
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix effects of carbon and bromine in inductively coupled plasma optical emission spectrometry†
Helmar
Wiltsche
*,
Monika
Winkler
and
Paul
Tirk
Graz University of Technology, Institute of Analytical Chemistry and Food Chemistry, Graz, Austria. E-mail: helmar.wiltsche@tugraz.at
First published on 19th August 2015
Abstract
In inductively coupled plasma (ICP) based techniques the signal enhancing effect of carbon on some elements like arsenic or selenium is well documented. However, there is a large spread in the reported magnitude of this effect and whether it can be observed for other elements too. In this investigation we studied the effect of larger amounts of carbon on a total of 157 emission lines of 36 elements. A strong instrument dependence of the “carbon enhancement effect” was encountered in inductively coupled plasma optical emission spectrometry (ICP-OES), despite the use of the same sample solutions and the same sample introduction system. Several potential enhancement sources (carbon in the form of methanol, phenylalanine and CO2 as well as bromine) were compared. By tapping the high voltage power supply of the RF generator, current and voltage fed to the power oscillator could be recorded simultaneously with the emission line signal. From these data it was concluded that the carbon-based matrix effect is a combination of five factors: (1) depending on the source of carbon, changes in the sample nebulization; (2) carbon induced charge exchange reactions; (3) plasma impedance changes caused by the introduction of large quantities of carbon into the ICP: depending on the RF generator used, this effect causes power regulation problems and results in higher RF power coupled to the discharge; (4) thermal pinch effect – the ICP discharge shrinks and becomes smaller; (5) the state of matter (gaseous or liquid) of the introduced carbon sources is relevant to the magnitude of the carbon enhancement effect.
1. Introduction
The introduction of larger quantities of carbon into the inductively coupled plasma (ICP) instrument is reported to increase the sensitivity of elements with the ionization potential (IP) in the range of about 9 to 12 eV.1–14 This enhancement has been observed in both inductively coupled plasma optical emission spectrometry (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS). Although this “carbon enhancement effect” was reported nearly 25 years ago,1 the understanding of the underlying mechanism is still unsatisfactory as apparent from the many conflicting results published since then.By far the largest number of studies describing the “carbon enhancement effect” have been performed using ICP-MS as the speciation analysis of As and Se received great attention and these two elements exhibit in general the largest enhancement by carbon. The differences in the attained level of enhancement are striking: whereas some authors reported a carbon-induced suppression of the As and Se signal in the range of 50–70% (1350 W RF power, 2% v/v methanol),8 most authors observed a signal enhancement by a factor of 2–5 (e.g.ref. 5–7, 9, 13, 15 and 16). Even an 11-fold enhancement of the Se-signal has been reported.14 Indeed, it is difficult to compare these data as different sources of carbon were used. Several authors13,15,16 concluded that the change in nebulization efficiency caused by C-sources such as methanol cannot explain the carbon induced signal enhancement, particularly when considering that the same effect is caused by glycerin,1,16 glucose,15 ammonium acetate15 and urea.17 By using methane as the carbon source Allain et al.1 confirmed that differences in the nebulization efficiency are not the sole reason for the “carbon enhancement effect”. Despite the different carbon sources used, most authors1,2,4,5,7–9,13 found that in ICP-MS the As and Se signal initially increases with increasing carbon content of the sample solution, and then reaches a plateau and decreases again. The maximum enhancement of As and Se appears to be a function of the carbon source and of the instrument used.
The reports on the enhancement or suppression of other elements by carbon are even more confusing. Whereas for Au (IP 9.23 eV)1,13,18 and Be (IP 9.32 eV)4 carbon induced signal enhancement is reported, for Zn (IP 9.39 eV), S (IP 10.36 eV), Hg (IP 10.44 eV) and I (IP 10.45 eV) both carbon induced enhancement1,4,5,7,13,15–17 and the absence of this effect1,12,16 are reported.
Fewer authors have so far investigated the carbon enhancement effect in ICP-OES. Machát et al.11 found that the small spectral interference on the Se I 196.026 nm emission line is caused by CO band emission. The authors noted an instrument dependent enhancement of Se by 20 to 58% when 10 g L−1 C (glycerol; data corrected for nebulization efficiency changes) was introduced into the ICP and that As and Te were enhanced too. Moreover, they found that S, P and Br (from H2SO4, H3PO4 and HBr, respectively) enhanced the signals of As, Se and Te significantly. Grindlay et al.3 investigated the carbon-related matrix effect and found As and Se enhanced by up to 30% in the presence of 20 g L−1 C from citric acid. Simultaneously low excitation energy atomic lines (EP < 6 eV) were suppressed by up to 15%, while ionic lines remained unaffected.
A thorough discussion about the potential mechanism behind the “carbon enhancement effect” is given only in few publications. Abou-Shakra et al.19 proposed a charge exchange mechanism between C+ and selenium atoms. According to this hypothesis, excited Se+ (4p3 2D0) is formed whose upper level energy of 1098 kJ mol−1 is sufficiently close to the IP of carbon (1086 kJ mol−1). Thereby, both the enhancement in ICP-OES and ICP-MS could be explained. Grindlay et al.16 extended and refined this theory further, as a charge transfer reaction requires not only the minimum energy difference between the involved levels, but obliges the fulfillment of the Wigner spin conservation rule. The authors determined by ICP-MS that the dominant carbon species in the ICP are C+ and CO+ and concluded that a high charge transfer probability exists for As, Au, Hg, I, Ir, Os, P, Pt, S, Sb, Se, Te, and Pd. Experimentally Grindlay et al. found a carbon induced signal enhancement only for As, Au, Hg, I, Sb, Se, and Te.
As listed above, reports on the carbon enhancement effect presented in the literature rely heavily on ICP-MS data. Particularly for mechanistic studies, the lack of additional information on fundamental plasma parameters such as electron number densities or ionization temperature is unsatisfying. The processes happening in the interface region are another source of potential bias for mechanistic studies: just behind the sample cone a supersonic jet is formed, wherein the broad velocity distribution of atoms from the ICP is turned into a much narrower one, centered around the velocity of Ar.20 Moreover, this zone is a source of collisions and reactions between the constituents of the expanding gas. It seems not entirely unlikely that, in parts, the large differences in the signal enhancement of As and Se observed for different ICP-MS instruments are caused in this region, as carbon is added in a very high concentration. Another factor that could cause additional bias when studying the carbon enhancement effect is the reported shift of the region of maximum ion density in the ICP9,21 that necessitates the readjustment of the sampling depth. Particularly for ICP-MS instruments that alter the sampling depth by changing the nebulizer gas flow, additional bias from different nebulized sample masses might be introduced.
The situation is different for ICP-OES: once the radiation is emitted from an excited state, the factors that could affect the signal thereafter are minute compared to the expansion of a hot atmospheric pressure plasma into the high vacuum of the mass spectrometer, the associated supersonic expansion and charge separation. Particularly for axially viewed ICP-OES slight changes in the zone of maximum excitation will not affect the overall signal intensity as much as in ICP-MS, as the observed region inside the normal analytical zone will be averaged by the optical setup.
The aim of this work is to provide insights into the factors that affect the signal enhancement of high IP elements by carbon in the ICP in order to distinguish between spectroscopic and instrumental effects. As apparent by the foregoing discussion, ICP-OES was mainly used for this purpose.
2. Experimental
2.1. Instrumentation
An axially viewed ICP-OES (CIROS Vision EOP, Spectro, Germany) with a fixed viewing depth was used in this work. The instrument dependence of the investigated effect was also studied using two other ICP-OES (Optima 3000 XL and Optima 5300 DV; both PerkinElmer, USA) under instrument conditions comparable to those provided in Table 1 as well as one ICP-MS (Elan DRC+, PerkinElmer, USA). The ICP-OES operating conditions listed in Table 1 are compromise conditions for multi-element analysis. They were optimized for aqueous (3% v/v HNO3) solutions for the highest signal to background ratio for the observed emission lines of As, Ca, Cl, Fe, P, S, Se, and Zn.| Spectrometer | Paschen–Runge mount with22 charge-coupled device (CCD) line-detectors |
| RF generator | 27 MHz, free running Huth–Kühn22 type power oscillator; also known as the tuned plate tuned grid oscillator23 |
| Plasma power, W | 1350 |
| Outer gas flow, L min−1 | 12 |
| Intermediate gas flow, L min−1 | 0.6 |
| Nebulizer gas flow, L min−1 | 0.83 |
| Sample flow rate, mL min−1 | 1.8 |
| ICP torch | Standard torch with 2.5 mm ID injector; fixed (one piece) torch |
| Nebulizer | Cross-flow |
| Spray chamber | Scott type |
| Number of measurements | 4 |
| Integration time, s | 24 |
A 4-turn probe coil connected to a frequency counter (TF930, TTi Thurlby Thandar Instruments Limited, UK) recorded the frequency response of the free running RF generator. Details of this setup were published previously.24
Current and voltage delivered to the RF generator were constantly recorded by taping the low voltage control lines between the high voltage power supply (HV-PSU) and the generator control board with analog to digital converters (ADCs). These control lines provide voltages proportional to the output current (1 V ≡ 200 mA) and output voltage (1 V ≡ 1000 V) of the HV-PSU. Four 16 bit ADCs (ADS1115, Texas Instrument) controlled by a microcontroller (ATSAM3X8E on an “Arduino Due – open source electronics prototyping platform”; https://www.arduino.cc) delivered 475 samples/second for each channel to the control PC. The schematic of the ADC board is shown in the ESI† of this article. As the ADS1115 includes a programmable gain amplifier, no operational amplifier buffer stage was deemed necessary and the connection to the HV-PSU control lines was made directly with a 10 kΩ resistor. The ADC board fulfilled the noise-, gain- and drift-specifications given in the ADS1115 datasheet. However, due to the lethal voltage present at the HV-PSU output, no attempt was made to check the factory calibration between the high voltage output and the low voltage control lines.
