
Locally controlling mesenchymal stem cell morphogenesis by 3D PDGF-BB gradients towards the establishment of an in vitro perivascular niche†
P. S.
Lienemann‡
ab,
Y. R.
Devaud‡
ab,
R.
Reuten
c,
B. R.
Simona
d,
M.
Karlsson
e,
W.
Weber
e,
M.
Koch
c,
M. P.
Lutolf
b,
V.
Milleret
a and
M.
Ehrbar
*a
aLaboratory for Cell and Tissue Engineering, Department of Obstetrics, University Hospital Zurich, Schmelzbergstr. 12, 8091 Zurich, Switzerland. E-mail: martin.ehrbar@usz.ch
bInstitute of Bioengineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), Station 15, Bld AI 1109, 1015 Lausanne, Switzerland
cInstitute for Dental Research and Oral Musculoskeletal Biology, Center for Biochemistry, University of Cologne, 50931 Cologne, Germany
dLaboratory of Biosensors and Bioelectronics, Institute for Biomedical Engineering, University and ETH Zurich, F76 Gloriastr. 35, 8092 Zurich, Switzerland
eFaculty of Biology and BIOSS Centre for Biological Signalling Studies, University of Freiburg, Schänzlestr. 18, 79104 Freiburg, Germany
First published on 28th October 2014
Abstract
The perivascular niche is a complex microenvironment containing mesenchymal stem cells (MSCs), among other perivascular cells, as well as temporally organized biochemical and biophysical gradients. Due to a lack of conclusive phenotypic markers, MSCs' identity, heterogeneity and function within their native niche remain poorly understood. The in vitro reconstruction of an artificial three-dimensional (3D) perivascular niche would offer a powerful alternative to study MSC behavior under more defined conditions. To this end, we here present a poly(ethylene glycol)-based in vitro model that begins to mimic the spatiotemporally controlled presentation of biological cues within the in vivo perivascular niche, namely a stably localized platelet-derived growth factor B (PDGF-BB) gradient. We show that 3D-encapsulated MSCs respond to soluble PDGF-BB by proliferation, spreading, and migration in a dose-dependent manner. In contrast, the exposure of MSCs to 3D matrix-tethered PDGF-BB gradients resulted in locally restricted morphogenetic responses, much as would be expected in a native perivascular niche. Thus, the herein presented artificial perivascular niche model provides an important first step towards modeling the role of MSCs during tissue homeostasis and regeneration.
Insight, innovation, integrationThe in vitro reconstruction of natural microenvironments represents a promising approach to investigate complex biological phenomena under highly reproducible and defined conditions. Therefore, technologies are sought after that allow for closely mimicking key determinants of cell behavior in the natural situation. Such a key determinant is the spatial distribution of growth factors. Here, we report on the combination of materials engineering and a microfluidic approach to control the distribution of growth factors in a three dimensional (3D) biomimetic matrix. The developed technology is applied to investigate the morphogenetic behavior of mesenchymal stem cells in a stable 3D PDGF-BB gradient as it is suggested to occur in the microenvironment of the perivascular niche. |
Introduction
Mesenchymal stem cells (MSCs) have recently been described to reside in perivascular niches of several human tissues.1,2 The de novo formation of these niches, which consist of an endothelium that is surrounded by perivascular cells, such as MSCs, relies on the spatiotemporal controlled presentation of biological cues.3 Whereas metabolic demand, low oxygen tension, or healing wounds control the sprouting of endothelial cells by an elevated expression of vascular endothelial growth factor (VEGF),4 the mobilization of perivascular cells and their stable retention has been described to rely on factors released and presented from endothelial cells.5 One of these biological cues is platelet derived growth factor-B (PDGF-BB), which has been shown to promote chemotaxis, proliferation, and spreading of various perivascular cells.6 Its secretion at relatively large quantity by endothelial tip cells during vascular sprouting is required for perivascular cells to spread and proliferate along the vascular wall, initiating the indispensable vessel stabilization.7 Moreover, the distribution of endothelial cell secreted PDGF-BB is spatiotemporally controlled through its interaction with heparin sulfate proteoglycans (HSPG) of the extracellular matrix (ECM). The affinity binding of PDGF-BB to ECM components results in an altered presentation of the molecule to MSCs, which will face matrix tethered growth factors rather than soluble factors. In addition, continuous ECM immobilization results in creation of a stable PDGF-BB gradient that descends from the vessel. This gradient is in turn sensed by perivascular cells and steers their localized assembly along the surface of the vessel (Fig. 1A).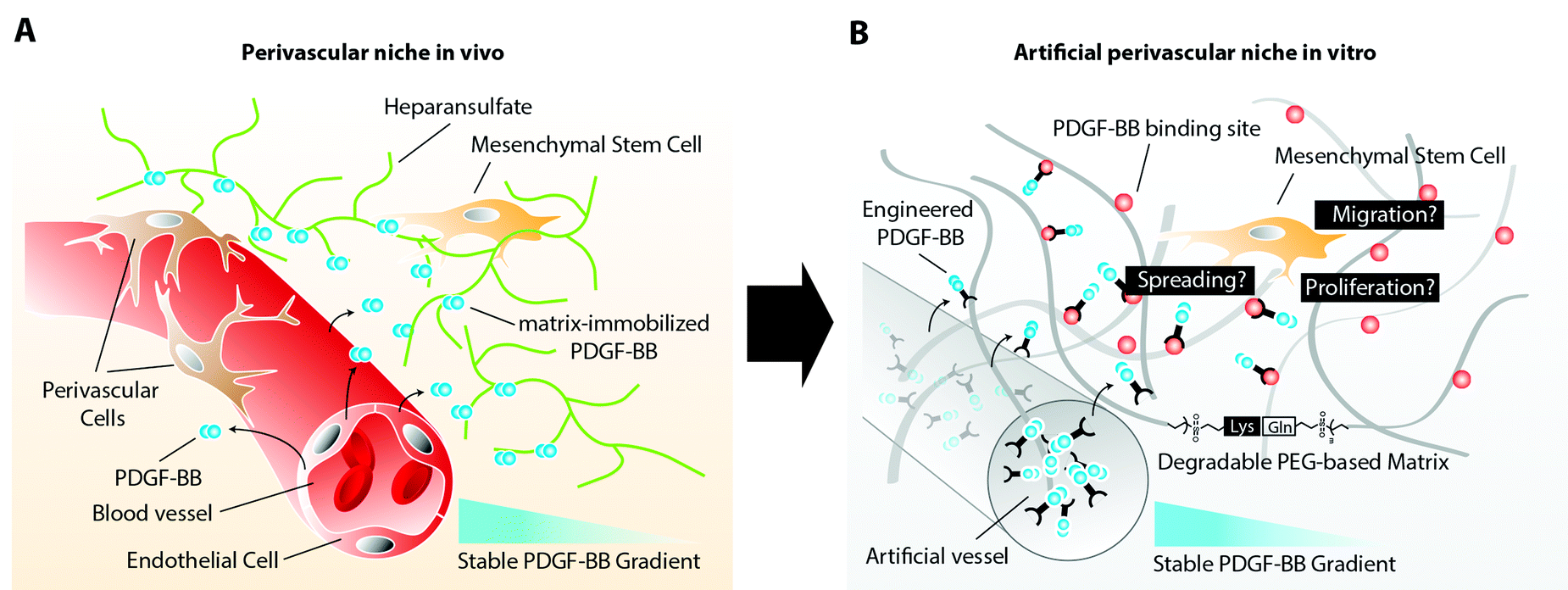 | ||
| Fig. 1 Schematic representation of the perivascular niche. (A) Native perivascular niche. The native perivascular niche is composed of endothelial cells, perivascular cells (pericytes and smooth muscle cells), MSCs and ECM matrix containing matrix associated and soluble cues. Endothelial cell derived PDGF-BB binds with high affinity to HSPG of the ECM and thereby form a stable localized gradient. (B) Engineered perivascular niche. The engineered perivascular niche, analogous to its in vivo counterpart, consists of an artificial vessel, a proteolytically degradable matrix with integrin binding sites, and a stable gradient of PDGF-BB. The fate of MSCs can be evaluated within the defined, positional microenvironments. | ||
MSCs are operatively defined by their potential to differentiate into cells of the osteogenic, adipogenic, and chondrogenic lineage after in vitro expansion. However, the complexity of their in vivo localization in the perivascular niche and the lack of MSC-specific markers hamper our further understanding of their identity, heterogeneity, and functional role in adult tissue homeostasis and regeneration.8 Therefore, there is great need for three dimensional (3D) in vitro models, which adequately recapitulate the in vivo arrangement of cells, ECM components and other biomolecules of the perivascular niche. Such models would allow for elucidation of biological processes that control the fate and localization of MSCs in the perivascular niche during vessel assembly, quiescence, or injury induced activation.
