
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutralizing antibodies against West Nile virus identified directly from human B cells by single-cell analysis and next generation sequencing†
Konstantinos
Tsioris‡
a,
Namita T.
Gupta‡
b,
Adebola O.
Ogunniyi
a,
Ross M.
Zimnisky
a,
Feng
Qian
cd,
Yi
Yao
d,
Xiaomei
Wang
d,
Joel N. H.
Stern
efg,
Raj
Chari
h,
Adrian W.
Briggs
i,
Christopher R.
Clouser
i,
Francois
Vigneault
hi,
George M.
Church
h,
Melissa N.
Garcia
j,
Kristy O.
Murray
j,
Ruth R.
Montgomery‡
d,
Steven H.
Kleinstein‡
bk and
J. Christopher
Love‡
*a
aDepartment of Chemical Engineering, Koch Institute of Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Bldg. 76-253, Cambridge, MA 02139, USA. E-mail: clove@mit.edu; Tel: +1-617-324-2300
bInterdepartmental Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06511, USA
cState Key Laboratory of Genetic Engineering and Ministry of Education Key Laboratory of Contemporary Anthropology, School of Life Sciences, Fudan University, Shanghai, 200438, China
dDepartment of Internal Medicine, Yale University School of Medicine, New Haven, CT 06520, USA
eDepartment of Science Education, Hofstra North Shore-LIJ School of Medicine, Hempstead, NY 11549, USA
fDepartment of Neurology, Hofstra North Shore-LIJ School of Medicine, Hempstead, NY 11549, USA
gDepartment of Neurology, Yale University School of Medicine, New Haven, CT 06520, USA
hDepartment of Genetics, Harvard Medical School, Boston, MA 02115, USA
iAbVitro, Inc., Boston, MA 02210, USA
jBaylor College of Medicine, Houston, TX 77030, USA
kDepartment of Pathology and Department of Immunobiology, Yale University School of Medicine, New Haven, CT 06520, USA
First published on 8th October 2015
Abstract
West Nile virus (WNV) infection is an emerging mosquito-borne disease that can lead to severe neurological illness and currently has no available treatment or vaccine. Using microengraving, an integrated single-cell analysis method, we analyzed a cohort of subjects infected with WNV – recently infected and post-convalescent subjects – and efficiently identified four novel WNV neutralizing antibodies. We also assessed the humoral response to WNV on a single-cell and repertoire level by integrating next generation sequencing (NGS) into our analysis. The results from single-cell analysis indicate persistence of WNV-specific memory B cells and antibody-secreting cells in post-convalescent subjects. These cells exhibited class-switched antibody isotypes. Furthermore, the results suggest that the antibody response itself does not predict the clinical severity of the disease (asymptomatic or symptomatic). Using the nucleotide coding sequences for WNV-specific antibodies derived from single cells, we revealed the ontogeny of expanded WNV-specific clones in the repertoires of recently infected subjects through NGS and bioinformatic analysis. This analysis also indicated that the humoral response to WNV did not depend on an anamnestic response, due to an unlikely previous exposure to the virus. The innovative and integrative approach presented here to analyze the evolution of neutralizing antibodies from natural infection on a single-cell and repertoire level can also be applied to vaccine studies, and could potentially aid the development of therapeutic antibodies and our basic understanding of other infectious diseases.
Insight, innovation, integrationWest Nile virus (WNV) infection is a mosquito-borne disease that can cause neurological illness and there is no available treatment or vaccine. We integrated single-cell analysis by microengraving with next-generation sequencing to analyze the humoral response to WNV from single cells to the repertoire level. Through this approach, we identified four novel human-derived neutralizing antibodies from a cohort WNV-infected individuals. While the antibody response does not predict clinical severity of WNV infection, our data provide insight into the long-term persistence of the humoral response to WNV and these antibodies have potential use as therapeutics against WNV infection. |
Introduction
West Nile virus (WNV) is a mosquito-borne, enveloped positive-strand RNA virus that can lead to severe neurological disease. The virus belongs to the family flaviviridae, which includes yellow fever, hepatitis C virus, and dengue viruses.1,2 The emergence of WNV in North America was first documented in 1999 in New York, USA. WNV has now become established throughout the USA and has spread into Canada, Mexico, and the Caribbean. Reports from the CDC indicate infection of more than 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people to date, including more than 1700 fatalities.3 The cumulative incidence of WNV infection may reach 3 million people.2,4 Currently, there is no approved treatment or vaccine against WNV.1,2,5
000 people to date, including more than 1700 fatalities.3 The cumulative incidence of WNV infection may reach 3 million people.2,4 Currently, there is no approved treatment or vaccine against WNV.1,2,5
Passive administration of neutralizing antibodies (NAbs) is one possible route to treat viral infections6,7 and could have therapeutic value in the context of severe flavivirus infection.8–11 WNV-specific NAbs have been derived by phage and yeast display libraries from both humans and mice.12–16 Murine antibodies have shown potency in mouse models, but the epitopes targeted by this class of antibodies comprise only a minor component of the neutralizing response in humans. These antibodies have limited utility as therapeutics to date.17 Other disadvantages of library-based methods to derive NAbs are the random pairing of heavy- and light-chains, which obscures the natural humoral response, involves time-consuming assays, and identifies antibodies with limited neutralization function.18 In contrast, recent studies have identified a large number of highly potent human immunodeficiency virus-1 (HIV-1) specific NAbs directly derived from HIV-infected patients using flow cytometry to sort memory B cells (MBCs) based on their affinity to HIV antigens.19,20 For potential vaccine strategies, WNV-specific NAbs directly derived from humans could also reveal information about naturally targeted epitopes on the virus. NAbs, however, are only a part of the humoral immune response to WNV. For a better understanding of antibody-mediated mechanisms involved, such as disease outcome or persistence of antibodies and virus,21 our analysis of the humoral response should span from single cells to the level of the antibody repertoire.
In contrast to HIV or influenza, the prevalence of WNV is low and many cases are undiagnosed, making it challenging to assemble a large WNV cohort. Despite these obstacles, we effectively discovered and evaluated four novel NAbs against WNV directly from rare WNV-specific human B cells isolated from a set of recently infected and post-convalescent subjects. For this purpose, we combined microengraving,22–24 an integrated multiparameter single-cell analysis method and next-generation sequencing (NGS). We analyzed activated memory B cells (MBCs) and antibody-secreting cells (ASCs) from blood on a single-cell level and characterized WNV-specific clones within the circulating repertoire of B cells (Fig. 1). Despite a low frequency of WNV-specific B cells (mean <24 Ag+ events per 100000 PBMCs), these methods for integrated analysis allowed us to obtain NAbs and enabled analysis of the humoral response to WNV on a single-cell and repertoire level. The single-cell analysis revealed rare but persistent WNV-specific MBCs and ASCs in post-convalescent subjects. Furthermore, the results presented here indicate that the antibody response is independent of an asymptomatic vs. symptomatic disease outcome, as we have noted previously.25 Using the nucleotide coding sequences for WNV specific antibodies discovered from single cells, we also revealed expanded WNV specific clones in the repertoires of recently infected subjects through NGS and bioinformatic analysis.
