chambers and asymmetric connecting microchannels.Results
No-flow chamber design
We refer here to a no-flow chamber as a confined space in which fluid flow is absent or comparable to thermal agitation (Fig. 1A). Our aim was to design such a chamber in 2D with a minimum number of inlets/outlets to facilitate chamber manufacturing and use. We defined few key criteria for the design of the chamber. The no-flow chamber should contain one or several side channels for the supply of new solutes. Pressures should be balanced in order to restrict fluid flow to these side channels (Fig. 1B). The size of the openings between the side channels and the chamber should protect the chamber from fluid entry. However, these openings should allow solute diffusion from the side channels to the chamber. Finally, the chamber size should allow fluid renewal by diffusion within a few minutes (Fig. 1C).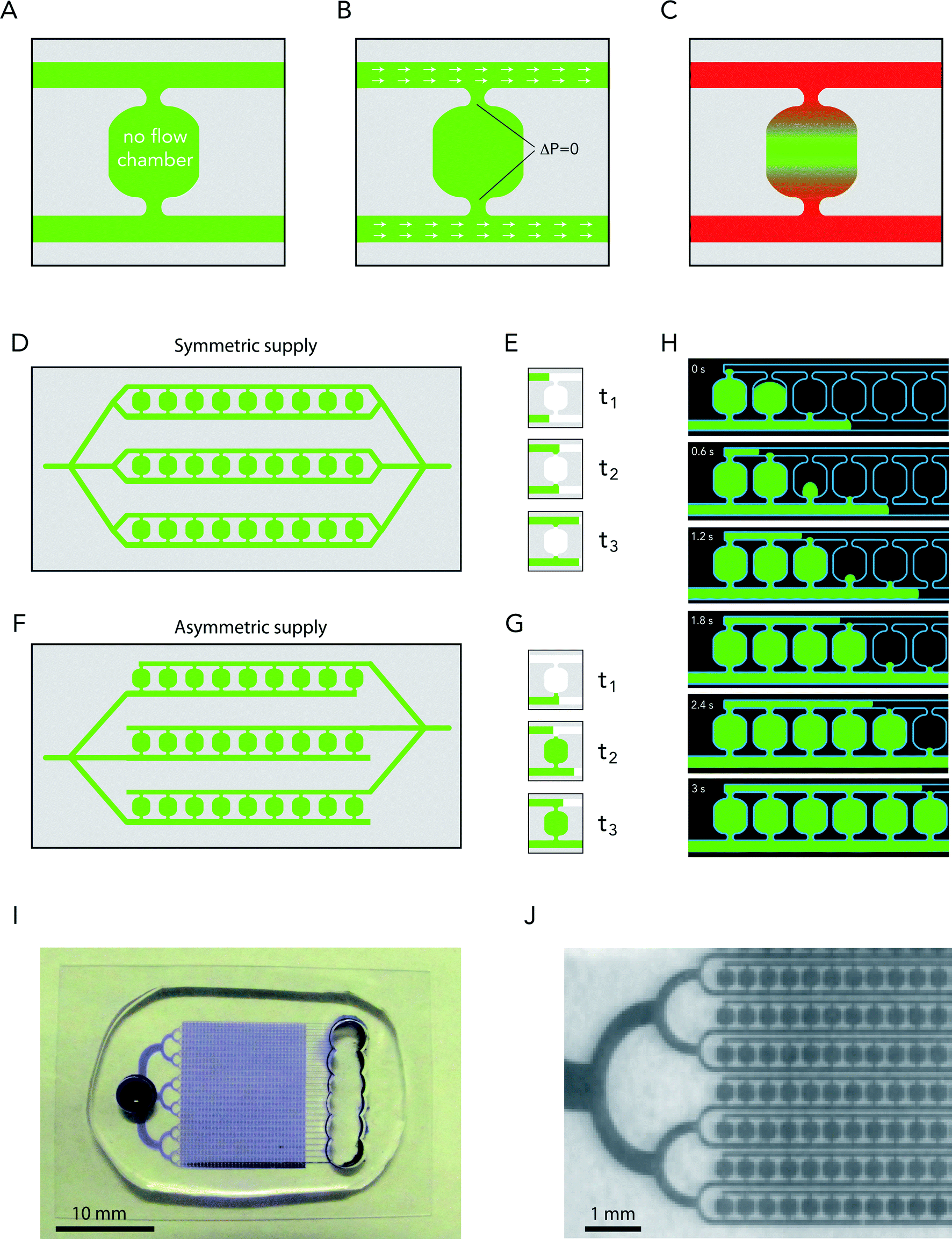 | ||
| Fig. 1 No-flow chamber design. (A) Design principle showing the central chamber and the side channels allowing medium supply. (B) Illustration of the pressure balance condition required for protecting the central chamber from the flow in the side channels (shown as white arrows). (C) Illustration of medium renewal based on solute diffusion through chamber openings. (D) Array of no-flow chambers supplied with symmetric side channels. (E) Illustration of no-flow chamber filling from symmetric side channels. The liquid is shown in green, the air is in white. Air is trapped in the chamber. (F) Array of no-flow chambers supplied with asymmetric side channels. (G) Illustration of no-flow chamber filling from asymmetric side channels. The liquid is shown in green, the air is in white. The delay between the liquid progressions in the two channels prevents air trapping in the chamber. (H) Video recording of chamber filling with water containing Alexa-488 fluorophore as a marker (shown in green). Blue lines highlight the channel shapes. Time is in seconds. (I) Picture of the PDMS chip. (J) Zoomed picture of the PDMS chip. | ||