Recently, the systematic deconstruction of stem cell niches and their reconstruction under defined conditions has been proposed as a mean to understand individual parameters of complex niche signaling networks.9 Indeed, great progress has been made towards the development of ECM-mimicking synthetic matrices, potentially allowing for the in vitro emulation of stem cell niches by the generation of 3D microenvironments.9–11 Such hydrogel matrices, as they are based on biologically inert materials such as poly(ethylene glycol) (PEG), can be formed with defined stiffness and degradability. We have described synthetic, transglutaminase cross-linked hydrogels (TG–PEG hydrogels), which are enzymatically degradable and contain the cell adhesion site RGD12,13 and at low stiffness allow 3D cell adhesion, spreading and migration.14 Their modular design, additionally to the flexible immobilization of engineered building blocks such as growth factors or enzymes,15,51 allowed the formation of structured hydrogels by locally controlling the enzymatic gel formation.16 Of note, such PEG-based matrices, in contrast to biologically derived materials such as the widely used Matrigel™ or collagen, are highly defined and do not exert any biological response, allowing the study of cell behavior on a blank canvas.17–19
Here, in order to elucidate MSC behavior during PDGF-controlled formation of the perivascular niche, we created a 3D biomimetic model, that mimics the naturally occurring stable, matrix-immobilized PDGF-BB gradient, which is present in the perivascular niche. By placing MSCs within a 3D-structured TG–PEG hydrogel,13 we describe an in vitro bottom-up approach for the recapitulation of physical as well as PDGF-presenting properties of the perivascular niche (Fig. 1B). In this model we first confirmed the specific ability of soluble PDGF-BB but not VEGF121 to activate MSCs, leading to 3D morphogenetic processes such as cell proliferation, spreading, and migration. Furthermore, we assessed the activity of matrix tethered PDGF on MSCs and finally we created stable 3D PDGF-BB gradients, which resulted in spatiotemporal control of MSC spreading and motility, as described during the naturally occurring investment of endothelium with perivascular cells. The herein described biomimetic niche model is a promising first step to study the role of MSCs during vascular morphogenesis, tissue healing and regeneration in vitro.
Materials and methods
Cell culture
Sampling of bone marrow was done during orthopedic surgical procedure on healthy donors (average age 28 y.o.) after informed consent during orthopaedic surgical procedures in accordance with the local ethical committee (University Hospital Basel; Prof. Dr Kummer; approval data 26/03/2007 Ref Number 78/07). Cells were isolated as described previously.20 Human bone marrow MSCs were cultured in minimal essential medium alpha (MEMalpha, Gibco Life Technologies, cat. no. 22571-020) supplemented with 10% (v/v) fetal calf serum (FCS, Gibco Life Technologies, cat. no. 10500), 1% (v/v) penicillin–streptomycin solution (Gibco Life Technologies, cat. no. 15140-122) and 5 ng ml−1 FGF-2 (Peprotech, cat. no. 100-18B). For experiments, MSCs were cultured in serum-free DMEM/F-12 + GlutaMAX™ (Gibco Life Technologies, cat. no. 31331-028) supplemented with 1% (v/v) penicillin–streptomycin solution. MSCs between passages 2 and 4 were used for all of the experiments.Preparation of TG–PEG hydrogels
Matrix metalloproteinase (MMP)-sensitive TG–PEG hydrogels were prepared as described previously.21 In brief, eight-arm PEG precursors containing the pending factor XIIIa substrate peptides glutamine acceptor (n-PEG-Gln) or lysine donor with an additional MMP-sensitive linker (n-PEG–MMPsensitive–Lys) were mixed stoichiometrically (final dry mass content 1.5%) in Tris-buffer (50 mM Tris, pH 7.6) containing 50 mM calcium chloride. 50 μM Gln-RGD peptide and indicated amounts of Gln-ZZ were added to the precursor solution prior to initiation of cross-linking by 10 U ml−1 thrombin-activated factor XIIIa and vigorous mixing.Microtissue formation
For microtissue formation, indicated cell suspensions (final. conc. 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells ml−1) were suspended in the respective medium for the experiment and supplemented with 0.2% (w/v) methyl cellulose (Sigma-Aldrich, cat. no. M0512). Droplets of 30 μl were placed in non-adhesive cell culture dishes (Greiner bio-one, cat. no. 633180) and cultured overnight as hanging drops. The resulting spheroids (approx. 750 cells) were harvested in cell culture medium and washed once with cell culture medium.
000 cells ml−1) were suspended in the respective medium for the experiment and supplemented with 0.2% (w/v) methyl cellulose (Sigma-Aldrich, cat. no. M0512). Droplets of 30 μl were placed in non-adhesive cell culture dishes (Greiner bio-one, cat. no. 633180) and cultured overnight as hanging drops. The resulting spheroids (approx. 750 cells) were harvested in cell culture medium and washed once with cell culture medium.
Encapsulation of single cells and microtissues in TG–PEG hydrogels
For encapsulating single dispersed cells or microtissues in TG–PEG gels, cell or microtissue suspensions were diluted in the respective medium and added to the complete TG–PEG solution. To form disc shaped hydrogels of approximately 5 mm diameter, 20 μl of this mixture were then sandwiched between sterile hydrophobic glass microscopy slides (obtained by treatment with SigmaCote, Sigmacote, Sigma-Aldrich, cat. no. SL2) separated by spacers (ca. 1 mm thickness) and clamped with binder clips. To prevent sedimentation of cells or microtissues, the forming matrices were slowly rotated at room temperature (RT) until the onset of gelation and then incubated for additional 30 min at 37 °C. The hydrogels were thereafter released and transferred into a 24-well plate. Finally, 500 μl of the respective medium was added to the wells with the gels and the plates were incubated at 37 °C and 5% CO2 in a humidified atmosphere.Cell labeling
After the experiments, the samples were fixed with 4% paraformaldehyde. After stopping the reaction with 0.1 M Glycine in PBS (phosphate buffered saline, pH 7.2), the samples were washed twice with PBS. Permeabilization was performed for 20 min at RT with 0.2% Triton X-100 (Sigma-Aldrich, cat. no. T-8787) in PBS followed by 2 washing steps with PBS. For F-actin staining, the samples were incubated over night at 4 °C with rhodamine-labeled phalloidin (Life technologies, cat. no. R415). Afterwards, the samples were washed 3 times with PBS and stained for cell nuclei using Hoechst (1:1000, Life technologies, cat. no. H3570). Subsequently, the samples were washed again 3 times with PBS before analysis with either confocal laser scanning microscopy or fluorescent microscopy.
Microtissue migration assay
MSC microtissues were encapsulated in TG–PEG hydrogels (final conc.: approx. 250 microtissues ml−1) and cultured for 20 hours. The samples were fixed and stained for F-actin. Z-stack images of microtissues were acquired at 10× magnification using a confocal microscope (Leica TCS SP5). Cell migration was quantified by an automated image analysis script written in MATLAB (R2013a, MathWorks Inc, USA) that measures the distance of stained pixels from the microtissue center. Data is displayed as histogram depicting the amount of pixels relative to the migration distance. Pixels with a distance larger than 100 μm from the spheroid center were considered in the statistical analysis.Single cell proliferation, spreading, and migration assays
MSCs were embedded in TG–PEG hydrogels (final conc.: 0.5 × 106 ml−1) and cultured for 5 days. For migration assays, three random positions were selected using an inverted microscope (Leica DMI6000 B) equipped with a motorized focus and stage. Cell migration was followed for 22.5 hours using the manual tracking plugin in ImageJ. For proliferation and spreading assays, after cell fixation, cells were stained for nuclei as well as F-actin and Z-stack images (total stack size: 500 μm) were acquired at 10× magnification using a confocal microscope (Leica TCS SP5). Cell proliferation was analyzed by measuring in 3D the numbers of F-actin stained objects using the ImageJ plugin ‘Object Counter3D’. For circularity ratio, a Z-stack projection from F-actin Z-stack images (total stack size: 150 μm) was made and cell circularity ratio was calculated using ImageJ. Cell circularity ratio is the ratio between a cell's shape and a circle with the same perimeter. It therefore is a dimensionless number that is greater than 0 and less or equal to 1. The more elongated a cell is the closer the circularity ratio is to 0. Cells with a circularity ratio below 0.25 were considered as elongated.Recombinant proteins
Untagged growth factors were purchased from Peprotech (VEGF121, cat. no. 100-20A; PDGF-BB, cat. no. 100-14B). Linker protein Gln-ZZ was recombinantly produced in E. coli Bl21 as described previously.25 Fc-tagged PDGF-BB (PDGF-BBFc) and VEGF-A (VEGF-AFc) were cloned, expressed and purified as follows: PDGF-B and VEGF-A from Homo sapiens (PDGF-B: NP_002599.1, aa: 21-190 and VEGF-A: NP_001020539.2, aa: 207-371) were cloned into a modified PCEP vector with an N-terminal double Strep-tag II followed by Igh protein from Mus musculus (AAH03435.1, aa: 245-463). A pool of HEK293 cells were stably transfected followed by screening for high level expression. Secreted PDGF-BBFc and VEGF-AFc were purified by Streptavidin beads (IBA). The purified PDGF-BBFc and VEGF-AFc were then dialyzed against 1× phosphate buffer saline (PBS) pH 7.4. Labeling of PDGF-BBFc was done using DyLight 550 Fluorophore (Thermo Fisher Scientific Inc.) following the manufacturer's instructions.Solid phase binding assay between Gln-ZZ and PDGF-BBFc
Gln-ZZ peptide was coated at 10 μg ml−1 (500 ng well−1) overnight at 4 °C onto 96-well plates (Nunc Maxisorb). After washing with TBS, plates were blocked for 2 hours at RT with TBS containing 3% bovine serum albumin. Ligand (PDGF-BBFc) was diluted to concentrations from 0.03 nM to 500 nM and incubated for 2 hours at room temperature. After extensive washing with Tris-buffered saline (TBS), bound ligands were detected with a streptavidin horseradish peroxidase (HRP)–conjugate (IBA) 1:5000. HRP was detected by Pierce TMB ELISA Substrate (Thermo Scientific™). Absorption was measured at 450 nm after stopping the reaction with 2 M sulfuric acid. A Blank value corresponding to BSA coated wells was automatically subtracted.
