
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferentiation capacity and maintenance of differentiated phenotypes of human mesenchymal stromal cells cultured on two distinct types of 3D polymeric scaffolds†
A. M.
Leferink
ab,
D.
Santos
a,
M.
Karperien
c,
R. K.
Truckenmüller
ab,
C. A.
van Blitterswijk
ab and
L.
Moroni‡
*ab
aDepartment of Tissue Regeneration and MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Drienerlolaan 5, 7522NB, Enschede, The Netherlands
bDepartment of Complex Tissue Regeneration, Faculty of Health, Medicine and Life Sciences, Maastricht University, Universiteitssingel 40, 6229ER, Maastricht, The Netherlands
cDepartment of Developmental Bioengineering, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Drienerlolaan 5, 7522NB, Enschede, The Netherlands
First published on 6th November 2015
Abstract
Many studies have shown the influence of soluble factors and material properties on the differentiation capacity of mesenchymal stromal cells (MSCs) cultured as monolayers. These types of two-dimensional (2D) studies can be used as simplified models to understand cell processes related to stem cell sensing and mechano-transduction in a three-dimensional (3D) context. For several other mechanisms such as cell–cell signaling, cell proliferation and cell morphology, it is well-known that cells behave differently on a planar surface compared to cells in 3D environments. In classical tissue engineering approaches, a combination of cells, 3D scaffolds and soluble factors are considered as the key ingredients for the generation of mechanically stable 3D tissue constructs. However, when MSCs are used for tissue engineering strategies, little is known about the maintenance of their differentiation potential in 3D scaffolds after the removal of differentiation soluble factors. In this study, the differentiation potential of human MSCs (hMSCs) into the chondrogenic and osteogenic lineages on two distinct 3D scaffolds, additive manufactured electrospun scaffolds, was assessed and compared to conventional 2D culture. Human MSCs cultured in the presence of soluble factors in 3D showed to differentiate to the same extent as hMSCs cultured as 2D monolayers or as scaffold-free pellets, indicating that the two scaffolds do not play a consistent role in the differentiation process. In the case of phenotypic changes, the achieved differentiated phenotype was not maintained after the removal of soluble factors, suggesting that the plasticity of hMSCs is retained in 3D cell culture systems. This finding can have implications for future tissue engineering approaches in which the validation of hMSC differentiation on 3D scaffolds will not be sufficient to ensure the maintenance of the functionality of the cells in the absence of appropriate differentiation signals.
Insight, innovation, integrationThe scientific research presented in our manuscript shows the response of human bone marrow stromal cells (hMSCs) on differentiation-inducing-soluble factors in two distinct 3D polymeric scaffolds. Our work contributes to further understanding of the maintenance of differentiated hMSC phenotype. Cell populations which appeared to be successfully differentiated towards the chondrogenic or osteogenic lineage did not always maintain this differentiated state upon soluble factor removal. From these results, we conclude that in vitro differentiation will not necessarily result in sufficient functionality of these tissue constructs, for example, upon implantation in vivo. This also indicates that the conventional assessment of the differentiated phenotype will not be sufficient to be able to predict the cells’ functionality in clinical applications. |
1. Introduction
Multipotent cell types are promising candidates for classical tissue engineering and regenerative medicine approaches in which a synthetic or biological material is combined with autologous cells to construct a mechanically stable implant to replace damaged or lost tissue. The use of multipotent cell types is opted since many other differentiated cell types have limited availability and several cell types have shown to lose their phenotype upon expansion by a process known as de-differentiation.1–4 This can result in a decreased functionality of the cells, which lowers their potential to secrete specific tissue components within the construct. A cell type that can circumvent this problem is human adult stem cells from bone marrow stroma, referred to as human mesenchymal stem or stromal cells (hMSCs). MSCs were initially defined as a heterogeneous population of fibroblast-like cells, from which a subset of the population has great potential to proliferate and differentiate.5–8 More recent definitions consist of minimal criteria to identify multi-potent hMSCs, which include specific surface antigen expression and multipotent differentiation potential into osteoblasts, adipocytes and chondrocytes.9–11 Recent studies showed that by culturing hMSCs on biomaterials with controlled properties, or by adding certain soluble factors, depending on the donor and isolation method, differentiation towards chondrogenic,12–14 osteogenic,15–17 neurogenic,18 endothelial19 and myogenic20 lineages can be achieved.A large number of studies have shown the influence of soluble factors and material properties on the differentiation capacity of MSCs in two-dimensional (2D) culture systems.21 For example, Engler et al. reported on the influence of gel stiffness on the fate of stem cells.22,23 In other studies, nano- and micro-topographies were introduced to investigate the influence of physical properties like roughness of the material on cell differentiation.24–27 These types of studies can be used as simplified models to understand cell processes related to stem cell sensing and mechano-transduction in three-dimensional (3D) environments. However, cells in a tissue, organ or organism are exposed to complex biological environments. In contrast to classic culture systems, these environments are not flat, but 3D. Consequently, 2D tissue culture models used to translate a new therapy to the clinics are far from accurate. For several other mechanisms like cell–cell signalling, cell proliferation and cell morphology, it is well-known that cells behave differently on a 2D flat surface compared to cells in 3D systems.28 Although the lineage of MSCs, plasticity and therapeutic potential have been widely studied in 2D, little is known about these combined properties in 3D.29 In particular, plasticity is the ability of mature cells to switch or differentiate among phenotypes that are different from the tissue of origin or different from their phenotype after considerable differentiation.30,31 Rottmar et al. recently showed that hMSCs retain their plasticity after 18 days of 2D in vitro culture under osteogenic culture conditions.32 To our knowledge, phenotypic plasticity of hMSCs after osteogenic or chondrogenic differentiation has not yet been assessed in 3D scaffolds for tissue engineering approaches.
In this study, the differentiation potential of hMSCs into the chondrogenic and osteogenic lineages on two distinct 3D scaffolds, both of which have been opted as potential scaffolds for bone and cartilage tissue engineering applications,33,34 was assessed and compared to the differentiation capacity in 2D monolayer cultures and in chondrogenic pellet-culture. The maintenance of the induced differentiated phenotype was assessed after the removal of soluble factors to investigate the role of the 3D culture system in phenotypic alterations of cells. For both scaffold architectures, a previously optimized cell number for the estimated available surface area was applied. However, the cell seeding density was kept constant to allow for comparable cell adherence conditions.35 After 7 days of cell adherence and culture in proliferation medium, differentiation inducing media were added to the cultures. As commonly used to steer hMSC differentiation, 10 nM dexamethasone (dex) was supplemented to the medium for osteogenic differentiation, while 100 nM of dex and 10 ng mL−1 of TGF-β3 were introduced for chondrogenic differentiation.
After 14 days of differentiation, the soluble factors were removed (de-differentiation phase) and the cellular phenotype was assessed after 3 or 14 days of potential de-differentiation. We hypothesize that after the removal of differentiation inducing soluble factors, the stability of hMSC phenotype may be altered in 3D similarly to what was reported for hMSC phenotype in 2D culture.29,36 Ultimately, this may have an influence on developing stem cell based regenerative medicine strategies in which 3D constructs loaded with hMSCs are implanted in vivo subsequent to culture in vitro. In these approaches, the traditional assessment of the differentiation potential hMSCs in 3D constructs will not be sufficient to predict the functionality of hMSCs upon implantation without assessing the stability of the differentiated phenotype of hMSCs.
2. Materials and methods
2.1. Cell culture and culture media
Basal medium (BM) consisted of α-minimal essential medium (Life Technologies) complemented with 10% heat-inactivated fetal bovine serum (FBS; Lonza), 0.2 mM ascorbic acid (Asap; Life Technologies), 2 mM L-glutamine (Life Technologies), 100 units per mL penicillin (Life Technologies), and 10 μg mL−1 streptomycin (Life Technologies). Proliferation medium (PM) consisted of BM supplemented with a 1 ng mL−1 basic fibroblast growth factor (FGF; Instruchemie). Osteogenic medium (OM) consisted of BM supplemented with 10 nM dex. Bone-gla-protein (BGP) was omitted, since it is known to aid in the differentiation of osteoblasts into mineral forming osteocytes and not necessarily in the early phase of osteoblastic differentiation. In this study, we only looked at early indicators of differentiation and not at mineralization. Chondrogenic medium (CM) consists of Dulbecco's modified Eagle medium (D-MEM, Life Technologies) complemented with 0.2 mM ascorbic acid, 100 units per mL penicillin (Life Technologies), 10 μg mL−1 streptomycin (Life Technologies), 50 μg mL−1 ITS-premix, 100 μg mL−1 sodium pyruvate, 10 ng mL−1 TGFβ-3 and 100 nM dex. For the studies presented here, two different types of hMSC populations were utilized. For all experiments, hMSCs selected based on their potential to propagate and differentiate (referred to as donor 1) were utilized unless stated differently.These pre-selected hMSCs (male, age 22) were retrieved from the Institute of Regenerative Medicine (Temple, Texas).8,37 Briefly, a bone marrow aspirate was drawn and mononuclear cells were separated by density centrifugation. The cells were plated to obtain adherent hMSCs, which were harvested when cells reach 60–80% confluence. These were considered passage zero (P0) cells. These P0 cells were expanded, harvested and frozen at passage 1 (P1) for distribution.