Compartmentalized microenvironments protected from fluid flow by linear arrays of micropillars have been designed to confine multicellular clusters of adherent cells.18 The underlying concept is brilliant and has proved to be quite efficient to confine these cell groups.19 Numerical simulations revealed that in such a device a residual flow would persist and affect loosely adherent cells such as HSCs (Fig. S1A†). Physical barriers perpendicular to this residual flow in the chambers should reduce it (Fig. S1B†). Reducing the number of openings between the side channels and the main chamber should also reduce internal flow. Eventually, square chambers with two side openings, as shown in Fig. 1A, appeared optimal (Fig. S1D†).
A single opening should even be more efficient than two openings to protect the chamber from fluid entry. Dead-end chambers with a single opening have been proven to be efficient in confining cells.20–22 However, they require the use of vacuum and porous materials to remove the trapped air bubbles in the chambers and suck the cells inside.
Actually, trapped air bubbles constitute the most critical problem in no-flow devices, since they tend to form particularly in regions of interest where the flow is reduced. Intuitive symmetric designs, in which no-flow chambers are flanked with two side channels (Fig. 1D), induce the trapping of air bubbles in most chambers. Indeed, the synchrony of fluid filling in the two symmetric side channels immediately balances the pressure between the two openings and stops liquid flow in the central chamber, leading to air bubble trapping, as illustrated in the sequence described in Fig. 1E. We designed asymmetric supply channels to circumvent this problem (Fig. 1F). With asymmetric supply channels, fluid arrival close to the upper opening can be delayed so that the fluid fills the chamber from the bottom opening (Fig. 1G). The most straightforward way to ensure that the delay corresponds exactly to the time it takes to fill a chamber is to add one chamber as a delay line to the upper supply channel (Fig. 1F). In the time sequence shown in Fig. 1H, fluorescently labeled proteins were used to show the filling of no-flow chambers with the asymmetric supply design (Movie S1†). Almost no bubbles were trapped with these asymmetric side channels. We also found that rounded chambers, rather than square chambers with right angles, improved chamber filling without trapping air bubbles (Fig. S2†).
The dimensions of the optimal chamber design are shown in Fig. S3A.† Importantly, our design offers the possibility to align chambers in series and to load many of those lines in parallel with a single inlet. We could thus design a 1 cm2 chip with a single inlet distributing the fluid to 800 chambers (Fig. 1I, J and S3B†).