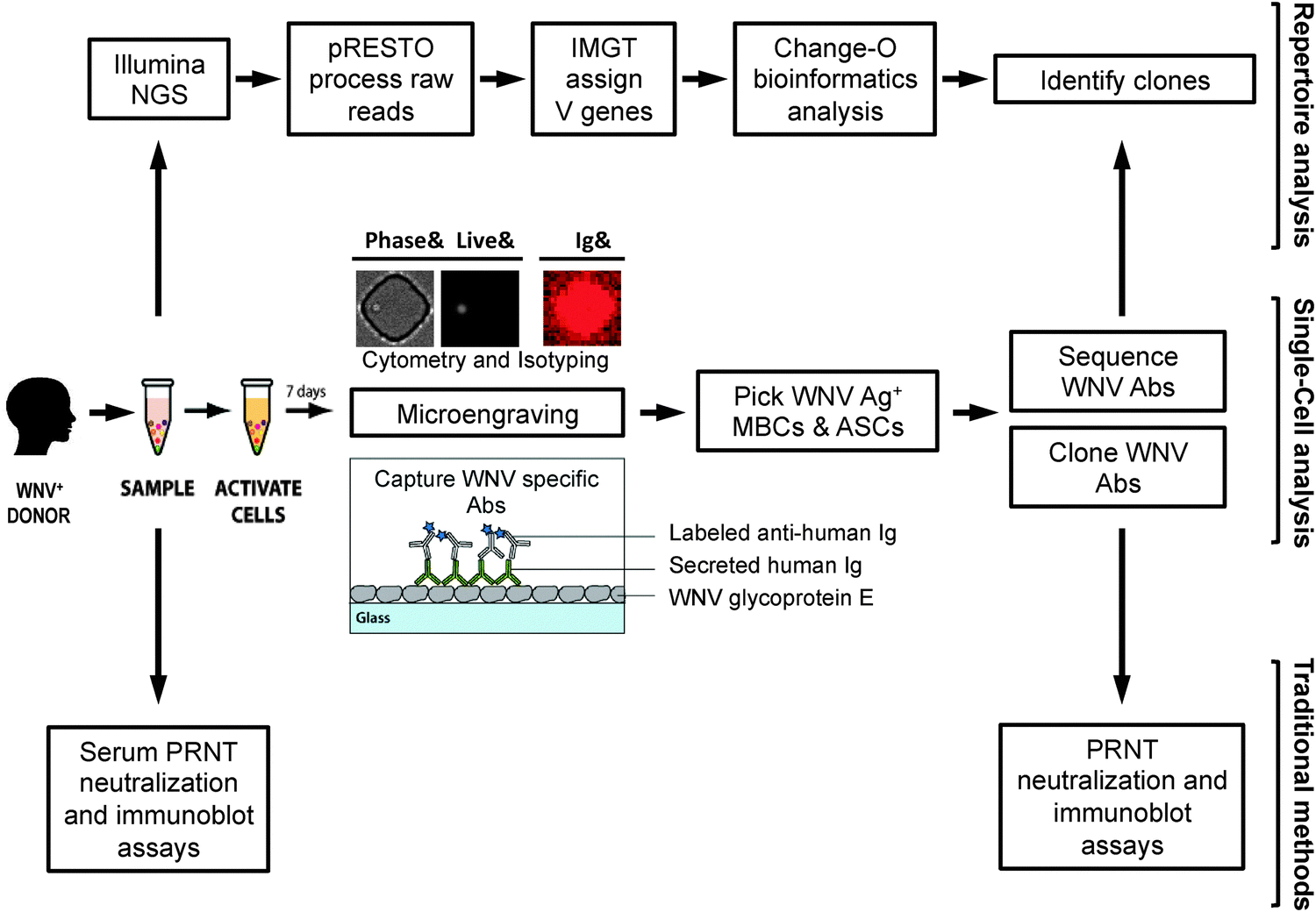 | ||
| Fig. 1 Analysis workflow. Blood is collected from individuals with a history of infection with WNV and PBMCs are isolated for further analysis. In parallel subject serum is analyzed using neutralization and immunoblot assays to determine the individual's overall response to WNV. PBMC samples are analyzed by microengraving, an integrative single-cell analysis process. WNV-specific antibodies are captured on a protein microarray from single antibody-secreting cells (ASCs) and single stimulated memory B cells (MBCs). Subsequently, individual WNV-specific MBCs and ASCs identified by microengraving are recovered and the sequences coding the variable region of the antibody heavy and light chains of their corresponding antibodies are obtained. Paired heavy and light chain coding sequences are used to clone the WNV-specific antibodies. The cloned antibodies are evaluated by neutralization and immunoblot assay. In parallel, the subject antibody repertoire is analyzed by next generation sequencing (NGS). The NGS data is processed by an integrated bioinformatics pipeline (pRESTO and Change-O) to identify clones of WNV-specific antibodies. | ||
Results
Identification of study subjects with high serum neutralizing antibody titers and profiling of their West Nile virus-specific humoral response
To examine developing humoral responses to WNV, we recruited a cohort of recently infected subjects during the 2012 WNV outbreak in Houston, Texas (Table 1). Blood samples were collected at two time points, 1.9 ± 0.8 months (range 0.8–2.9; n = 7) and 3.2 ± 1.2 months (range 2–4.7; n = 6, one subject lost to follow-up) after infection. Both time points correspond to samples of the recently infected period following exposure to WNV. To identify post-convalescent subjects with high serum neutralizing antibody titers, we screened our cohort of more than 160 WNV subjects whom we have monitored since 2002.26 We initially selected individuals with a history of exposure from asymptomatic and severe infection groups less than 2 years after infection with WNV (total n = 17, enrolled n = 4). We quantified anti-WNV antibodies by immunoblot and an antibody neutralization assay from four post-convalescent subjects; all four exhibited high serum neutralizing antibodies (1:80), with the highest neutralization score found (1:640) in an asymptomatic subject. In addition, we measured the serum antibody isotype concentrations among the recently infected subjects and found no deviation from standard values (IgM 40–230 mg dL−1, IgA 70–400 mg dL−1, and IgG 700–1600 mg dL−1),27 indicating a normal antibody response. Furthermore, immunophenotyping of surface markers28 showed no significant difference between the frequencies of naïve B cells, MBCs, and plasmablasts of recently infected and post-convalescent subjects in this study (Fig. 2). Notably we found significant differences comparing the recently infected subjects to a separate cohort of post-convalescent subjects from a previously published study (ImmPort study identification number SDY58).29 The current cohort of post-convalescent subjects with high serum neutralizing antibody titers showed slightly different frequencies of transitional B cells, plasmablasts and memory B cells, which may be due to the shorter convalescent period or the smaller sample size between the two groups of post-convalescent subjects. In total, we identified 11 subjects from our cohort of varying age, gender, disease outcome, and disease stage for our study.