The nebulization efficiency and mass flow of samples introduced into the ICP were determined using the well-established method of continuous weighing.25 Contrary to the setup employed previously for this purpose26 two beakers located at the balance were used rather than one: the first beaker contained the sample solution whereas the second was used to collect the liquid returned from the spray chamber. Thereby the density of each solution could be measured independently with a density meter (DMA4500, Anton Paar, Austria).
An external thermal-based mass flow controller (100 sccm, Mass-Flo 1179, MKS Instruments, Germany) controlled the flow of CO2. The appropriate gas correction factor was used to correct the response of the nitrogen calibrated mass flow controller for the different physical properties of CO2 as recommended by the manufacturer.
2.2. Reagents
Purified water (18 MΩ cm, Barnstead Nanopure, Thermo Fisher Scientific, USA) and high purity acids (HNO3, purified by subboiling) were used throughout. Standard solutions were prepared from a 100 mg L−1 multi-element stock solution (Al, Ag, As, B, Ba, Be, Bi, Ca, Cd, Co, Cr, Cu, Fe, K, Li, Mg, Mn, Mo, Na, Ni, Pb, Sb, Se, Sr, Ti, Tl, V, and Zn, Roth, Germany), several 1 g L−1 single element stock solutions (As, Au, B, Bi, Hg, Pb, Se: Merck, Germany; Br, S, P: SCP Science, USA) and in the case of I from KIO3 (pa, Merck, Germany) by dilution with 3% HNO3 (v/v). The ICP excitation temperature was determined using a 300 mg L−1 Fe solution containing 3% HNO3 (v/v) prepared from a 10 g L−1 Fe single element stock solution (Alfa Aesar, Germany). Water saturated with bromine was prepared by equilibration of high purity water with Br2 (Sigma Aldrich, ACS reagent, Germany) for one week in a dark glass bottle. Methanol (Normapur, VWR, Germany), NaCl (Fluka, Germany), and phenylalanine (f. biochemistry, Merck, Germany) dissolved in 3% (v/v) HNO3 were used to study plasma based matrix effects. Argon was of 5.0 quality (Messer, Austria) and carbon dioxide of Biogen C quality (Linde, Austria).2.3. Optical emission-based plasma diagnostics
The magnesium ion to atom ratio (plasma robustness criteria27) was calculated from the Mg II 280.270 nm/Mg I 285.213 nm emission line intensity ratio using a 10 mg L−1 Mg solution.The Boltzmann plot method28 was used to determine the Fe(I) excitation temperature Texc. Details on the emission lines used and fundamental constants are reported elsewhere.24
2.4. Experimental procedure and processing of the spectra
In order to record a consistent set of data, the effects of gaseous (CO2) and liquid reagents (Br2, aqueous solutions (3% v/v HNO3) of NaCl or phenylalanine, or methanol) were investigated using a standardized procedure with five test standards: after recording the blank spectra, standard solutions 1 to 4 were measured. Between the samples, the sample introduction system was rinsed with diluted nitric acid (3% v/v) for 60 seconds. The elements and their concentrations in each standard solution were: solution 1: 2 mg L−1 of Al, Be, Ca, Cd, Co, Cr, Cu, Fe, K, Li, Mg, Mn, Mo, Na, Ni, Sb, Sr, Ti, Tl, V, Zn, 10 mg L−1 of B, Bi, Pb, and 20 mg L−1 As and Se; solution 2: 10 mg L−1 of Au, Br, Hg, P, S; solution 3: 30 mg L−1 I; solution 4: 300 mg L−1 Fe.Gases were added either to the intermediate gas stream of the ICP torch using a small T-piece, or to the spray chamber through a 2 mm ID PTFE tube. For experiments with liquid reagents, the aforementioned four test standards and a blank solution were prepared from the appropriately diluted reagent (methanol, water saturated with Br2, solutions of NaCl or phenylalanine) and the relevant single or multi-element stock solution.
All analyte signals were normalized to the aqueous standard (3% v/v HNO3) using the following procedure: background correction was applied to all spectra. In the first step of the data treatment, the emission signals of the blank solutions for each reagent concentration level were compared with the emission signals of the respective analyte-containing standard. If the signal in the blank solution was larger than 10% of the uncorrected standard, the line was rejected for further processing, as a spectral interference could not be excluded. Otherwise, if the emission signal passed this criterion, the blank signal was subtracted from the analyte signal and the data were then normalized to the respective aqueous signal (3% v/v HNO3).
3. Results and discussion
3.1. Repeatability of the excitation temperature determination
The interpretation of the results presented in this work relies heavily on the excitation temperature of Fe(I) determined by the Boltzmann plot method. Particularly when comparing data recorded over a period of about one month, the repeatability of the excitation temperature (Texc) is of vital importance. It is important to note that the Texc reported here represents an average temperature of the region in the ICP that is observed by the spectrometer.The excitation temperature of Fe(I) was determined on four consecutive days. Before recording the emission spectra the RF generator was allowed to “warm up” for 30 min.24 The excitation temperature was 6470 ± 15 K, 6450 ± 20 K, 6450 ± 10 K, 6460 ± 10 K (mean value ± s; n = 5) using the plasma conditions listed in Table 1. Then the ICP torch was removed, acid cleaned, reinstalled and the excitation temperature was again recorded on the following days: 6530 ± 20 K, 6540 ± 20 K, 6520 ± 20 K, 6520 ± 10 K. After installing the torch again, the observation position of the spectrometer was tuned following the instrument manufacturer's recommendations and similar signal intensities were obtained for the Mn tuning solution. Simultaneously with the observed small change of the excitation temperature, the plasma robustness increased slightly from 6.8 ± 0.1 to 7.0 ± 0.1 when the torch position was slightly altered in the cause of the cleaning process.
The significant difference in the excitation temperature before and after the torch cleaning can be attributed to small changes in the position of the ICP torch within the load coil. Clearly, any investigation on the effect of an external parameter must be recorded without removing or changing the ICP torch, as otherwise an uncontrolled experimental bias is introduced. It is important to note that in the instrument used there is no mechanical indicator for the torch alignment but only the recommendation to introduce the torch into the load coil until the distance between the inner glass tube and the first turn of the load coil is 1 mm. Although for the experiment reported above the exact position of the torch was not determined, a later investigation with a caliper indicated a positioning repeatability of about ±0.2 mm.
It seems also important to note that the precision of the excitation temperature determination was very high: for five independent recordings the standard deviation was always found to be between 10 and 20 K. The uncertainty of the slope of the least square fitted line in the Boltzmann plot was about 100 K. As both the short term (5 repetitions) as well as the long term (4 consecutive days) precision of the excitation temperature determination were much higher, it must be concluded that the uncertainty of the slope in the Boltzmann plot is dominated by the uncertainty of the transition probability data. Consequently, the absolute value of the reported excitation temperatures might be biased, but relative changes can be expected to be identified correctly.