No-flow chamber test
The fluid flow in no-flow chambers was first estimated with numerical simulations. Fluid flow was present in the first three and the last three chambers of the line because of the asymmetric supply but was almost absent from the 30 chambers in between (Fig. S4A, B†). In those chambers, the simulated flow in the supply channels barely entered the chambers (Fig. S4C†). This was further confirmed experimentally by looking at the motion of the fluorescent beads (Fig. 2A, Movie S2†). Long exposure during image acquisition revealed local bead displacements. Time-lapse acquisitions of such images revealed bead trajectories. Beads appeared almost immobile in the no-flow chambers, whereas they were rapidly transported in the supply channels (Fig. 2A). Measurements of bead displacement within chambers depending on their position along the supply channels confirmed that the 30 chambers at the center displayed no fluid flow even when the flow in the supply channel reached 1500 μm s−1 (Fig. 2B, C). | ||
| Fig. 2 Flow measurement in the no-flow chambers. (A) One-micron polystyrene beads were used as fiducial markers to reveal fluid flow. Images were taken with 200 ms exposure time (see Movie S2†) to record bead trajectories. The displayed image is the overlay of 10 images taken with a 500 ms time interval in order to reveal the field lines. The beads do not enter the no-flow chambers. (B) Bead motion movies were recorded at various positions in the chip, at the entry (left), in the middle (center) and at the exit (right) of the microchannels, and used to record bead velocities in the chambers. (C) Bead velocities were measured in all chambers along the microchannels for two inlet fluid velocities: 150 μm s−1 (left) and 1500 μm s−1 (right). Data showed that flow was null in the 30 central chambers. | ||
The possibility of renewing the medium in no-flow chambers was tested by switching the inlet from green to red fluorophores (Fig. 3A, Movie S3†). Since the new fluid could not flow through the chambers, the renewal was entirely supported by diffusion. We measured the green and the red fluorescence signals in the chamber (position A in Fig. 3B) and in the supply side channels (position B). The chamber content appeared completely renewed 7 minutes after the fluid switch. Larger chambers would have allowed larger observation fields but would have made this renewal time much longer.
 | ||
| Fig. 3 Medium renewal. (A) The chip was filled with 1 μM Alexa-488 (green fluorescence emission) in water. The inlet was then switched to 1 μM Alexa-568 (red fluorescence emission) in water. Green and red fluorescence in the chambers were video-recorded. Time is indicated in min:s. (B) Green and red fluorescence emission signals were measured in the no-flow chamber (position A) and in the side channel (position B). Intensity values were normalized to the initial and final values for green and red, respectively. Dashed lines show the fluorescence in the side channel. Full lines show the fluorescence in the no-flow chambers. It took about 7 minutes after inlet switch to renew completely the medium in the chambers. | ||
HSC loading and monitoring