Donor 2, 3, and 4 hMSCs (female, age 55; male, age 75; and female, age 76; respectively) were obtained by bone marrow aspiration from patients who had given written informed consent. These hMSCs were isolated and proliferated as previously described.38 In short, aspirates from the donors were resuspended using a 20-gauge needle, plated at a density of 5 × 105 cells per cm2 and cultured in PM. Cells were grown at 37 °C in a humidified atmosphere with 5% CO2. Medium was refreshed twice per week and cells were used for further subculturing or cryopreservation on reaching near confluence.
2.2. Fabrication of 3D scaffolds from PEOT/PBT by fused deposition modelling (FDM)
Scaffolds were fabricated from 300PEOT55PBT45 (300/55/45) (PolyVation, The Netherlands), a block copolymer composed of poly(ethylene oxide terephthalate) (PEOT) and poly(butylene terephthalate) (PBT) with a weight ratio of 55 to 45 for the two components, respectively, and a molecular weight of the starting poly(ethylene glycol) (PEG) segments of 300 Da used in the co-polymerization process. Fused deposition modeling was used to fabricate 3D grids using a bioscaffolder (SysENG, Germany), as described before.33 Grids had a height of 3 mm, a fiber-to-fiber distance of 800 μm (XY spacing), a fiber diameter of approximately 200 μm, and a layer thickness of 150 μm (Z spacing). Cylindrical scaffolds with a diameter of 4 mm and a height of 3 mm were punched from the plotted grids. The morphology of the fabricated scaffold is shown (in the z-direction) in Fig. 1A and C. | ||
| Fig. 1 Two distinct types of scaffolds were used to assess hMSC differentiation capacity in vitro in 3D. (A and C) Cylindrical FDM-scaffolds were fabricated of 300/55/45 with a diameter of 4 mm and a height of 3 mm. (B and D) ESP scaffolds were fabricated of the same polymer as FDM-scaffolds and punched out from a large electrospun sheet to fit in a 12-well plate. | ||
2.3. Fabrication of 3D scaffolds from PEOT/PBT by electrospinning (ES)
For ES scaffolds, a 28% (w/v) solution of 300/55/45 was prepared in a mixture of chloroform (CHCl3)–1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) with a ratio of 78–22% v/v. The solution was left for stirring at room temperature overnight. Subsequently, the solution was loaded into a syringe and pumped at a controlled flow rate using a syringe pump (KDS 100, KD Scientific). PTFE tubing was used to connect the syringe and a needle to which a high voltage was applied (Gamma High Voltage Research Inc., FL, USA). A stainless steel collector, covered by an aluminium foil, was used to collect the spun scaffold. Scaffolds were produced using a voltage of 12 kV applied to a 1.2 mm needle and placed at a distance of 25 cm from the collector. The flow rate was set at 15 mL h−1, temperature to 25 °C, and humidity to 30%. The morphology of the fabricated scaffold is shown in Fig. 1B and D.2.4. Cell culture in 2D on tissue culture polystyrene (TCPS)
The differentiation potential of the hMSCs without the presence of a 3D scaffold system was determined by culture of hMSCs as a monolayer in BM and OM and as pellet-culture in CM.7 The cells from all donors were seeded with a density of 5000 cells per cm2 on TCPS 6-well plates (NUNC) and cultured for 3, 7 or 14 days in BM as a negative control on differentiation and in OM to induce osteogenic differentiation. For the chondrogenic differentiation in pellet-culture, 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hMSCs per well were added in a U-bottom 96-well plate for suspension culture (NUNC), centrifuged for 3 min at 800g and cultured for 3, 7 or 14 days in CM.
000 hMSCs per well were added in a U-bottom 96-well plate for suspension culture (NUNC), centrifuged for 3 min at 800g and cultured for 3, 7 or 14 days in CM.
2.5. Cell culture on FDM-scaffolds
Sterilization of the scaffolds was performed in 70% ethanol twice for 30 minutes, subsequently washed in PBS first for 5 minutes and additionally twice for another 30 minutes each time and finally incubated in BM overnight prior to cell culture. Scaffolds were dried by medium aspiration and placed in a non-treated 48-well plate (NUNC). Passage 3 hMSCs were harvested from monolayer expansion, and seeded on the scaffolds with a density of 125000 cells in 50 μL of PM, which has shown to be in the optimal range of cell densities for a high cell seeding efficiency.35 After 1.5 hour incubation to let the cells adhere, the medium was filled up to 1 mL and culture was continued for 7 days to allow the cells to spread and proliferate on the scaffold. After 7 days of culture in PM, the media were changed to BM, CM or OM. After 14 days of culture in CM or OM, culture in CM or OM was continued up to 28 days or the soluble factors were removed by replacing CM and OM with BM (referred to as CB and OB respectively).
2.6. Cell culture on ES-scaffolds
The scaffolds were washed with sterile PBS to remove any remaining traces of ethanol and then placed into a non-treated 24-well plate (NUNC). Non-treated plates were used to minimize cell attachment to the bottom of the plates. The constructs were then incubated at 37 °C in a humid atmosphere with 5% CO2 overnight in BM to pre-wet them. To prevent the scaffold discs from floating, rubber O-rings (Eriks BV, The Netherlands) were used to hold the discs in place. After removing the medium used for incubation, 75000 cells were seeded in 30 μL of PM onto each electrospun scaffold. The cell number was chosen as such to maintain the cell seeding concentration similar to on the FDM scaffolds, yet preventing over-confluence of the top-layer of the ESP-scaffold. After 1.5 h of incubation to let the cells adhere, the medium was filled up to 1 mL and culture was continued for 7 days to allow the cells to spread and proliferate on the scaffold. After 7 days of culture in PM, the media were replaced with BM, CM and OM to induce differentiation and the culture was continued as depicted in Fig. 1. After 14 days of culture in CM or OM, culture in CM or OM was prolonged up to 28 days or the soluble factors were removed by replacing CM and OM with BM (referred to as CB and OB respectively).
2.7. Gene expression analysis of hMSCs
For gene expression analysis, samples were taken from culture after the medium was carefully aspirated. All samples were transferred to 2 mL Eppendorf tubes and 500 μL of TRIzol (Invitrogen) was added prior to preservation at −80 °C. RNA isolation was performed by using a Bioke RNA II nucleospin RNA isolation kit (Macherey-Nagel). The samples were disrupted mechanically by crushing using RNA isolation pestles (Kimble Kontes). Samples for basal gene expression analysis were seeded in T25 tissue culture flasks (NUNC) in BM at a cell density of 5000 cells per cm2. After 1 day of culture, the medium was aspirated and 500 μL of TRIzol was added after which the cell/TRizol suspension was transferred to an Eppendorf tube. Subsequently, 200 μL of CHCl3 was added to all samples, both from 2D as well as from 3D culture, and mixed by vigorously shaking the tubes. The TRizol/CHCl3 mixture was centrifuged at 12000g for 15 minutes at 4 °C. The aqueous phase was transferred to a new Eppendorf tube and mixed 1:1 with 70% ethanol. The mixture was transferred to filter columns from the kit and RNA isolation was continued following the manufacturer's protocol. RNA concentrations and purity were determined by using an ND1000 spectrophotometer (Nanodrop Technologies, USA). CDNA was synthesized from 240, 320 and 600 ng of RNA for the samples from FDM-scaffolds, ES-scaffolds and 2D respectively, using iScript™ (BIO-RAD) according to the manufacturer's protocol. Quantitative polymerase chain reaction (qPCR) was performed on the obtained cDNA by using the iQ SYBR Green Supermix (Bio-Rad) and the primers as listed in the ESI,† in Table S1.
PCR reactions were carried out on the MyiQ2 Two-Color Real-Time PCR Detection System (Bio-Rad) under the following conditions: cDNA was denatured for 10 min at 95 °C, followed by 40 cycles, consisting of 15 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C. For each reaction, a melting curve was generated to test primer dimer formation and nonspecific priming. The cycle threshold (CT) values were determined using the Bio-Rad iQ5 optical system software, in which a threshold value was set for the fluorescence signal at the lower log-linear part above the baseline. CT values were normalized to the B2M housekeeping gene and ΔCT ((average of CTcontrol) − CTvalue). Results are expressed as relative mRNA expression calculated as fold induction (FI) equal to 2−ΔCT and subsequently normalized to the basal gene expression levels determined after one day in 2D culture (n = 6). All values representing down-regulation (fold induction (FI) < 1) are represented as −1/FI and error bars represent standard deviations which are determined accordingly.
2.8. DNA assay
After the samples were lysed for alkaline phosphatase (ALP) activity, the lysate was mixed in a 1:1 volume ratio with 1 mg mL−1 proteinase K (Sigma-Aldrich), 18.5 μg mL−1 iodoacetamine (Sigma Aldrich) and 1 μg mL−1 pepstatin A (Sigma Aldrich) in Tris/EDTA buffer (pH 7.6) and incubated for 16 hours at 56 °C. Quantification of total DNA was done using the CyQuant™ DNA assay (Molecular Probes) and a spectrophotometer (excitation 480 nm, emission 520 nm) (Victor 3, Perkin Elmer).
2.9. ALP activity assay
After culture, all scaffolds were washed gently in PBS, dried by aspirating the PBS, cut in pieces and stored at −80 °C for at least 24 hours. After thawing, the constructs were incubated in a cell lysis buffer with a pH of 7.8 composed of 0.1 M KH2PO4, 0.1 M K2HPO4 and 0.1% Triton X-100, (all Acros Chemicals) for 1 hour at room temperature. Subsequently, the relative ALP activity was determined by chemo-luminescence using CDP-star® (Roche) according to the manufacturer's protocol. The ALP activity was normalized to DNA quantity per sample and the averages per condition were presented as a relative value compared to the respective culture system in BM at day 3.2.10. GAG assay
The amount of GAG was determined spectrophotometrically after reaction with dimethylmethylene blue dye (DMMB, Sigma-Aldrich) in a solution consisting of 16 mg L−1 DMMB, 10 mM hydrochloric acid, 3.04 g L−1 of glycine and 2.37 g L−1 of NaCl (pH 3). A micro plate reader (Multiskan GO, Thermo Fisher) was used to determine the absorbance at 525 nm. The amount of GAG was calculated using a standard of chondroitin sulfate (Sigma-Aldrich). The GAG production was normalized to DNA quantity per sample.2.11. Scanning electron microscopy (SEM) analysis
Cell morphology, attachment and distribution were characterized by SEM analysis using a Philips XL 30 ESEM-FEG. Samples were fixed for 30 minutes in 10% formalin. Subsequently, the samples were dehydrated in sequential ethanol series and critical point dried from liquid carbon dioxide using a Balzers CPD 030 Critical Point Dryer. The constructs were gold sputter-coated (Cressington) prior to SEM analysis. SEM images were obtained under high vacuum with an acceleration voltage of 30 kV and a working distance of 10 mm.2.12. Statistical analysis
Results are presented as mean ± standard deviation, and compared using one-way ANOVA with Dunnett's post-test. Statistical significance between the control group and the experimental groups is indicated with (#) which represents a p-value < 0.05, (@) which represents a p-value < 0.01, and (*) which represents a p-value < 0.001.2.13. Ethics statement
Donor 1 hMSCs were isolated and provided by the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White in compliance with relevant laws and institutional ethics guidelines.Donor 2, 3, and 4 hMSCs were isolated from human bone marrow from donors after written informed consent. This study was carried out in strict accordance with the recommendations of Medisch Ethische Toetsings Commissie Twente (Medical Ethical Research Committee Twente) and was approved by this Committee in compliance with relevant laws on the application of biological materials from a human source for research purposes.
3. Results
To first gain insight into the differentiation potential of hMSCs when cultured on the selected 3D polymeric scaffolds, before looking into phenotype maintenance, differentiation was induced by soluble factors. All results presented in the main figures were retrieved from donor 1, referred to as pre-selected hMSCs, unless stated otherwise. Donor 1 hMSCs have been selected after being tested positive for the capacity to form colonies (CFUs) and the differentiation potential of these cells in the colonies towards the osteogenic and adipogenic lineages according to protocols reported previously.8 To be able to compare these results with the results from a more clinically relevant heterogeneous population of non-tested hMSCs (donor 2, 3 and 4), every experiment was carried out for at least one of those donors for which the results can be found in the ESI.† An overview of the whole data-set can be found in Table S2 in the ESI.†3.1. Differentiation potential of hMSCs in 3D scaffolds by qPCR
The differentiation potential of hMSCs was assessed by mRNA expression analysis and normalized to the expression levels after 1 day of monolayer culture (2D) in the basal medium. RT-QPCR analysis shows the relative mRNA expression of Alkaline Phosphatase (ALP) as an early marker for osteogenesis and Sox9 as an early marker for chondrogenesis (Fig. 2). A significant up-regulation of ALP over time was found for hMSCs cultured as a monolayer in OM on TCPS. In CM in 2D culture an initial down-regulation of ALP was found; after 7 and 14 days of culture the ALP expression was too low to be detected and was therefore not displayed. Comparing these results to the gene expression levels in 3D in the two scaffolding systems, it could be seen that also in 3D in both scaffold types only OM consistently induced a significant up-regulation of ALP. CM seemed to slightly down-regulate the expression of ALP on ES-scaffolds similarly as in 2D. In BM, the ALP expression did not show any significant changes over time in 2D or in any of the scaffold systems. When looking at the relative expression levels, it could be seen that in 2D a 10-fold increase of ALP expression was found after 14 days of culture, which is in the same order as the fold increase in FDM and ES-scaffolds after 14 days. In ES-scaffolds this up regulation of ALP expression is increasing even more than a 40-fold increase after 28 days of culture. | ||
| Fig. 2 RT-QPCR analysis was carried out to determine the relative mRNA expression of ALP and Sox9 for pre-selected hMSCs cultured in 2D as monolayers (BM and OM) or pellets (CM) on TCPS and in 3D in two different scaffold systems. The mRNA expression levels were normalized and compared statistically to hMSCs cultured for 1 day as a monolayer in BM on TCPS. ALP gene expression was significantly up-regulated over time for hMSCs in 2D and in both systems in 3D when cultured in OM. A significant down-regulation of Sox9 expression was found after monolayer or pellet culture in BM, OM and CM. Sox9 expression was significantly up-regulated after 7 and 28 days of culture in CM on FDM-scaffolds. On ES-scaffolds an initial significant increase for Sox9 expression was found after 7 days of culture in CM while the gene seemed to be down-regulated at day 14 and 28 (n = 5, # p < 0.05, @ p < 0.01, * p < 0.001 Dunnett's posttest with BM 2D day 1 as control). | ||
Sox9 expression was significantly down-regulated in 2D culture under nearly all conditions but most profoundly in OM. In 3D, there was a significant up-regulation observed for Sox9 expression after 7 and 28 days of culture in CM on FDM-scaffolds. On ES-scaffolds an initial significant increase of Sox9 expression was found after 7 days of culture in CM while the gene seemed to be down-regulated at day 14 and 28.
Collagen type-1 (collagen-1) and collagen type-2 (collagen-2) were assessed as markers for ECM production (Fig. 3). In 2D on TCPS, the expression of both collagen-1 and collagen-2 was significantly down-regulated in CM after 3 and 7 days. However, for collagen-2 this change in mRNA expression levels was not statistically significant. In 3D, the expression of both genes followed similar trends over time per scaffolding system. A down-regulation of both collagen-1 and collagen-2 was found in BM after 3 days of culture on FDM scaffolds in comparison to hMSCs in BM after 1 day of 2D culture. The expression of collagen-1 and collagen-2 fluctuated over-time and remained down-regulated after 28 days of culture in BM in both scaffold types. In CM, both genes showed up-regulation after 3 and 7 days of culture, thus suggesting an influence at early culture times of chondrogenic soluble factors on the expression of collagen type-1 and collagen type-2. However, similar to that in BM, in CM also the gene expression levels fluctuated for later time-points. Finally, hMSCs cultured in OM showed non-significant changes in the fluctuating gene expression levels, except for ES-scaffolds after 21 days of culture. This implied that the addition of osteogenic soluble factors did not directly correlate with the expression of collagen type-1 and collagen type-2 genes on these 3D scaffolds.
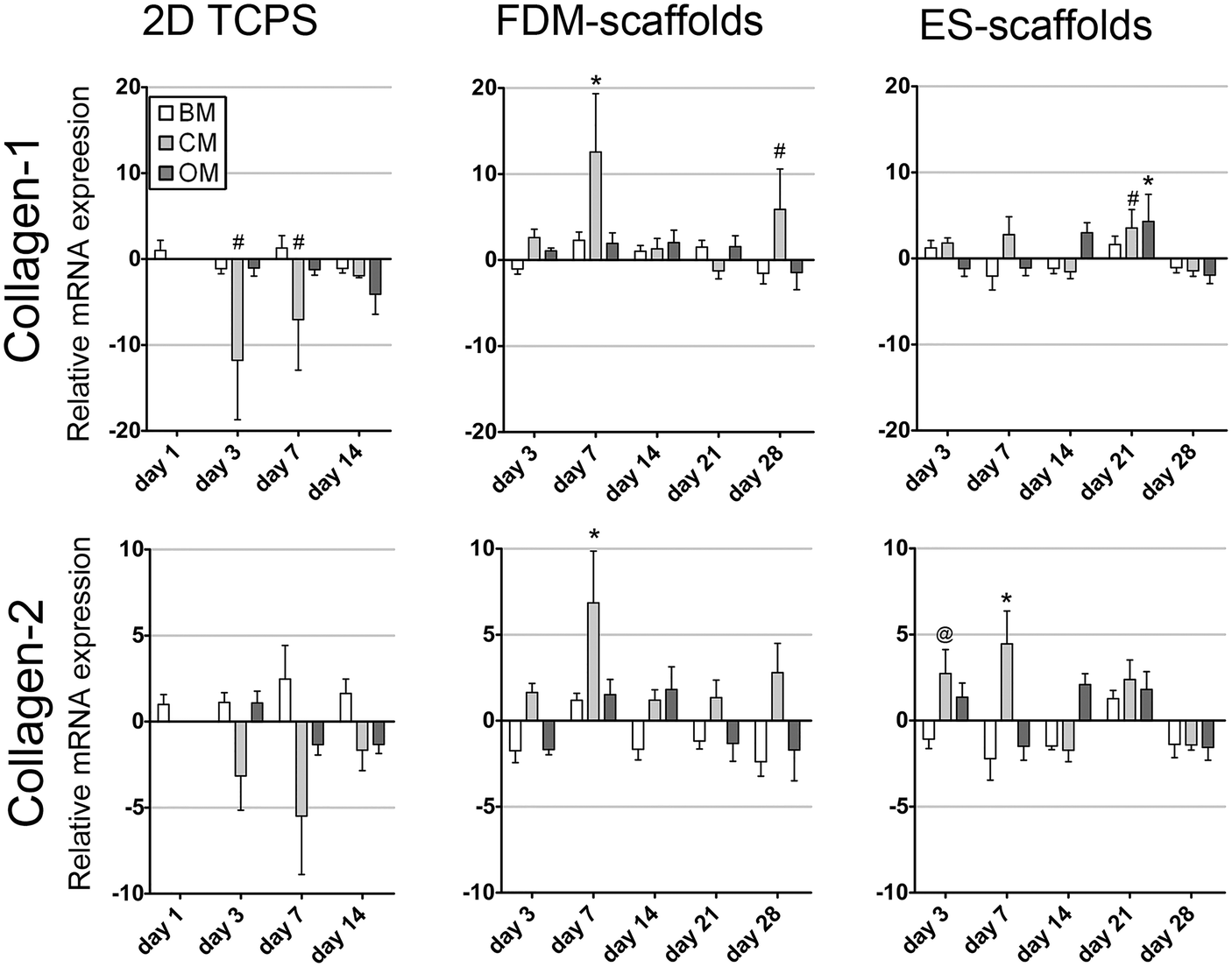 | ||
| Fig. 3 Relative mRNA expression of collagen-1 and collagen-2 for pre-selected hMSCs cultured in 2D as a monolayer (BM and OM) or a pellet (CM) on TCPS and in 3D on two different scaffold systems. Collagen type-1 and collagen type-2 expression followed similar trends over time in 2D and in 3D per scaffolding system. The collagen type-1 and collagen type-2 expression fluctuated over-time and remained down-regulated after 28 days of culture in BM in both scaffold types (n = 5, # p < 0.05, @ p < 0.01, * p < 0.001, Dunnett's posttest with BM 2D day 1 as control). | ||
Another set of genes related to hMSC differentiation was assessed for donor 1 hMSCs cultured in 2D as a monolayer and in 3D on FDM-scaffolds and reported in the ESI,† Fig. S1. Aggrecan (ACAN), which is involved in chondrogenesis, showed a significant up-regulation in OM after 3 days of culture on TCPS. On FDM-scaffolds ACAN showed a significant up-regulation after 7 and 28 days of culture on FDM-scaffolds in CM. BMP2 was assessed as a gene involved in osteogenesis. Culture in BM in 2D resulted in a significant 5- to 6-fold down-regulation of BMP2 expression after 3, 7 and 14 days. In contrast to the results in 2D, a significant up-regulation of BMP2 was found in BM after 7, 14 and 21 days of culture on FDM-scaffolds, whereas in CM and OM, no significant change in BMP2 expression was found. The activated leukocyte cell adhesion molecule (ALCAM), related to the cell surface marker CD166, is often described as an indicator of the clonogenic potential of hMSCs.39 It can be seen that ALCAM was significantly down-regulated a two-fold in all time-points under nearly all conditions on FDM-scaffolds. Similar results were found in BM in 2D culture in which a significant down-regulation of 1.6, 2.3 and 3.5 fold was observed after 3, 7 and 14 days, respectively.
To compare the results of the pre-selected hMSCs in 2D to the results of the more heterogeneous population of hMSCs from donor 2 (ESI,† Fig. S2) and donor 4 (ESI,† Fig. S3), these cells were also cultured on 2D scaffolds for 1 and/or 3, 7 and 14 days. In 2D culture, it could be seen that similar to the results for pre-selected hMSCs the ALP expression was down-regulated in CM for both donors. In OM, Sox9 expression was also down-regulated, while the expression of collagen-1 and collagen-2 showed similar trends per donor and seemed to fluctuate between a 2-fold up- and down-regulation without a consistent change in both donors. In contrast to the results on ACAN expression for pre-selected hMSCs (donor 1) in 2D, hMSCs from donor 4 showed an increase in ACAN expression after 14 days of culture in CM.
To compare the expression levels of pre-selected hMSCs on 3D scaffolds to the more heterogeneous population of hMSCs from donor 2 and donor 3, cells were cultured for 3, 7, 14, 21 and 28 days on FDM- (ESI,† Fig. S4) and on ES-scaffolds (ESI,† Fig. S5) for these two donors, respectively. Donor 2 generally showed no profound gene expression level changes between the differentiation inducing culture media (OM and CM) and BM for all genes. Also donor 3 on ES-scaffolds showed no apparent differences in gene expression levels for ALP, ACAN and ALCAM between the different culture media. Compared to 2D culture at day 1, all conditions of donor 3 showed a significant down-regulation of ALP at all time-points and a significant up-regulation of ALCAM. Sox9 was down-regulated in CM at all time-points and in BM for all time-points after 7 days of culture. Collagen-1 expression was initially significantly up-regulated in BM and OM. After 14 days the expression stabilized at the same level as the 2D control after 1 day of culture. In both FDM-scaffolds and ES-scaffolds, a more profound up-regulation of chondrogenic or osteogenic marker expression after culture in CM and OM, respectively, was found for pre-selected hMSCs than for the more heterogeneous populations of hMSCs from donors 2 and 3.
3.2. Differentiation potential of hMSCs in 3D scaffolds by protein expression analysis
To investigate whether or not up-regulation of osteogenic or chondrogenic genes resulted in a functional change in the behaviour of the cells behavior, protein expression analysis was carried out (Fig. 4). ALP activity was determined as an indicator of osteogenic behavior and glycosaminoglycan (GAGs) secretion was determined as an indicator of chondrogenesis. Both ALP activity and GAG secretion were first normalized to the number of cells per sample, which was determined by DNA quantification. The results of DNA quantification can be found in the ESI,† Fig. S6, from which it can be seen that there are just slight changes in DNA quantities over time and between different culture media. No direct correlation between the cell number and the ALP activity per cell or GAG production per cell was observed (Fig. 4 and ESI,† Table S3).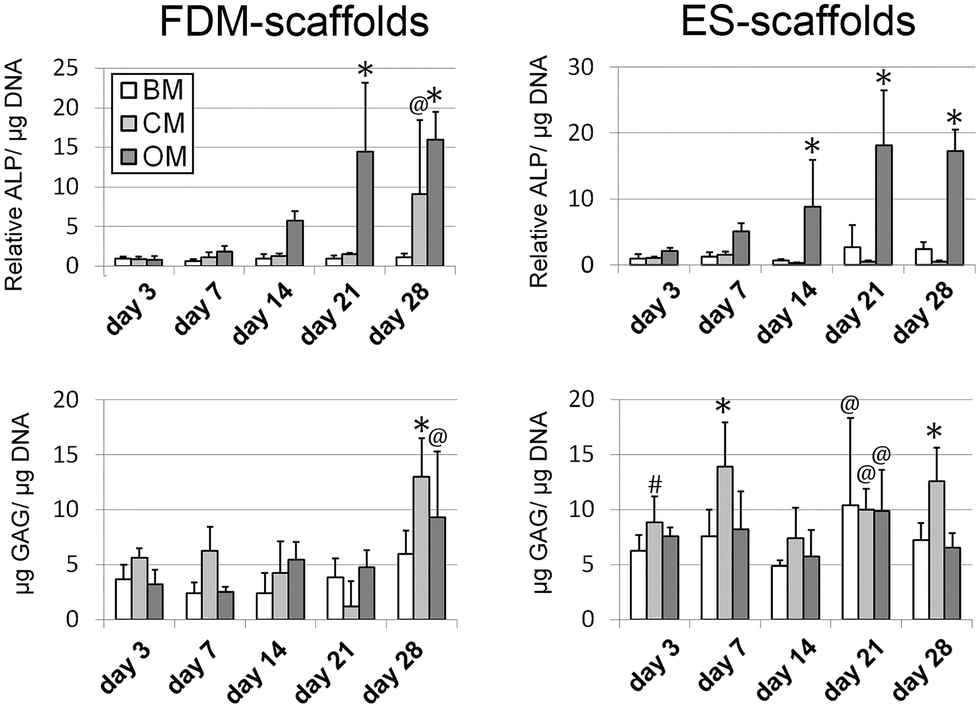 | ||
| Fig. 4 Protein expression analysis showed an increase in ALP activity over time for hMSCs in both scaffold types when cultured in OM, whereas in BM and CM the ALP activity was maintained at a basal level. A significant increase in GAG production was observed for hMSCs cultured in CM after 28 days on FDM-scaffolds and after 3, 7, 21 and 28 days on ES-scaffolds (n = 5, # p < 0.05, @ p < 0.01, * p < 0.001, Dunnett's posttest with BM 2D day 4 as control). | ||
ALP activity increased significantly over time for hMSCs in both scaffold types when cultured in OM, whereas in BM and CM it was minimally produced. A significant increase in GAG production was observed for hMSCs cultured in CM after 28 days on FDM-scaffolds, and after 3, 7, 21 and 28 days on ES-scaffolds. Furthermore, a significant increase in GAG production was found after 28 days in OM on FDM-scaffolds and after 21 days in OM and BM on ES-scaffolds. To compare the expression levels of pre-selected hMSCs on 3D scaffolds to a more heterogeneous population of hMSCs, protein expression levels were determined for cells cultured for 3, 7, 14, 21 and 28 days on FDM-scaffolds (donor 2) and on ES-scaffolds (donor 3) (ESI,† Fig. S7). For both assays the results of DNA quantification are given in the ESI,† Fig. S8. For both donors a positive correlation between the ALP activity and the application of OM was found. The up-regulation of ALP activity on ES-scaffolds was observed at an earlier time-point than for FDM-scaffolds, which was also observed with the pre-selected donor (Fig. 4). GAG expression was significantly up-regulated in the presence of CM on FDM-scaffolds after 7 days of culture and on ES-scaffolds after 7, 14, 21 and 28 days of culture. In BM and OM, GAG production was also up-regulated after 14, 21 and 28 days, and after 21 and 28 days, respectively.
3.3. SEM analysis of ECM production after soluble factor induced differentiation
The distribution and morphology of the cells and ECM throughout the 3D scaffolds were assessed by SEM (Fig. 5). In FDM-scaffolds, CM resulted often in lower cell numbers and less ECM production than BM and OM. However, the ECM that was secreted showed collagen-like ECM fibers with a tendency to align into bundles, whereas in BM and OM this orientation appeared more random. Furthermore, in CM, vacuole-like structures were observed in the ECM (Fig. 5B indicated with an arrow). On ES-scaffolds, no profound differences were observed at the micro-scale and all culture media resulted in a dense cellular layer on top of the sheets with cells penetrated in between the scaffold fibers (Fig. 5G–I).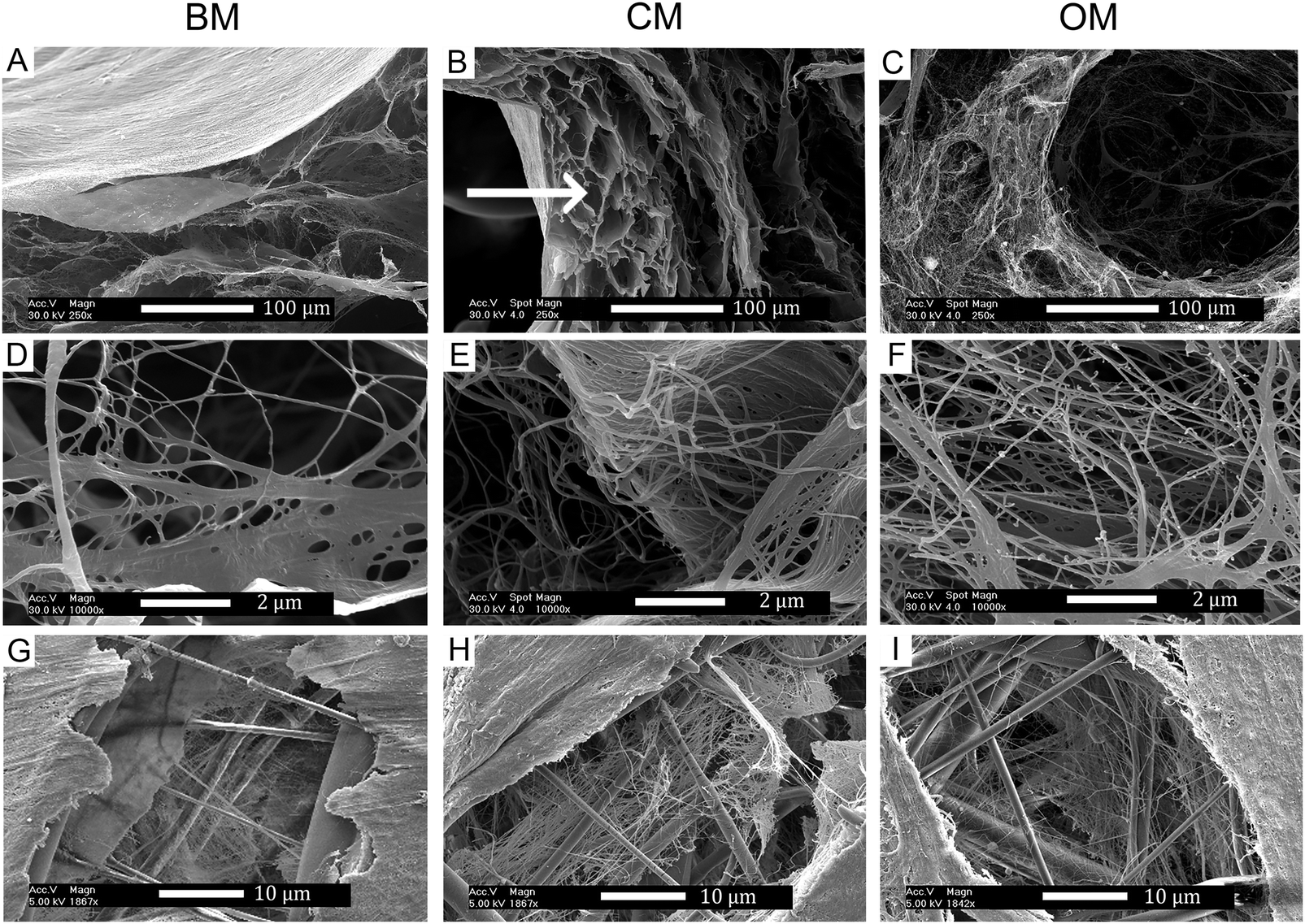 | ||
| Fig. 5 SEM analyses after 28 days of culture on FDM scaffolds (A–F) and ES-scaffolds (G–I). Under all three culture conditions BM (A, D and G), CM (B, E and H) and OM (C, F and I) collagen-like ECM matrix deposition was found. The scaffolds showed a dense outer layer of cells under all conditions. In FDM-scaffolds cultured with hMSCs in CM vacuole-like structures, which show comparable tissue morphology as in native cartilage, were found. | ||
3.4. Loss of the differentiated state of hMSCs in FDM- and ES-scaffolds upon the removal of soluble factors
The stability of differentiated phenotype was assessed for pre-selected hMSCs as well as for more heterogeneous populations of hMSCs from donor 2, 3 and 4 on FDM- and ES-scaffolds. Soluble factors were removed by replacing the culture media with BM after 14 days of inducing differentiation into the chondrogenic (dex and TGF-β3) and osteogenic (dex) lineages on FDM- and ES-scaffolds, a procedure that will further be referred to as the de-differentiation phase. The culture period was prolonged for another 3 or 14 days to investigate if changes in gene expression levels and protein production were observed with respect to the specimens that were further cultured in differentiation media. Fig. 6A shows the gene expression levels for BM, CM and OM for day 14 and day 28, as already presented in Fig. 2 and 3, to enable visual comparison to the results of the de-differentiation phase introduced here. As can be observed in Fig. 6, the ALP expression significantly increased in OM after 14, 17 and 28 days in both scaffold types compared to the levels for BM after 14 days of culture. After 3 days of potential de-differentiation, the ALP expression levels in OB were still significantly up-regulated compared to day 14 in BM; however, a small decrease was observed compared to OM at the same time-point. After 14 days of potential de-differentiation, there was no significant difference in ALP expression levels between samples in BM and OB, whereas in OM the expression remained up-regulated.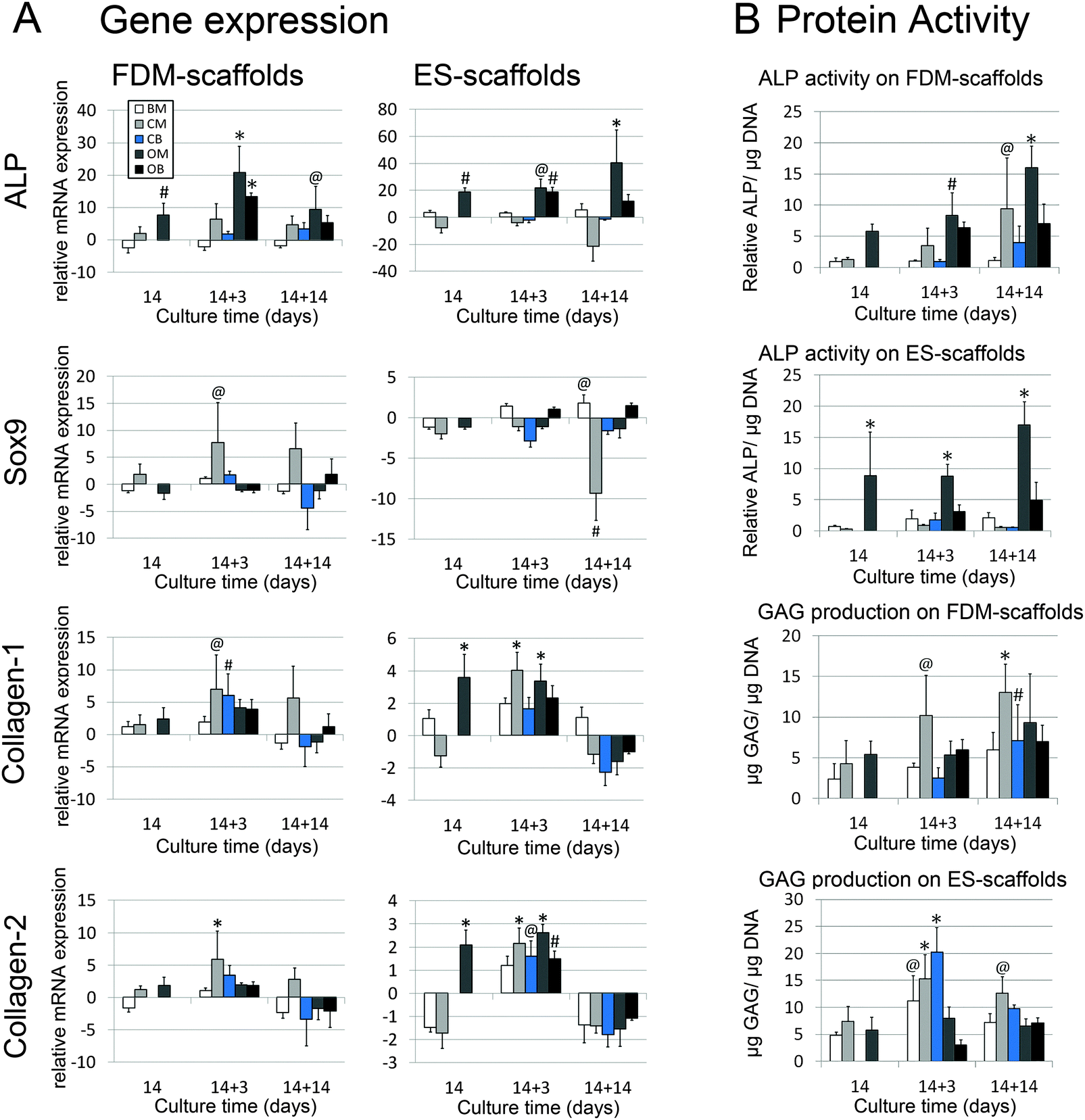 | ||
| Fig. 6 (A) RT-QPCR results after 14 days of soluble-factor-induced differentiation followed by 3 or 14 days of de-differentiation. ALP gene expression on FDM-scaffolds significantly increased in OM after 14, 17 and 28 days in both scaffold types compared to the levels for BM after 14 days of culture. After 14 days of de-differentiation there was no significant difference between BM and OB, whereas OM remained up-regulated. In FDM-scaffolds a significant increase in Sox9 expression was found only after 17 days of culture in CM, whereas in ES-scaffolds a significant down-regulation was found after 28 days in CM. In CB no differences were observed after 3 days or 14 days of de-differentiation compared to BM after 14 days of culture. The expression collagen type-1 and collagen type-2 was slightly or under some conditions even significantly up-regulated after 17 days of culture, whereas a down-regulation was found under all conditions after 28 days of culture. (B) ALP activity showed an up-regulation in OM over time as observed before, yet the ALP activity in OB did not change in FDM-scaffolds compared to OM at day 14. In ES-scaffolds the ALP activity was decreased compared to OM at day 14. GAG expression showed a significant up-regulation in CM after 17 and 28 days of culture in both scaffold types. In CB this up-regulation was found to be even higher after 17 days on ES-scaffolds whereas a decrease in GAG production was found in FDM-scaffolds at the same time-point ((A) n = 5, (B) n = 4, # p < 0.05, @ p < 0.01, * p < 0.001, Dunnett's posttest with BM 3D of respective scaffold type day 14 as control). | ||
In FDM-scaffolds, a significant increase in Sox9 expression was found only after 17 days of culture in CM, whereas in ES-scaffolds a significant down-regulation was found after 28 days in CM. In CB, no differences were observed after 3 days or 14 days of de-differentiation compared to BM after 14 days of culture. Similarly to the results presented in Fig. 3, the expression of collagen-1 and collagen-2 expression showed the same trends per scaffold type. In the two scaffold types, the expression of collagen-1 and collagen-2 was slightly or under some conditions even significantly up-regulated after 17 days of culture, whereas down-regulation was found under all conditions after 28 days of culture.
The stability of gene expression levels after differentiation was also assessed for donor 2 on FDM-scaffolds, for donor 3 on ES-scaffolds, and for donor 4 on FDM-scaffolds (ESI,† Fig. S9, S10 and S11, respectively). In the FDM-scaffolds, it was observed that Sox9, collagen-1 and collagen-2 generally showed similar trends per donor, which was also observed for donor 1 on FDM-scaffolds. There seemed to be no correlation between the gene expression of ALP, ACAN and ALCAM per donor. Donors 2 and 4 did not show a consistent positive correlation between ALP gene expression and the presence of OM. Also for the markers of chondrogenesis, Sox9, ACAN and collagen-2, no positive correlation was found between the gene expression levels and the presence of CM. For donor 3 hMSCs on ES-scaffolds, ALP was significantly down-regulated under most of the conditions compared to BM day 14. Only after 14 days of de-differentiation after osteogenic differentiation, the gene expression levels were restored to comparable levels as for hMSCs in BM after 14 days of culture. Sox9 and ACAN showed a consistent but not significant down-regulation in CM compared to BM at day 14 on ES-scaffolds. Also this down-regulation was lost during the de-differentiation phase.
To investigate the functional response of hMSCs upon de-differentiation, protein expression levels were analyzed (Fig. 6B). BM, CM and OM after 14 and 28 days of differentiation culture are based on the same results as presented in Fig. 4. ALP activity in OM showed an up-regulation over time as observed before. Yet, the ALP activity in OB did not change in FDM-scaffolds compared to OM at day 14. In ES-scaffolds, the ALP activity was decreased compared to OM at day 14 and showed no significant difference with BM at day 14. GAG expression showed a significant up-regulation in CM after 17 and 28 days of culture in both scaffold types. In CB, this up-regulation showed to be even higher after 17 days on ES-scaffolds whereas a decrease in GAG production was found in FDM-scaffolds at the same time-point. After 14 days of de-differentiation GAG production was significantly increased in CB compared to BM at day 14. However, the GAG amount was lower than that for CM after 28 days of culture. ESI,† Fig. S12 shows the protein production levels for donors 2 and 4 on FDM-scaffolds and for donor 3 on ES-scaffolds. It can be observed that only donor 2 and 3 hMSCs showed a significant increase in ALP activity when cultured in OM, and this activity was lost upon the removal of soluble factors. With respect to GAG production, only donor 4 showed a response in CM, whereas for the other 2 donors no profound changes in GAG production were found. In just a few cases where a change in the protein production levels was observed for heterogeneous populations of hMSCs after differentiation, de-differentiation resulted in a decrease of these protein production levels. For pre-selected hMSCs, this effect of the de-differentiation phase on protein production levels was found to be much stronger.
4. Discussion
Several studies have been reported on the differentiation potential of hMSCs when cultured on 3D scaffolds with or without the addition of soluble factors.40,41 However, as far as we know, there is limited knowledge available on the stability of the differentiated phenotype after in vitro culture of hMSC on such 3D scaffolds. In many tissue engineering applications, scaffolds cultured in vitro with hMSCs ultimately have to be implanted in a defect site. Therefore, it is of high importance to assess whether the results achieved in vitro have the potential to be maintained upon implantation.42Here, differentiation towards the osteogenic and chondrogenic lineages was induced by the introduction of well-known soluble factors in the culture media. Basic medium (BM) served as a negative control on differentiation, assuming that similar to that in 2D also in 3D no differentiation will be induced by the media components in BM. As expected, the two different scaffold types did not seem to change the cellular phenotype of pre-selected hMSCs towards the osteogenic or chondrogenic lineage without the presence of differentiation inducing media. In BM, there were no significant differences in mRNA expression levels in both scaffold systems and in 2D culture for ALP, collagen-1 and collagen-2 after 3, 7, 21 or 28 days of culture compared to the control group in BM in 2D after 1 day of culture. Also ALP activity and GAG production did not show consistent changes over time for hMSCs in BM in any of the scaffold systems for both the colony-picked hMSCs and the three more heterogeneous hMSCs populations. Overall, there is a strong indication that none of the two scaffold-types consistently and significantly induced osteogenic or chondrogenic differentiation in both pre-selected hMSCs and in the more heterogeneous populations of hMSCs in the absence of differentiation inducing soluble factors. Similar results were recently reported by Manferdini et al. who showed soluble-factor dependency of the differentiation of hMSCs on biomimetic gradient composite scaffolds.43 From their results on differentiation, it could be concluded that the type of media used dominated over the potential influence of the properties of scaffolds. In our study, the over-time fluctuations in gene expression levels in BM found in nearly all experiments in 3D were in the same order as the fluctuations found in 2D culture and therefore the properties of scaffolds are not expected to contribute to this inconsistency in gene expression.
To assess whether the two scaffolds in this study support soluble-factor induced hMSC differentiation, differentiation towards osteogenic and chondrogenic lineages was assessed. As an indication of osteogenic differentiation capacity, both scaffold systems showed an up-regulation of ALP expression and ALP activity over time when the pre-selected hMSCs were cultured in OM. The corresponding fold increases were comparable to the levels observed in 2D over time. This soluble-factor-induced change in cellular phenotype was partly lost when OM was replaced by BM after 14 days of differentiation, which again suggests that the scaffold by itself does not induce or promote hMSC differentiation towards the osteogenic lineage.
The induced chondrogenic response on CM in pellet-culture (presented as 2D) was not as profound as the osteogenic response on OM. Sox9, an early marker for chondrogenesis, did not consistently increase over time. In contrast, in 2D and pellet-culture it showed a direct decrease in BM and OM, and CM, respectively. In 3D, a positive correlation between CM and Sox9 gene expression was only found in FDM-scaffolds. A down-regulation of Sox9 in combination with an up-regulation of osteogenic markers was observed in OM both in 2D and in 3D and could be associated with differentiation towards the osteogenic lineage.44
A negative response in Sox9, collagen-2 and ACAN was found for hMSCs from donor 3 cultured in CM on ES-scaffolds. The differentiation potential towards the chondrogenic lineage for this heterogeneous population of hMSCs cultured in pellets could not be assessed since the quantities of isolated RNA were too low to run further analysis. For this donor, it could be concluded that these hMSCs lack differentiation potential towards the chondrogenic lineage under traditional chondrogenesis inducing culture conditions. ES-scaffolds might still be favorable as a 3D support for chondrogenic differentiation, in case the hMSC population of interest shows a high potential to differentiate into the chondrogenic lineage in pellet-culture. Low RNA quantities were not observed for the pellet-cultures of the other donors and hence could be an effect of donor variation.37,45 In summary, gene expression levels for pre-selected hMSCs showed a more profound response to osteogenic factors than the hMSCs from the other donors, probably due to the heterogeneity of those cell populations. Furthermore, pre-selected hMSCs showed a positive correlation between ALP gene expression levels and ALP activity, as well as between Sox-9 gene expression and GAG production. For the more heterogeneous populations of hMSCs, a positive and consistent response on differentiation inducing factors was only found in protein expression. The discrepancy between the fluctuating gene expression levels and the more consistent protein expression levels can be related to a faster response of RNA transcription processes on extra and intracellular signals than the response of more down-stream post-transcriptional modifications and protein translation processes. Protein expression cumulates in the cells and in the ECM, whereas RNA transcription can be regulated at a faster rate. This could be the case when a pathway for the production of a specific protein is activated; this pathway could be inhibited by a negative feedback when a sufficient amount of protein has been synthesized. The corresponding gene expression could show down-regulation because RNA transcription is inhibited, whereas the produced protein remains active and detectable. To the best of our knowledge, the influence of the solid 3D scaffold culture environment on these specific differentiation related pathways in hMSCs has not yet been explored thoroughly. More studies on the molecular level should aid in unravelling these differentiation pathways in complex environments, taking into account the heterogeneity of the cell populations and the observed donor variation for multiple time-points.46 Even though heterogeneous populations of hMSCs showed fluctuating gene expression levels, the use of these heterogeneous populations from older patients is more clinically relevant since in future therapies pre-selection of hMSCs might be too costly and time-consuming. For example, in the context of skeletal regeneration one could think of combining 3D scaffolds with heterogeneous hMSCs being retrieved from the subchondral bone marrow after performing microfracture,47 the latter being the current standard as first-line therapy for cartilage repair.48
With respect to de-differentiation, generally, it was observed that when hMSCs showed a positive response on CM or OM towards the chondrogenic or osteogenic lineage, respectively, the differentiated phenotype was lost within 3 or 14 days after the removal of soluble factors. Both the response on differentiation inducing factors and the loss of the differentiated phenotype upon de-differentiation were found to be stronger in pre-selected hMSCs than in hMSCs from donors 2, 3 and 4. From these results, it can be concluded that the soluble-factor-induced differentiated phenotype of pre-selected hMSCs showed to be dependent on the presence of such soluble factors. The phenotypic stability of more heterogeneous populations of hMSCs also showed to be limited, since the increased protein expression levels were lost upon the removal of soluble factors under most of the conditions similarly to the results for pre-selected hMSCs.
With the results presented herein, we would like to emphasize that for future in vitro tissue engineering it is important to bear in mind that successful soluble factor induced differentiation of any population of hMSCs does not necessarily result in a stable commitment of the hMSCs to the targeted lineage when such soluble factors are not available anymore. Therefore, the development of differentiation inductive scaffolds that will retain their intrinsic instructive cues over long time-periods could be vital for future tissue engineering applications.
5. Conclusions
In this study, soluble-factor-induced differentiation of hMSCs was assessed for its stability on two distinct polymeric scaffold types and compared to their differentiation capacity in 2D. Firstly, we showed that soluble-factor-induced differentiation was neither completely inhibited nor dominantly promoted by the scaffold properties, which makes them potentially suitable non-inductive supports for several tissue engineering applications. Secondly, after the removal of soluble factors, the possibly differentiated phenotype of the hMSCs was found to be lost and the mRNA expression levels of the majority of the assessed genes returned to comparable levels as for hMSCs that were cultured in non-inductive media during the full time-period of the study. From these results, we conclude that although some scaffold types might be opted as promising candidates to support hMSC differentiation and tissue growth, these scaffolds do not always guarantee a stable differentiated phenotype when soluble factors are not available anymore. In future tissue engineering approaches, when combining scaffolds with hMSCs to be differentiated into a certain lineage in vitro, the stability of the differentiation phenotype should be assessed to better predict the functionality of constructs upon implantation.Acknowledgements
The authors gratefully acknowledge the funding from the Netherlands Institute for Regenerative Medicine (NIRM) through the grant number FES0908. This project/research has been also made possible with the support of the Dutch Province of Limburg. Some of the materials employed in this work were provided by the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through a grant from NCRR of the NIH (Grant # P40RR017447).Notes and references
- B. Ma, J. C. H. Leijten, L. Wu, M. Kip, C. A. van Blitterswijk, J. N. Post and M. Karperien, Osteoarthr Cartilage, 2013, 21, 599–603 CrossRef CAS PubMed.
- T. Kluba, T. Niemeyer, C. Gaissmaier and T. Grunder, Spine, 2005, 30, 2743–2748 CrossRef PubMed.
- A. G. Kayali, L. E. Flores, A. D. Lopez, B. Kutlu, E. Baetge, R. Kitamura, E. Hao, G. M. Beattie and A. Hayek, Diabetes, 2007, 56, 703–708 CrossRef CAS PubMed.
- E. Buzhor, O. Harari-Steinberg, D. Omer, S. Metsuyanim, J. Jacob-Hirsch, T. Noiman, Z. Dotan, R. S. Goldstein and B. Dekel, Tissue Eng., Part A, 2011, 17, 2305–2319 CrossRef CAS PubMed.
- K. Lee, M. K. Majumdar, D. Buyaner, J. K. Hendricks, M. F. Pittenger and J. D. Mosca, Mol. Ther., 2001, 3, 857–866 CrossRef CAS PubMed.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143–147 CrossRef CAS PubMed.
- A. M. Mackay, S. C. Beck, J. M. Murphy, F. P. Barry, C. O. Chichester and M. F. Pittenger, Tissue Eng., 1998, 4, 415–428 CrossRef CAS PubMed.
- C. M. DiGirolamo, D. Stokes, D. Colter, D. G. Phinney, R. Class and D. J. Prockop, Br. J. Haematol., 1999, 107, 275–281 CrossRef CAS PubMed.
- D. Menicanin, P. M. Bartold, A. C. Zannettino and S. Gronthos, Stem Cell Rev., 2009, 5, 36–50 CrossRef CAS PubMed.
- M. Dominici, K. Le Blanc, I. Mueller, I. Slaper-Cortenbach, F. C. Marini, D. S. Krause, R. J. Deans, A. Keating, D. J. Prockop and E. M. Horwitz, Cytotherapy, 2006, 8, 315–317 CrossRef CAS PubMed.
- V. Rasini, M. Dominici, T. Kluba, G. Siegel, G. Lusenti, H. Northoff, E. M. Horwitz and R. Schafer, Cytotherapy, 2013, 15, 292–306 CrossRef CAS PubMed.
- C. K. Abrahamsson, F. Yang, H. Park, J. M. Brunger, P. K. Valonen, R. Langer, J. F. Welter, A. I. Caplan, F. Guilak and L. E. Freed, Tissue Eng., 2010, 16, 3709–3718 CrossRef CAS PubMed.
- J. Hu, K. Feng, X. Liu and P. X. Ma, Biomaterials, 2009, 30, 5061–5067 CrossRef CAS PubMed.
- J. M. Silva, N. Georgi, R. Costa, P. Sher, R. L. Reis, C. A. Van Blitterswijk, M. Karperien and J. F. Mano, PLoS One, 2013, 8, e55451 CAS.
- L. T. H. Nguyen, S. Liao, C. K. Chan and S. Ramakrishna, Nanomedicine, 2012, 7, 1561–1575 CrossRef CAS PubMed.
- A. M. Barradas, V. Monticone, M. Hulsman, C. Danoux, H. Fernandes, Z. Tahmasebi Birgani, F. Barrere-de Groot, H. Yuan, M. Reinders, P. Habibovic, C. van Blitterswijk and J. de Boer, Integr. Biol., 2013, 5, 920–931 RSC.
- A. Nandakumar, Z. T. Birgani, D. Santos, A. Mentink, N. Auffermann, K. van der Werf, M. Bennink, L. Moroni, C. van Blitterswijk and P. Habibovic, Biofabrication, 2013, 5, 015006 CrossRef CAS PubMed.
- A. V. Shakhbazau, N. V. Petyovka, S. M. Kosmacheva and M. P. Potapnev, Bull. Exp. Biol. Med., 2011, 150, 547–550 CrossRef CAS PubMed.
- K. Janeczek Portalska, A. Leferink, N. Groen, H. Fernandes, L. Moroni, C. van Blitterswijk and J. de Boer, PLoS One, 2012, 7, e46842 Search PubMed.
- H. Tian, S. Bharadwaj, Y. Liu, H. Ma, P. X. Ma, A. Atala and Y. Zhang, Biomaterials, 2010, 31, 870–877 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- H. V. Unadkat, M. Hulsman, K. Cornelissen, B. J. Papenburg, R. K. Truckenmuller, A. E. Carpenter, M. Wessling, G. F. Post, M. Uetz, M. J. T. Reinders, D. Stamatialis, C. A. van Blitterswijk and J. de Boer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5905 Search PubMed.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS PubMed.
- P. Decuzzi and M. Ferrari, Biomaterials, 2010, 31, 173–179 CrossRef CAS PubMed.
- L. Zhang, L. P. Peng, N. Wu and L. P. Li, Chin. Med. J., 2012, 125, 1650–1655 Search PubMed.
- T. Schilling, U. Noth, L. Klein-Hitpass, F. Jakob and N. Schutze, Mol. Cell. Endocrinol., 2007, 271, 1–17 CrossRef CAS PubMed.
- J. E. Grove, E. Bruscia and D. S. Krause, Stem Cells, 2004, 22, 487–500 CrossRef PubMed.
- R. O. Oreffo, C. Cooper, C. Mason and M. Clements, Stem Cell Rev., 2005, 1, 169–178 CrossRef CAS PubMed.
- M. Rottmar, M. Hakanson, M. Smith and K. Maniura-Weber, J. Mater. Sci.: Mater. Med., 2010, 21, 999–1004 CrossRef CAS PubMed.
- L. Moroni, J. R. de Wijn and C. A. van Blitterswijk, J. Biomed. Mater. Res., 2005, 75, 957–965 CrossRef CAS PubMed.
- L. Moroni, R. Licht, J. de Boer, J. R. de Wijn and C. A. van Blitterswijk, Biomaterials, 2006, 27, 4911–4922 CrossRef CAS PubMed.
- A. M. Leferink, W. J. Hendrikson, J. Rouwkema, M. Karperien, C. A. van Blitterswijk and L. Moroni, J. Tissue Eng. Regener. Med., 2013 DOI:10.1002/term.1842 [Epub ahead of print].
- L. Song and R. S. Tuan, FASEB J., 2004, 18, 980–982 CAS.
- D. G. Phinney, G. Kopen, W. Righter, S. Webster, N. Tremain and D. J. Prockop, J. Cell. Biochem., 1999, 75, 424–436 CrossRef CAS PubMed.
- J. D. de Bruijn, I. van den Brink, S. Mendes, R. Dekker, Y. P. Bovell and C. A. van Blitterswijk, Advances in Dental Research, 1999, 13, 74–81 CrossRef CAS PubMed.
- G. Siegel, T. Kluba, U. Hermanutz-Klein, K. Bieback, H. Northoff and R. Schafer, BMC Med., 2013, 11, 146 CrossRef CAS PubMed.
- G. Wang, L. Zheng, H. Zhao, J. Miao, C. Sun, N. Ren, J. Wang, H. Liu and X. Tao, Tissue Eng., 2011, 17, 1341–1349 CrossRef CAS PubMed.
- J. Zhang, Y. Wu, T. Thote, E. H. Lee, Z. Ge and Z. Yang, Biomed. Mater., 2014, 9, 035011 CrossRef PubMed.
- S. Boeuf and W. Richter, Stem Cell Res. Ther., 2010, 1, 31 CrossRef PubMed.
- C. Manferdini, C. Cavallo, B. Grigolo, M. Fiorini, A. Nicoletti, E. Gabusi, N. Zini, D. Pressato, A. Facchini and G. Lisignoli, J. Tissue Eng. Regener. Med., 2013 DOI:10.1002/term.1723 [Epub ahead of print].
- N. Giuliani, G. Lisignoli, M. Magnani, C. Racano, M. Bolzoni, B. Dalla Palma, A. Spolzino, C. Manferdini, C. Abati, D. Toscani, A. Facchini and F. Aversa, Stem Cells Int., 2013, 2013, 312501 Search PubMed.
- R. Siddappa, R. Licht, C. van Blitterswijk and J. de Boer, J. Orthop. Res., 2007, 25, 1029–1041 CrossRef CAS PubMed.
- S. R. Husain, Y. Ohya, J. Toguchida and R. K. Puri, Tissue Eng., Part B Rev, 2014, 20, 189 CrossRef PubMed.
- B. Wei, C. Jin, Y. Xu, X. Du, C. Yan, C. Tang, M. Ansari and L. Wang, Tissue Eng., 2014, 20, 2646–2655 CrossRef CAS PubMed.
- D. Saris, A. Price, W. Widuchowski, M. Bertrand-Marchand, J. Caron, J. O. Drogset, P. Emans, A. Podskubka, A. Tsuchida, S. Kili, D. Levine, M. Brittberg and S. S. Grp, Am. J. Sport Med., 2014, 42, 1384–1394 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00177c |
| ‡ Department of Complex Tissue Regeneration, MERLN Institute, Maastricht University, Universiteitssingel 40, 6229ER Maastricht, The Netherlands. E-mail: l.moroni@maastrichtuniversity.nl |
| This journal is © The Royal Society of Chemistry 2015 |