Preparation of the PDMS frames
Polydimethylsiloxane (PDMS) frames were made as described previously.16 In short, the silicon elastomer and the curing agent (Sylgard 184, Dow Corning Corporation, USA) were mixed (10:1 in mass) at 2000 rpm for 3 min in a ARE-250 mixer (Thinky Corporation, Japan). The mixture was subsequently poured into poly(methyl methacrylate) (PMMA) molds, where 500 μm in diameter stainless steel wires were positioned to create the holes for the future electrodes. The mixture was subsequently degassed for 30 min in a vacuum chamber and baked for 4 h at 60 °C. The stainless steel wires and the PDMS forms were removed from the PMMA molds. The forms were rinsed with isopropanol (IPA), Oxygen plasma cleaned (1 min at 300 W, Plasma-System 100, Technics Plasma GmbH, Germany) and finally pressed onto plasma cleaned microscope glass cover slips. Straightened Tungsten wires (W, 500 μm in diameter, Advent Research Materials Ltd, UK) were inserted in the PDMS forms and connected to a potentio-galvanostat in a two electrode setup (PGU-10V-1A-IMP-S and ECMwin computer interface, Elektroniklabor Peter Schrems, Germany).
Channel and PDGF-BB gradient formation in TG–PEG hydrogels
Perfusable channels with a diameter of 500 μm were produced in TG–PEG hydrogels as described previously.16 Briefly, 60 μl of complete TG–PEG hydrogel mixture containing 0.5 × 106 ml−1 MSCs were poured before the onset of gelation into the PDMS frame accommodating a straightened Tungsten wire. The polymerization of the TG–PEG was allowed to progress during 6 minutes while the Tungsten wire was anodically polarized (2 V DC, versus a Platinum auxiliary electrode). Subsequent removal of the anodically polarized Tungsten wires lead to reproducible formation of regularly shaped channels within the TG–PEG gel. Stable or evolving PDGF gradients were formed by perfusing the channel with 1 μl PDGF-BBFc or fluorescently labeled PDGF-BBFc (180 ng μl−1) in presence or absence of 20 μM Gln-ZZ in the surrounding TG–PEG gel, respectively. Images of fluorescently labeled PDGF-BBFc gradients were taken after 0.5, 24, and 48 hours using an inverted fluorescence microscope (Leica DMI6000 B). Fluorescence intensity was measured on a line perpendicular to the channel using the PlotProfile function in ImageJ.Analysis of MSC spreading and migration within PDGF-BB gradients
MSCs were encapsulated in TG–PEG hydrogels (final conc.: 0.5 × 106 ml−1) within the PDMS mold and cultured overnight. The day after, Tungsten wires were removed and indicated amounts of PDGF-BBFc were loaded into the channel. For migration assays, two positions adjacent to the channel and two positions 3 mm away from the channel were selected using an inverted microscope (Leica DMI6000 B) equipped with a motorized focus and stage. Cell migration was followed for 22.5 hours using the manual tracking plugin in ImageJ. For spreading analysis, cells were fixed after 48 hours in culture. After F-actin staining, Z-stack images (total stack size: 150 μm) of the complete constructs were acquired at 10× magnification using a confocal microscope (Leica TSC SP5) equipped with a long distance 10× objective. Z-stack projections were made thereof, stitched together and the image was divided into 3 equally sized segments each having a width of 0.5 mm. For quantification, cell circularity ratio was analyzed in each part using ImageJ.Statistical analysis
All mean values were compared by one-way analysis of variance (ANOVA) using MATLAB (R2013a, MathWorks Inc, USA) followed by a Tukey–Kramer post hoc test to judge statistical significance. Statistical significance was accepted for p < 0.05. One asterisks reports p < 0.05 and two asterisks p < 0.01.Results
PDGF-BB modulates 3D MSC functions in vitro
In order to study the role of PDGF-BB on MSCs during morphogenesis in the perivascular space, we established a 3D microtissue-based in vitro assay.MSC microtissues were embedded within a recently developed polyethylene glycol (PEG) hydrogel, termed ‘TG–PEG hydrogel’, that recapitulates biochemical and biophysical properties of the microenvironment MSCs face in their anatomical location.12,13 This synthetic hydrogel was formulated to be matrix MMP-1-degradable, have low cross-link density (94 ± 25 Pa shear modulus)14 and provide cell adhesion sites (50 μM RGD) in order to offer the MSCs a hydrated microenvironment in which proliferation, spreading and migration are possible. 4 hours after stimulation of the MSC microtissues with PDGF-BB, MSCs started to migrate from the microtissues into the surrounding synthetic environment. Quantification of migration showed that in absence of PDGF-BB cells remained in a quiescent state within the microtissue, whereas upon 20 hours of exposure to PDGF-BB (5 nM) cells had invaded the artificial ECM up to 500 μm (Fig. 2A and B). When MSC microtissues were treated with equal amounts of VEGF121 no significant outgrowth was observed as compared to the controls. Taken together, this data confirmed that PDGF-BB specifically controls morphogenesis related functions of MSCs and that its role can be studied in vitro within the designed artificial environment.