In order to test the design of the chamber which was optimized for fluid filling, flow blocking and medium renewal, we introduced living cells in the no-flow chambers. As a first test, we used the Jurkat cell line, which are easy-to-use non-adherent human T lymphocytes. They could survive and proliferate in the no-flow chambers for several days. We then switched to human HSCs. Cells were collected from human umbilical cord blood, sorted based on the expression of CD34 (ref. 23) and immediately frozen in liquid nitrogen. CD34 is a cell surface glycoprotein from the family of sialomucins that is commonly used as a marker of stemness during hematopoiesis.24HSCs were thawed, resuspended in the growth medium and loaded in no-flow chambers during initial chip filling. Cell density was adapted to obtain 1 to 10 cells per chamber (Fig. 4A). The cell culture medium in side channels was renewed every 6 hours. Cell growth and movements were monitored by phase contrast microscopy over several days using multiposition time-lapse acquisitions (Fig. 4B, Movie S4†). It appeared that HSC growth rates were highly variable: some cells divided up to six times in three days, whereas others remained quiescent. To gain further insights into the link between this variability and cell profile diversity, we took advantage of the key feature of our new device which is to change the cell culture medium without detaching non-adherent cells and introduced fluorescently labeled antibodies at the end of the acquisition (Fig. 4C). These antibodies targeted various surface receptors known to reveal the cell differentiation state. CD38 and CD34 were used as markers of non-differentiated cells,24 whereas CD33 was used as a marker of early engagement in the myeloid lineage.25
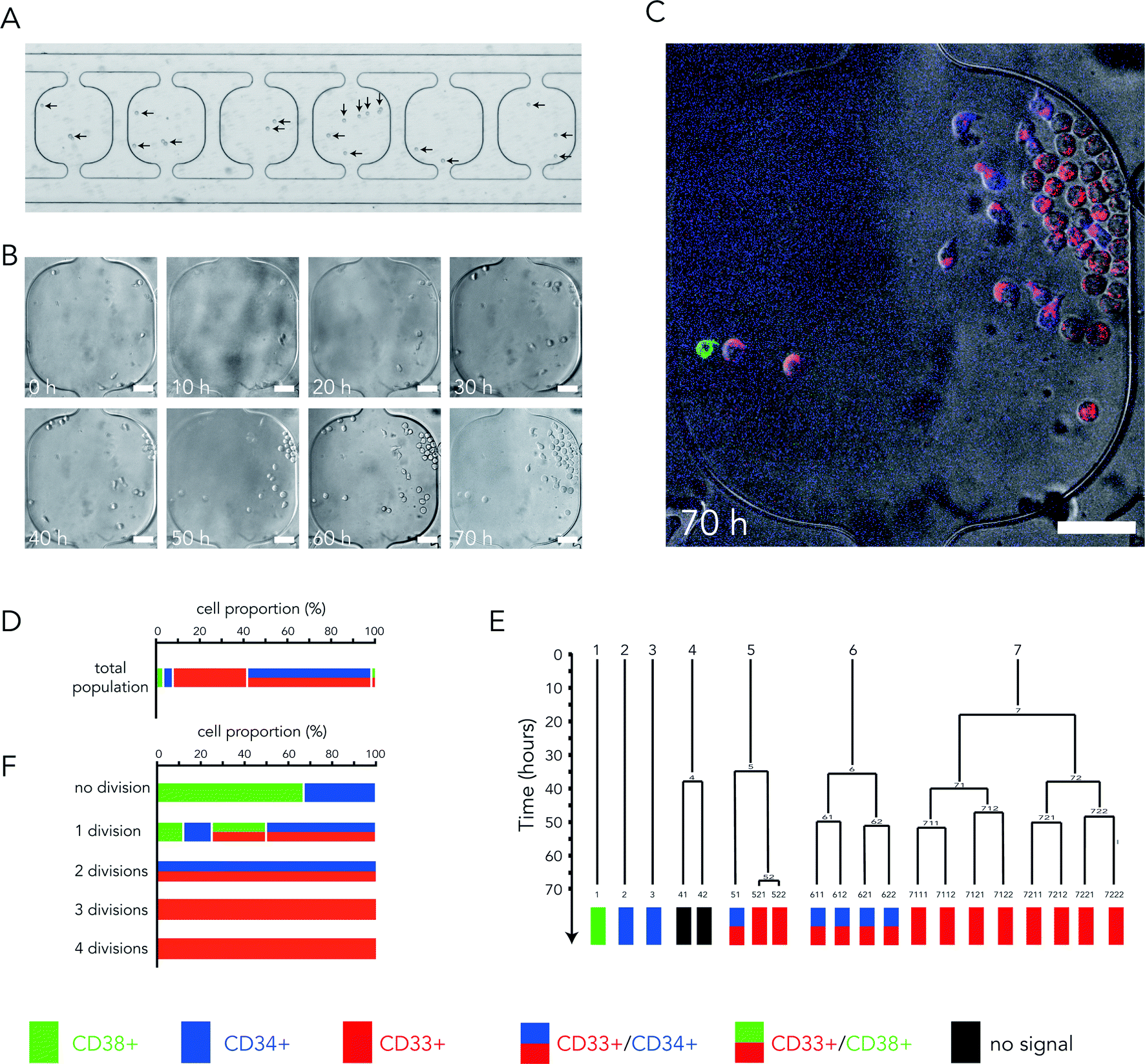 | ||
| Fig. 4 HSC growth in no-flow chambers. (A) Observation of HSCs (arrows) under transmitted light after no-flow chamber filling. (B) Video recording of HSCs in a no-flow chamber under transmitted light. Time is in hours. Scale bar is 50 μm. (C) Live HSC staining with fluorescent primary antibodies against CD38 (green), CD34 (blue) and CD33 (red) at the end of the video recording. Antibodies were added to the culture medium in the side channels. Scale bar is 50 μm. (D) Quantification of the proportion of cells expressing the various makers after 3 days of culture in no-flow chambers. Color code is shown at the bottom. N = 257 cells, n = 17 chambers. (E) Example of a lineage tree obtained after visual tracking of HSCs in the no-flow chambers. Bottom line shows cell staining at the end of the movie. Color code is shown at the bottom. (F) Quantification of the surface receptor expression profiles depending on the number of cell division during three days of observation. N = 257 cells, n = 17 chambers. | ||
Three days after cell collection and loading in the no-flow chambers, most cells had started to express CD33, few cells were CD38 positive and only 60% were still expressing the stemness marker CD34. Thus the entire population appeared quite heterogeneous (Fig. 4D). Several hypotheses could account for this diversity. It could be the consequence of asymmetric HSC divisions, leading to daughter cells with distinct protein expression profiles. It could also be due to differences in the proliferation rates of distinct sub-populations. Clues could be obtained by tracking individual cells and reconstituting cell lineage trees (Fig. 4E). Although some asymmetric divisions were observed (cell #5 in Fig. 4E for example), leading to daughter cells with distinct expression profiles, they were quite rare and most of the clones displayed identical surface markers. This favored the differential growth rate hypothesis. To further test it, we counted the number of division the cells went through for all combinations of surface markers. Cells that were still expressing CD38 after three days did not divide or divided only once. Cells expressing CD34 went up to two rounds of division. In contrast, cells expressing CD33 had divided two to four times (Fig. 4F). These data accounted for the massive presence of CD33 cells after three days and suggested that population heterogeneity was mainly due to differences in cell proliferation capacities.