| ID | Age at onset/blood draw | Disease status | Diagnosis | WNV E IgG immunoblot | PRNT serum titer |
|---|---|---|---|---|---|
| a Age of the donor at the time of blood draw. WNE, encephalitis; WNF, febrile illness; WNM, meningitis; Asymp, asymptomatic infection. PRNT indicates the dilution of serum needed to achieve 90% reduction of WNV growth in Vero cells. Immunoblot scale maximum is five +++++, corresponding to the reactivity with a known positive WNV patient serum. | |||||
| PC1 | 48a | Post-conv. | WNM | +++++ | 1:80 |
| PC2 | 44a | Post-conv. | Asymp | ++++ | 1:640 |
| PC3 | 26a | Post-conv. | WNF | +++ | 1:80 |
| PC4 | 21a | Post-conv. | WNF | +++++ | 1:80 |
| RI1 | 67 | Recent | WNE | ++ | 1:80 |
| RI2 | 30 | Recent | Asymp | +++++ | 1:40 |
| RI3 | 64 | Recent | WNE | +++++ | 1:40 |
| RI4 | 65 | Recent | Asymp | +++ | 1:40 |
| RI5 | 37 | Recent | WNE | +++++ | 1:40 |
| RI6 | 31 | Recent | WNF | +++++ | 1:80 |
| RI7 | 59 | Recent | Asymp | +++++ | 1:40 |
 | ||
| Fig. 2 B lymphocyte frequencies quantified by flow cytometry. (A) Plot of frequencies of transitional B cells (CD3−, CD19+, CD20+, CD24hi, CD38hi). A significant difference was found between the frequencies of transitional B cells in recently infected and post-convalescent individuals. A significant difference was found between the frequencies of transitional B cells in recently infected and post-convalescent individuals from a previous study (SDY58). (B) Plot of frequencies of naïve B cells (CD3−, CD19+, CD20+, CD27−). No significant differences were found between the frequencies of naïve B cells in recently infected and post-convalescent individuals. No significant differences were found between the frequencies of naïve B cells in recently infected and post-convalescent individuals from a previous study (SDY58). (C) Plot of frequencies of MBCs (CD19+, CD20+, CD27+). No significant differences were found between the frequencies of MBCs in recently infected and post-convalescent individuals. A significant difference was found between the frequencies of MBCs in recently infected and post-convalescent individuals from a previous study (SDY58). (D) Plot of frequencies of plasmablasts (CD3−, CD19+, CD20−, CD27hi, CD38hi). No significant differences were found between the frequencies of plasmablasts in recently infected and post-convalescent individuals. A significant difference was found between the frequencies of plasmablasts in recently infected and post-convalescent individuals from a previous study (SDY58). Multiple blood draws from our cohort of post-convalescent and recently infected individuals were included in the flow cytometry analysis. Frequencies are determined as percent of total B lymphocyte parent population. Statistical analysis was performed by Student's t-test. | ||
West Nile virus neutralizing antibodies identified by single-cell analysis
Using microengraving23,24 (Fig. 1), we recovered more than 90 WNV-specific single MBCs and ASCs (Fig. S1, ESI†). From those cells, we recovered 19 antibody-coding sequences where both heavy (V1, V3, V4 V region family) and light (V1, V2, V3 V region family) chains were present after PCR amplification. These 19 paired antibody-coding sequences were used for cloning and further evaluation (Fig. S2, ESI†). We found no significant difference (p > 0.05) in the amount of selection pressure between MBCs and ASCs by analyzing the mutations in the complementarity-determining (CDR) and framework (FWR) regions.30 Of the 19 recovered antibodies, twelve expressed with sufficient amounts of protein for further evaluation by immunoblot (Fig. S3, ESI†), plaque reduction neutralization assay (PRNT), and protein binding assays to determine their relative affinities (Table 2). As controls, we used serum from WNV positive (HS+) and WNV negative (HS−) subjects, and an antibody (2G12) with known affinity to HIV-1. Of the WNV-specific antibodies, five were from post-convalescent and seven from recently infected subjects. Three expressed antibodies showed reactivity to WNV envelope (E) protein by immunoblot and four showed efficient neutralization of WNV. Interestingly, three of the neutralizing antibodies did not show reactivity to WNV E protein by immunoblot, suggesting that they recognize conformation-dependent epitopes. Antibody MIT89 was positive both by immunoblot and PRNT. Furthermore, we observed a positive trend between the relative antibody affinities for WNV E protein and WNV neutralization. All neutralizing antibodies were recovered exclusively from post-convalescent subjects.| Antibody ID | Subject ID | Disease state | WNV E IgG Immunoblot | PRNT neutralizationa | Relative affinity (Ag/Ig) to WNV E protein |
|---|---|---|---|---|---|
| a Neutralization cut off value: 90%; 2G12 – HIV specific antibody control; HS – human serum controls. | |||||
| MIT 87 | PC3 | Post-conv. | + | +++ | |
| MIT 89 | PC3 | Post-conv. | + | + | +++ |
| MIT 90 | PC4 | Post-conv. | + | ++++ | |
| MIT 91 | PC1 | Post-conv. | +++ | ||
| MIT 95 | PC2 | Post-conv. | + | +++++ | |
| MIT 180 | RI7 | Recent infect. | +++++ | ||
| MIT 181 | RI7 | Recent infect. | ++++ | ||
| MIT 182 | RI7 | Recent infect. | + | ||
| MIT 183 | RI7 | Recent infect. | ++ | ||
| MIT 184 | RI7 | Recent infect. | + | ||
| MIT 185 | RI1 | Recent infect. | No data | ||
| MIT 187 | RI1 | Recent infect. | + | No data | |
| 2G12 | NA | HIV | NA | NA | |
| HS+ (1:20) |
+++ | + | |||
| HS− (1:20) |
|||||
West Nile virus-specific B cells in circulation are persistent and disease severity is independent of the humoral immune response
To determine whether elevated levels of WNV-specific B cells persist in post-convalescent subjects, we used the results obtained by single-cell analysis to determine the frequencies of WNV-specific MBCs and ASCs (Fig. 3) in circulation. The overall mean frequency and SEM across all subjects for the MBCs and the ASCs was 23.4 ± 9.7 and 8.4 ± 3.4 per 100000 PBMCs, respectively. We found no significant difference between recent and post-convalescent WNV-infected individuals in the frequencies of either PBMC-derived MBCs (p = 0.61) or ASCs (p = 0.64). Nevertheless, we observed a lower mean frequency (per 100000 PBMCs) in the MBC post-convalescent population (Δμ = 11). These results suggest that the frequencies of WNV-specific MBCs and ASCs decrease after the acute phase of infection, but still persist in circulation. These data provide a direct measure of WNV-specific B lymphocyte persistence in circulation of subjects with a history of WNV infection. These findings are consistent with our results of frequencies of B lymphocytes detected by flow cytometry of cells from recently infected and post-convalescent subjects (Fig. 2). These results are also consistent with previous reports of long-term persistence of WNV specific antibodies in serum.26,31,32
 | ||
| Fig. 3 Persistence and independence of disease severity of West Nile virus-specific B cell frequencies quantified by single-cell analysis. (A) Plot of frequencies of WNV-specific MBCs. No significant differences were found between the frequencies of WNV-specific MBCs in recently infected and post-convalescent individuals. (B) Plot of frequencies of WNV-specific MBCs in symptomatic and asymptomatic individuals, no significant differences were found between the two groups. (C) Plot of frequencies of WNV-specific ASCs by disease status (recent infected vs. post-convalescent). No significant differences were found between the frequencies of antigen specific ASCs in recently infected and post-convalescent individuals. (D) Plot of frequencies of WNV-specific ASCs by disease severity (symptomatic vs. asymptomatic). No significant differences were found between the frequencies of antigen specific ASCs in symptomatic vs. asymptomatic individuals. All WNV antigen specific single-cell events were determined by microengraving, and shown as events per 100K PBMCs. | ||