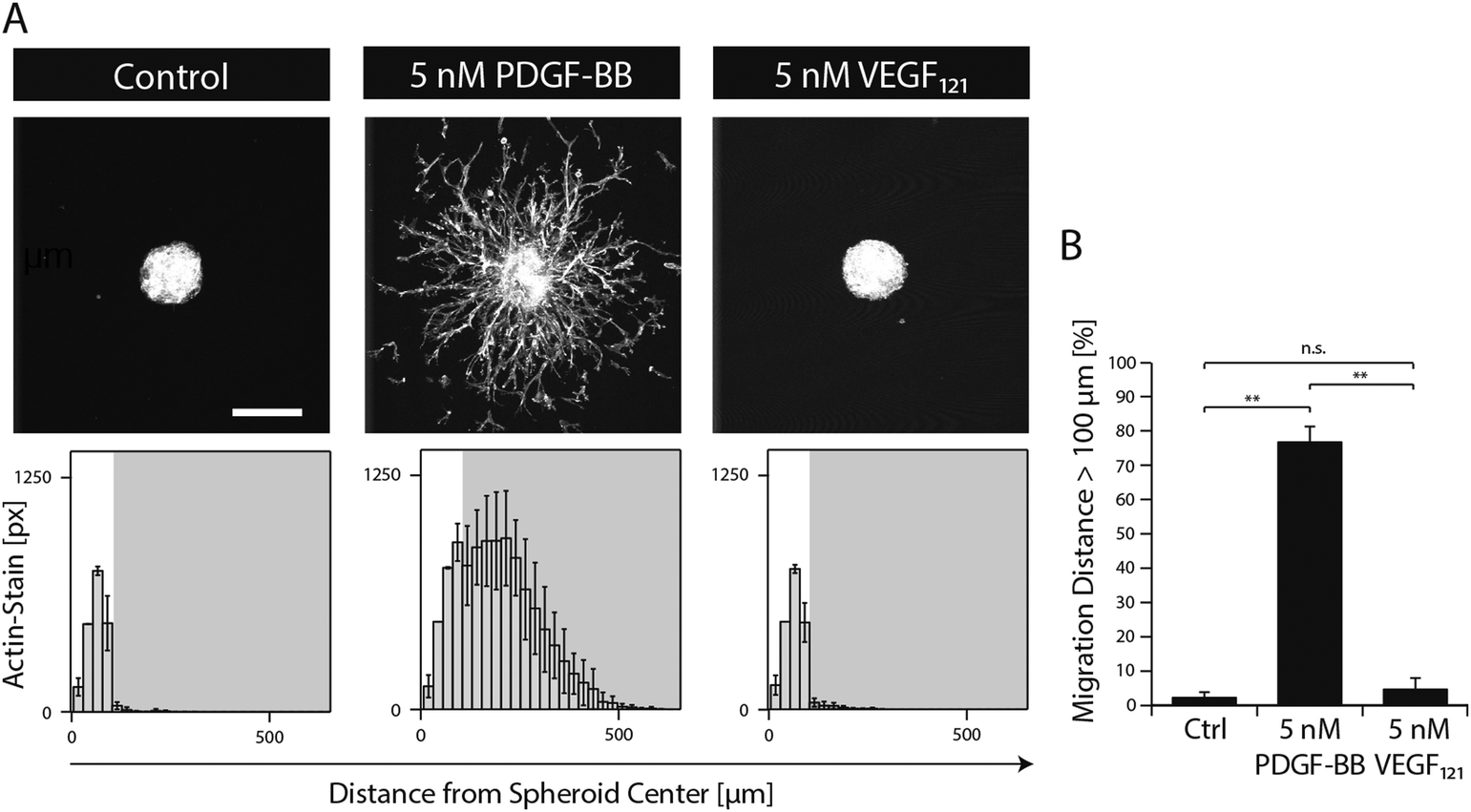 | ||
| Fig. 2 PDGF-BB induces 3D MSC morphogenesis in biomimetic microenvironments. MSC microtissues consisting of approx. 750 cells were embedded in synthetic matrices and stimulated with the indicated growth factor for 20 hours under serum-free conditions. (A) Z-stack projections of F-actin stained MSCs (upper panels, scale bar = 200 μm). Histograms showing the distribution of MSCs from microtissue center when stimulated with the respective growth factor (lower panels). (B) Bar graph illustrating the percentage of F-actin-stained pixels being located at a distance larger than 100 μm from the centre of the microtissue. Data is depicted as mean ± SD (n ≥ 4, **, p < 0.01, n.s., not significant). | ||
Discrimination of 3D morphogenetic processes by single cell assay
To elucidate the effect of PDGF-BB on individual cellular processes, single dispersed cells were embedded in TG–PEG hydrogels and cultured for several days in the absence or in the presence of 5 and 25 nM PDGF-BB. Confocal images (z-stacks) of cytoskeleton stained cells after 5 days of culture revealed that presence of PDGF-BB had a dose dependent effect, which resulted in the formation of relatively dense cellular networks (Fig. 3A). Automated quantification of confocal images was applied to analyze cell number, cell circularity ratio as a measure of cell elongation, and single cell migration. PDGF-BB enhanced MSC numbers on average by 45% (5 nM PDGF-BB) and 81% (25 nM PDGF-BB) (Fig. 3B). Moreover, MSCs' circularity ratio decreased because of their increased elongation after treatment with PDGF-BB (Fig. 3C). Based on the evaluation of the cell populations' circularity ratio histograms, a cell that had a circularity ratio below 0.25 was considered as elongated. In the untreated cell population only 13 ± 4% of the cells were elongated whereas in the PDGF-BB treated conditions 32 ± 9% (5 nM PDGF-BB) and 63 ± 5% (25 nM PDGF-BB) of the cells were elongated (Fig. 3D). Also, cell motility was increased in a dose dependent manner as found by tracking cells for 22.5 hours (Fig. 3E). Whereas the mean distance that cells migrated during this time in the control sample was 47 ± 11 μm, cells that were exposed to 5 nM or 25 nM PDGF-BB migrated 173 ± 20 μm or 246 ± 14 μm, respectively. Taken together, this data indicates that PDGF-BB is an important player in influencing multiple facets of MSC morphogenesis.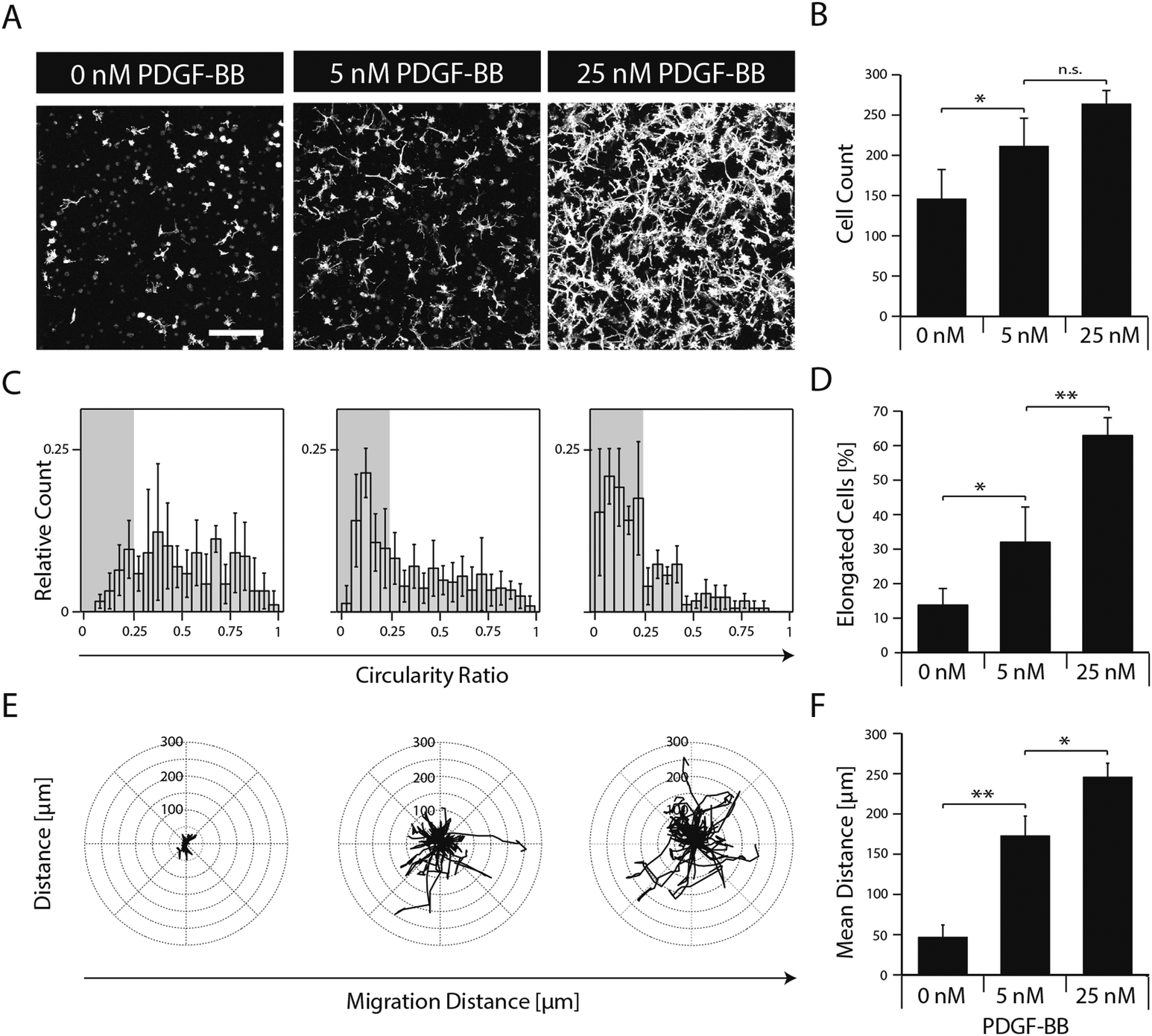 | ||
| Fig. 3 PDGF-BB induces proliferation, spreading and migration of 3D encapsulated MSCs. MSCs were encapsulated in TG–PEG hydrogels (0.5 × 106 cells ml−1) and cultured under serum-free conditions in presence of the indicated amounts of PDGF-BB. (A) Z-stack projections (total stack size 500 μm) of F-actin stained MSCs after 5 days of culture (scale bar = 200 μm). (B) MSC numbers as analysed by counting connected F-actin objects in 3D by an automated image analysis script as described in materials and methods. (C) Histogram depicting the MSC population's circularity ratio (4π × (cell area)/(cell perimeter2)). (D) Bar graph depicting percentage of elongated cells. Cells with a circularity ratio below 0.25 were considered as elongated. (E) Cell tracks of MSCs were acquired and overlaid with a common starting point. The tracks represent a period of 22.5 hours (18 × 75 min) starting 24 hours after first growth factor exposure. (F) Bar graph depicting mean migration distance. Data is depicted as mean ± SD (n ≥ 4, *, p < 0.05, **, p < 0.01, n.s., not significant). | ||
Matrix-mediated presentation of PDGF-BB
In order to mimic the naturally occurring interactions between heparin sulphates of the ECM and PDGF-BB and to control the spatiotemporal distribution of PDGF-BB within the in vitro niche model, the fragment crystallisable (Fc) region of antibodies was added to the N-terminus of PDGF-BB. This new variant (PDGF-BBFc) enables its high-affinity immobilization in TG–PEG hydrogels provided that during hydrogel formation a recently developed bifunctional linker protein (GlnZZ)15 was incorporated in the TG–PEG backbone. Gln-ZZ can be incorporated into the TG–PEG hydrogel backbone via its C-terminal domain (Gln) and has high affinity for Fc-tagged proteins through its N-terminal Z domains. Specific binding of the PDGF-BBFc fusion protein to Gln-ZZ was confirmed by a solid-phase binding assay (Fig. S1A, ESI†). To test the biological activity of PDGF-BBFc microtissues were embedded in TG–PEG gels and cultivated in medium containing 5 or 25 nM soluble PDGF-BBFc (no Gln-ZZ linker). Fig. S1A and B (ESI†) show cell migration distance and attest bioactivity of the engineered Fc-tagged protein.As for testing the activity of bound PDGF-BBFc, microtissues were embedded in TG–PEG gels containing 20 μM Gln-ZZ incorporated in the hydrogel backbone. These microtissues were exposed to admixed 0, 5 and 25 nM PDGF-BBFc. The excess of Gln-ZZ over PDGF-BBFc assures that the majority of PDGF-BBFc molecules are present in their matrix immobilized form. In the absence of growth factors, MSCs fully remained in the microtissue, with as little as 3% of actin stained areas (pixels) belonging to cells that had left the microtissue (Fig. 4). On the other hand, cells that were exposed to hydrogels containing 5 and 25 nM PDGF-BBFc displayed clear morphogenesis related functions with 41% and 67% of F-actin stained pixels detached from the microtissue, respectively (Fig. 4). This significant difference indicates that MSC activity can be modulated in a dose dependent manner. In addition, the data suggests that PDGF-BBFc immobilization does not significantly alter its activity. This is shown by the similar morphogenetic activity observed between 5 nM immobilized (20 μM Gln-ZZ) and free diffusing (0 μM Gln-ZZ) PDGF-BBFc in Fig. 4A.