Cell monitoring revealed that most HSCs acquired elongated and polarized leukocyte-like shapes soon after being loaded in the chip.26,27 These cells developed dynamic membrane protrusions at their leading edge and displayed slow amoeboid-like migration throughout the no-flow chamber (Movie S4†). In contrast, static cells remained round. Here we also took advantage of the possibility of associating cell monitoring with fluorescence immunostaining to reveal the protein expression profiles of static and migrating cells. Three days after cell loading, the profiles of the static cells were heterogeneous: 60% were CD33+ and 30% were CD33+/CD34+ (Fig. 5A). Strikingly, 95% of migrating cells were CD33+/CD34+ (Fig. 5A). Thus the co-expression of CD34 and CD33 did not seem to be specific to but characteristic of all moving cells. To further understand the relationship between the CD33/CD34 co-expression and the acquisition of cell motility, we looked at receptor localization at the cell surface. In static cells, we could observe several patches of CD33, whereas in moving cells CD33 receptors were clustered at the cell rear in the uropod (Fig. 5B). These observations were further quantified by the measurement of the cell shape factor and surface marker distribution. Migrating cells appeared much more elongated than static cells (Fig. 5C). Surface marker polarization was quantified by measuring the position of the center of mass of the fluorescence signal with respect to the cell center. CD34 appeared evenly distributed in both static cells and migrating cells, whereas CD33 was strongly polarized toward the rear of migrating cells (Fig. 5C). Backward polarization of CD33 in the uropod appeared as a specific signature of migrating CD34+ HSCs.
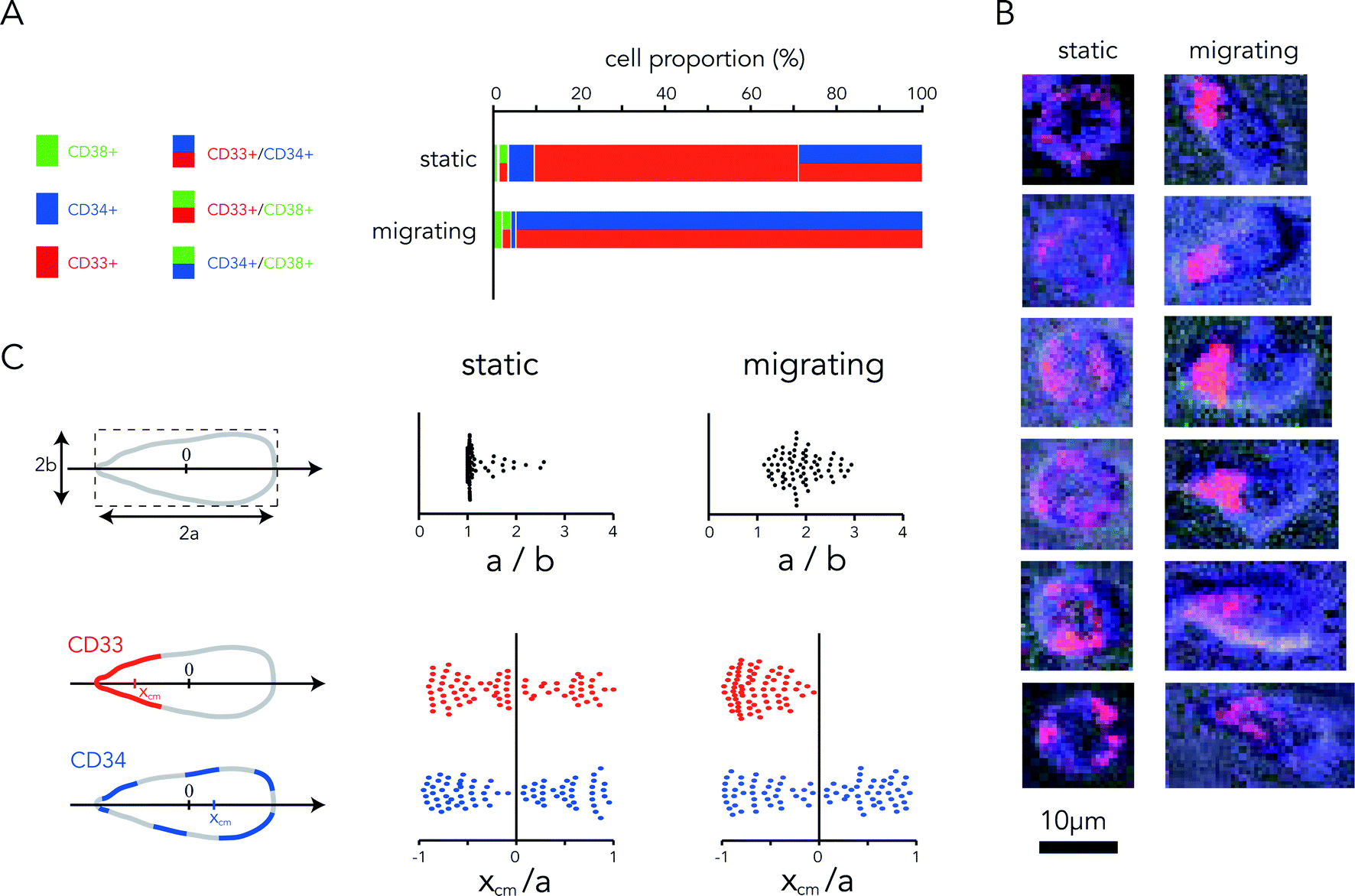 | ||
| Fig. 5 HSC polarization. (A) Quantification of the surface receptor expression profiles in static and migrating cells. N = 257 cells, n = 17 chambers. (B) Images showing the spatial distribution of CD34 (blue) and CD33 (red) at the surface of static and migrating HSCs after three days of culture in no flow-chambers. (C) Measurement of cell aspect ratios (top line) and localization of surface markers (bottom line) in static and migrating HSCs. The cell aspect ratio was measured by fitting the cell shape with an ellipse and quantifying the ratio of the short and long axes. Migrating cells appeared more elongated. Surface marker localization was quantified by measuring the coordinate of the center of mass of the fluorescence signal along the long cell axis (xcm) and normalizing it to the length of this axis. CD33 appeared systematically polarized in the rear of migrating HSCs. | ||