We tested whether there were significant differences between MBC or ASC frequencies according to disease outcome (severely infected vs. asymptomatic) (Fig. 3) or according to age (<40 vs. 40–60 vs. >60 years) (Fig. S4, ESI†). There were no significant differences (p = 0.92) between the frequencies of antigen-specific MBCs in symptomatic (24.2 ± 14.7, n = 7) and asymptomatic individuals (22.1 ± 10.8, n = 4), providing evidence that the frequency of B cells was not associated with the severity of WNV disease. We did, however, detect a significant increase in the frequencies of MBCs for subjects >60 years compared to the other age groups (p < 0.05). The results presented here suggest persistence of WNV-specific antibodies in post-convalescent subjects. Furthermore, the data indicate that the WNV-specific antibody response does not predict the clinical outcome of asymptomatic or symptomatic disease as noted previously.25,29
Distribution of isotypes corresponds to those in serum
We quantified the distribution of isotypes (IgM, IgA, IgG1, and IgG3) from activated MBCs and ASCs of recently infected subjects by microengraving (Fig. S5, ESI†). Overall, no significant differences were detected between severely infected and asymptomatic subjects among either MBCs or ASCs. We found elevated ratios of IgM to IgG1 in three of the four severely infected subjects (Fig. S5A, ESI†). As expected, we also found significantly higher levels of IgG1 compared to the other three isotypes among MBCs (Fig. S5B–D, ESI†). We observed slightly increased percentages of IgG3 among ASCs. Overall, the distributions of antibody isotypes we observed by microengraving were as expected based on the isotype distributions determined from serum found in other viral infections.33Next generation sequencing of B cell repertoires reveals clonal expansion in individuals recently infected with West Nile virus
To gain a better understanding of the overall variance among the B cells in circulation, we sequenced the B cell heavy chains from PBMCs, naïve, and memory B cell populations of seven recently infected subjects (Fig. S6, ESI†). The distributions of isotypes determined by NGS of PBMCs and MBCs showed no significant differences between asymptomatic and symptomatic recently infected subjects, confirming the results measured from individual cells by microengraving (Fig. S7, ESI†). To identify WNV-specific clones in the repertoire, the individual WNV-specific antibody sequences obtained by the single-cell analysis (“queries”) were combined with the NGS data and groups of clonally-related sequences were identified using Change-O as described in methods.34 This method identified three expanded clones that include one of the query sequences. To identify additional WNV-specific clones, the sequences from the NGS were plotted based on their level of mutation and similarity to each of the query antibody sequences (Fig. 4 and Fig. S9, ESI†). The three WNV-specific clones were clearly identifiable as outlier groups of sequences with higher similarity to the queried WNV-specific antibody sequences compared to other sequences with a similar level of mutation (Fig. 4B and Fig. S9, ESI†). Manual inspection of these plots identified two additional WNV-specific clones. The WNV-specific clones showed evidence of expansion, and sequences were found among both PBMCs and memory B cells with both IgA and IgG isotypes. Overall, five WNV-specific clones were identified through integration of the single-cell analysis and NGS data. The frequency of all expanded clones and degree of mutations in recently infected subjects is shown in Fig. S8 (ESI†). | ||
| Fig. 4 Trunk length analysis of West Nile virus-specific clones in the immunoglobin repertoire indicates unlikely previous exposure to the virus. (A) Size of WNV-specific clones found in recently infected subjects. (B) Representative plot of sequence similarity of heavy chain (same V and J segment) identified within the repertoire as a function of distance of the sequence from germline (x-axis) and the corresponding “query” WNV-specific sequence (y-axis). An outlier cluster representing a putative WNV-specific clone is circled. (C) Maximum parsimony lineage tree for one WNV-specific clone (MIT187). Each node represents a unique sequence, with size representing the number of duplicate reads observed. Edge lengths correspond to the number of mutations between sequences. Shading of the node represents the compartment in which the sequence was found. The nodes are labeled with the isotypes of the observed sequence (A: IgA, G: IgG, Q: WNV-specific query sequence from single-cell screening). (D) Plot of observed trunk lengths (number of mutations between germline and most recent common ancestor of clone). Emerging WNV-specific clones have significantly fewer trunk mutations (Student's t-test) compared to size-matched clones found in the same subjects, and to expanded clones (at least 0.05% of the repertoire) from subjects who received an influenza vaccination. | ||
Since WNV is a recent pathogen to North America, the WNV-specific clones we identified are likely the result of a primary immune response originating from naïve B cells, rather than a recall response from memory B cells. Therefore, we hypothesized that the WNV-specific clonal expansion we observed would have initiated from less affinity-matured cells than those that generate antigen-specific clones after a recall response to re-occurring infections or vaccinations to viruses such as influenza. To investigate this hypothesis, we approximated the sequence of the initiating B cell for each clone as the most recent common ancestor in a maximum parsimony lineage tree (Fig. 4C and Fig. S10, ESI†). Maximum parsimony lineage trees minimize the number of mutation events in each clone and infer sequences that may have existed between observed antibodies and the germline sequence. The trunk length of the lineage tree (i.e., the number of mutations in the most recent common ancestor compared with the germline sequence) approximates the maturation state of the initiating B cell for each clone. In comparison to other similarly sized B cell clones in the recently infected WNV subjects, the trunks in WNV-specific clones had significantly (p < 0.05) fewer mutations (Fig. 4D). It is possible, however, that some of the similarly-sized clones in this cohort are WNV-specific, which could confound the comparison. To address this issue, we also compared the trunk lengths in WNV-specific clones with expanded (at least 0.05% of the repertoire) clonal lineages from three responses following influenza vaccination, obtained from publicly available data.35 We found that the WNV-specific clones once again had trunks that were significantly (p < 0.05) less mutated than those found from the influenza response. The close mutational distance of sequences that give rise to WNV-specific B cell clones to their respective germlines supports our hypothesis that these subjects have not previously been exposed to WNV and have experienced primary affinity maturation rather than a recall response. In the case of two clones (MIT187 and MIT180) (Fig. S10, ESI†), the query sequence was not a terminal leaf; other sequences with additional mutations were observed in the lineage tree. Antibodies both more and less mutated than the query sequences could also show high affinity and neutralizing activity to WNV. This general approach of using specific antibodies found by microengraving as query sequences to reveal similar antibodies at the repertoire level could have wide applications for a larger scale search for therapeutic neutralizing antibodies specific to many other diseases.