3.2. Instrumental dependence of the effect of carbon on the signal of Se
When comparing the degree of enhancement of Se by carbon reported in the literature, the differences in the magnitude of the enhancements are striking, though the various experimental conditions and reagents make a direct comparison difficult. The effect of methanol (0–10% m/m) on selenium was investigated on three different ICP-OES and one ICP-MS instruments. As the torch geometries of these instruments are different, it was not possible to maintain similar outer- and intermediate gas flows. Consequently, these flow rates were selected to match typical conditions used for the analysis of aqueous (diluted HNO3) samples: both Optima instruments were operated at 15 L min−1 outer gas flow and 0.5 L min−1 intermediate gas flow; the ICP-MS used 15 L min−1 outer gas flow and 1.3 L min−1 intermediate gas flow. The RF power, nebulizer gas flow and sample flow rate were kept constant as listed in Table 1, as well as the nebulizer and spray chamber. Thereby comparable experimental conditions were achieved as evident from the similar plasma robustness (Mg II 280.270 nm/Mg I 285.213 nm ratio in 3% v/v HNO3) that were 7.0 ± 0.1, 6.8 ± 0.1 and 6.5 ± 0.1 for the Spectro CIROS Vision, PerkinElmer Optima 3000 XL and Optima 5300 DV (the data of the two Optima instruments were corrected for differences in the spectrometers' echelle grating efficiency by applying the correction factor 1.85 to the Mg-ratio29,30).For the ICP-MS experiment, the nebulizer gas flow was optimized for maximum signal intensity in an aqueous solution (3% v/v HNO3).
The differences in the instrument dependent effect of carbon are shown in Fig. 1. In ICP-MS the 78Se signal first increased by a factor of up to 2.3 at 1.5% (m/m) methanol and then decreased again. This pattern is consistent with ICP-MS data reported by other authors,2,4,9,15 though the maximum enhancement factor and the corresponding methanol concentration appear to differ from instrument to instrument.
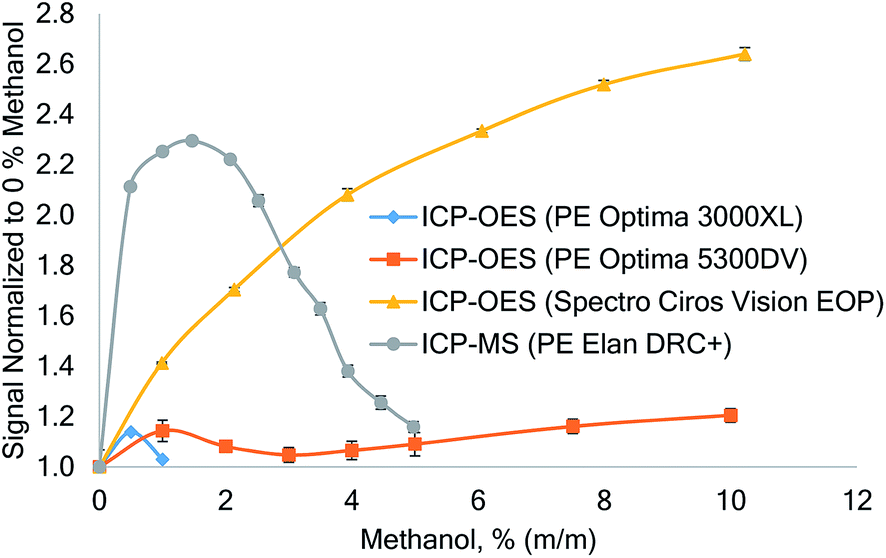 | ||
| Fig. 1 Effect of increasing methanol concentration on the signal of Se in ICP-OES (Se 203.985 nm) and ICP-MS (m/z = 78) recorded on different instruments. The Se concentration was 10 mg L−1 in ICP-OES and 10 μg L−1 in ICP-MS. No internal standard was used; the Einzel-lens of the ICP-MS was tuned for aqueous solutions. Error bars: standard deviation, n = 5. | ||
A different behavior was recorded on the three ICP-OES instruments investigated: PerkinElmer Optima 3000XL showed little tolerance to increased methanol load to the plasma and above 1% (m/m) methanol the ICP extinguished. Interestingly, only a rather small Se signal enhancement of 14% was recorded on this instrument at 0.5% (m/m) methanol. Using a PerkinElmer Optima 5300DV (axially viewed), the ICP discharge was stable up to a maximum methanol concentration of 10% (using a 1.8 mm ID injector tube), but the enhancement of selenium was only 20% at the highest methanol concentration tested.
Using the Spectro Ciros Vision EOP ICP-OES, a steady and large signal enhancement of Se was encountered. At 10% (m/m) methanol a signal enhancement by a factor of 2.6 was observed for Se I 203.985 nm. This enhancement was consistent on all investigated Se emission lines and factors of 2.5 and 2.2 were recorded for Se I 196.090 nm and Se I 207.479 nm, respectively.
Machát et al.11 also encountered different Se enhancement factors on different ICP-OES instruments, but different sample introduction systems were used.
3.3. Effect of methanol
As the Spectro Ciros Vision ICP-OES showed the largest enhancement of Se and allowed the simultaneous acquisition of the entire relevant spectrum, this instrument was used for further investigations. A total of 157 emission lines of 36 elements were investigated. Table 2 lists the effect of 10% (m/m) methanol on selected lines. The data can be classified broadly into four groups.| Emission line, nm | Total line energy, eV | Methanol 10% | Phenylalanine 8 g L−1 C | 15 sccm CO2 added to the aerosol gas stream | 15 sccm CO2 added to the intermediate gas flow | Br2 34 g L−1 | NaCl 30 g L−1 |
|---|---|---|---|---|---|---|---|
| Al I 396.152 | 3.14 | 1.00 | 0.98 | 0.76 | 1.27 | 1.06 | 0.79 |
| Al II 167.078 | 13.41 | 1.50 | 1.02 | 1.04 | 1.83 | 0.95 | 0.69 |
| Ar I 404.442 | 14.69 | 1.97 | 0.98 | 1.36 | 2.49 | 0.88 | 0.90 |
| As I 197.262 | 6.29 | 1.92 | 1.08 | 1.47 | 1.98 | 1.03 | 0.84 |
| As I 189.042 | 6.56 | 1.97 | 1.08 | 1.52 | 2.08 | 1.04 | 0.83 |
| As I 193.759 | 6.40 | 1.90 | 1.07 | 1.48 | 2.01 | 1.04 | 0.84 |
| Au I 267.595 | 4.63 | 1.09 | 1.00 | 0.89 | 1.17 | 1.02 | 0.82 |
| Au I 242.795 | 5.11 | 1.18 | 1.00 | 0.92 | 1.25 | 1.03 | 0.81 |
| B I 249.773 | 4.96 | 1.16 | 0.97 | 0.94 | 1.26 | 1.01 | 0.76 |
| Be I 234.861 | 5.28 | 1.18 | 0.99 | 0.93 | 1.28 | 0.96 | 0.75 |
| Be II 313.042 | 13.28 | 1.42 | 1.08 | 1.07 | 1.40 | 1.05 | 0.66 |
| Br I 148.845 | 8.33 | 1.57 | 0.98 | 0.93 | 1.59 | ND | ND |
| Br I 144.99 | 8.55 | Int | 0.98 | 1.05 | 1.73 | ND | ND |
| Cd I 228.802 | 5.42 | 1.31 | 1.02 | 0.98 | 1.44 | 0.97 | 0.79 |
| Cd II 214.438 | 14.77 | 1.57 | 1.04 | 1.07 | 1.85 | 0.91 | 0.67 |
| Cl I 134.724 | 9.20 | 1.77 | 0.99 | 1.20 | 2.03 | 0.83 | Int |
| Co II 230.786 | 13.75 | 1.19 | 1.02 | 0.85 | 1.40 | 0.90 | 0.65 |
| Cr II 205.552 | 12.80 | 1.20 | 1.03 | 0.86 | 1.36 | 0.95 | 0.67 |
| Hg I 253.652 | 4.89 | 0.97 | 0.99 | 0.84 | 1.12 | 0.99 | 0.83 |
| Hg I 184.95 | 6.70 | 1.45 | 1.00 | 1.16 | 1.79 | 0.98 | 0.85 |
| Hg II 194.227 | 16.82 | 1.41 | 1.01 | 1.05 | 1.77 | 0.91 | 0.77 |
| I I 183.038 | 6.77 | 1.52 | 1.04 | 1.20 | 1.86 | Int | 0.76 |
| I I 178.276 | 6.95 | 1.69 | 1.05 | 1.61 | 2.07 | Int | 0.83 |
| Mg I 285.213 | 4.35 | 1.00 | 0.96 | 0.73 | 1.28 | 0.95 | 0.79 |
| Mg II 280.27 | 12.07 | 1.14 | 1.00 | 0.84 | 1.35 | 0.95 | 0.61 |
| P I 213.618 | 7.21 | 2.09 | 1.03 | 1.63 | 2.20 | 1.14 | 0.86 |
| P I 169.403 | 8.73 | 2.08 | 1.05 | 1.71 | 2.21 | 1.13 | 0.81 |
| P I 138.147 | 8.97 | 2.00 | 1.03 | 1.65 | 2.16 | 1.13 | 0.88 |
| S I 180.731 | 6.86 | 1.59 | 0.97 | 1.18 | Int | 1.18 | 0.94 |
| S I 182.034 | 6.86 | 1.64 | 0.98 | 1.17 | 1.67 | 1.19 | 0.91 |
| Se I 207.479 | 5.97 | 2.24 | 1.22 | 1.65 | 2.37 | 1.85 | 0.71 |
| Se I 196.09 | 6.32 | 2.51 | 1.20 | 1.98 | 2.89 | 1.86 | 0.78 |
| Se I 203.985 | 6.32 | 2.64 | 1.22 | 2.02 | 2.97 | 1.83 | 0.78 |
| Tl I 276.787 | 4.48 | 1.06 | 0.95 | 0.79 | 1.27 | 1.04 | 0.81 |
| Tl II 132.171 | 15.49 | 2.13 | 1.03 | 1.36 | 2.68 | Int | 0.72 |
| Zn I 213.856 | 5.80 | 1.28 | 1.01 | 0.94 | 1.41 | 0.93 | 0.77 |
| Zn II 202.548 | 15.51 | 1.81 | 1.05 | 1.23 | 2.10 | 0.91 | 0.69 |
| Plasma robustness | 8.2 ± 0.1 | 7.5 ± 0.1 | 7.8 ± 0.1 | 7.2 ± 0.1 | 7.1 ± 0.1 | 5.4 ± 0.1 | |
| T exc, K | 6980 ± 10 | 6590 ± 10 | 6800 ± 10 | 6840 ± 20 | 6550 ± 20 | 6510 ± 20 |
The first group of emission lines was suppressed by about 10–20% and includes some ion lines of Ba and Sr as well as some atom lines of Mn, Na and Ni. These lines have a very low excitation potential (2–3 eV) in common.