Discussion
Here we described the design and use of a new no-flow chamber. Sophisticated microfluidic 3D chambers with several layers of channels and valves were used to screen multiple compounds on a chip.15 The goal here was simply to protect the biological sample from fluid flow in a large array of chambers. The chamber shape and chip circuitry have been designed for routine use in biology. The aim was to minimize the manufacturing difficulties and to make the chip robust and easy-to-use. The outcome of our work is a single-layer 2D chip with a single inlet and 800 chambers per cm2 (the complete chip is shown in Fig. S3†). The circuitry has been optimized to allow the fluid loading without formation of air bubbles. Since vacuum is not required to eliminate air bubbles, the chip can be produced in plastic or glass as an alternative to PDMS. Finally, the chamber shape and size allow rapid medium exchange by diffusion. This last feature is particularly useful since it is almost impossible to change the fluid on top of flow-sensitive biological samples under classical experimental conditions.No-flow chambers are suited for biological systems that are sensitive to or could be damaged by fluid flow. For example, several biochemical assays are based on the detection of a specific protein or DNA strand in a sample due to its interaction with a surface-bound probe. These assays can be limited by the affinity between the probe and the ligand, particularly during the washing steps. Reducing fluid shear stress in no-flow chambers could improve the sensitivity of these assays. Another example relates to the cell types, such as endothelial or epithelial cells, which can be oriented in space or physiologically modified by fluid shear stress.13 No flow chambers could be used to prevent such stress and get rid of this bias. Here we took advantage of the absence of flow to monitor HSCs, which are loosely adherent cells and therefore often displaced by fluid convection under classical cell culture conditions.
As HSCs were used to validate the suitability of no-flow chambers for monitoring their behavior, we made two interesting observations that may have important implications in our understanding of stem cell physiology. First, the early rise in population heterogeneity seemed to involve mainly the differences in the proliferation rates of various cell types. Second, HSC migration appeared to be coupled with cell shape polarization and rear clustering of CD33 receptors. These two observations fully validated the relevance of the no-flow chambers for the study of HSC physiology and opened new questions that remained to be investigated.