Discussion
In this study, we analyzed PBMCs from individuals with recent or post-convalescent WNV infections. We effectively identified WNV-specific antibodies by single-cell analysis. We next cloned and expressed the antibodies identified by single-cell analysis and performed PRNT to find WNV-specific NAbs. Four of the NAbs identified also showed high affinity to WNV E protein as determined by protein microarray. In contrast, only one NAb was also positive in the immunoblot assay. These results may indicate that three of the discovered NAbs recognize conformation dependent epitopes on the WNV E protein that could have been denatured during the immunoblot assay.36 Similarly, for antibodies to the related Flavivirus family member dengue virus, a quaternary epitope was required to neutralize the virus,9 indicating that denaturing of the higher order antibody protein structure could result in a loss of function. We also observed that neutralizing activity was positively correlated with increased antibody affinity to WNV E protein. This trend suggests that the development of NAbs in WNV infection requires a high degree of affinity maturation. In support of this hypothesis, we found NAbs exclusively in the post-convalescent subjects, which could indicate that the detected antibodies developed later during the course of infection, and that they require a prolonged exposure to antigen to promote affinity maturation. Chronic or repeated exposure to viral antigens such as HIV-1 can elicit affinity maturation beyond the degree observed here.19 In contrast to previously identified NAbs derived using murine phage and yeast display libraries,12–16 the antibodies here were directly derived from human B cells obtained from individuals previously infected with WNV. These antibodies therefore reflect the natural selection and maturation of WNV-specific NAbs in humans. The challenge here exists in identifying the rare B cells producing NAbs. For instance, in infections with the dengue virus, a related flavivirus, only a small fraction of the antibodies expressed strongly neutralized the virus8 although recent studies have identified an epitope that may be relevant for effective neutralization.37 This outcome highlights the importance of utilizing single-cell analysis methods to identify rare B cells of interest. Future studies should further elucidate the epitopes and mechanisms of action of the NAbs identified here. In addition, developing and validating a small animal model and testing these antibodies could provide evidence for potential therapeutic use.This study marks the first time that the frequencies of circulating WNV-specific MBCs and ASCs were determined. We observed no significant difference between the frequencies in post-convalescent subjects compared to recently infected subjects, indicating potential long-term persistence of antigen specific MBCs and ASCs in WNV infection (Fig. 3). These results have limited power due to the relatively small sample size of four post-convalescent subjects and seven recently infected subjects. However, they are supported by previous studies where WNV-specific serum antibodies were detected in subjects up to 8 years post-infection.26,31,32,36,38 Persistence of serum antibodies specific to other viral infections and vaccines (e.g. dengue, influenza, and measles) have also been previously observed8,39–41 and are well documented in the case of the related hepacivirus hepatitis C.42,43 Repeated acute exposure to WNV is unlikely due to the low prevalence of this flavivirus.2,4 Chronic exposure to the viral envelope from an unknown reservoir has been suggested by recent studies.21,26
Though previous longitudinal studies of the immune response to influenza vaccination indicate that clonal expansion can be observed in blood seven days post-vaccination,35,44,45 the recently infected subjects in this study had been infected with WNV at least four weeks prior to the first blood draw. The expansion of WNV-specific clones in comparison with post-convalescent subjects exposed to WNV years prior to sampling implies that the antibody-mediated response may persist longer than in the case of other infections or vaccines. Chronic infections have been shown to lead to expanded B cell clones in the case of cytomegalovirus (CMV) or clones with higher levels of mutations in the case of Epstein-Barr virus (EBV).46
To date, it has not been thoroughly described how the nature and role of the humoral response to WNV affects clinical manifestation of the disease, although we have recently identified a transcriptional signature of susceptibility to severe disease.29 The results presented here (Fig. 3) support our previous results that the levels of antibodies to WNV were not significantly different between asymptomatic and severely infected subjects.25 These findings show that although WNV infection induces a humoral response, the level of the anti-viral antibody titers is not a distinguishing feature of disease severity. This observation has also been noted for antibody levels in humans infected with a related hepacivirus, hepatitis C virus, for which antibody responses also fail to predict the clearance of infecting virus.42,43 Our data suggest that the pathways needed to mount a successful antibody response remain intact and that the mechanisms leading to severe disease are independent of these pathways. Further supporting this hypothesis, all study subjects showed serum isotype (IgM, IgA, IgG) titers in the normal range, indicating that B cell function and class switching were intact. Interestingly, we found increased levels of activated IgA MBCs in severely infected subjects. Class switching following the progression of WNV infection has been described as an initial increase in IgM and IgA, followed by a decrease in IgM and an increase in IgG.26,47 We postulate that our observed increased ratio of IgM/IgG1 in severely infected subjects could therefore indicate a delayed immune response to WNV infection and that the kinetics of this lag could trend with clinical severity.
In summary, we effectively identified novel human-derived NAbs and analyzed the humoral response to WNV infection by integrating single-cell analysis, repertoire-level next generation sequencing, and conventional methods. Detecting persistent antigen-specific MBCs and ASCs in the periphery of post-convalescent subjects can reveal the origin of these antibodies and potentially provide clues about the persistence of WNV in the body. We believe that WNV provides an excellent model to examine primary naïve responses of human B cells, given the disease's well-defined clinical history for the onset of infection, the low occurrence of infection by this or others flaviviruses (e.g., dengue) in North America, which allows the characterization of naïve responses to a recently introduced pathogen with unlikely re-exposure. Our results comparing the distances of inferred clonal founder cells from their respective germlines support the hypothesis that WNV-specific antibodies develop from naïve B cells during a primary infection, rather than a recall response from an affinity matured memory B cells. These results highlight the importance of analyzing the immune response to infection with integrated single-cell data, serum analysis, and whole repertoire analysis. These methods to analyze the humoral response can also be applied to other infectious disease research and in vaccine trials.
Material and methods
Recruitment of human subjects and sample validation
Human subjects exhibiting various WNV infection phenotypes were recruited from a well-characterized cohort of volunteers. Blood was obtained with written informed consent under the guidelines of the Human Investigations Committee of Baylor College of Medicine, which approved the study. Investigation of coded samples was approved by Yale University and Massachusetts Institute of Technology in compliance with HIPAA guidelines. The diagnosis of WNV infection was determined following CDC guidelines48 and subjects were stratified by CDC definitions to a severe neuroinvasive phenotype, mild fever-only phenotype, or asymptomatic infection as described previously.48 Infection was validated by qualitative rapid nucleic acid test at the blood bank (cobas®TaqScreen West Nile virus Test, Roche Molecular Systems, Pleasanton, CA), positive immunoblot as described previously,25 or IgM ELISAs. Subjects (n = 11) were 45.5% female and 90.9% white and included asymptomatic, mild, and severe subjects. Subjects from the asymptomatic, mild, and severely infected groups were not statistically different for age, gender, or race in this study.Preparation of blood cells, B cell sorting, and immunophenotyping
Blood samples were collected in CPT tubes (Becton Dickinson and Co., Franklin Lakes, NJ) according to the manufacturer's instructions (including centrifugation within 2 hours of collection) and were processed the following day.Serum was collected separately and stored at −80 °C until assay.25 Peripheral blood mononuclear cells (PBMCs) were sorted fresh to obtain B cell subsets.
For B cell subset sorting, fresh PBMCs were enriched using anti-CD20 magnetic beads (Stemcell Technologies Inc., Vancouver, BC, Canada). PBMCs were stained on ice with anti-human antibodies (Becton Dickinson and Biolegend) and directly coupled to either CD19-Pacific Blue, CD21-allophycocyanin (APC), IgM-fluorescein-isothiocyanate (FITC), CD10-phycoerythrin-Cy7 (PE-Cy7), CD27 (PuCL45.5), CD138-phycoerythrin (PE), to distinguish among individual B cell subpopulations.49,50 B cells were sorted on a FACSAria (Becton Dickinson). Cells were pelleted and frozen in lysate buffer (Ambion mirVana™ miRNA Isolation Kit, Life Technologies, Grand Island, NY) and RNA was isolated according to the manufacturer's instructions. For multiparameter robotic immunophenotyping, frozen PBMCs were thawed in one batch, washed, dispensed into wells, and incubated with antibody panels using a custom programmed BioMek robotic platform prior to FACS analysis using the LSR Fortessa (BD BioSciences) as described.29 Antibody markers included 8 panels of which 5 were defined by the Human Immunophenotyping Consortium (HIPC; http://www.immuneprofiling.org/hipc/page/show).28 The B lymphocyte panel consists of CD24, CD19, CD27, CD38, CD20, CD3 and IgD.