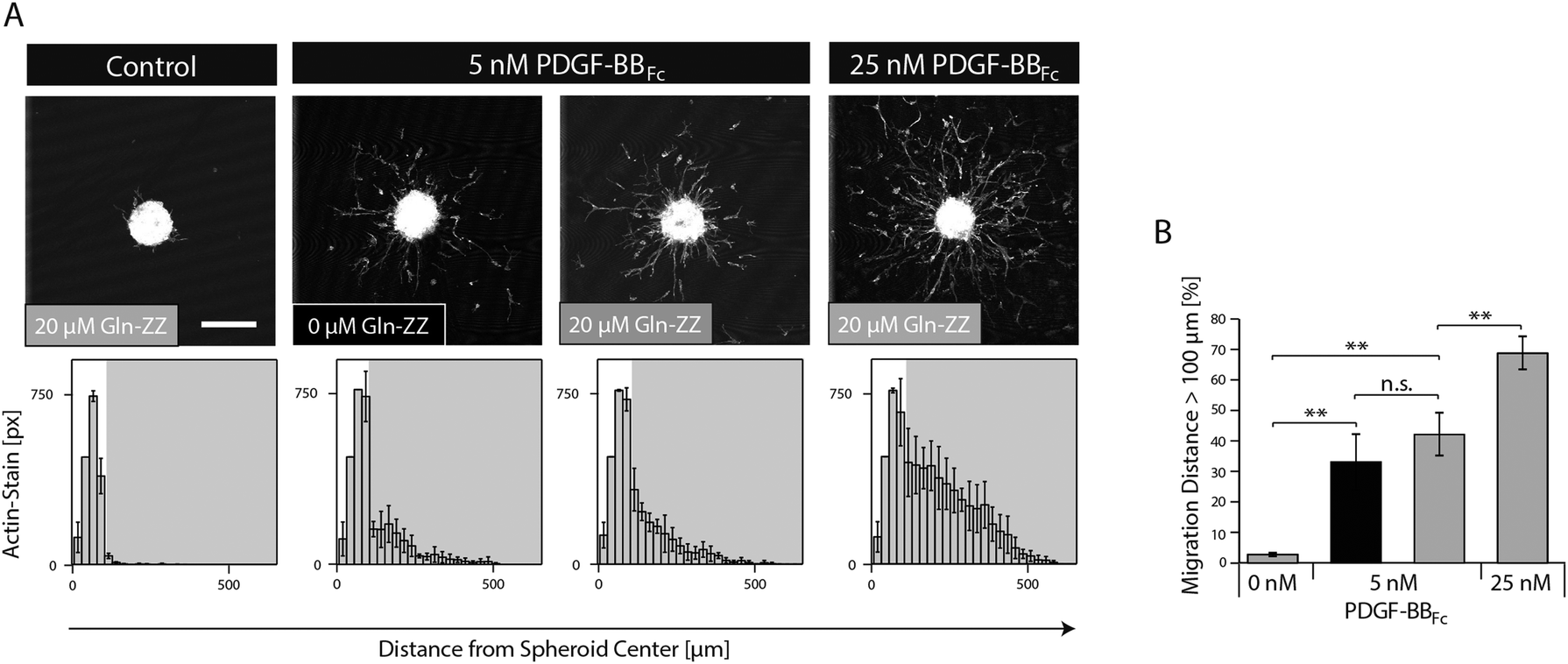 | ||
| Fig. 4 MSC migration in synthetic PDGF-BBFc-presenting cell-instructive matrices. MSC microtissues consisting of approx. 750 cells were encapsulated in TG–PEG hydrogels containing 0 μM (black) or 20 μM (grey) Gln-ZZ and 0, 5, or 25 nM PDGF-BBFc. (A) Representative Z-stack projections of F-actin stained MSCs cultured for 20 hours under serum-free conditions (upper panels, scale bar = 200 μm). Histograms showing the distribution of MSCs from the microtissue center when embedded within the indicated matrix (lower panels). (B) Bar graph shows the percentage of F-actin-stained pixels being located at a distance larger than 100 μm from the centre of the microtissue. Data is depicted as mean ± SD (n ≥ 3, **, p < 0.01, n.s., not significant). | ||
Microfluidic approach for the creation of 3D PDGF-BB gradients in TG–PEG hydrogels
As opposed to traditional cell culture assays, biomolecules within the body are not present in uniform concentrations, but rather in concentration gradients that descend from the site of biomolecule expression.21 In order to mimic in vitro the 3D PDGF-BB gradient that occurs in the perivascular niche around blood vessels, we applied a recently developed electrochemistry-based technique to form artificial vessels in the middle of the hydrogel by spatially inhibiting hydrogel polymerization.16 Such channels were loaded with controlled amounts of PDGF-BBFc, which could freely diffuse from the channel into the surrounding hydrogel and reach the MSCs (Fig. 5A). To monitor PDGF-BB diffusion into MSC containing hydrogels, fluorescently labeled PDGF-BBFc was loaded in a channel and its distribution in the gel was analysed (Fig. S2, ESI†). After 48 hours the PDGF-BBFc gradient had leveled out and PDGF-BBFc was almost homogenously distributed within the hydrogel (Fig. 5B). In order to fabricate a more sophisticated model that takes into account the interaction of PDGF-BB with the ECM, we made channels in Gln-ZZ containing hydrogels.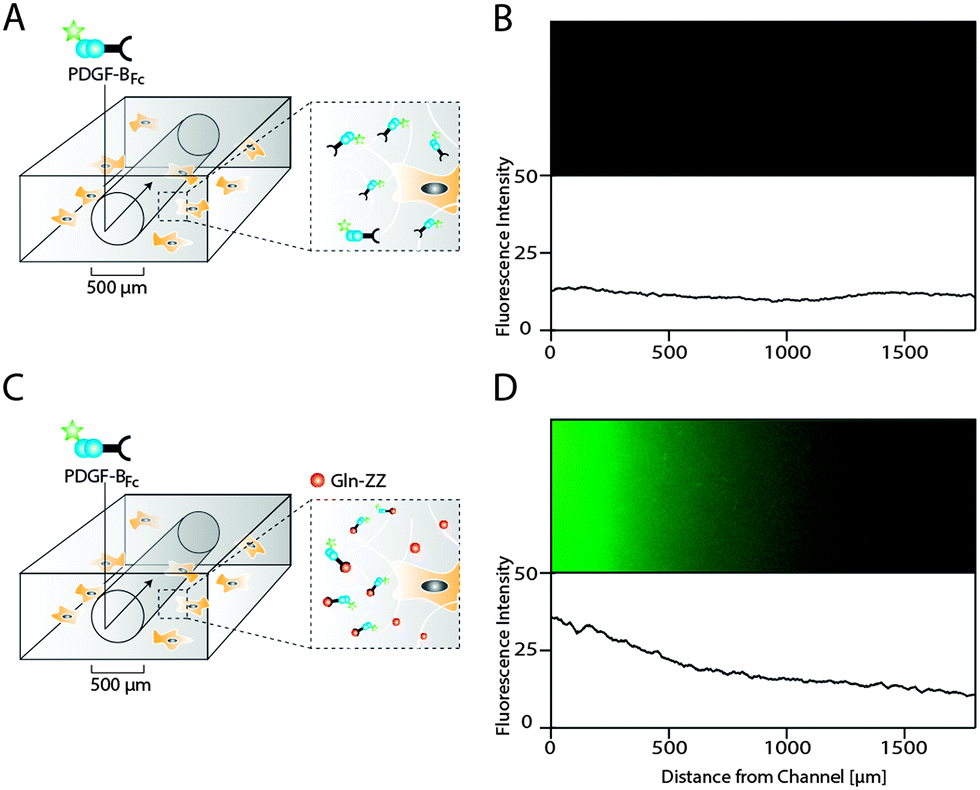 | ||
| Fig. 5 Hydrogel microfluidics for the creation of evolving and stable PDGF-BB gradients in presence of cells. Scheme of PDGF-BBFc delivery to 3D encapsulated MSCs using a perfusable channel in standard (A) and 20 μM Gln-ZZ modified (C) TG–PEG hydrogels. Hydrogel channels were formed by electrochemical inhibition of hydrogel polymerization as described previously.16 (B and D) Fluorescent microscopy images showing the area around the respective hydrogel channels 48 hours after perfusion with DyLight 550-labeled PDGF-BBFc (upper panel) and the corresponding fluorescent intensity plots. | ||
To test if the previously described interaction between PDGF-BBFc and Gln-ZZ would indeed lead to the establishment of the graded 3D immobilization of PDGF-BB, we perfused the fluorescently labeled PDGF-BBFc through the channel into the Gln-ZZ presenting and MSC containing hydrogel (Fig. 5C). 48 hours after perfusion, a stable 3D PDGF-BBFc gradient originated from the channel into the hydrogel, indicating a stable interaction between PDGF-BBFc and the matrix in presence of MSCs (Fig. 5D). 1.5 mm from the channel the level of fluorescently labelled PDGF-BBFc had decreased to background level.