Human primary HSCs were initially purified from umbilical cord blood samples by immunosorting based on the expression of CD34 according to the classical method that is commonly used in therapy protocols. The yield is known to be quite high and the initial proportion of CD34+ cells is around 90%. How this proportion decreased to 60% in three days is still not fully explained. CD34−/CD33+ cells proliferate quite fast and tend to predominate in the entire population. CD34+/CD33+ proliferated slightly slower and CD34+/CD33− divided rarely. This suggests that the expression of CD33 and the loss of expression of CD34 tend to be correlated with the increased proliferation capacities. CD33+ cells are engaged in the myeloid lineage and somehow impair the regeneration capacities of bone marrow transplant. Further investigations are required to identify the mechanism underlying the early appearance of this marker and the loss of CD34 in order to prevent them and maintain the regeneration capacities of the transplant. The occurrence of asymmetric divisions in the very early stages of the cell culture is an attractive hypothesis that could account for the evolution we observed.
HSCs have been shown to display sometimes a polarized organization of cell surface receptors.28,29 Several receptors, such as ICAMs, mucins, CD44 and integrins, were shown to cluster in the cholesterol-rich membrane of the uropod at the cell rear.28,30 Our study showed that CD33 should be added to this list. More importantly, the long-term HSC monitoring that was enabled by the absence of flow in the chambers revealed the tight coupling between this polarization and cell migration. This suggests that the early mechanisms involved in the initiation of cell migration are probably coupled with receptor clustering and cell polarization. Further studies are required to investigate the original symmetry break in HSCs. The association with cell migration suggests that the interplay between cell adhesion and contractility could be a key element in this event.31 No-flow chambers offer the best-suited experimental conditions to study these basic and fundamental questions and improve our understanding of HSC biology.
Acknowledgements
MT and JL acknowledge support from the European Research Council (starting grant ERC-310472) and the Agence National pour la Recherche (ANR-05PRIB01103, ANR-10-IBHU-0002). TC received a PhD fellowship from the president of Grenoble University.References
- M. A. Ostrowski, N. F. Huang, T. W. Walker, T. Verwijlen, C. Poplawski, A. S. Khoo, J. P. Cooke, G. G. Fuller and A. R. Dunn, Biophys. J., 2014, 106, 366–374 CrossRef CAS PubMed.
- C. Boehlke, F. Kotsis, V. Patel, S. Braeg, H. Voelker, S. Bredt, T. Beyer, H. Janusch, C. Hamann, M. Gödel, K. Müller, M. Herbst, M. Hornung, M. Doerken, M. Köttgen, R. Nitschke, P. Igarashi, G. Walz and E. W. Kuehn, Nat. Cell Biol., 2010, 12, 1115–1122 CrossRef CAS PubMed.
- K. Yamamoto, T. Takahashi, T. Asahara, N. Ohura, T. Sokabe, A. Kamiya and J. Ando, J. Appl. Physiol., 2003, 95, 2081–2088 Search PubMed.
- F. le Noble, D. Moyon, L. Pardanaud, L. Yuan, V. Djonov, R. Matthijsen, C. Bréant, V. Fleury and A. Eichmann, Development, 2004, 131, 361–375 CrossRef CAS PubMed.
- J. G. Goetz, E. Steed, R. R. Ferreira, S. Roth, C. Ramspacher, F. Boselli, G. Charvin, M. Liebling, C. Wyart, Y. Schwab and J. Vermot, Cell Rep., 2014, 6, 799–808 CrossRef CAS PubMed.
- F. Moazzam, F. A. DeLano, B. W. Zweifach and G. W. Schmid-Schönbein, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 5338–5343 CrossRef CAS.
- L. Adamo, O. Naveiras, P. L. Wenzel, S. McKinney-Freeman, P. J. Mack, J. Gracia-Sancho, A. Suchy-Dicey, M. Yoshimoto, M. W. Lensch, M. C. Yoder, G. García-Cardeña and G. Q. Daley, Nature, 2009, 459, 1131–1135 CrossRef CAS PubMed.
- A. Mammoto, T. Mammoto and D. E. Ingber, J. Cell Sci., 2012,(13), 3061–3073 CrossRef CAS PubMed.