WNV immunoblot and neutralization assays
Antibody titer from human sera was quantified by immunoblot against WNV recombinant envelope (E) protein and by quantitative virus-specific plaque-reduction neutralization test (PRNT) assay as described.51 WNV studies were conducted in a Biosafety Level 3 facility, licensed by the State of Connecticut and Yale University. Virulent WNV (CT-2741) provided by Dr John Anderson, Connecticut Agricultural Experiment Station, New Haven, CT52 was passaged once in Vero cells (ATCC CCL-81).Single-cell analysis by microengraving
Actively secreting and memory B cell populations in PBMC samples obtained from subjects were analyzed by microengraving22,23 to determine the distributions of secreted antibody isotypes and the frequencies of WNV E specific antibodies. There was a range of cells from our donors with a median of 149090 antibody secreting cells (range 51888–375028) and a median of 145601 memory B cells (range 56862–385436). Frozen PBMCs were thawed and maintained in complete RPMI media containing IL-6 (20 ng mL−1, Peprotech) and Chk2 inhibitor II (2 μg mL−1, Sigma-Aldrich). After resting or stimulation, cells were stained for viability (Calcein violet AM) and for expression of CD19 (Brilliant Violet® 605), CD20 (Alexa Fluor® 488), CD27 (PerCP-efluor® 710), CD38 (PerCP-efluor® 710) and CD138 (APC), loaded into nanowells, then imaged on an epifluorescence microscope. Separate microarrays were generated by sealing nanowells with capture slides functionalized with donkey anti-human Ig (25 μg mL−1, Jackson Immunoresearch) or WNV E protein (50 μg mL−1, Aviva Systems Biology). We used a flu specific antibody secreting cell line39 to determine non-specific binding events for subsequent background frequency subtraction.23 Isotype-specific information was obtained by staining microarrays with a panel of anti-human IgG1, IgA, IgG3 and IgM detection antibodies (BD Biosciences and Invitrogen). Microarrays from WNV E-coated slides were probed with mouse anti-human Igκ and Igλ (BD Biosciences) detection antibodies. Microarrays were scanned on a GenePix® 4200AL Autoloader (Molecular Devices), and median fluorescence intensities (MFIs) from bound species were extracted with GenePix® Pro 6.0 software. Isotype-specific data was analyzed as previously described.23 Antibodies were considered WNV-E specific if microarray elements were from wells with viable, CD19+/CD20+/− cells that also satisfied the following criteria: % saturation ≤2, signal-to-noise ratio ≥1, ≥40% of pixels above background +1 standard deviation, CV ≤ 70 and background corrected MFI ≥ 1500. A fraction of cells from such wells were recovered with an automated micromanipulator (AVISO CellCellector™, ALS GmbH), and heavy and light chain variable genes were amplified by single-cell RT-PCR.23 In short, the cells were recovered using a glass capillary mounted on an automated micromanipulator and a microscope. The glass capillary was placed over the nanowell to recover the desired MBC or ASC by applying a vacuum. The cell was subsequently released into a well of a 96 well plate containing water. To subsequently recover antibody heavy and light chain variable genes, a nested PCR was performed using the primers and protocol previously published by Stollar et al.53 These procedures have been described in more detail by Ogunniyi et al.24
Expression and validation of WNV specific antibodies
The protocols to clone, express and validate antigen specific antibodies has been previously described.23,54 In brief, paired heavy and light chain coding sequences were cloned into vectors for expression as human IgG1. We utilized human embryonic kidney (HEK) 293 T cells (ATCC) to transiently express the cloned antibodies. Antibodies in culture supernatants were evaluated for binding on a custom protein microarray consisting of WNV E protein. Microarrays were scanned on GenePix ® 4200AL and analyzed with GenePix ® Pro 6.0. Subsequently, the expressed WNV specific antibodies were evaluated by immunoblot and PRNT assay as described above.Library preparation and next generation sequencing
To evaluate B cell subsets from recently infected subjects listed, fresh PBMCs were sorted to isolate plasma cells, naïve and memory B cells, and RNA was isolated for sequencing. The number of sorted B cells from each subject was a median of 536792 (range 51232–2108051). UID barcoded NGS libraries55 were prepared by AbVitro, Inc from 250 ng of the extracted RNA of both sorted B cells as well as unsorted PBMCs. NGS libraries were subsequently sequenced at the MIT BioMicrocenter on the Illumina Miseq platform using the Illumina 2 × 300 bp sequencing kit according to the manufacturer specifications. PBMCs from post-convalescent subjects were directly processed without sorting, and libraries were sequenced using the Illumina 2 × 150 bp sequencing kit.
High-throughput antibody repertoire sequence analysis
Preprocessing was carried out using pRESTO version 0.4 (http://clip.med.yale.edu/pRESTO)55 and involved removing primer sequences, filtering based on sequence quality and annotating sequences with sample information. Following preprocessing, V(D)J germline segments were determined using IMGT/HighV-QUEST.56 Functional V(D)J sequences of each subject were combined with WNV-specific sequences and then Change-O34 was used to divide the sequences into clonally-related groups by a two-step procedure: (1) sequences were partitioned based on common V gene, J gene, and junction region length, and (2) within these larger groups, sequences differing from one another by a distance of less than 0.01 were defined as clones. Distance was measured as the number of point mutations weighted by a nucleotide substitution probability previously described.57 These data will be available through controlled access from the database of Genotypes and Phenotypes (dbGaP).Clones for antibodies MIT185 and MIT186 as portrayed in Fig. S8 (ESI†) were identified by manual inspection of Fig. S8 (ESI†). To generate Fig. S8 (ESI†), for each WNV-specific query sequence, the NGS data was filtered for sequences with the same V and J germline gene segment. For each of these sequences, similarity to the query (y-axis) is the Needleman–Wunsch distance on the entire sequence without end-gap penalties divided by the length of the alignment. The distance from germline (x-axis) is the Hamming distance (with ambiguous characters not counting as a mismatch) between the sequence and its germline divided by the number of non-N nucleotides in the sequence.
The smallest observed WNV-specific clone composed 0.01% of the repertoire. Selection pressures were quantified using BASELINe30,58 with the S5F somatic hypermutation targeting model.57
Generation and analysis of lineage trees
Lineage trees were constructed for each clonal group by removing indel positions and then using maximum parsimony with the dnapars application of PHYLIP.59 This was followed by recursively replacing inferred ancestors in each tree with descendants having a Hamming distance of zero from their inferred parent. The trunk length of a clone is defined to be the branch length from the germline root node to the most recent common ancestor. Comparisons were made to Ig repertoire sequencing of three individuals who received an influenza vaccination and had blood drawn at several time-points (8 days, 2 days, and 1 hour pre-vaccination; 1 hour, 1 day, 3 days, 7 days, 2 weeks, 3 weeks, and 4 weeks post-vaccination). Clones were defined in the same way as described above. For the vaccination data, trunk lengths were only measured for expanded clones, or clones that at some time-point compose at least 0.05% of the Ig repertoire. Comparisons between trunk lengths were made using a two-tailed Student's t-test.Author contributions
A.O.O., K.T., N.T.G., J.C.L, S.H.K., R.R.M. designed research; A.O.O., K.T., R.M.Z, F.Q., Y.Y., X.W., J.N.H.S., F.V. performed research; M.G., K.O.M recruited subjects; S.H.K., F.V., A.W.B., C.R.C., R.C., G.M.C. contributed new reagents or analytic tools; N.T.G., K.T., A.O.O., A.K. analyzed data; and K.T., N.T.G., R.R.M., S.H.K., J.C.L. wrote the paper.Acknowledgements
The authors are grateful to Stephanie Argraves, Shu Chen, Barbara Siconolfi, and Sui Tsang for expert research support, and to the Human Immunology Project Consortium (HIPC) for insightful discussions. This work was supported in part by the National Institutes of Health (U19AI089992, AI091816, R01AI104739, 1F32AI112359-01, T15LM07056), a pilot project from the HIPC (http://www.immuneprofiling.org), and the Gillson Longenbaugh Foundation. This work was also supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, and we thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the BioMicroCenter. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy And Infectious Diseases or the National Institutes of Health. J.C.L. is a Camille Dreyfus Teacher-Scholar.References
- M. S. Suthar, M. S. Diamond and M. Gale Jr, Nat. Rev. Microbiol., 2013, 11, 115–128 CrossRef CAS PubMed.
- T. M. Colpitts, M. J. Conway, R. R. Montgomery and E. Fikrig, Clin. Microbiol. Rev., 2012, 25, 635–648 CrossRef CAS PubMed.
- R. R. Montgomery and K. O. Murray, Expert Rev. Anti-Infect. Ther., 2015, 13, 317–325 CrossRef CAS PubMed.
- L. R. Petersen, P. J. Carson, B. J. Biggerstaff, B. Custer, S. M. Borchardt and M. P. Busch, Epidemiol. Infect., 2013, 141, 591–595 CrossRef CAS PubMed.
- S. K. Austin and K. A. Dowd, Viruses, 2014, 6, 1015–1036 CrossRef PubMed.
- W. A. Marasco and J. Sui, Nat. Biotechnol., 2007, 25, 1421–1434 CrossRef CAS PubMed.
- F. Klein, H. Mouquet, P. Dosenovic, J. F. Scheid, L. Scharf and M. C. Nussenzweig, Science, 2013, 341, 1199–1204 CrossRef CAS PubMed.
- R. de Alwis and A. M. de Silva, Dengue, Springer, 2014, pp. 27–39 Search PubMed.
- R. de Alwis, S. A. Smith, N. P. Olivarez, W. B. Messer, J. P. Huynh, W. M. Wahala, L. J. White, M. S. Diamond, R. S. Baric and J. E. Crowe, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7439–7444 CrossRef CAS PubMed.
- K. A. Dowd and T. C. Pierson, Virology, 2011, 411, 306–315 CrossRef CAS PubMed.
- Y. P. de Jong, M. Dorner, M. C. Mommersteeg, J. W. Xiao, A. B. Balazs, J. B. Robbins, B. Y. Winer, S. Gerges, K. Vega and R. N. Labitt, Sci. Transl. Med., 2014, 6, 254ra129 CrossRef PubMed.
- T. Oliphant, M. Engle, G. E. Nybakken, C. Doane, S. Johnson, L. Huang, S. Gorlatov, E. Mehlhop, A. Marri and K. M. Chung, Nat. Med., 2005, 11, 522–530 CrossRef CAS PubMed.
- T. C. Pierson, Q. Xu, S. Nelson, T. Oliphant, G. E. Nybakken, D. H. Fremont and M. S. Diamond, Cell Host Microbe, 2007, 1, 135–145 CAS.
- M. Throsby, C. Geuijen, J. Goudsmit, A. Q. Bakker, J. Korimbocus, R. A. Kramer, M. Clijsters-van der Horst, M. de Jong, M. Jongeneelen and S. Thijsse, J. Virol., 2006, 80, 6982–6992 CrossRef CAS PubMed.
- M. R. Vogt, B. Moesker, J. Goudsmit, M. Jongeneelen, S. K. Austin, T. Oliphant, S. Nelson, T. C. Pierson, J. Wilschut and M. Throsby, J. Virol., 2009, 83, 6494–6507 CrossRef CAS PubMed.
- L. H. Gould, J. Sui, H. Foellmer, T. Oliphant, T. Wang, M. Ledizet, A. Murakami, K. Noonan, C. Lambeth and K. Kar, J. Virol., 2005, 79, 14606–14613 CrossRef CAS PubMed.
- T. Oliphant, G. E. Nybakken, S. K. Austin, Q. Xu, J. Bramson, M. Loeb, M. Throsby, D. H. Fremont, T. C. Pierson and M. S. Diamond, J. Virol., 2007, 81, 11828–11839 CrossRef CAS PubMed.
- C. M. Hammers and J. R. Stanley, J. Invest. Dermatol., 2014, 134, e17 CrossRef CAS PubMed.
- J. F. Scheid, H. Mouquet, N. Feldhahn, M. S. Seaman, K. Velinzon, J. Pietzsch, R. G. Ott, R. M. Anthony, H. Zebroski and A. Hurley, Nature, 2009, 458, 636–640 CrossRef CAS PubMed.
- J. F. Scheid, H. Mouquet, B. Ueberheide, R. Diskin, F. Klein, T. Y. K. Oliveira, J. Pietzsch, D. Fenyo, A. Abadir and K. Velinzon, Science, 2011, 333, 1633–1637 CrossRef CAS PubMed.
- K. Murray, C. Walker, E. Herrington, J. A. Lewis, J. McCormick, D. W. Beasley, R. B. Tesh and S. Fisher-Hoch, J. Infect. Dis., 2010, 201, 2–4 CrossRef CAS PubMed.
- J. C. Love, J. L. Ronan, G. M. Grotenbreg, A. G. van der Veen and H. L. Ploegh, Nat. Biotechnol., 2006, 24, 703–707 CrossRef CAS PubMed.
- A. O. Ogunniyi, B. A. Thomas, T. J. Politano, N. Varadarajan, E. Landais, P. Poignard, B. D. Walker, D. S. Kwon and J. C. Love, Vaccine, 2014, 32, 2866–2873 CrossRef CAS PubMed.
- A. O. Ogunniyi, C. M. Story, E. Papa, E. Guillen and J. C. Love, Nat. Protoc., 2009, 4, 767–782 CrossRef CAS PubMed.
- F. Qian, J. Thakar, X. Yuan, M. Nolan, K. O. Murray, W. T. Lee, S. J. Wong, H. Meng, E. Fikrig and S. H. Kleinstein, Viral Immunol., 2014, 27, 39–47 CrossRef CAS PubMed.
- K. O. Murray, M. N. Garcia, C. Yan and R. Gorchakov, Am. J. Trop. Med. Hyg., 2013, 89, 996–1000 CrossRef PubMed.
- F. Dati, G. Schumann, L. Thomas, F. Aguzzi, S. Baudner, J. Bienvenu, O. Blaabjerg, S. Blirup-Jensen, A. Carlström and P. Hyltoft-Petersen, Eur. J. Clin. Chem. Clin. Biochem., 1996, 34, 517–520 CAS.
- H. T. Maecker, J. P. McCoy and R. Nussenblatt, Nat. Rev. Immunol., 2012, 12, 191–200 CAS.
- F. Qian, G. Goel, H. Meng, X. Wang, F. You, L. Devine, K. Raddassi, M. N. Garcia, K. O. Murray and C. R. Bolen, Clin. Vaccine Immunol., 2015, 22, 6–16 CrossRef PubMed.
- G. Yaari, M. Uduman and S. H. Kleinstein, Nucleic Acids Res., 2012, 40, e134 CrossRef CAS PubMed.
- A. Papa, K. Danis, A. Athanasiadou, M. Delianidou and T. Panagiotopoulos, J. Med. Virol., 2011, 83, 1857–1860 CrossRef PubMed.
- J. T. Roehrig, D. Nash, B. Maldin, A. Labowitz, D. A. Martin, R. S. Lanciotti and G. L. Campbell, Emerging Infect. Dis., 2003, 9, 376 CrossRef CAS PubMed.
- A. Ferrante, L. J. Beard and R. G. Feldman, Pediatr. Infect. Dis. J., 1990, 9, 516–524 CrossRef.
- N. T. Gupta, J. A. Vander Heiden, M. Uduman, D. Gadala-Maria, G. Yaari and S. H. Kleinstein, Bioinformatics, 2015, 20, 3356–3358 CrossRef PubMed.
- U. Laserson, F. Vigneault, D. Gadala-Maria, G. Yaari, M. Uduman, J. A. Vander Heiden, W. Kelton, S. Taek Jung, Y. Liu, J. Laserson, R. Chari, J.-H. Lee, I. Bachelet, B. Hickey, E. Lieberman-Aiden, B. Hanczaruk, B. B. Simen, M. Egholm, D. Koller, G. Georgiou, S. H. Kleinstein and G. M. Church, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4928–4933 CrossRef CAS PubMed.
- S. Chabierski, L. Barzon, A. Papa, M. Niedrig, J. L. Bramson, J. M. Richner, G. Palù, M. S. Diamond and S. Ulbert, BMC Infect. Dis., 2014, 14, 246 CrossRef PubMed.
- W. Dejnirattisai, W. Wongwiwat, S. Supasa, X. Zhang, X. Dai, A. Rouvinsky, A. Jumnainsong, C. Edwards, N. T. H. Quyen and T. Duangchinda, Nat. Immunol., 2015, 16, 170–177 CrossRef CAS PubMed.
- P. J. Carson, H. E. Prince, B. J. Biggerstaff, R. Lanciotti, L. H. Tobler and M. Busch, J. Clin. Microbiol., 2014, 52, 57–60 CrossRef PubMed.
- X. Yu, T. Tsibane, P. A. McGraw, F. S. House, C. J. Keefer, M. D. Hicar, T. M. Tumpey, C. Pappas, L. A. Perrone and O. Martinez, Nature, 2008, 455, 532–536 CrossRef CAS PubMed.
- I. J. Amanna, N. E. Carlson and M. K. Slifka, N. Engl. J. Med., 2007, 357, 1903–1915 CrossRef CAS PubMed.
- S. A. Smith, Y. Zhou, N. P. Olivarez, A. H. Broadwater, A. M. de Silva and J. E. Crowe, J. Virol., 2012, 86, 2665–2675 CrossRef CAS PubMed.
- G. L. Armstrong, A. Wasley, E. P. Simard, G. M. McQuillan, W. L. Kuhnert and M. J. Alter, Ann. Intern. Med., 2006, 144, 705–714 CrossRef PubMed.
- F. Qian, C. R. Bolen, C. Jing, X. Wang, W. Zheng, H. Zhao, E. Fikrig, R. D. Bruce, S. H. Kleinstein and R. R. Montgomery, Clin. Vaccine Immunol., 2013, 20, 146–155 CrossRef CAS PubMed.
- N. Jiang, J. He, J. A. Weinstein, L. Penland, S. Sasaki, X.-S. He, C. L. Dekker, N.-Y. Zheng, M. Huang and M. Sullivan, Sci. Transl. Med., 2013, 5, 171ra19 Search PubMed.
- C. Vollmers, R. V. Sit, J. A. Weinstein, C. L. Dekker and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13463–13468 CrossRef CAS PubMed.
- C. Wang, Y. Liu, L. T. Xu, K. J. Jackson, K. M. Roskin, T. D. Pham, J. Laserson, E. L. Marshall, K. Seo and J.-Y. Lee, J. Immunol., 2014, 192, 603–611 CrossRef CAS PubMed.
- H. E. Prince, L. H. Tobler, M. Lapé-Nixon, G. A. Foster, S. L. Stramer and M. P. Busch, J. Clin. Microbiol., 2005, 43, 4316–4320 CrossRef CAS PubMed.
- M. S. Nolan, J. Schuermann and K. O. Murray, Emerging Infect. Dis., 2013, 19, 137 CrossRef PubMed.
- H. Wardemann, S. Yurasov, A. Schaefer, J. W. Young, E. Meffre and M. C. Nussenzweig, Science, 2003, 301, 1374–1377 CrossRef CAS PubMed.
- T. Tiller, M. Tsuiji, S. Yurasov, K. Velinzon, M. C. Nussenzweig and H. Wardemann, Immunity, 2007, 26, 205–213 CrossRef CAS PubMed.
- K.-F. Kong, K. Delroux, X. Wang, F. Qian, A. Arjona, S. E. Malawista, E. Fikrig and R. R. Montgomery, J. Virol., 2008, 82, 7613–7623 CrossRef CAS PubMed.
- J. F. Anderson, T. G. Andreadis, C. R. Vossbrinck, S. Tirrell, E. M. Wakem, R. A. French, A. E. Garmendia and H. J. Van Kruiningen, Science, 1999, 286, 2331–2333 CrossRef CAS PubMed.
- X. Wang and B. D. Stollar, J. Immunol. Methods, 2000, 244, 217–225 CrossRef CAS PubMed.
- T. Tiller, E. Meffre, S. Yurasov, M. Tsuiji, M. C. Nussenzweig and H. Wardemann, J. Immunol. Methods, 2008, 329, 112–124 CrossRef CAS PubMed.
- J. A. Vander Heiden, G. Yaari, M. Uduman, J. N. Stern, K. C. O'Connor, D. A. Hafler, F. Vigneault and S. H. Kleinstein, Bioinformatics, 2014, 13, 1930–1932 CrossRef PubMed.
- E. Alamyar, P. Duroux, M.-P. Lefranc and V. Giudicelli, Methods Mol. Biol., 2012, 882, 569–604 CAS.
- G. Yaari, J. A. Vander Heiden, M. Uduman, D. Gadala-Maria, N. Gupta, J. N. H. Stern, K. C. O'Connor, D. A. Hafler, U. Laserson, F. Vigneault and S. H. Kleinstein, Front. Immunol., 2013, 4, 358 CrossRef PubMed.
- M. Uduman, G. Yaari, U. Hershberg, J. A. Stern, M. J. Shlomchik and S. H. Kleinstein, Nucleic Acids Res., 2011, 39, W499–W504 CrossRef CAS PubMed.
- J. Felsenstein, Department of Genome Sciences University Washington, 2005.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ib00169b |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2015 |