MSC morphogenesis in evolving versus stable 3D PDGF-BB gradients
In order to elucidate MSCs' morphogenetic response to being exposed to evolving and stable gradients of PDGF-BBFc, MSCs were encapsulated in TG–PEG hydrogels containing an artificial vessel for biomolecule perfusion. As expected, perfusion of the channel with control medium without PDGF-BBFc did not lead to a morphogenetic response as shown by the cellular circularity ratio two days after perfusion within 3 consecutive segments each spanning 1.5 mm (Fig. S3, ESI†). Percentage of cells that were elongated (circularity ratio < 0.25) remained around 10% in all segments. This coincides with the value that was previously observed for the control in the static condition (Fig. 3C and D). On the other hand, perfusion of 1.8 μM PDGF-BBFc, which created a short-term-evolving gradient of PDGF-BBFc as shown before (Fig. 5B), led to cell spreading (decrease in circularity ratio) similarly to the previous static experiments (Fig. 3C and D). After 2 days, however, only minor spatial differences in cell spreading were observed due to the levelling out of the soluble PDGF-BBFc concentrations throughout the sample (Fig. 6A). Percentage of cells that were elongated (circularity ratio < 0.25) was roughly between 20% and 40% (Fig. 6C), which correlated with the elongation that was seen in the static condition in presence of 10 nM PDGF-BB (Fig. 3C and D).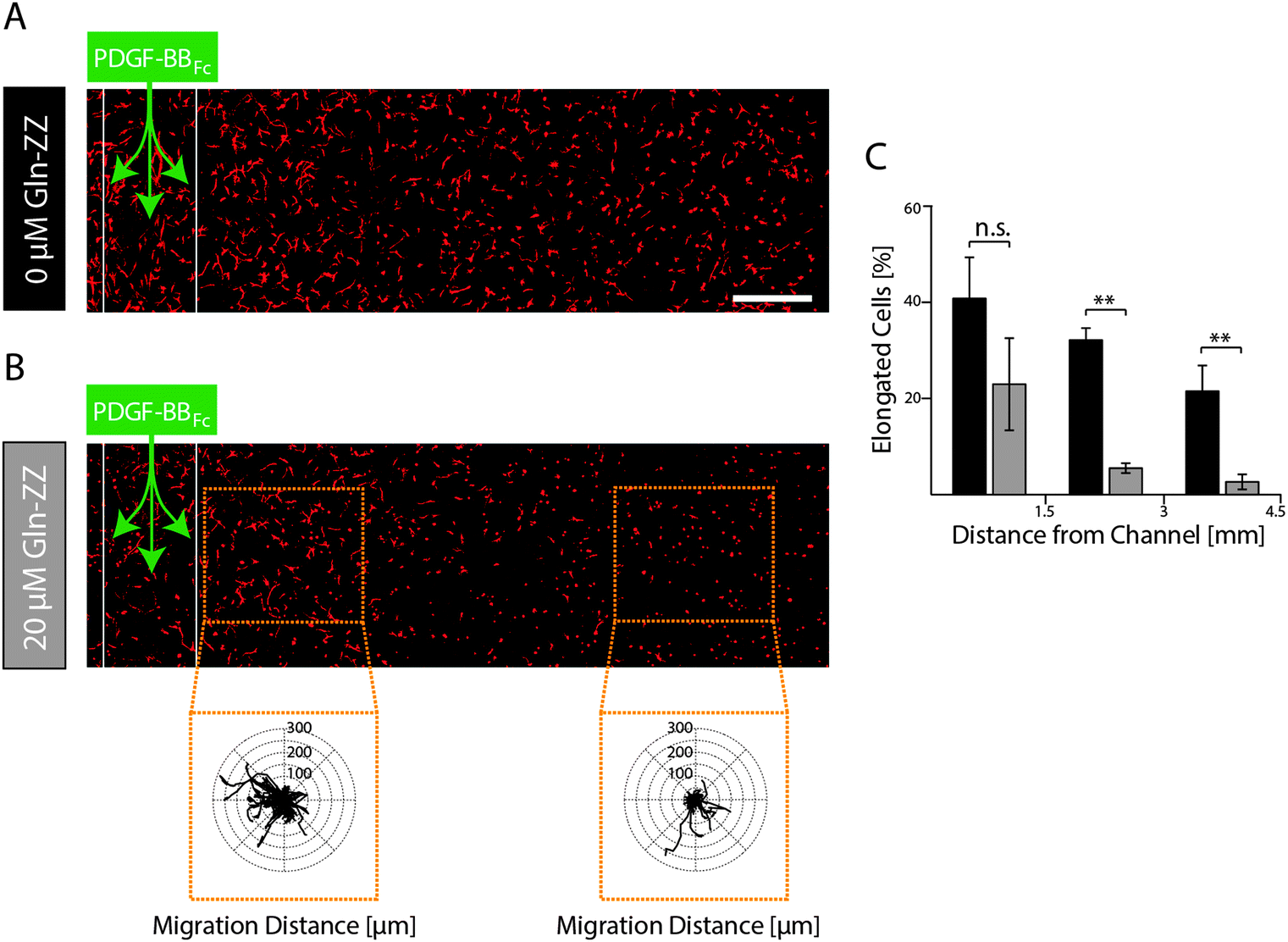 | ||
| Fig. 6 Evaluation of MSCs in a model of the perivascular niche microenvironment mimicking evolving and stable PDGF-BB gradients. MSCs were encapsulated (0.25 × 106 cells ml−1) in TG–PEG hydrogels and channels for biomolecule perfusion were formed by electrochemical inhibition of hydrogel polymerization. Data is shown for both conditions after 48 hours of culture as representative Z-stack images (total stack size 500 μm) of F-actin stained MSCs (scale bar = 500 μm). Channels in unaltered (A) and 20 μM Gln-ZZ containing (B) TG–PEG hydrogels were perfused with 1.8 μM PDGF-BBFc. Inlays display overlaid tracks of migrating MSCs at the indicated positions of the sample. The tracks represent a period of 22.5 hours (18 × 75 min) starting 24 hours after PDGF-BBFc perfusion. (C) Histogram depicting percentage of cells with a circularity ratio below 0.25 relative to distance to channel in gels with 0 μM (black) or 20 μM Gln-ZZ (grey). Statistical comparison is made between the two groups of each region. Data is depicted as mean ± SD (n ≥ 4, n.s., not significant, **, p < 0.01). | ||
In contrast, if a stable PDGF-BB gradient was formed by perfusion of Gln-ZZ (20 μM) modified TG–PEG hydrogels with 1.8 μM PDGF-BBFc the morphogenetic response of encapsulated MSCs was locally restricted to the vicinity (0–1500 μm) of the artificial vessel (Fig. 6B). Due to the stable retention of PDGF-BBFc only in the first segment 20% of the cells were elongated (circularity ratio < 0.25) whereas cells further away were not affected by the growth factor presence (Fig. 6C). Moreover, the bound growth factor was locally inducing MSC motility as shown by the cell tracks of cells at 0–1 mm versus 2.5–3.5 mm distance from the channel (Fig. 6B). These results emphasize the role of biomolecules within complex in vitro models and even more so the importance of controlling them spatiotemporally by mimicking the processes occurring in nature.