The effect of methanol was small on the second group of elements (enhancement by a factor of 0.9 to 1.1). In general, the excitation energies of atom lines in this group were between 3 and 5 eV. For ion lines the total line energy (ionization + excitation energy) was between 9 and 11 eV.
The third group of emission lines was enhanced by about 20–50%. The excitation energy of these lines was in general between 5 and 6 eV and the total line energy of ion lines was between 11 and 14 eV.
Signal enhancements compared to an aqueous solution (3% v/v HNO3) of more than 50% were observed for atom lines with an excitation potential greater than 6 eV and for some high energy ion lines in the region of about 15.5 eV. This fourth group included the atom lines of Ar, As, Br, Cl, I, P, S, and Se as well as some high energy ion lines of Ag, Cd, Tl, and Zn.
It is important to note that the listed energy ranges of the four groups are not sharply defined and that some emission lines do not follow the listed general trend. Moreover, there is no clear correlation between the total line energy (excitation potential for atom lines) and the enhancement, but only a trend.
The power drawn by the RF generator plotted as a function of the methanol concentration nebulized during the measurement is shown in Fig. 2. The Huth–Kühn type power oscillator used is current controlled. For every RF power level that can be selected in the instrument software, a corresponding current is set by the HV power supply. The current is derived from a factory power calibration. The power oscillator behaves purely resistive during normal operation – Ohm's law therefore applies and the power drawn by the power oscillator is the product of current (RMS) and voltage (RMS) read directly from the HV power supply.
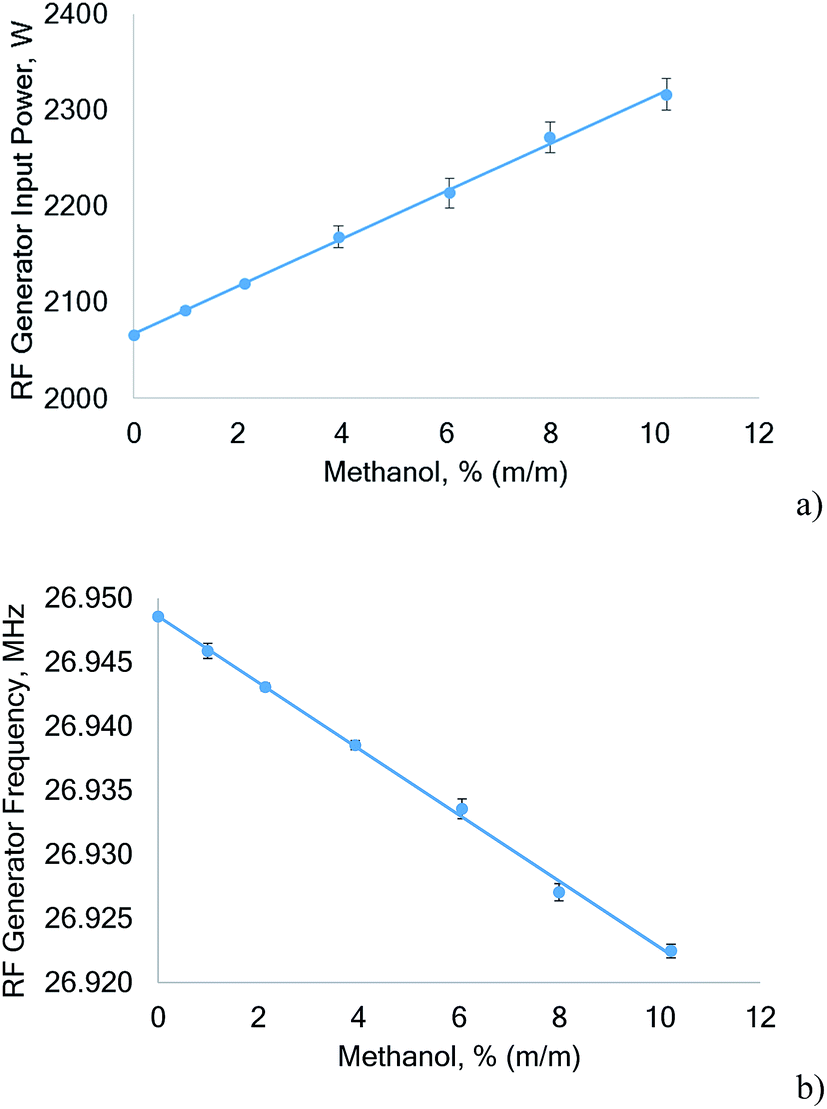 | ||
| Fig. 2 Power drawn by the RF generator for a nominal output of 1350 W (a) and frequency response of the generator (b) as a function of the methanol concentration nebulized during measurement. The error bars represent the standard deviation of five independent experiments. | ||
From Fig. 2a it is evident that with increasing methanol concentration the power drawn by the RF generator increases significantly (about 250 W when changing from diluted nitric acid (3% v/v HNO3) to 10% methanol). As expected from a current controlled power oscillator, the power vacuum tube plate current remained constant, but the plate voltage increased from 3565 V to 4000 V when the sample solution was switched from 3% (v/v) HNO3 to 10% (m/m) methanol. At the same time the frequency of the power oscillator decreased by 26.1 kHz. It seems interesting to note that for aqueous aerosols an increase of the RF generator power is accompanied by a small increase in the frequency. The observed linear decrease of the RF generator frequency (Fig. 2b) and the simultaneous linear increase in the power uptake when the methanol concentration in the sample solution is increased indicates destabilization of the plasma as also observed previously.24 In addition, this can also be concluded from the rising SDs of the plate voltage that increased the SDs of the generator input power when the methanol concentration rose.
The reason for the observed increase of power drawn by the RF generator can be expected to be a significant impedance change of the plasma caused by the large quantities of carbon introduced. However, only a part of the additionally drawn power will be coupled to the plasma, while the rest is converted to heat in the RF generator.
The reported increase of power uptake of the RF generator has, to the best of our knowledge, not been reported in the literature. However, Larsen and Stürup7 noted that “the upper practical RF power input was 1320 W” because otherwise the cooling capacity of the ICP-MS interface would have been exceeded. As aqueous solutions could indeed be analyzed at higher RF power levels, this finding could be attributed to a higher power uptake of the RF generator.
When increasing the methanol concentration in the nebulized sample solution, the ICP was found to shrink in size and withdraw from the injector tip. This thermal pinch effect31 results in a smaller, brighter discharge. From Fig. 3 it is evident that by nebulizing 10% (m/m) methanol, the plasma completely withdrew from the injector region and the plasma base was located just after the second turn of the load coil. Yet, the quantification of this effect is difficult, as the load coil hides the most important region of the ICP.
 | ||
| Fig. 3 Effect of methanol on plasma volume. (a) Introduction of 3% v/v HNO3; (b) introduction of 10% (m/m) methanol. The positions of the three turns of the load coil and the injector are represented with white bars for clarity. Both images were recorded under identical photographic conditions (aperture setting, exposure time). Note that in image (b) the plasma withdrew from the space between the first and the second turn of the load coil. | ||
The shifting of the plasma base away from the injector can also be expected to affect the recorded analyte emission intensities: although the plasma was axially viewed and the viewing depth was not changed, the zone within the ICP imaged onto the entrance slit of the spectrometer will follow the shift of the plasma base to some extent. Simultaneously the plasma power density changed the behavior of the emission lines, which is not easily predictable. Chan and Hieftje32 showed that the magnitude of matrix effects in the ICP is not constant when changing the observed location within the ICP. However, the large focal length of the spectrometer's entrance optics might reduce this effect when compared to the plasma cross-sections investigated by Chan and Hieftje or radial viewing.
In conclusion, the large quantities of carbon introduced by nebulizing 10% methanol altered plasma impedance and thereby led to an increase of the power vacuum tube's plate voltage. This in turn increased the RF power delivered to the ICP. Simultaneously, the plasma decreased in size and became hotter as evident from the rise in the excitation temperature from 6610 ± 20 K (3% v/v HNO3) to 6980 ± 10 K (10% methanol). It is apparent that an increase of RF power delivered to the ICP combined with a decrease of the plasma volume is associated with a change in the analyte emission signal intensity.
While all analyte emission lines that were significantly enhanced by carbon showed a steady signal increase with rising methanol concentration, plasma robustness only increased up to 6% (m/m) methanol (peak value 8.8) and then decreased again to 8.2 at 10% (m/m) methanol.
One of the inherent problems of using methanol as the source of carbon is the difficulty of quantifying the exact amount of carbon introduced into the ICP. The density of all methanol containing solutions was measured prior to the introduction into the spray chamber and in the waste solution that left the spray chamber after system equilibration. Moreover, the mass flow of aerosol leaving the spray chamber was determined by the method of continuous weighing. From these data a mass balance was calculated assuming a constant liquid flow rate from the peristaltic pump of 1.82 ml min−1: by subtracting the mass flow of methanol in the drained solution from the mass flow of methanol pumped to the spray chamber the mass flow of methanol introduced into the ICP was calculated. Using this method corrects for differences in aerosol formation and preferential evaporation of methanol from the surface of the spray chamber and aerosol droplets. Mora et al.33 found that the D50 (droplet distribution diameter below which 50% of the cumulative aerosol volume is found) of the primary aerosol of a water–methanol mixture decreases with increasing methanol content of the mixture. However, this effect was found to be rather small (D50 changed by 3%) when comparing water with 10% methanol.
The carbon mass flow introduced into the ICP increased almost linearly with rising methanol concentration in the sample solution fed to the nebulizer. However, below 4% (m/m) methanol the function bends, indicating less pronounced vaporization of methanol in the spray chamber. At the maximum concentration of 10% (m/m) methanol, 8.4 mg min−1 carbon was introduced into the ICP. The uncertainty of this calculation was found to be dominated by the method of continuous weighing: whereas the removal of the liquid from the sample beaker was constant with time, the draining of the waste solution was discontinuous, as individual droplets formed on the lower end of the Scott type spray chamber inner tube. Thereby a fluctuating signal was created. The error of the slope was below 6% for the used recording time of 10 minutes.
It is interesting to note that without the described correction for the preferential vaporization of methanol in the spray chamber the mass flow of carbon introduced into the ICP would have been underestimated by a factor of 1.7.
3.4. Effect of phenylalanine and CO2
Methanol is a volatile liquid that is introduced into the ICP not only as a liquid contained in the aerosol but also as vapor. Though the total mass flow of methanol transported to the ICP was quantified, we attempted to separate the effects of these two phases on the analyte signal. Firstly, carbon was introduced in the form of a polar, well-soluble but non-volatile substance – phenylalanine was chosen for this purpose. Secondly, a gaseous carbon source (CO2) was investigated, whereby any nebulization-induced bias could be excluded. CO2 was added either to the aerosol or to the intermediate gas stream.As shown in Table 2 the effects of these three carbon sources on the investigated analytes are quite different: 8 g L−1 carbon introduced into the ICP as phenylalanine did not change the signal intensity of most investigated emission lines by more than 5%. The only exceptions were As, Be(II), and Se with signal enhancements of 10–20%. It is also interesting to note that of all carbon containing reagents investigated only phenylalanine did not alter the emission intensity of the Ar I 404.442 nm emission line. Phenylalanine neither changed the excitation temperature significantly nor did it alter the RF generator frequency appreciably (about 1 kHz). The power vacuum tube's plate voltage also remained constant. Combined, these facts suggest that carbon introduced as phenylalanine did not alter the plasma impedance and the enhancement of As, Be(II), and Se is not caused by changes in the ICP itself, as observed when using methanol as the carbon source. It is important to note that the enhancement of As, Be(II), and Se increased with greater phenylalanine concentration in the solution, reaching its maximum at the highest tested reagent concentration (close to the limit of solubility).
In contrast to phenylalanine, CO2 as the gaseous carbon source showed a markedly different behavior, as listed in Table 2. When introduced into the aerosol gas stream, a signal enhancement pattern comparable to that of methanol was recorded, although the signal enhancement factors were between 20 and 40% lower than when using methanol as the carbon source. The excitation temperature was lower too (6800 ± 10 K vs. 6980 ± 10 K). Similar to methanol, a pronounced thermal pinch effect was observed when increasing the flow of CO2. The plasma impedance was also found to change, as evident from the higher power vacuum tube plate voltage (at 15 sccm a plate voltage of 3993 V was recorded). The frequency of the power oscillator decreased by 28.9 kHz when switching from 0 to 15 sccm CO2.
When CO2 was introduced into the intermediate gas flow of the ICP, 5–20% higher signal enhancement factors were encountered when compared to methanol, as listed in Table 2. The excitation temperature was slightly higher and the plasma robustness was lower. Just as with methanol, when CO2 was introduced into the aerosol gas stream, a strong thermal pinch effect was observed, which again was accompanied by high plate voltage and frequency excursion (3950 V and 27.4 kHz).
In Fig. 4a the carbon emission signal (C I 193.091 nm) of the investigated carbon sources is plotted as a function of the carbon mass flow delivered to the ICP. This mass flow was calculated from the determined nebulization efficiency in the case of phenylalanine, or by using the ideal gas law for CO2. For methanol, the method described above that also includes the preferential vaporization was used. The emission signals did not increase in a linear fashion as the mass flow of carbon was extremely high. For CO2 the C emission signal was surprisingly similar, despite the different gas flows it was introduced into. Although below 5.5 mg min−1 carbon from CO2, the C emission signal was higher when introducing the gas into the aerosol stream rather than into the intermediate stream, the difference was only about 30% at 1 mg min−1 and 9% at 2.7 mg min−1. From Fig. 4b it can be deduced that CO2 introduced into the intermediate gas stream of the ICP acts stronger on the plasma core than when introduced into the aerosol gas stream, as the power vacuum tube's plate voltage rises faster due to the impedance changes of the plasma core. As already noted, higher C-loading of the ICP caused the base of the discharge – the region where the aerosol gas stream enters the plasma – to withdraw from the region between the end of the injector tube and the first turn of the load coil (“normal position”) to the space between the first and the second turn of the load coil (thermal pinch effect). The increased distance between the injector tube and the base of the ICP can be expected to allow a higher diffusional exchange between the aerosol and the intermediate gas stream. Thereby the carbon loading is not restricted to one of these streams, explaining the observed effects.
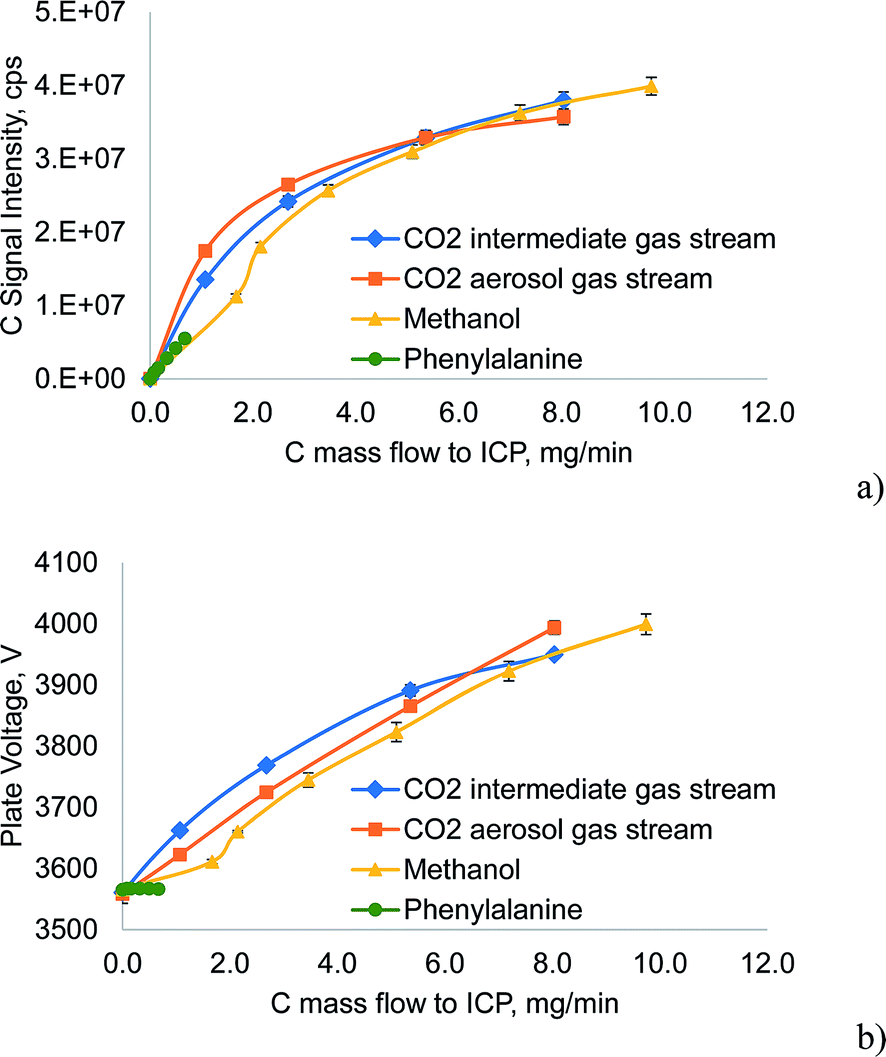 | ||
| Fig. 4 Signal intensity of the C I 193.091 nm emission line and power vacuum tube plate voltage as a function of the carbon mass flow introduced into the ICP for the investigated carbon sources. The error bars represent the standard deviation of five independent experiments. | ||
Another interesting feature shown in Fig. 4 is the effect of low concentrations of methanol on the ICP. Whereas high C loading resulting from methanol causes comparable effects on the plate voltage to similar mass flows of C from CO2, low concentrations of methanol showed significantly less effect on the plate voltage than in the case of CO2. This trend was even stronger for phenylalanine, where no effect on the plate voltage was found. These effects could be explained by considering that phenylalanine is introduced only in the form of an aerosol. Carbon is released only after decomposition inside the analyte channel and consequently the diffusion of C into the plasma core can be expected to start much later than in the case of a similar mass flow of CO2. Thereby, the mass of C reaching the plasma core is much lower and the plate voltage reflects this trend. Following this hypothesis, two processes must be considered in the case of methanol: the vapor phase behaves similarly to gaseous reagents (CO2) whereas the methanol inside the liquid droplets of the aerosol behaves like phenylalanine. At low methanol concentrations the evaporation of methanol in the spray chamber is not as dominant as at higher concentrations; the lower C emission signal and the smaller change in plate voltage for 1.7 mg min−1 C from methanol can be related to this mechanism.
Generally speaking, the enhancement factors of As and Se reported in the literature for non-volatile carbon sources (glucose, citric acid, oxalic acid or glycerol) were always lower than for more volatile reagents such as methanol or even for gases (CH4), both in ICP-OES3,10,11 and in ICP-MS.1,2,4,6,7,9,13–15
3.5. Effect of bromine
As already noted many attempts to explain the signal enhancing effect of carbon rely on the hypothesis of a charge transfer between C+ and the high IP element. If carbon with an IP of 11.26 eV is behaving in this manner, Br with an IP of 11.81 eV can be expected to show similar effects.In contrast to previous reports11 we wanted to avoid variations in the nebulization efficiency and therefore used water saturated with bromine rather than HBr. Seven concentration levels (0–34 g L−1 Br2) were prepared from bromine-saturated water. The Br2 concentration was calculated assuming a solubility of Br2 in water34 of 33.56 g L−1. All bromine concentrations given below correspond to the mass of bromine dissolved in water. Due to the toxicity of bromine, we have not attempted to quantify the mass flow of bromine reaching the plasma.
Bromine had little to no effect on most of the investigated emission lines. The signal intensities recorded in a 34 g L−1 Br2 solution differed by less than ±10% from the signal recorded from an aqueous solution (3% v/v HNO3). The only exceptions were P, S and Se. For P and S the signal intensities initially rose quickly with increasing Br2 concentration reaching a plateau at about 3.4 g L−1 Br2 with signal intensities of about 13% and 20% (P and S respectively) higher than in an aqueous standard. For Se the signal increased almost linearly up to 6.7 g L−1 Br2 (67% higher signal than in diluted nitric acid) and then leveled off, reaching about 85% signal enhancement at 34 g L−1 Br2. It is most interesting to note that neither As was enhanced by Br2, nor were Au or Hg.
Plasma robustness and the excitation temperature remained constant when switching from an aqueous solution (3% v/v HNO3) to 34 g L−1 Br2 and neither the plate voltage nor the power drawn by the RF generator changed significantly. The frequency of the RF generator dropped slightly but significantly by 2.6 kHz.
From these findings it must be concluded that the introduction of bromine did not affect the RF generator and it can be assumed that the power delivered to the ICP remained unchanged. The signal enhancement of P, S and particularly Se cannot be attributed to changes in the nebulization efficiency as other elements would have been affected too. Therefore, the effect of bromine on P, S and Se appears to be a true matrix effect of bromine on these three elements.
If a charge transfer process is the underlying mechanism of the observed effects, two conditions must be met: firstly, the Wigner spin conservation rule35 must be fulfilled; secondly, the reaction energy defect should be minimal. Grindlay et al.16 assumed that this type of reaction is likely to happen if the involved energy levels differ by between −1.0 eV and +0.5 eV.
From the data listed in Table 3 we can draw the conclusion that the Wigner spin conservation rule is fulfilled for every examined element. However, the energy difference between the involved levels appears to explain most of the observed effects of bromine: with the exception of Au, I, P, S, and Se the energy difference between the involved levels is too large. Iodine was not investigated, as the Br2 used was slightly contaminated with this element. For P, S and Se a signal enhancement was observed that could consequently be attributed to a charge transfer reaction. However, for Au no significant signal increase was encountered when using Br2, though the energy difference of the involved levels was only slightly larger than in the case of Se. This finding is unexpected, as the Au signal was enhanced in the presence of C.
| Element | Atom ground state term | Energy level of the electronic state closest to the IP of Br | ΔE (eV) | Spin | ||
|---|---|---|---|---|---|---|
| Term | Energy (eV) | S i | S p | |||
| As | 4S | 1D | 11.04 | 0.77 | 5/2, 3/2, 1/2 | 1/2 |
| Au | 2S | 3D | 11.41 | 0.40 | 3/2, 1/2 | 3/2, 1/2 |
| Be | 1S | 2P | 13.28 | −1.47 | 1, 0 | 1, 0 |
| Cd | 1S | 2P | 14.47 | −2.66 | 1, 0 | 1, 0 |
| Cl | 2P | 3P | 12.97 | −1.16 | 3/2, 1/2 | 3/2, 1/2 |
| Hg | 1S | 2S | 10.44 | 1.37 | 1, 0 | 1, 0 |
| I | 2P | 1D | 12.15 | −0.34 | 3/2, 1/2 | 1/2 |
| P | 4S | 1D | 11.59 | 0.22 | 5/2, 3/2, 1/2 | 1/2 |
| S | 3P | 2D | 12.20 | −0.39 | 2, 1 | 1, 0 |
| Se | 3P | 2D | 11.46 | 0.35 | 2, 1 | 1, 0 |
| Zn | 1S | 2S | 9.39 | 2.42 | 1, 0 | 1, 0 |
3.6. Effect of NaCl
The presence of high concentrations (>1 g L−1) of easily ionizable elements (EIEs) such as Na is reported to cause plasma related signal variations37 in the emission line intensities of other elements. Consistent with our previous work24 we encountered a small, but significant decrease of the RF generator frequency (2.2 kHz) and a severe drop of plasma robustness (from 7.0 to 5.4 when increasing the NaCl concentration in the sample solution from 0 g L−1 to 30 g L−1). However, the excitation temperature decreased only to 6510 ± 20 K over the same interval. Dennaud et al.38 calculated the theoretical plasma robustness as a function of the temperature and found a linear correlation between these two factors, assuming local thermal equilibrium (LTE) in the plasma discharge. When plotting the excitation temperature vs. plasma robustness for all reagents reported in Table 2 bromine and all carbon based reagents follow a general linear trend. However, for NaCl the excitation temperature strongly deviates from this trend. As the measured decrease of the plasma robustness agrees well with data reported in the literature (e.g. Brenner et al.39 found that at 1350 W RF power plasma robustness decreased from 9.1 to 7.4 when increasing the NaCl concentration from 0 to 10 g L−1), the surprisingly high excitation temperature might stem from a stronger deviation of the iron atom level population from the Boltzmann distribution and the associated departure from the LTE. However, the Boltzmann plots of the Fe(I) lines were not significantly curved (calculation based on an f-test of the residual standard deviations of the Boltzmann plot using either a liner fit or a second order polynomial fit) as reported by Kitagawa and Horlick.40The investigated emission lines were in general all suppressed by large quantities of NaCl (30 g L−1) as shown in Table 2. In contrast to the effect of C, the power vacuum tube's plate voltage remained constant up to about 6 g L−1 and then dropped almost linearly by 50 V towards the highest investigated NaCl concentration of 30 g L−1. Though this drop is significant, the suppression of the analyte emission signals cannot be related to this reduction in RF power, as the drop of 50 V corresponds to a reduction of the RF generator input power of only about 30 W.
3.7. Differentiating between the factors contributing to the carbon enhancement effect
From the foregoing discussion it can be concluded that the signal enhancing effect of carbon on several elements is a combination of changes in the excitation equilibrium (e.g. caused by a charge transfer reaction) and higher plasma power density. The latter stems from RF generator control issues and the thermal pinch effect.In an attempt to discriminate between these two effects, solutions containing As, Au, Be, Br, Cl, Hg, P, S, and Se (concentration of each element similar to the previously used standard solutions) in either 3% (v/v) HNO3 or 10% (m/m) methanol together with the respective blank solutions (only diluted nitric acid or 10% methanol) were measured at constant torch gas flows (as listed in Table 1) but at different RF power levels (1300–1495 W; 50 W increments; setting a higher power level than 1495 W was not possible, as the instrument control software then increased the outer gas flow in an attempt to protect the torch).
As expected, a rising RF power level resulted, in both 3% (v/v) HNO3 and methanol, in an increase of the blank signal corrected analyte emission intensity. However, distinct differences were encountered between the investigated emission lines: methanol had little effect on low excitation energy (4–5 eV) atom lines (e.g. Au I 267.595 or Hg I 253.652 nm) as the signal intensity was similar to that in 3% (v/v) HNO3. This finding is consistent with data presented in Table 2. For emission lines of higher excitation energy the analyte emission signal in methanol was always higher than in the corresponding aqueous standard for all RF power levels. This effect became more pronounced with increasing total emission line energy, although only a general trend was encountered but no function was found to sufficiently fit all data points. When plotting the signal intensity for each emission line as a function of the RF power level used, straight lines were obtained (R2 > 0.999). The slope of these lines was always lower in methanol when compared to aqueous solutions. From this finding it can be concluded that the magnitude of the carbon enhancement effect is also dependent on the RF power level used. A similar observation has been reported by Kralj and Veber.15
To separate the effects of increased plasma power density and changes in the emission line excitation equilibrium on the analyte signal, the RF power vs. emission line intensity plots can be used. When assuming that there is no change in the excitation equilibrium by methanol (e.g. by a charge transfer reaction), any change in the emission signal caused by methanol would be dependent on the power density in the ICP discharge. Consequently, “apparent” RF power levels can be calculated from the RF power vs. emission line intensity plots of aqueous standards (3% v/v HNO3) and the corresponding analyte signal in 10% methanol at a fixed RF power level (e.g. 1350 W). These “apparent” RF power levels describe the hypothetical RF power necessary for obtaining the same emission signal for an aqueous (3% v/v HNO3) solution as in 10% methanol if the change in plasma power density was the only effect of methanol on the ICP. In Table 4 these “apparent” RF power levels are listed for 1350 W. There is no clear correlation between these “apparent” RF power levels and the total energy of the emission line.
| Element and emission line, nm | Total line energy, eV | “Apparent” RF power, W |
|---|---|---|
| Ar I 404.442 | 14.69 | 1540 |
| Ar I 430.010 | 14.51 | 1540 |
| As I 189.042 | 6.56 | 1660 |
| As I 193.759 | 6.40 | 1640 |
| As I 197.262 | 6.29 | 1660 |
| Au I 201.200 | 7.30 | 1480 |
| Au I 242.795 | 5.11 | 1430 |
| Au I 267.595 | 4.63 | 1400 |
| Be I 234.861 | 5.28 | 1410 |
| Be II 313.042 | 13.28 | 1480 |
| Be II 313.107 | 13.28 | 1470 |
| Br I 144.990 | 8.55 | 1540 |
| Cl I 134.724 | 9.20 | 1540 |
| Cl I 135.165 | 9.28 | 1720 |
| Cl I 136.345 | 9.20 | 1540 |
| Hg I 184.950 | 6.70 | 1480 |
| Hg II 194.227 | 16.82 | 1480 |
| Hg I 253.652 | 4.89 | 1320 |
| Hg I 435.835 | 7.73 | 1440 |
| P I 138.147 | 8.97 | 1620 |
| P I 169.403 | 8.73 | 1630 |
| P I 177.495 | 6.99 | 1640 |
| P I 178.287 | 6.95 | 1620 |
| P I 213.618 | 7.21 | 1650 |
| P I 214.914 | 7.18 | 1660 |
| S I 142.503 | 8.70 | 1530 |
| S I 180.731 | 6.86 | 1560 |
| S I 182.034 | 6.86 | 1590 |
| Se I 196.090 | 6.32 | 1830 |
| Se I 203.985 | 6.32 | 1900 |
The tapping of the RF generator's HV power supply revealed, as reported above, that in the presence of 10% methanol the power drawn by the RF generator increased by about 250 W. The efficiency of the RF generator (power delivered to the ICP as set in the instrument control software divided by the generator input power) was about 60%. Thereby it can be estimated that the power level in the ICP discharge in the presence of methanol was roughly 1350 W + 250 W × 0.6 = 1500 W. Simultaneously, the thermal pinch effect decreased the volume of the plasma. Using the method of Greenfield and McGeachin41 the decrease of the plasma volume estimated from Fig. 3 was 20–30%. This value represents only a rough estimate, as the boundaries of the ICP are not sharply defined and the plasma can only be approximated as a geometrical combination of a cylinder and a cone. It appears questionable that these two mechanisms for increasing the power density of the ICP are simply additive. Moreover, it is not clear whether the impedance changes of the ICP are a consequence of the plasma's constriction or a change in the total gas composition, or a combination of both effects. In conclusion, too many uncertainties and unknowns are present to even estimate the true power level in the ICP discharge when loaded with methanol and only calorimetric means appear to be a viable method for providing reliable data. However, the power fed to the ICP can be approximated using voltage and current drawn from the HV power supply: at 1350 W the set current is 0.58 A. As previously described, the current remains constant but the voltage fed to the RF generator increases with increasing carbon load of the ICP. Consequently, at the maximum rating of the HV power supply of 4200 V the generator can draw 4200 V * 0.58 A = 2436 W power. Using again an efficiency of 60% results in about 1460 W that the RF generator can drive into the load – the ICP – under worst-case conditions.
Using this 1460 W, the “apparent” power levels listed in Table 4 might allow us to distinguish between plasma power density related signal enhancement and changes of the excitation equilibrium: if the “apparent” power level of an emission line is much higher than 1460 W, a secondary process must be involved. When assuming that a difference of 200 W is significant, the following emission lines can be expected to be enhanced by other processes rather than increased RF power: As I 189.042 nm, As I 193.759 nm, As I 197.262 nm, Cl I 135.165 nm, P I 138.147 nm, P I 169.403 nm, P I 177.495 nm, P I 178.287 nm, P I 213.618 nm, P I 214.914 nm, Se I 196.090 nm, and Se I 203.985 nm. The excitation energy of these atom lines is between 6.3 and 9.3 eV. It is interesting to note that all investigated emission lines of As, P and Se follow this trend but only one chlorine line (135.165 nm) follows this trend.
On the other hand, the enhancement of emission lines with an “apparent” power level of ≤1460 W can be explained solely on the basis of the increased RF power coupled into the ICP. This group involves Au I 242.795 nm, Au I 267.595 nm, Be I 234.861 nm, Hg I 253.652 nm, Hg I 435.835 nm and potentially also Au I 201.200 nm, Be II 313.042 nm, Be II 313.107 nm, Hg I 184.950 nm, and Hg II 194.227 nm (the “apparent” power of these lines is only 20 W above the calculated one). The energy sum of these lines spans from 4.6 to 16.8 eV.
For the remaining emission lines (Ar I 404.442 nm, Ar I 430.010 nm, Br I 144.990 nm, Cl I 134.724 nm, Cl I 136.345 nm, S I 142.503 nm, S I 180.731 nm, and S I 182.034 nm) listed in Table 4 no clear trend can be defined.
4. Conclusion
Carbon-based matrix effects in ICP-OES and ICP-MS arise not only from a single cause but appear to be a combination of several factors: (1) depending on the source of carbon used, changes of the sample transport efficiency in the nebulizer and spray chamber due to variations in viscosity, surface tension or density; (2) matrix-induced excitation of certain energy levels by mechanisms such as the charge transfer reaction; (3) thermal pinch effect that results in a constriction of the ICP and consequently a higher power density of the remaining discharge; (4) RF generator related effects due to changes in plasma impedance. Effects 3 and 4 can be expected to be different for every type of RF generator as the impedance changes will act differently on each specific circuit arrangement (free-running generators or crystal stabilized ones with an additional impedance matching network). Particularly the power control algorithm that regulates current and voltage fed to the power electronics can be expected to have a great influence on the stability of the RF generator when loading the ICP with large amounts of carbon. Even the dimensions of the torch or the operating frequency might cause differences in the effect of carbon. As already reported, slight changes of the torch position were found to alter the excitation temperature. The frequency on the other hand determines the skin depth of the RF current in the ICP and is often associated with either a three or a four turn load coil (27 or 40 MHz respectively). In conclusion, the response of the RF generator to plasma impedance changes appears to be the dominant reason for the large spread of the enhancement factors reported in the literature.Another factor that should be considered in this context is the fact that not the concentration of carbon in the sample solution determines the degree of matrix effect, but the mass flow of carbon that reaches the ICP and its state of matter. Gaseous reagents affect the plasma core more readily than non-volatile liquid ones.
Based on the findings presented here, the charge transfer reaction combined with the higher power density in the ICP discharge appears to explain most of the element enhancements by C or Br. However, the mechanism fails to explain that in the presence of sufficient carbon elements that cannot be expected to be enhanced by a charge transfer reaction (very high reaction energy defect) also were strongly enhanced (e.g. some emission lines of Cl). We can only but agree with Grindlay et al.16 that “additional studies are required to explain these anomalies”.
References
- P. Allain, L. Jaunault, Y. Mauras, J. M. Mermet and T. Delaporte, Anal. Chem., 1991, 63, 1497–1498 CrossRef CAS.
- B. Gammelgaard and O. Jøns, J. Anal. At. Spectrom., 1999, 14, 867–874 RSC.
- G. Grindlay, L. Gras, J. Mora and M. T. C. de Loos-Vollebregt, Spectrochim. Acta, Part B, 2008, 63, 234–243 CrossRef.
- Z. Hu, S. Hu, S. Gao, Y. Liu and S. Lin, Spectrochim. Acta, Part B, 2004, 59, 1463–1470 CrossRef.
- M. Kovačevič and W. Goessler, Spectrochim. Acta, Part B, 2005, 60, 1357–1361 CrossRef.
- M. Kovačevič, W. Goessler, N. Mikac and M. Veber, Anal. Bioanal. Chem., 2005, 383, 145–151 CrossRef.
- E. H. Larsen and S. Stürup, J. Anal. At. Spectrom., 1994, 9, 1099–1105 RSC.
- S. Liu and D. Beauchemin, Spectrochim. Acta, Part B, 2006, 61, 319–325 CrossRef.
- I. Llorente, M. Gómez and C. Cámara, Spectrochim. Acta, Part B, 1997, 52, 1825–1838 CrossRef.
- J. Machát, V. Kanický and V. Otruba, Anal. Bioanal. Chem., 2002, 372, 576–581 CrossRef.
- J. Machát, V. Otruba and V. Kanický, J. Anal. At. Spectrom., 2002, 17, 1096–1102 RSC.
- M. Pettine, B. Casentini, D. Mastroianni and S. Capri, Anal. Chim. Acta, 2007, 599, 191–198 CrossRef CAS.
- I. Rodushkin, P. Nordlund, E. Engström and D. C. Baxter, J. Anal. At. Spectrom., 2005, 20, 1250–1255 RSC.
- E. Warburton and H. Goenaga-Infante, J. Anal. At. Spectrom., 2007, 22, 370–376 RSC.
- P. Kralj and M. Veber, Acta Chim. Slov., 2003, 50, 633–644 CAS.
- G. Grindlay, J. Mora, M. de Loos-Vollebregt and F. Vanhaecke, Spectrochim. Acta, Part B, 2013, 86, 42–49 CrossRef CAS.
- T. Nakazawa, D. Suzuki, H. Sakuma and N. Furuta, J. Anal. At. Spectrom., 2014, 29, 1299–1305 RSC.
- A. S. Al-Ammar, R. K. Gupta and R. M. Barnes, Spectrochim. Acta, Part B, 1999, 54, 1849–1860 CrossRef.
- F. R. Abou-Shakra, M. P. Rayman, N. I. Ward, V. Hotton and G. Bastian, J. Anal. At. Spectrom., 1997, 12, 429–433 RSC.
- H. Niu and R. S. Houk, Spectrochim. Acta, Part B, 1996, 51, 779–815 CrossRef.
- J. Goossens, F. Vanhaecke, L. Moens and R. Dams, Anal. Chim. Acta, 1993, 280, 137–143 CrossRef CAS.
- E. F. Huth and L. Kühn, Ger. Pat., 310152, 1917.
- P. Wetzl and O. Zehnder, in Industrieröhren in Hochfrequenzgeneratoren, BBC AG, Baden, Switzerland, 1980 Search PubMed.
- H. Wiltsche, F. Moradi and G. Knapp, Spectrochim. Acta, Part B, 2012, 71–72, 48–53 CrossRef CAS.
- F. J. Maessen, P. J. H. Seeverens and G. Kreuning, Spectrochim. Acta, Part B, 1984, 39, 1171–1180 CrossRef.
- H. Wiltsche, F. Moradi, P. Tirk and G. Knapp, J. Anal. At. Spectrom., 2014, 29, 1767–1777 RSC.
- J. M. Mermet, Anal. Chim. Acta, 1991, 250, 85–94 CrossRef CAS.
- M. W. Blades, in Inductively Coupled Plasma Emission Spectroscopy, ed. P. W. J. M. Boumans, Wiley Interscience, New York, 1987, vol. 90, pp. 387–420 Search PubMed.
- I. B. Brenner and A. T. Zander, Spectrochim. Acta, Part B, 2000, 55, 1195–1240 CrossRef.
- J. C. Ivaldi and J. F. Tyson, Spectrochim. Acta, Part B, 1995, 50, 1207–1226 CrossRef.
- D. G. Weir and M. W. Blades, J. Anal. At. Spectrom., 1994, 9, 1311–1322 RSC.
- G. C. Y. Chan and G. M. Hieftje, Anal. Chem., 2013, 85, 58–65 CrossRef CAS.
- J. Mora, V. Hernandis and A. Canals, J. Anal. At. Spectrom., 1991, 6, 573–579 RSC.
- Reaxys Database (Beilstein, Gmelin), Reed Elsevier Properties SA, 2015.
- C. S. Enos, A. G. Brenton and A. R. Lee, Int. J. Mass Spectrom., 1992, 122, 361–376 CrossRef CAS.
- A. E. Kramida, Y. Ralchenko, J. Reader and NIST ASD Team, in NIST Atomic Spectra Database (ver. 5.2 Online), National Institute of Standards and Technology, Gaithersburg, 2014 Search PubMed.
- J. L. Todolí, L. Gras, V. Hernandis and J. Mora, J. Anal. At. Spectrom., 2002, 17, 142–169 RSC.
- J. Dennaud, A. Howes, E. Poussel and J.-M. Mermet, Spectrochim. Acta, Part B, 2001, 56, 101–112 CrossRef.
- I. B. Brenner, M. Zischka, B. Maichin and G. Knapp, J. Anal. At. Spectrom., 1998, 13, 1257–1264 RSC.
- K. Kitagawa and G. Horlick, J. Anal. At. Spectrom., 1992, 7, 1221–1229 RSC.
- S. Greenfield and H. M. McGeachin, Anal. Chim. Acta, 1978, 100, 101–119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ja00237k |
| This journal is © The Royal Society of Chemistry 2015 |