- J.-W. Shin, J. Swift, I. Ivanovska, K. R. Spinler, A. Buxboim and D. E. Discher, Differentiation, 2013, 86, 77–86 CrossRef CAS PubMed.
- A. Gratwohl, H. Baldomero and J. Passweg, Curr. Opin. Hematol., 2013, 20, 485–493 CrossRef CAS PubMed.
- A. Trounson, R. G. Thakar, G. Lomax and D. Gibbons, BMC Med., 2011, 9, 52 CrossRef PubMed.
- E. A. Copelan, N. Engl. J. Med., 2006, 354, 1813–1826 CrossRef CAS PubMed.
- W. J. Polacheck, R. Li, S. G. M. Uzel and R. D. Kamm, Lab Chip, 2013, 13, 2252–2267 RSC.
- W. Liu, L. Li, X. Wang, L. Ren, X. Wang, J. Wang, Q. Tu, X. Huang and J. Wang, Lab Chip, 2010, 10, 1717–1724 RSC.
- V. Lecault, M. Vaninsberghe, S. Sekulovic, D. J. H. F. Knapp, S. Wohrer, W. Bowden, F. Viel, T. McLaughlin, A. Jarandehei, M. Miller, D. Falconnet, A. K. White, D. G. Kent, M. R. Copley, F. Taghipour, C. J. Eaves, R. K. Humphries, J. M. Piret and C. L. Hansen, Nat. Methods, 2011, 8, 581–586 CrossRef CAS PubMed.
- J. J. VanDersarl, A. M. Xu and N. A. Melosh, Lab Chip, 2011, 11, 3057–3063 RSC.
- T. M. Keenan, C. W. Frevert, A. Wu, V. Wong and A. Folch, Lab Chip, 2010, 10, 116–122 RSC.
- Y.-C. Toh, C. Zhang, J. Zhang, Y. M. Khong, S. Chang, V. D. Samper, D. van Noort, D. W. Hutmacher and H. Yu, Lab Chip, 2007, 7, 302–309 RSC.
- C. Zhang, Z. Zhao, N. A. Abdul Rahim, D. van Noort and H. Yu, Lab Chip, 2009, 9, 3185–3192 RSC.
- L. Wang, X.-F. Ni, C.-X. Luo, Z.-L. Zhang, D.-W. Pang and Y. Chen, Biomed. Microdevices, 2009, 11, 679–684 CrossRef CAS PubMed.
- C. Luo, L. Jiang, S. Liang, Q. Ouyang, H. Ji and Y. Chen, Biomed. Microdevices, 2009, 11, 981–986 CrossRef PubMed.
- M. Kolnik, L. S. Tsimring and J. Hasty, Lab Chip, 2012, 12, 4732–4737 RSC.
- L. Lu, M. Xiao, R. N. Shen, S. Grigsby and H. E. Broxmeyer, Blood, 1993, 81, 41–48 CAS.
- L. W. Terstappen, S. Huang, M. Safford, P. M. Lansdorp and M. R. Loken, Blood, 1991, 77, 1218–1227 CAS.
- R. G. Andrews, B. Torok-Storb and I. D. Bernstein, Blood, 1983, 62, 124–132 CAS.
- J. S. Nielsen and K. M. Mcnagny, J. Cell Sci., 2008, 4145, 3683–3692 CrossRef PubMed.
- A.-V. Fonseca, D. Freund, M. Bornhäuser and D. Corbeil, J. Biol. Chem., 2010, 285, 31661–31671 CrossRef CAS PubMed.
- F. Sánchez-Madrid and J. M. Serrador, Nat. Rev. Mol. Cell Biol., 2009, 10, 353–359 CrossRef PubMed.
- A.-V. Fonseca and D. Corbeil, Commun. Integr. Biol., 2011, 4, 201–204 CrossRef CAS PubMed.
- N. Vannini, A. Roch, O. Naveiras, A. Griffa, S. Kobel and M. P. Lutolf, Cell Cycle, 2012, 11, 1535–1543 CrossRef CAS PubMed.
- J. Shin, A. Buxboim, K. R. Spinler, J. Swift, D. A. Christian, C. A. Hunter, C. Léon, C. Gachet, P. C. D. P. Dingal, I. L. Ivanovska, F. Rehfeldt, J. A. Chasis and D. E. Discher, Cell Stem Cell, 2014, 14, 81–93 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00807c |
| This journal is © The Royal Society of Chemistry 2015 |