Discussion
A prevalent limitation to 2D and 3D cell culture assays has long been the lack of spatial and temporal control over the presentation of biomolecules, which especially for the engineering of complex milieus such as the stem cell niche is an evident prerequisite. We here present a biomimetic approach that recapitulates the localized ECM immobilization of PDGF-BB in the perivascular niche. By combining an electrochemistry-based hydrogel patterning approach16 with engineering of growth factor matrix interactions and diffusion controlled establishment of 3D gradients, human MSCs can readily be exposed to selected micro-environmental conditions as would be expected to occur in the surrounding of a blood vessel. This experimental setup offers several advantages to existing models: (i) it is based on engineered ECM with defined and tunable physical, chemical and biological properties (ii) growth factors can be promiscuously immobilized via affinity interactions and thus can be loaded independently of hydrogel formation and cell encapsulation (iii) the use of channels allows the sequential loading of multiple engineered and native growth factors (iv) due to the transparent character of the hydrogel system, cell behavior can be monitored throughout the length of the experiment so that morphogenetic parameters can be followed and independently evaluated.Despite the fact that great efforts were devoted towards the development of models for studying cell fates in stable growth factor gradients in vitro, described approaches either do not take into account the three dimensionality of the microenvironment that surrounds cells in vivo22–26 or employ matrices with inherent biological properties.27–29
To the best of our knowledge, this is the first report on the implementation of a stable growth factor gradient within a fully defined synthetic hydrogel matrix. Moreover, as opposed to other studies that show stable 3D growth factor gradients,27–29 we here describe a method that allows for gradient formation in presence of cells, which is not only more closely mimicking the situation in nature, but as well omitting material interface problems that result from pre-fabrication of the gradient and post hoc seeding of the cells onto the gradient.
VEGFs and PDGFs are known to be jointly involved in the assembly and stabilization of new vessels.5 This was illustrated by the elimination of either VEGF signaling in endothelial cell or PDGF signaling in pericytes or by the deletion of either the ligands or the receptors, which has resulted in a largely defective development and function of vessels, leading to embryonic lethality.30–33In vitro PDGFs promote the migration of multiple mesenchymal cell types such as fibroblasts, smooth muscle cells, and MSCs. However their role in controlling cell behavior during vessel stabilization has not been characterized in detail. As opposed to other studies,34 we demonstrate the specific in vitro induction of morphogenetic processes in hydrogel encapsulated MSC microtissues by soluble PDGF-BB but not VEGF.
Importantly, the majority of expressed VEGF and PDGF molecules contain a positively charged C-terminal domain, which has been described to bind HSPGs of the extracellular matrix.35In vivo this affinity of growth factors to the ECM limits their diffusion away from the site of production and likely results in the formation of relatively stable positional cues. The consequence of such ECM immobilized cues for the formation of functional, properly structured vascular networks has been impressively shown by transgenic mice which either express only VEGF-variants that lack the C-terminal heparin binding domain (VEGF121) or only have high affinity for heparin (VEGF189).36,37 While in the first case due to loss of directionality during branching, resulting vessels were large in diameter and less branched in the latter case extensive filopodia formation, highly branched and thin vessels were observed.38 Similarly, the deletion of the C-terminal heparin binding motif of PDGF-BB resulted in abnormal coverage of pericytes, and their partial detachment from the vessel wall which for example manifests in defective vessels and consequently in retinal deterioration, or proteinuria.3,39 These data indicate a role for the ECM immobilization of PDGF-BB in the assembly of functional blood vessels. The C-terminal binding motif of PDGF-BBs largely restricts its distribution and availability in vivo near to the site of productions, which in the case of vessels is in the perivascular zone.3 In the presented in vitro model, a synthetic protein linker was applied to mimic this natural PDGF-BB ECM interaction. Additionally, fabrication of a perfusable channel in the hydrogel allowed for controlling the PDGF-BB source and thereby creation of evolving as well as, in the case of matrix-interacting PDGF-BB, stable 3D PDGF-BB gradients. Stable PDGF-BB gradients restricted PDGF-BB-induced spreading and migration to the vicinity of the growth factor source. In contrast, the freely diffusible growth factor led to a transient gradient rapidly leveling out throughout the gel. Similar observations were made by Nguyen et al.,40 who recently described that a gradient formed in a hydrogel from a channel, initially spans over 300 μm and covers over 900 μm after 1 h only. This short-term gradient of the growth factor led to homogenous biological response throughout the entire model. These data indicate that the native perivascular organization of cells can be recapitulated in vitro by appropriate presentation of PDGF gradients. In 2D setups, soluble PDGF gradients were shown to induce fibroblast polarization and chemotaxis.41,42 However, in the herein presented 3D setup, both soluble PDGF and the gradient of matrix-immobilized PDGF stimulated a chemokinesis related (stochastic) migration process. From these experiments it remains unclear whether during the formation of the gradient a short-term exposure of the MSCs to soluble PDGF, residual non-bound PDGF, or trace amounts of released PDGF were sufficient to induce non-directed migration or whether PDGF generally induces a migration process resembling chemokinesis. The high binding affinity of the ZZ-tag to Fc-domains (Kd = 4.8 × 10−8 M−1)43 would suggest, that the release of the growth factor would be mainly due to the very slow (not yet carefully characterized) degradation of the PEG-based matrix by cell-mediated cleavage of built-in MMP-sensitive linker sequences.14,44 Although MSC migration might not occur in a highly directional fashion the sum of the PDGF-mediated processes leads to the assembly and stabilization of a perivascular niche. A further limitation of the current study is that in this simplified model the direct contact of MSCs with the vascular basement membrane, which is a known regulator of various stem cells, is not reproduced.45,46 Together, these observations indicate that our in vitro model recapitulates some of the dynamics that are assumed to be present during vessel formation, including the diffusion of trace amounts of PDGF from the site of production before the capturing by the glycosaminoglycan components of the ECM. Enzymatic remodeling of the ECM by mobilized cells is also represented by the slow MMP-dependent degradation of the PEG-matrix and the consequent release of PDGF.
The steepness of the gradient and the protease-mediated release of PDGF from gradients can have a large influence on the amount of factor seen by a cell. As in this study these parameters were kept constant we cannot exclude that chemotactic migration would preferentially occur in steeper gradients or upon cell-mediated promotion of release. Based on the observed slow degradability and thus the low availability for cells under evaluated cell culture conditions it is tempting to speculate that the graded PDGF reservoir is only very slowly exploited during the observed culture period. Both faster release kinetics and longer observations could thus clearly reinforce MSC function and even lead to improved tissue morphogenesis.
This study was restricted to the description of the isolated mesenchymal compartment of the perivascular niche. Earlier in vitro studies have described models, which use both mesenchymal, and endothelial cells to create a perivascular niche.47,48 One approach which integrated microfluidic channels and 3D fibrin encapsulation of endothelial cells and MSCs revealed an important function of laminin to control the perivascular location of MSCs in an α6β1 integrin mediated manner.48 This finding correlates well with studies showing the perivascular location of neuronal stem cells46,49 and the critical function of vascular basal membrane laminin to control MSC function by α6β1 integrin and PDGF-BB signaling.48,50 The integration of endothelial cells with herein described perivascular niche model consisting of MSCs which are exposed to stable growth factor gradients might provide a promising platform for the studying of stem cells in their perivascular niche while varying individual cellular, matrix associated or soluble bio-molecular factors.
Conclusions
By combining advanced biomaterials with microfluidic technology we here present the creation of a spatially-defined tissue model recapitulating mechanical properties and stable PDGF gradients of the perivascular niche. We believe that this model and its further development by the incorporation of endothelial cells will help increasing our understanding of the role of MSCs in vascular function as well as the cell extrinsic cues that control their fate.Acknowledgements
We thank Aida Kurmanaviciene for technical assistance and Prof. Ivan Martin for kindly providing bone marrow MSCs. This work has been supported by funding from Swiss National Science Foundation (Nos. 31003A_141051, CR2313_143766, CR3213_125426), the german research foundation (CRC829) and the Center for Clinical Research, University Hospital Zurich and University of Zurich.References
- M. Crisan, C. W. Chen, M. Corselli, G. Andriolo, L. Lazzari and B. Peault, Ann. N. Y. Acad. Sci., 2009, 1176, 118–123 CrossRef CAS PubMed.
- B. Sacchetti, A. Funari, S. Michienzi, S. Di Cesare, S. Piersanti, I. Saggio, E. Tagliafico, S. Ferrari, P. G. Robey, M. Riminucci and P. Bianco, Cell, 2007, 131, 324–336 CrossRef CAS PubMed.
- A. Armulik, A. Abramsson and C. Betsholtz, Circ. Res., 2005, 97, 512–523 CrossRef CAS PubMed.
- N. Ferrara, H. P. Gerber and J. LeCouter, Nat. Med., 2003, 9, 669–676 CrossRef CAS PubMed.
- A. Armulik, G. Genove and C. Betsholtz, Dev. Cell, 2011, 21, 193–215 CrossRef CAS PubMed.
- C. H. Heldin and B. Westermark, Physiol. Rev., 1999, 79, 1283–1316 CAS.
- H. Gerhardt and C. Betsholtz, Cell Tissue Res., 2003, 314, 15–23 CrossRef PubMed.
- I. R. Murray, C. C. West, W. R. Hardy, A. W. James, T. S. Park, A. Nguyen, T. Tawonsawatruk, L. Lazzari, C. Soo and B. Peault, Cell. Mol. Life Sci., 2014, 71, 1353–1374 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- M. Prewitz, F. P. Seib, T. Pompe and C. Werner, Macromol. Rapid Commun., 2012, 33, 1420–1431 CrossRef CAS PubMed.
- M. Ehrbar, S. C. Rizzi, R. Hlushchuk, V. Djonov, A. H. Zisch, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomaterials, 2007, 28, 3856–3866 CrossRef CAS PubMed.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. S. Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS PubMed.
- M. Ehrbar, A. Sala, P. Lienemann, A. Ranga, K. Mosiewicz, A. Bittermann, S. C. Rizzi, F. E. Weber and M. P. Lutolf, Biophys. J., 2011, 100, 284–293 CrossRef CAS PubMed.
- P. S. Lienemann, M. Karlsson, A. Sala, H. M. Wischhusen, F. E. Weber, R. Zimmermann, W. Weber, M. P. Lutolf and M. Ehrbar, Adv. Healthcare Mater., 2013, 2, 292–296 CrossRef CAS PubMed.
- V. Milleret, B. R. Simona, P. S. Lienemann, J. Voros and M. Ehrbar, Adv. Healthcare Mater., 2014, 3, 508–514 CrossRef CAS PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Sci. Transl. Med., 2012, 4, 160ps24 Search PubMed.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- A. Papadimitropoulos, E. Piccinini, S. Brachat, A. Braccini, D. Wendt, A. Barbero, C. Jacobi and I. Martin, PLoS One, 2014, 9, e102359 Search PubMed.
- T. M. Keenan and A. Folch, Lab Chip, 2008, 8, 34–57 RSC.
- S. A. DeLong, J. J. Moon and J. L. West, Biomaterials, 2005, 26, 3227–3234 CrossRef CAS PubMed.
- T. A. Kapur and M. S. Shoichet, J. Biomater. Sci., Polym. Ed., 2003, 14, 383–394 CrossRef CAS PubMed.
- L. Liu, B. D. Ratner, E. H. Sage and S. Jiang, Langmuir, 2007, 23, 11168–11173 CrossRef CAS PubMed.
- T. J. Stefonek and K. S. Masters, Wound Repair Regen., 2007, 15, 847–855 CrossRef PubMed.
- T. J. Stefonek-Puccinelli and K. S. Masters, Ann. Biomed. Eng., 2008, 36, 2121–2133 CrossRef PubMed.
- Y. Aizawa and M. S. Shoichet, Biomaterials, 2012, 33, 5198–5205 CrossRef CAS PubMed.
- Y. Aizawa, R. Wylie and M. Shoichet, Adv. Mater., 2010, 22, 4831–4835 CrossRef CAS PubMed.
- R. G. Wylie, S. Ahsan, Y. Aizawa, K. L. Maxwell, C. M. Morshead and M. S. Shoichet, Nat. Mater., 2011, 10, 799–806 CrossRef CAS PubMed.
- N. Ferrara, K. Carver-Moore, H. Chen, M. Dowd, L. Lu, K. S. O'Shea, L. Powell-Braxton, K. J. Hillan and M. W. Moore, Nature, 1996, 380, 439–442 CrossRef CAS PubMed.
- M. Hellstrom, M. Kalen, P. Lindahl, A. Abramsson and C. Betsholtz, Development, 1999, 126, 3047–3055 CAS.
- P. Lindahl, B. R. Johansson, P. Leveen and C. Betsholtz, Science, 1997, 277, 242–245 CrossRef CAS.
- F. Shalaby, J. Rossant, T. P. Yamaguchi, M. Gertsenstein, X. F. Wu, M. L. Breitman and A. C. Schuh, Nature, 1995, 376, 62–66 CrossRef CAS PubMed.
- S. G. Ball, C. A. Shuttleworth and C. M. Kielty, J. Cell Biol., 2007, 177, 489–500 CrossRef CAS PubMed.
- J. Andrae, R. Gallini and C. Betsholtz, Genes Dev., 2008, 22, 1276–1312 CrossRef CAS PubMed.
- P. Carmeliet, Y. S. Ng, D. Nuyens, G. Theilmeier, K. Brusselmans, I. Cornelissen, E. Ehler, V. V. Kakkar, I. Stalmans, V. Mattot, J. C. Perriard, M. Dewerchin, W. Flameng, A. Nagy, F. Lupu, L. Moons, D. Collen, P. A. D'Amore and D. T. Shima, Nat. Med., 1999, 5, 495–502 CrossRef CAS PubMed.
- I. Stalmans, Y. S. Ng, R. Rohan, M. Fruttiger, A. Bouche, A. Yuce, H. Fujisawa, B. Hermans, M. Shani, S. Jansen, D. Hicklin, D. J. Anderson, T. Gardiner, H. P. Hammes, L. Moons, M. Dewerchin, D. Collen, P. Carmeliet and P. A. D'Amore, J. Clin. Invest., 2002, 109, 327–336 CrossRef CAS.
- C. Ruhrberg, H. Gerhardt, M. Golding, R. Watson, S. Ioannidou, H. Fujisawa, C. Betsholtz and D. T. Shima, Genes Dev., 2002, 16, 2684–2698 CrossRef CAS PubMed.
- P. Lindblom, H. Gerhardt, S. Liebner, A. Abramsson, M. Enge, M. Hellstrom, G. Backstrom, S. Fredriksson, U. Landegren, H. C. Nystrom, G. Bergstrom, E. Dejana, A. Ostman, P. Lindahl and C. Betsholtz, Genes Dev., 2003, 17, 1835–1840 CrossRef CAS PubMed.
- D. H. Nguyen, S. C. Stapleton, M. T. Yang, S. S. Cha, C. K. Choi, P. A. Galie and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6712–6717 CrossRef CAS PubMed.
- M. Martins, S. Warren, C. Kimberley, A. Margineanu, P. Peschard, A. McCarthy, M. Yeo, C. J. Marshall, C. Dunsby, P. M. French and M. Katan, J. Cell Sci., 2012, 125, 5758–5769 CrossRef CAS PubMed.
- I. C. Schneider and J. M. Haugh, J. Cell Biol., 2005, 171, 883–892 CrossRef CAS PubMed.
- U. K. Ljungberg, B. Jansson, U. Niss, R. Nilsson, B. E. Sandberg and B. Nilsson, Mol. Immunol., 1993, 30, 1279–1285 CrossRef CAS.
- A. Sala, P. Hanseler, A. Ranga, M. P. Lutolf, J. Voros, M. Ehrbar and F. E. Weber, Integr. Biol., 2011, 3, 1102–1111 RSC.
- B. Carrion, Y. P. Kong, D. Kaigler and A. J. Putnam, Exp. Cell Res., 2013, 319, 2964–2976 CrossRef CAS PubMed.
- Q. Shen, Y. Wang, E. Kokovay, G. Lin, S. M. Chuang, S. K. Goderie, B. Roysam and S. Temple, Cell Stem Cell, 2008, 3, 289–300 CrossRef CAS PubMed.
- B. M. Baker, B. Trappmann, S. C. Stapleton, E. Toro and C. S. Chen, Lab Chip, 2013, 13, 3246–3252 RSC.
- B. Carrion, C. P. Huang, C. M. Ghajar, S. Kachgal, E. Kniazeva, N. L. Jeon and A. J. Putnam, Biotechnol. Bioeng., 2010, 107, 1020–1028 CrossRef CAS PubMed.
- M. Tavazoie, L. Van der Veken, V. Silva-Vargas, M. Louissaint, L. Colonna, B. Zaidi, J. M. Garcia-Verdugo and F. Doetsch, Cell Stem Cell, 2008, 3, 279–288 CrossRef CAS PubMed.
- R. Z. Lin, R. Moreno-Luna, D. Li, S. C. Jaminet, A. K. Greene and J. M. Melero-Martin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10137–10142 CrossRef CAS PubMed.
- S. Metzger, P. S. Lienemann, C. Ghayor, W. Weber, I. Martin, F. E. Weber and M. Ehrbar, Adv. Healthcare Mater., 2014 DOI:10.1002/adhm.201400547.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ib00152d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |