
Riboregulation of the bacterial actin-homolog MreB by DsrA small noncoding RNA†
Bastien
Cayrol
ab,
Emilie
Fortas
ab,
Claire
Martret
ab,
Grzegorz
Cech
c,
Anna
Kloska
c,
Stephane
Caulet
abd,
Marion
Barbet
ab,
Sylvain
Trépout
ef,
Sergio
Marco
ef,
Aziz
Taghbalout
g,
Florent
Busi
dh,
Grzegorz
Wegrzyn
c and
Véronique
Arluison
*abd
aLaboratoire Léon Brillouin, CEA – Centre de Saclay, 91191 Gif-sur-Yvette, France. E-mail: Arluison@univ-paris-diderot.fr; Fax: +33 1 69 08 95 36; Tel: +33 1 69 08 32 82
bUMR 12 CEA/CNRS, 91191 Gif-sur-Yvette, France
cDepartment of Molecular Biology, University of Gdańsk, Gdańsk, Poland
dUniv Paris Diderot, Sorbonne Paris Cité, 75013 Paris, France
eInstitut Curie, Campus Universitaire d'Orsay, 91405 Orsay Cedex, France
fINSERM U759, Campus Universitaire d'Orsay, 91405 Orsay Cedex, France
gDepartment of Molecular Biology and Biophysics, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06032, USA
hUnité de Biologie Fonctionnelle et Adaptative (BFA) UMR 8251 CNRS, 75205 Paris, France
First published on 5th November 2014
Abstract
The bacterial actin-homolog MreB is a key player in bacterial cell-wall biosynthesis and is required for the maintenance of the rod-like morphology of Escherichia coli. However, how MreB cellular levels are adjusted to growth conditions is poorly understood. Here, we show that DsrA, an E. coli small noncoding RNA (sRNA), is involved in the post-transcriptional regulation of mreB. DsrA is required for the downregulation of MreB cellular concentration during environmentally induced slow growth-rates, mainly growth at low temperature and during the stationary phase. DsrA interacts in an Hfq-dependent manner with the 5′ region of mreB mRNA, which contains signals for translation initiation and thereby affects mreB translation and stability. Moreover, as DsrA is also involved in the regulation of two transcriptional regulators, σS and the nucleoid associated protein H–NS, which negatively regulate mreB transcription, it also indirectly contributes to mreB transcriptional down-regulation. By using quantitative analyses, our results evidence the complexity of this regulation and the tangled interplay between transcriptional and post-transcriptional control. As transcription factors and sRNA-mediated post-transcriptional regulators use different timescales, we propose that the sRNA pathway helps to adapt to changes in temperature, but also indirectly mediates long-term regulation of MreB concentration. The tight regulation and fine-tuning of mreB gene expression in response to cellular stresses is discussed in regard to the effect of the MreB protein on cell elongation.
Insight, innovation, integrationThe actin-like MreB protein determines the bacterial cell shape by directing cell-wall biosynthesis. In this paper we show that DsrA, a small noncoding RNA, is involved in the post-transcriptional regulation of mreB expression in Escherichia coli. Our quantitative analysis sheds light on the interplay between transcriptional and post-transcriptional regulation of mreB in response to cellular stresses. In particular, to detect subtle morphological changes associated with this regulation, we used our experience in cryo-transmission electron microscopy to obtain sharp measurements on frozen/hydrated E. coli K-12 whole cells, together with statistical analysis to unveil morphological differences in cell population. Our work provides, for the first time, an integrative analysis of mreB expression regulation during cellular stresses in light of the associated alterations in cell morphology. |
Introduction
Small regulatory noncoding RNAs (sRNA) range from ∼40 to 400 nucleotides and are used by bacteria as environmental response actors (for recent reviews see ref. 1–3). Most of these RNAs are trans-encoded and transcribed under cellular stresses. They act by an antisense mechanism via base-pairing to their target mRNA to form imperfect short duplexes that require the RNA chaperone Hfq protein for their stabilization and proper function in Gram negative bacteria.4,5 mRNA–sRNA interactions usually take place in the vicinity of the ribosome binding site (rbs) and thereby modulate translation efficiency and mRNA stability, for instance by exposing a cleavage site for RNaseE.3 One of the best-characterized Escherichia coli sRNA is DsrA, an 87 nucleotides RNA encoded by a gene located in the downstream region of rcsA, which encodes a transcriptional activator of polysaccharide capsule synthesis genes.6 DsrA was shown to sense environmental changes through its inherent temperature-sensitive transcription initiation and to stimulate the expression of the major stress regulator sigma factor σS.7rpoS mRNA (encoding σS) usually forms a secondary structure within its 5′-untranslated region, which results in poor translation due to weak ribosome accessibility to the rbs. Nevertheless, efficient rpoS translation takes place upon disruption of the secondary structure within the 5′ regulatory region by the Hfq-mediated DsrA-binding to an upstream sequence in the 5′-untranslated leader region of rpoS mRNA.8–10 Hence, transcription of σS-dependent genes is induced at low temperature in the exponential phase and during the stationary phase, a process that permits the cell to adapt to these stresses.11–14Many sRNAs have multiple targets and can therefore affect the translation of multiple mRNAs to coordinate or synchronize different cellular pathways. For example, DsrA binds to both hns and rpoS mRNAs and thus influences the expression of two global transcriptional regulators, leading to a reduction in cellular levels of the H–NS histone-like nucleoid structuring protein and to an accumulation of σS.6,15 Thus by acting on transcription factors, these sRNA-based regulation pathways result in important effects at the transcriptional level.16 In addition, the regulation of a specific mRNA by multiple sRNAs can also occur. For instance, at least four different sRNAs (DsrA, OxyS, RprA and ArcZ) have been shown to control rpoS expression under different environmental stresses.17,18
As many sRNAs accumulate during the stationary phase of growth when bacteria stop dividing,19 it was proposed that sRNAs could influence the expression of genes coding for cell division proteins.20 For example, the tubulin-like FtsZ that directs formation of the cytokinetic ring21 is thought to be regulated at the translational level by the DicF sRNA in a process that requires Hfq.20,22,23 Similarly, an Hfq-dependent negative post-transcriptional regulation of the actin-homolog MreB was previously reported but the putative sRNA(s) involved in the process was not identified.20
MreB plays an essential role in maintaining the rod shape of E. coli.24–26 Current models propose that MreB forms membrane-associated patches,27 helps to organize the membrane in domains28 and that together with the transmembrane proteins MreC, MreD or RodA, plays an important role in the peptidoglycan synthesis required for cell shape maintenance (for recent reviews see ref. 26 and 29). The interplay between MreB and the enzymes producing peptidoglycan helps to maintain the bacterial cell shape. When MreB concentration declines or when its polymerization is inhibited, the cell loses its rod shape and becomes round like cocci that naturally lack MreB.30
How mreB gene expression is regulated is not well understood. Three σ70-dependent promoters contribute to mreB expression in E. coli (Fig. S1, ESI†).31 The mreB gene, in many bacteria, is found in the same operon as mreC and mreD genes, which code for proteins required for coordinating peptidoglycan insertion.31 Most mreB transcripts of the mre operon are monocistronic, while polycistronic mreBCD mRNA seems to be scarce.31 Under stress response, BolA, whose expression is driven by the σS factor, binds mreB promoters and represses mreB transcription.32–34 This indicates that stresses, including entry into the stationary phase, lead to a reduced MreB protein level.34
In this study, we identify a newly discovered pathway of post-transcriptional regulation of MreB cellular levels, where Hfq-mediated interaction of the stress-induced DsrA with mreB transcript interferes with mreB expression. We propose a model for mreB gene regulation in which DsrA acts directly as an antisense sRNA to down-regulate the mreB mRNA translation and stability. Furthermore, DsrA is able to indirectly influence σS and H-NS-dependent inhibition of mreB transcription. We discuss here how this regulation affects the elongation of dividing E. coli cells.
Results
Hfq and DsrA downregulate the expression of mreB
DsrA, an E. coli sRNA, was previously shown to be transcribed under natural environmental changes. DsrA concentration increases when temperature drops off and during the stationary phase.7,13,35 Because we observed reduced MreB levels in cells grown at low temperature and during the stationary phase, we sought to determine the role of induced dsrA expression on MreB protein levels. We first performed quantitative Western Blot analyses of MreB on total protein extracts from wild-type cells and mutants that lacked DsrA or Hfq. This was tested in wild-type strains, and in dsrA or hfq mutants, since Hfq is a known cofactor for DsrA. This showed, and consistent with a previous report,20 that growth at low temperature or during the stationary phase is associated with decreased levels of MreB in wild-type cells (Fig. 1, WT columns). Exponentially growing cells showed normal concentration of MreB at 37 °C regardless of the presence of DsrA or Hfq. In contrast, at 16 °C and during the stationary phase, cells that lacked either DsrA or Hfq showed 30 to 50% more MreB than the respective wild-type cells (Fig. 1), suggesting that DsrA was responsible for part (stationary phase) or all (16 °C) of the decline.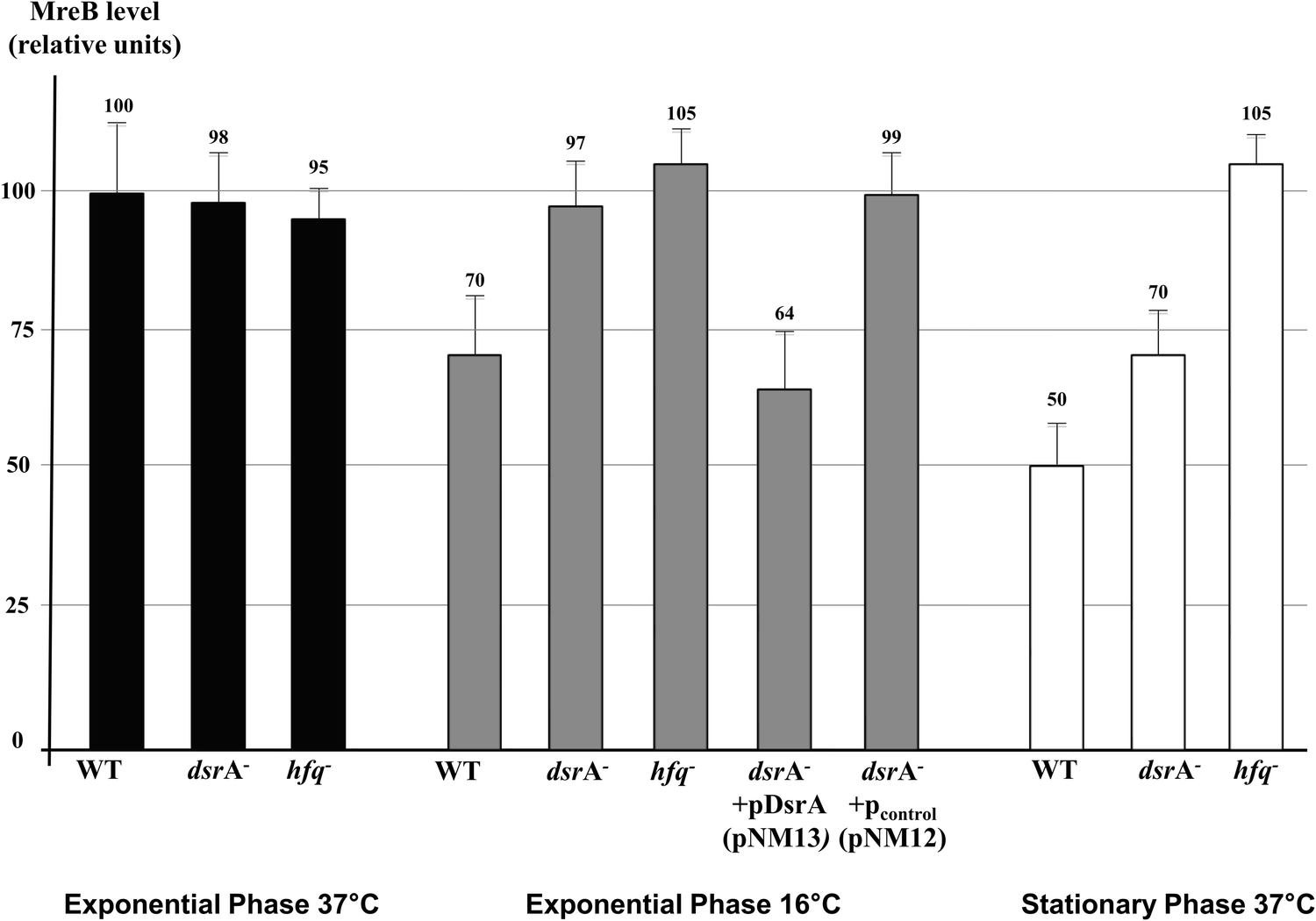 | ||
| Fig. 1 Influence of stresses on MreB cellular concentration. Quantitative Western Blot using a purified anti-MreB antibody and total cell extracts of the indicated strains is presented as relative MreB levels. Wild type and mutants were grown to the exponential phase at 37 °C (black) and 16 °C (grey), or to the stationary phase at 37 °C (empty). WT: DJ480; dsrA−: NM317 (DJ480ΔdsrA) and hfq−: DJ480 hfq::cm. Except otherwise indicated, the DJ480, NM317 and DJ480 hfq::cm were used. Standard deviation from 3 independent samples is shown in all experiments. | ||
Because the observed decrease in cellular MreB could result from other effects of cold or stationary phase-associated stress signals not necessarily mediated by dsrA expression, we tested the effect of heterologous expression of dsrA on MreB levels. The expression of dsrA in wild-type cells at 37 °C from either an arabinose inducible (PBAD) or a constitutive (PLlacO-1) promoters showed decrease in MreB levels (Fig. S2, ESI†). The fact that down-regulation of MreB levels was obtained strongly suggests that expression of dsrA leads to reduced MreB levels independent of other cellular stresses. Furthermore, the expression of the dsrA gene in ΔdsrA cells from an arabinose inducible promoter at 16 °C resulted in similar decrease in MreB concentration as seen in wild-type cells grown at 16 °C (Fig. 1). The dsrA gene expression from a PBAD arabinose induced promoter was carried out under conditions that give an rpoS-lacZ fusion activity comparable to that seen from the single-copy chromosomal dsrA.8
To test the effect of cold-induced dsrA expression on mreB transcription, we used plasmid born mreB-lacZ transcriptional fusions where one or more mreB promoters (P1mreB, P2mreB & P3mreB), and the native ribosome binding site and start codon (AUG) were fused to the lacZ coding region (see Fig. S1, ESI†).36 As previously reported, the transcription activity from the mreB promoter was reduced to approximately 50% in the absence of P3mreB or P2mreB and P3mreB, as indicated by β-galactosidase assays (Fig. S3, ESI,† columns 1, 6 and 10).36 Interestingly, the effects of cold-induced expression of dsrA at 16 °C on mreB expression were within 10–20%, independent of the presence of one or more mreB promoters (PmreB) (Fig. S3, ESI,† compare columns 1–2, 6–7 and 10–11). Furthermore, expression of dsrA under the control of the inducible promoter in ΔdsrA cells gave similar results as WT cells (Fig. S3, ESI,† compare columns 1–3, 6–8 and 10–12). This indicates that dsrA expression down regulates mreB expression and that the region containing P2mreB and P3mreB does not have a major effect for the observed DsrA-mediated effect on mreB expression in vivo.
To establish whether the predicted DsrA regulation affects mreB mRNA translation, we used an in-frame translational mreB-lacZ reporter fusion expressed under the control of a PBAD inducible promoter in wild-type cells and mutants that lacked DsrA or Hfq. The cellular levels of the MreB-lacZ protein would therefore be altered only by post-transcriptional regulatory event(s). The mreB-lacZ fusion includes only a P1mreB transcription initiation site as DsrA mediated down regulation of mreB expression was independent of P2mreB and P3mreB (see Fig. S1 and S3, ESI†). This showed, similar to the quantitative immunoblot of MreB (Fig. 1), no significant changes in MreB levels in ΔdsrA cells growing exponentially at 37 °C as indicated by the β-galactosidase levels (Fig. 2). However, at low temperature or in the stationary phase, when DsrA is efficiently produced in wild-type cells, deletion of dsrA increased the expression of the translational fusion by a factor 2 or 1.7, respectively (Fig. 2). The lack of Hfq resulted in a significant increase of mreB-lacZ mRNA translation in cells grown under normal and stress conditions, suggesting a possible additional negative effect of Hfq on mreB-lacZ mRNA stability and/or translation independent of DsrA. Although the expression of the fusion was significantly reduced during the stationary phase, the lack of DsrA or Hfq resulted in the same trend and led also to a significant increase of the mreB-lacZ expression. A possible reason for low activity during the stationary phase could be due to generally less efficient anabolic processes in bacteria in the stationary phase, thus experiencing nutritional limitations. However, the low activity was not due to consumption of an inducer as arabinose was added to harvested stationary phase cells.
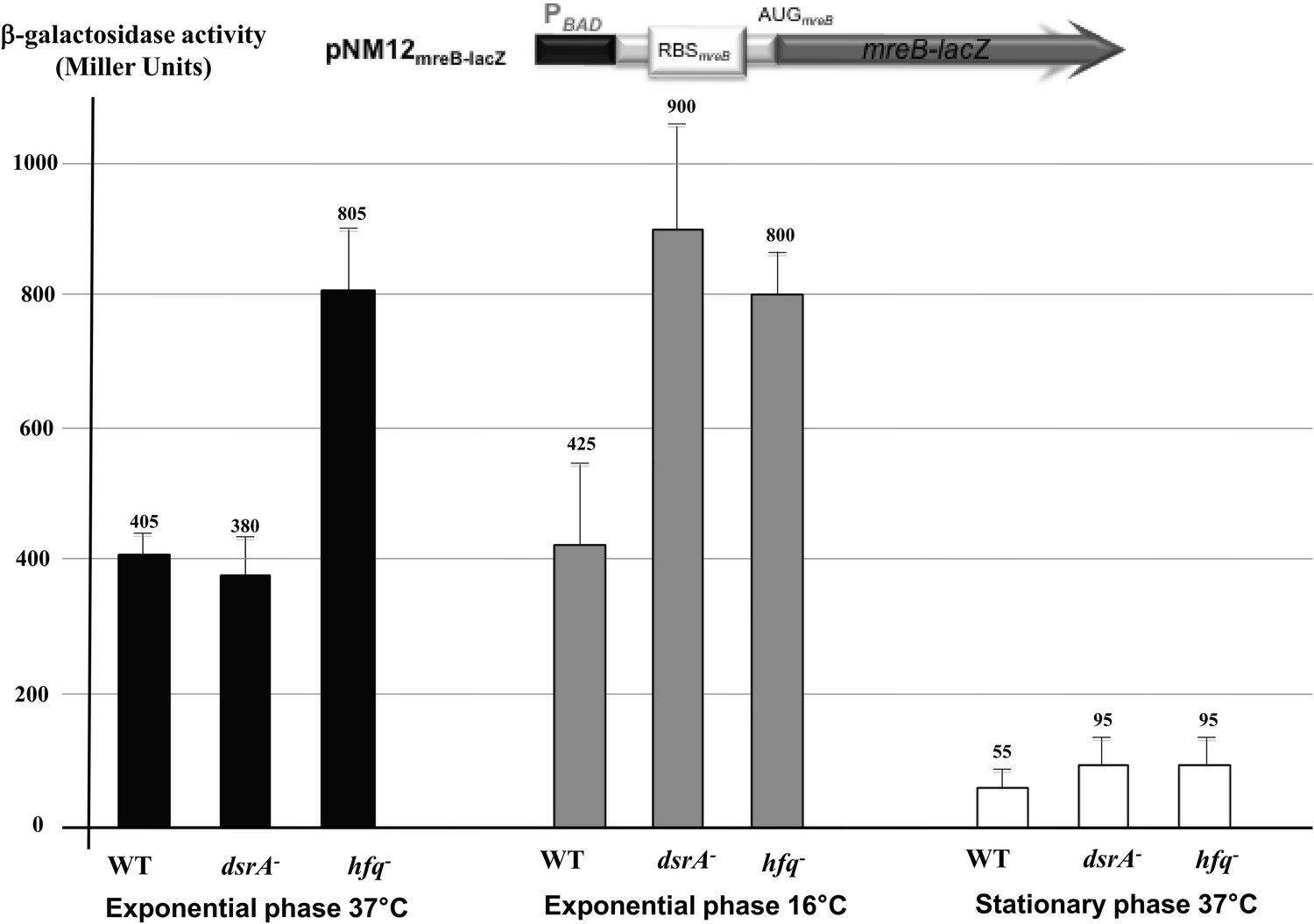 | ||
| Fig. 2 DsrA-mediated riboregulation on mreB-lacZ translational reporter fusion (drawing is a schematic representation of the mreB-lacZ reporter fusion). Transcription of the plasmid encoded mreB-lacZ fusion is driven by a PBAD promoter in the presence of 0.001% L-arabinose. WT: DJ480/pNM12mreB-lacZ; dsrA−: DJ480ΔdsrA/pNM12mreB-lacZ and hfq−: DJ480 hfq::cm/pNM12mreB-lacZ. β-galactosidase activities in extracts from cells that were grown to the exponential phase at 37 °C (black) and 16 °C (grey) or to the stationary phase at 37 °C (empty) are shown. | ||
DsrA regulates the expression of mreB through direct base pairing interaction
Evidence that DsrA, as a small noncoding RNA, could interfere with mreB expression through direct base pairing with mreB mRNA came from RNA duplex prediction obtained by using a RNAfold web server.37 This showed a predicted model for DsrA:mreB RNA duplex structure where DsrA interacts with site of mreB mRNA that includes mreB translation initiation codon (Fig. 3), suggesting that DsrA-binding could strongly interfere with initiation of mreB mRNA translation. This model supports the conclusion drawn from DsrA-mediated reduction of β-galactosidase activity in the mreB-lacZ fusions.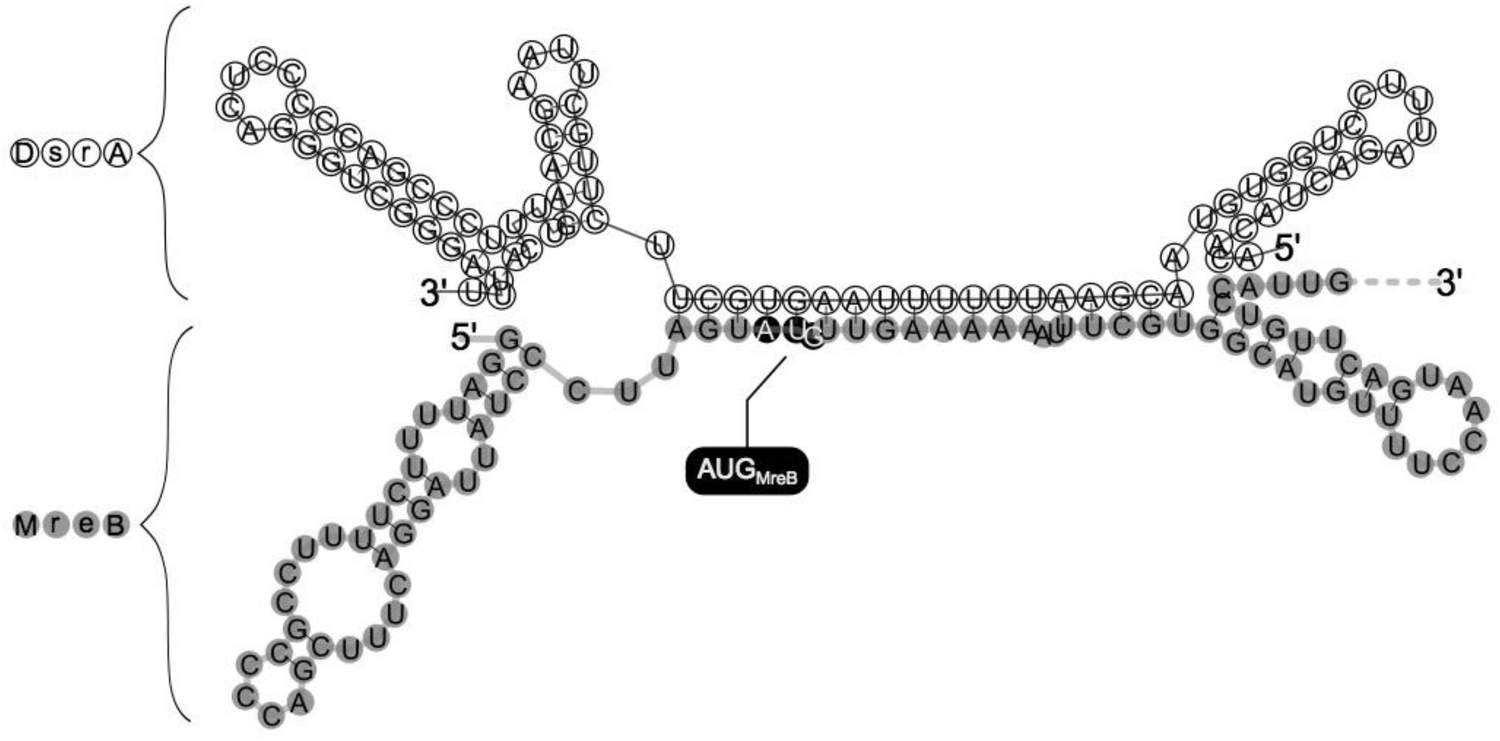 | ||
| Fig. 3 Model for the DsrA-MreB RNA duplex. The secondary structure model of the mreB:DsrA complex was predicted by using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). mreB mRNA is shaded in grey and mreB Start codon (AUG) is highlighted. Note that the region of mreB pairing within DsrA includes part of the region used for pairing with hns and rpoS.8,15 | ||
To test this model for direct mreB:DsrA annealing, we asked whether the DsrA-mediated regulation of MreB levels is affected by mutations in dsrA that alter mreB-DsrA annealing and by mutations in mreB that restore annealing to the mutant DsrA (Fig. S4, ESI†). We thus compared β-galactosidase activity in ΔdsrA cells that expressed a combination of plasmid encoded mreB-lacZ fusion and/or a dsrA allele under the control of PBAD promoters at 37 °C. As shown in Fig. 4, expression of mreBWT-lacZ was significantly reduced upon transcription of dsrAWT (repression of 90%), while the mutant derivative of DsrA where the region involved in the predicted annealing with mreB mRNA was mutated (DsrAmut) did not affect the β-galactosidase activity from mreBWT-lacZ fusion (repression of 9%, Fig. 4). Similarly, when mreB-lacZ fusion carrying compensatory mutation that restores annealing with DsrAmut was expressed, DsrAWT failed to alter mreBmut-lacZ expression whereas DsrAmut restored repression (repression of 70%). Together, these results clearly show that DsrA basepairs with mreB mRNA around the mreB start codon and affects MreB expression, confirming the direct annealing model presented in Fig. 3. The annealing could directly interfere with mreB translation by limiting ribosome access to the translation site. However, DsrA could also interfere with the mreB gene expression by altering mreB mRNA stability.
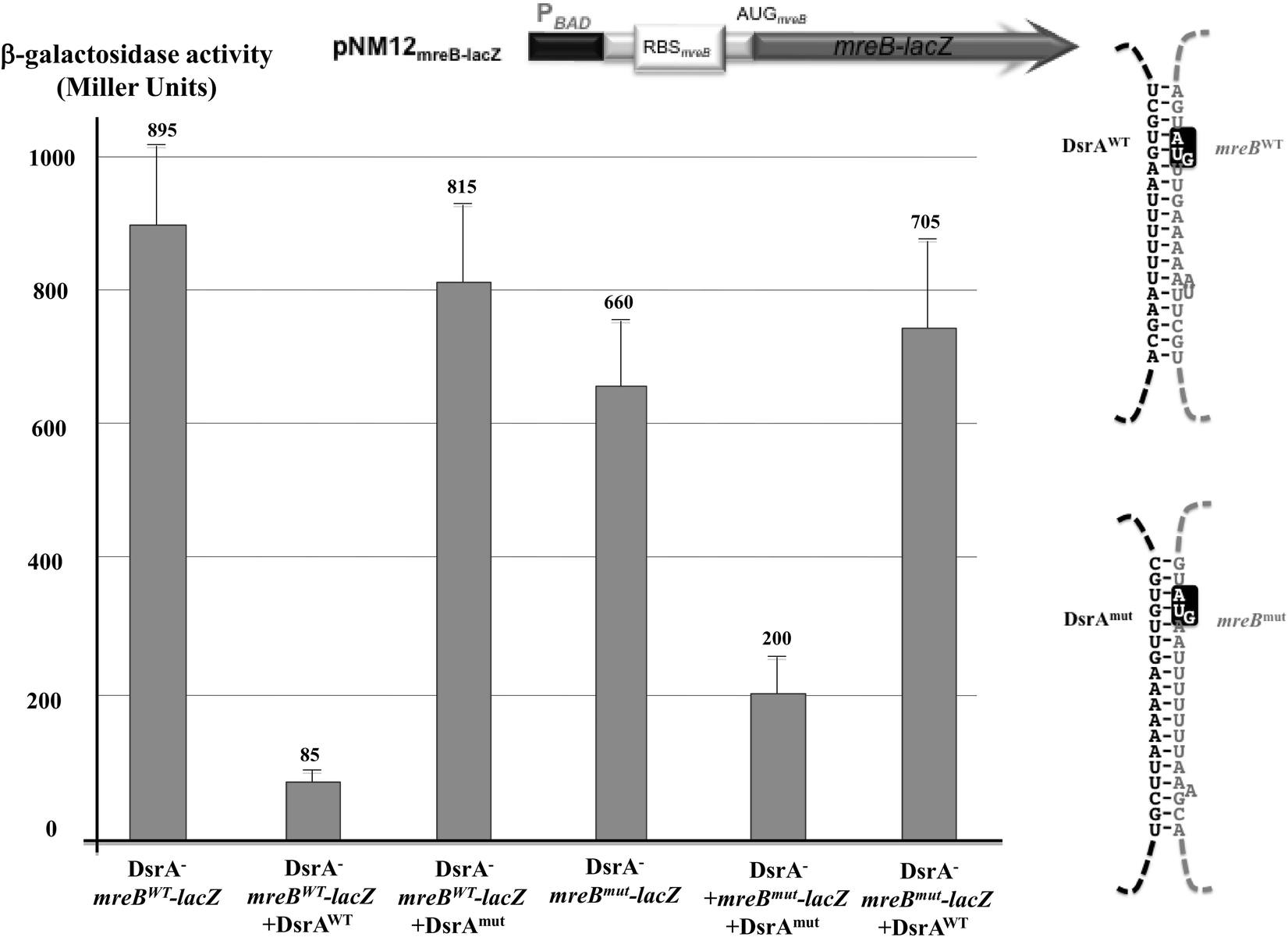 | ||
| Fig. 4 DsrA regulates mreB expression through direct base pairing interaction. Comparison of β-galactosidase activities in extracts from DrsA− cells (NM317) that expressed at 37 °C plasmid encoded wild-type translation reporter fusion (mreBwt-lacZ) or a mutant fusion (mreBmut-lacZ) that carries mutations predicted to prevent annealing with wild-type DsrA (DsrAWT). When indicated these cells also expressed plasmid encoded DsrAWT or a DsrA mutant (DsrAmut) that carries compensatory mutations predicted to restore annealing with mreBmut-lacZ. Drawings are schematic representation of the mreB-lacZ reporter fusions (top) and the expected mreB-DsrA duplex RNAs (right). Transcription of both lacZ and DsrA derivatives is driven by PBAD promoters in the presence of 0.002% L-arabinose. Observation of the activity and regulation required addition of twice as much inducer than in Fig. 2, explaining thus the relatively higher activity. Shown strains from left to right: column 1 (ΔdsrA/PBAD−mreBWT-lacZ); column 2 (ΔdsrA/PBAD−mreBWT-lacZ + PBAD−DsrAWT); column 3 (ΔdsrA/PBAD−mreBWT-lacZ + PBAD−DsrAmut); column 4 (ΔdsrA/PBAD−mreBmut-lacZ); column 5 (ΔdsrA/PBAD−mreBmut-lacZ + PBAD−DsrAmut); column 6 (ΔdsrA/PBAD−mreBmut-lacZ + PBAD−DsrAWT). | ||
DsrA affects the cellular concentration of mreB mRNA
To determine whether DsrA affects mreB mRNA cellular levels, we performed absolute quantifications of DsrA and mreB RNAs by using a reverse transcription quantitative-PCR (RT-qPCR) assay in wild-type cells and mutants that lacked DsrA or Hfq, as previously described.35 This showed in wild-type cells that DsrA was present as 4 copies per cell at 37 °C and that the DsrA level increased to 10 copies per cell at 16 °C (Fig. 5). These results slightly differ from absolute measurements made in the MC4100 strain,35 but are nevertheless in agreement with those reported previously.7,13,35,38 Consistent with a previous report, we also found that DsrA produced due to activity of the chromosomal gene is nearly absent in cells lacking Hfq, probably because of DsrA instability in the absence the Hfq protein.35,39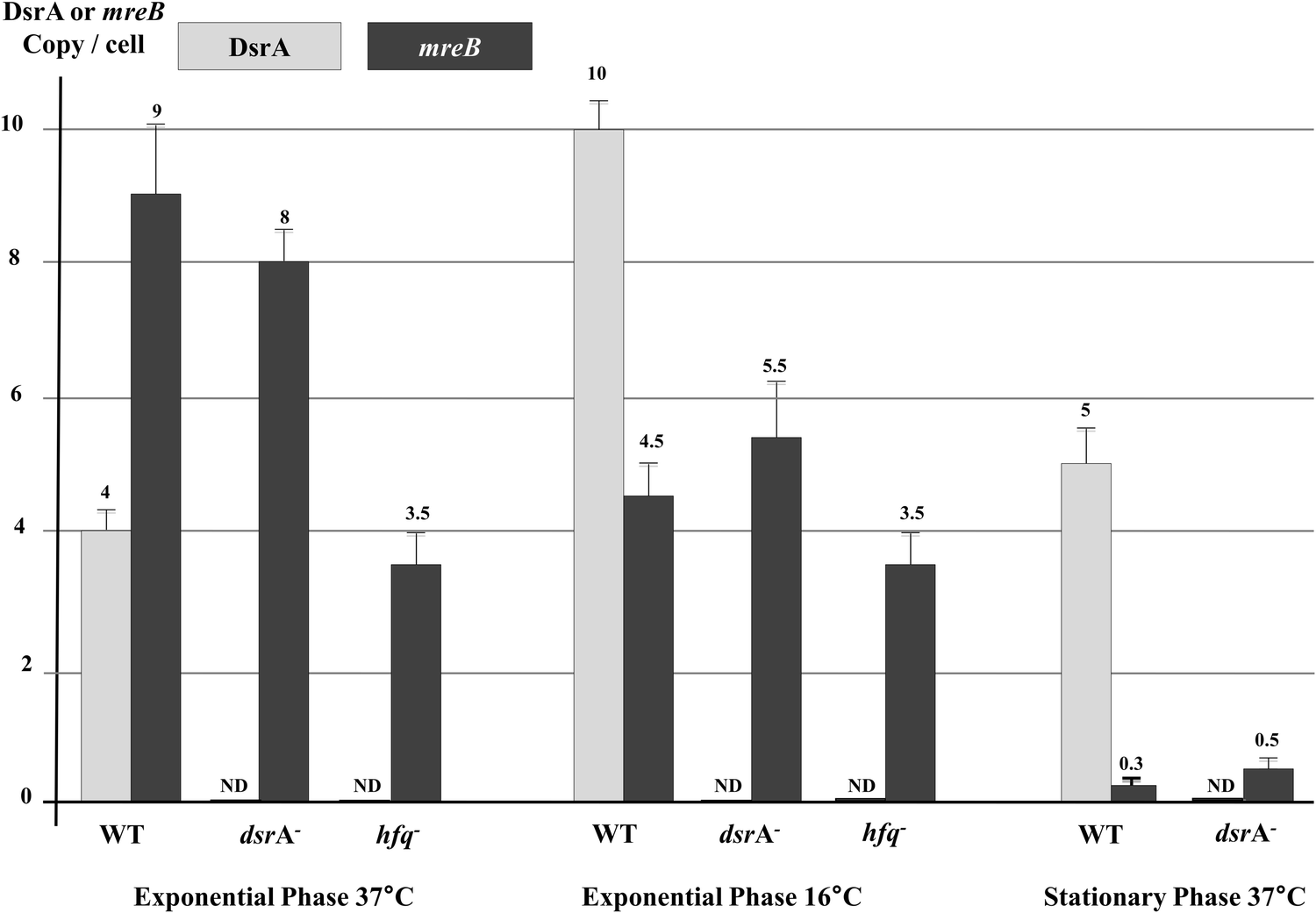 | ||
| Fig. 5 Effect of DsrA on mreB mRNA concentration. Absolute quantifications of DsrA and mreB RNAs were performed by using reverse-transcription quantitative PCR (RT-qPCR)35 and RNA extracted from wild type cells and mutants that lacked DsrA or Hfq. Cells were grown to the exponential phase at 37 °C and 16 °C or the stationary phase at 37 °C. Levels of DrsA (grey) and mreB mRNA (black) are shown. ND: non detectable. | ||
Measurements of cellular mRNA levels showed that the mreB mRNA was present as 9 copies per exponentially growing WT cell at 37 °C (Fig. 5), a level which decreased to less than 5 copies per cell at low temperature (16 °C), suggesting that DsrA negatively influences mreB mRNA concentration. However, cells that lacked DsrA only showed a moderate increase in mreB mRNA cellular abundance at low temperature (∼20% increase).
During the stationary phase, mreB mRNA cellular concentration was greatly reduced as it reached less than one copy per cell. This could reflect a cumulative effect of drsA expression and changes in mreB transcription during the stationary phase of growth (see Discussion). However, the lack of DsrA resulted in an increase of mreB mRNA abundance during the stationary phase (Fig. 5).
As mreB mRNA stability could be affected during cellular stress, we measured mreB mRNA half-life during growth at low temperature. For the stationary phase, mreB mRNA abundance was too low to accurately estimate its half-life (Fig. 5). As shown in Fig. 6, expression of DsrA at low temperature significantly destabilized the mreB transcript as indicated by the 40% decrease of its half-life in WT cells at low temperature. Thus, mreB mRNA was greatly destabilized when DrsA was induced at 16 °C and was significantly stabilized in cells that lacked DsrA.
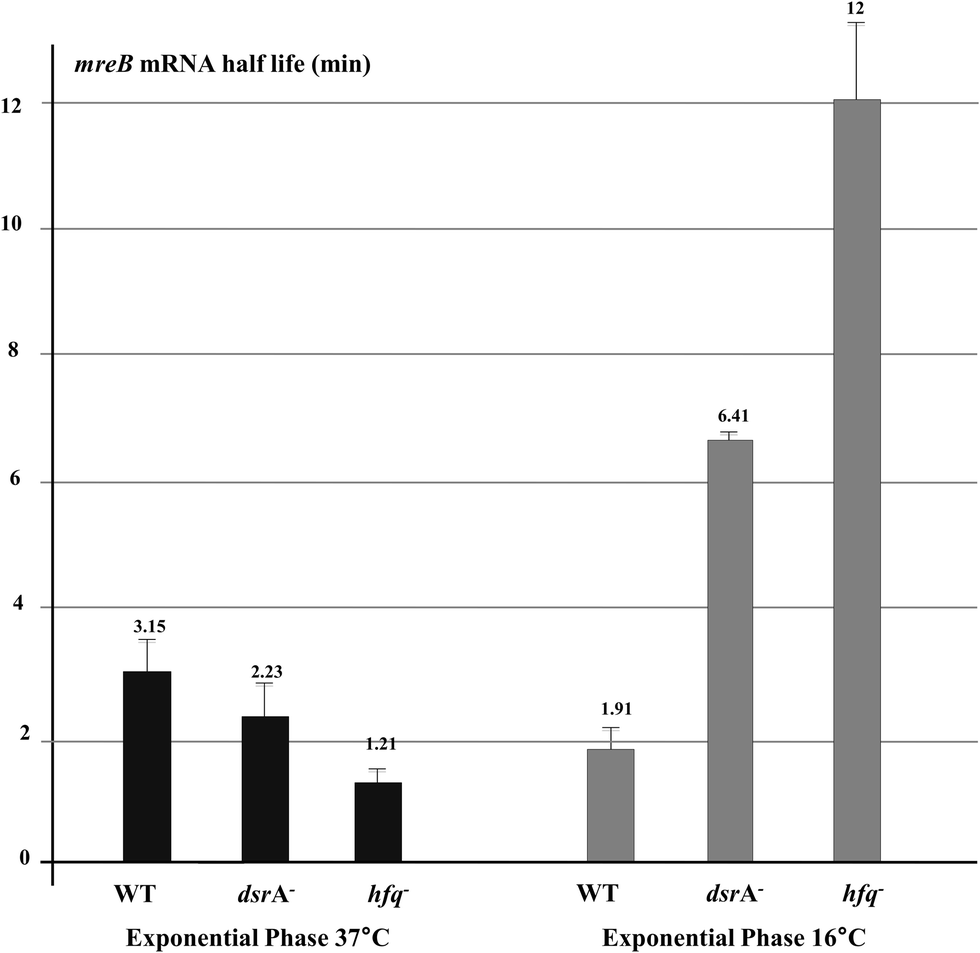 | ||
| Fig. 6 Effect of DsrA and Hfq on mreB mRNA half-life. Relative quantification of mreB mRNA were performed by RT-qPCR assay on RNA extracted from rifampicin-treated wild type cells and mutants that lacked DsrA or Hfq. DJ480 hfq::kan was used for this analysis. Half-life of mreB mRNA from cells grown to exponential phase at 37 °C (black) and 16 °C (grey) is shown. Note that due to the extremely low level of mreB mRNA under the stationary phase (less than 1 copy per cell), accurate half-life measurement for this condition was not possible. | ||
The absence of Hfq resulted in less mreB transcripts under normal exponential growth conditions when compared to wild-type cells (Fig. 5). This was consistent with the increased degradation rate of mreB mRNA as shown by the change of its half-life from 3.15 min in the WT to 1.21 min in the hfq mutant (Fig. 6), thus suggesting a DsrA-independent effect of the RNA chaperone Hfq that stabilizes mreB mRNA under exponential growth conditions at 37 °C. Indeed, it is likely that Hfq protects mreB transcript against RNaseE due to the overlapping cleavage sites and Hfq binding sites on the mRNA.40 Conversely, during exponential growth at 16 °C where the riboregulation by DsrA occurs, the absence of Hfq resulted in a completely different effect. mreB mRNA stability was greatly increased in cells that lacked DsrA or Hfq (Fig. 6). This result confirms that Hfq is required for the DsrA-mediated riboregulation during stress as the absence of Hfq, which enables the sRNA to anneal with its target, results in a stabilization of the mRNA. However, the lack of Hfq produced a significantly greater stabilization of mreB mRNA than the absence of DsrA, suggesting a role of other unidentified Hfq-dependent sRNAs. mreB mRNA destabilization might be mediated by an RNaseE-Hfq ribonucleoprotein complex as is the case of SgrS and RyhB sRNAs1,41 or a passive mechanism due to abortive translation.42
Stress regulation by DsrA influences the elongation of dividing cells
We showed that DsrA mediates posttranscriptional down-regulation of mreB expression, leading to a 30% reduction in cellular MreB (Fig. 1, the WT-exponential phase 37 °C vs. 16 °C). MreB is required for maintenance of the bacterial rod-shape as cells that lose MreB shorten and become round-shaped.26 We therefore investigated whether DsrA-mediated regulation of mreB expression is associated with morphological changes of the cells. We used high-resolution cryo-TEM technique to measure cell length (L) and width (w) for a population of about 50 cells in wild-type, and ΔdsrA cells and in each condition we determined the cell roundness as calculated from the cellular width to length ratio (w/L). The rod-shaped E. coli cell is approximately 1.5 μm long and 0.7 μm wide (roundness ∼0.47). As shown in Fig. 7, exponentially growing WT cells (DJ480) at 37 °C showed two cellular populations: one rod-shaped (∼70%) with a roundness of ∼0.36 (σ2 = 0.05) and the other short (∼30%) with a roundness of ∼0.48 (σ2 = 0.05). Intuitively, we can assume that the short population represents new-born cells whereas long rod population corresponds to cells that had elongated but did not divide. Similarly, exponentially growing WT cells at 16 °C (in which the MreB level is decreased by ∼30%, see Fig. 1) also showed rod-shaped and short populations with a roundness of ∼0.37 (σ2 = 0.05) and 0.49 (σ2 = 0.05), respectively. However, the short population is slightly more abundant than at 37 °C, a result in agreement with reduced MreB levels observed by Western Blot that should affect elongation of dividing cells. Conversely, cells that lacked DsrA (NM317) especially at 16 °C clearly showed mostly one population with a roundness of 0.45 (σ2 = 0.01) at 16 °C and 0.42 (σ2 = 0.01) at 37 °C. This result indicates that in the absence of DsrA, cells remain longer and more rod-shaped. Further evidence for the role of DrsA riboregulation of mreB expression in affecting cellular morphology came from complementation assays of ΔdsrA cells. Synthesis of plasmid-encoded DsrA (pNM13) was induced in ΔdsrA cells by using an arabinose concentration that gave an rpoS-lacZ fusion activity comparable to that seen from the single-copy chromosomal dsrA.8 The dsrA expression restored two cellular populations with a roundness of 0.33 (σ2 = 0.01) and 0.48 (σ2 = 0.01) (Fig. 7) and is consistent with Western Blot analysis (Fig. 1) and mreB-lacZ expression (Fig. S3, ESI†), which showed mreB expression at levels similar to that found in cells in the absence of stress.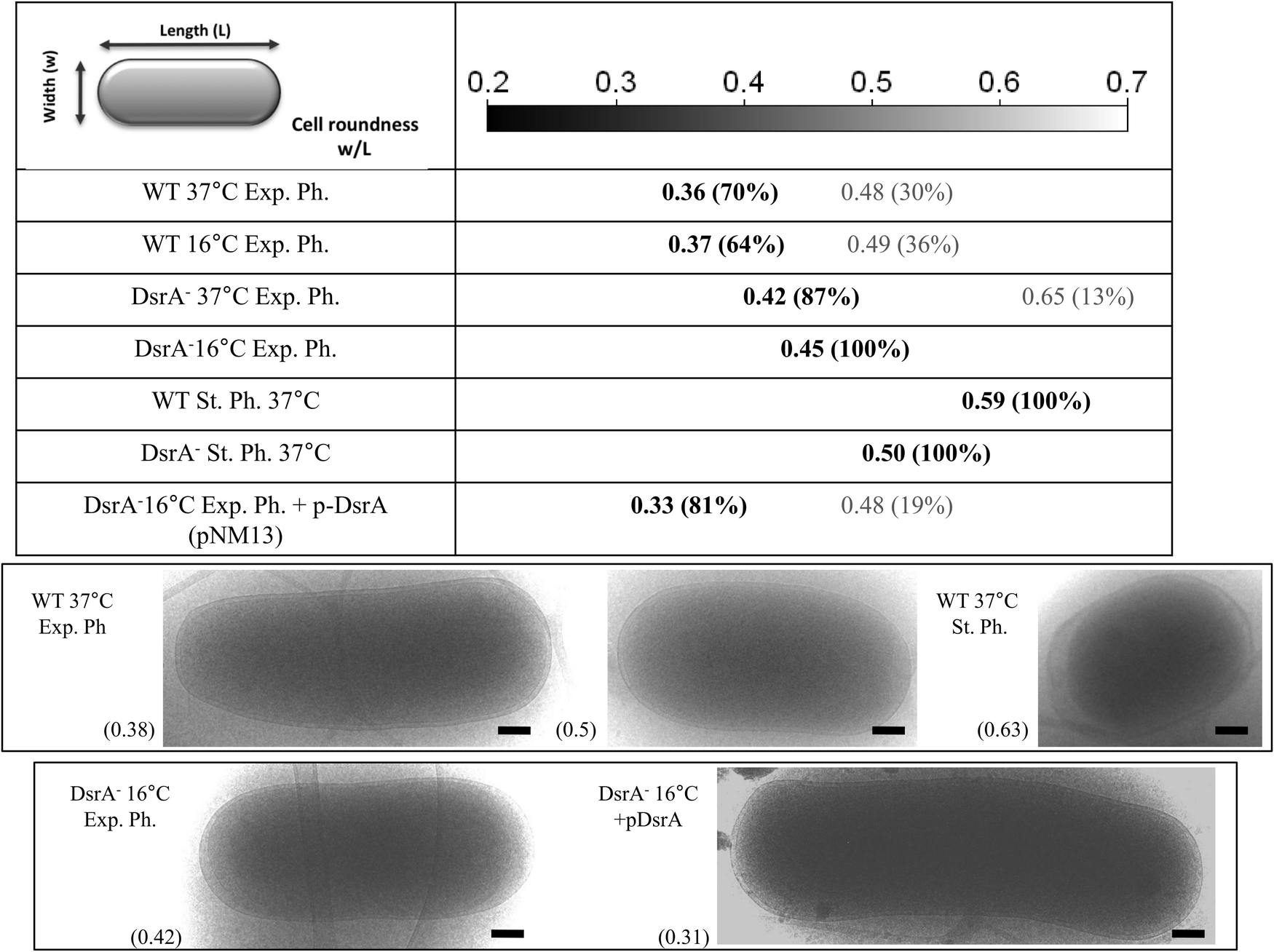 | ||
| Fig. 7 DsrA-mediated riboregulation changes the bacterial cell roundness under stress conditions. Cells were grown to reach the exponential phase at 37 °C and 16 °C or the stationary phase at 37 °C then analyzed by measuring the long and short axis from cryo-TEM images. The statistics made on roundness (defined as the ratio between the shortest and longest axis of the cell computed from cryo-TEM images) for the different cell populations are summarized in the upper part of the figure. Examples of cells are shown in the lower part, with respective roundness indicated into parenthesis. Exp. Ph.: exponential phase of growth; St. Ph.: stationary phase of growth. WT: DJ480; DsrA−: NM317 (DJ480ΔdsrA) and DsrA−p-DsrA: DJ480ΔdsrA/PBAD−dsrA. When indicated plasmid encoded DsrA was expressed in the presence of 0.001% L-arabinose. Scale bar 200 nm. | ||
During the stationary phase, both WT (DJ480) and ΔdsrA (NM317) strains form rounder cells. However ΔdsrA cells were less round than WT cells (roundness of 0.50 (σ2 = 0.01) vs. 0.59 (σ2 = 0.01)). This suggests that, in addition to DsrA riboregulation, other mechanisms affect cell morphology during the stationary phase.
In all, these results confirm that mreB expression regulation by DsrA affects the elongation of dividing E. coli cells during growth at low temperature and the stationary phase.
Because the observed difference in cell morphology could be due to a growth defect in ΔdsrA cells, we therefore compared the growth of wild type and the ΔdsrA mutant at low temperature. This showed that the doubling times were 33 and 32 min at 37 °C, and were 300 and 270 min at 16 °C for wild type and the ΔdsrA mutant, respectively. Consistent with previous reports, the growth rates did not change significantly for the mutant and the wild type strains at either temperature.13,14
Discussion
In this work we studied how DsrA, a small noncoding RNA, regulates the expression of the mreB gene during growth at low temperature and in the stationary phase and how the regulation affects the morphology of dividing E. coli cells (see Table 4 for a summary). Small regulatory RNAs and transcription factors are two means used by the bacterial cell for regulation of gene expression.43mreB was previously shown to be negatively regulated at the transcriptional level by the BolA transcription factor, which interacts with the mreB promoter and alters mreB transcription mainly during the stationary phase.34 Transcription of the bolA gene is driven by the σS factor.44,45 Although all sRNAs involved in rpoS riboregulation during the stationary phase have not been identified, positive regulation by sRNAs indeed results in an increase in BolA concentration, leading to a significant reduction of mreB transcription and consequently in the MreB protein level (Fig. 8), which causes rounder morphology.34,45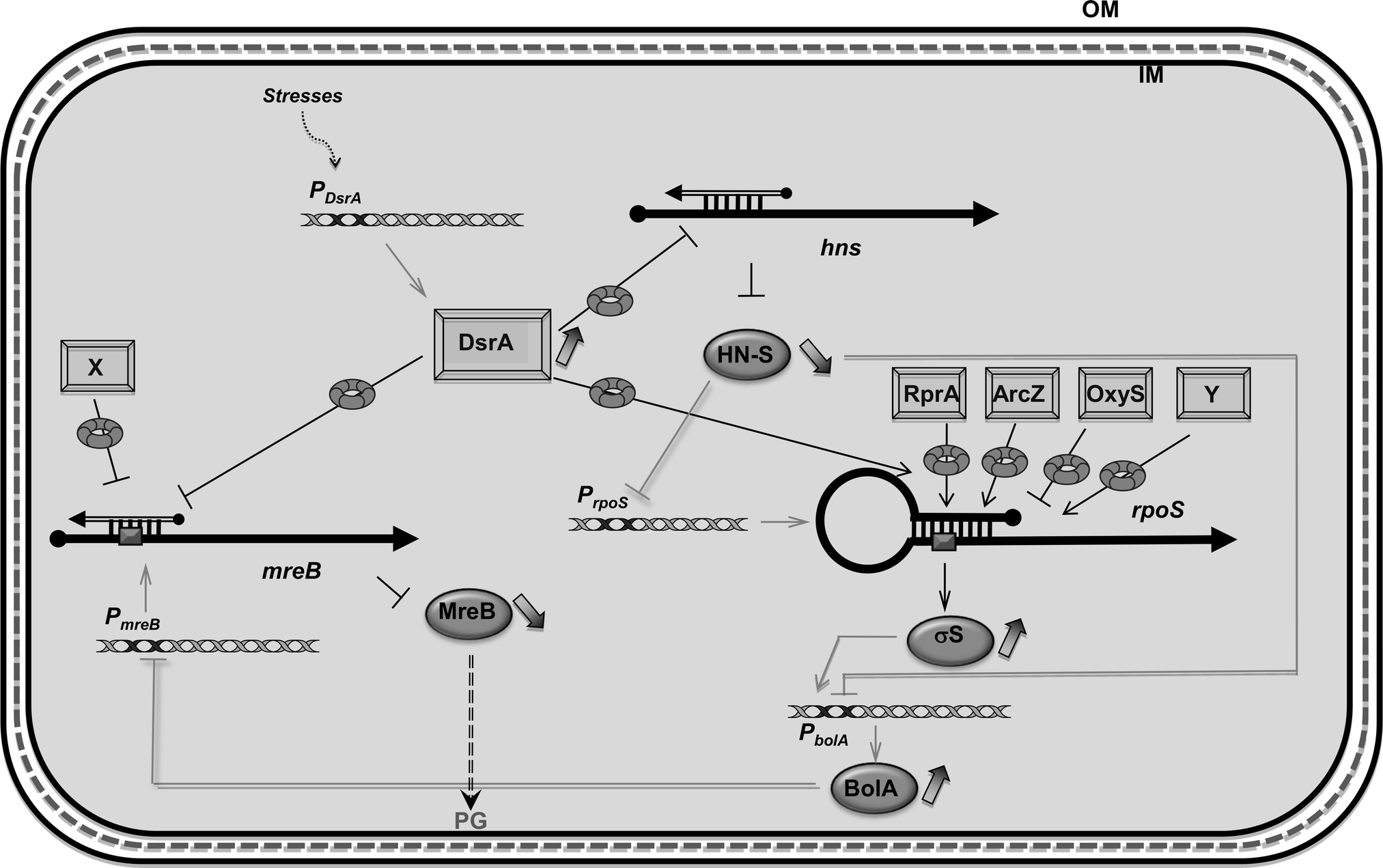 | ||
| Fig. 8 Network of DsrA-dependent regulations. mreB was previously shown to be regulated at the transcriptional level by the BolA transcription factor. As bolA gene is under the control of one main promoter driven by σS factor, its expression is itself dependent on sRNAs involved in rpoS post-transcriptional regulation in response to several stresses (DsrA, RprA, ArcZ and oxyS). Since the regulator and DNA organizer H-NS represses transcription of bolA and rpoS, DsrA also indirectly reduces the expression of mreB via repression of hns translation by DsrA (note that the nature of the DsrA:hns complex is still unclear and that it has been proposed that the 3′ UTR of hns could be required for its post-transcriptional regulation).65 Here we show that mreB translation is also directly repressed by the use of DsrA sRNA. sRNA regulators controlling mRNAs are depicted as rectangles and open arrows (X and Y, unidentified sRNAs); Hfq as a toroidal hexamer; σS, MreB and BolA proteins as grey ellipses, mRNAs as thick black lines; mRNA translation initiation region as a dark grey box; 5′ and 3′ of the mRNA are depicted by a “ball and arrow head”, respectively; transcriptional regulation are represented as thin grey lines; translational regulation as thin black lines; positive and negative regulation are indicated by arrows and horizontal bars, respectively; transcription promoters are indicated as Px; dotted line symbolizes Peptidoglycan (PG) between outer (OM) and inner (IM) membranes. | ||
Furthermore, bolA transcription is directly repressed by the global regulator and DNA organizer H-NS, which itself is downregulated by DsrA sRNA (Fig. 8).15,46 Indeed, as shown in Fig. 5, we observed that in the ΔdsrA strain, growth at low temperature resulted in a modest increase of mreB mRNA concentration (20% more relative to WT strain). Nevertheless, we also observed that during stationary phase, mreB concentrations are very low. The fact that DsrA concentration is only slightly increased during this phase of growth (see Fig. 5)35 suggests that other sRNAs might play an important role in σS production and consequently in bolA transcription.17,18
We show that mreB expression is negatively regulated at the post-transcriptional level by the stress-related noncoding small RNA DsrA, which is involved in rpoS and hns transcription regulation (Fig. 8). We also showed that DsrA regulates the expression of mreB through an Hfq-assisted base pairing that presumably prevents ribosome binding. A similar mechanism was shown for several negatively acting sRNAs.47 Consistent with this, we showed that mreB translational regulation is abolished in the absence of DsrA sRNA or Hfq. We also showed that DsrA and Hfq influence mreB mRNA concentration and turnover, suggesting a role for DsrA and Hfq in the control of mreB mRNA stability. However, the increase in mreB mRNA concentration associated with the absence of DsrA can result from both BolA-mediated mreB transcriptional regulation via σS and H-NS34 and changes in mreB mRNA stability (Fig. 8). The fact that DsrA regulation was observed with mreB-lacZ fusions whose expression is independent of BolA clearly shows that the observed DsrA-mediated down regulation of MreB synthesis includes a direct effect of DsrA on mreB translation and stability (Fig. 2 and 4).
This second point of control for MreB synthesis, which occurs subsidiarily to transcriptional control, is probably required for the cell as MreB is an abundant protein (in average ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 copies per cell).48 Nevertheless, MreB protein abundance is not necessarily correlated to the cellular concentration of its mRNA. If mreB mRNA is abundant as MreB, its translation and/or stability would unlikely be efficiently regulated by a sRNA of low abundance such as DsrA. Here we measured for the first time the mreB mRNAs steady-state level and showed that mreB mRNA is indeed present at a maximum of 9 copies per cell, a result in agreement with a possible regulation by DsrA. The copy number of mreB mRNA could appear too low for an abundant protein. Nevertheless, mRNA steady state copy number only reflects recent transcription events and can usually not be correlated with the protein concentration, which has a longer lifetime than the bacterial cell cycle and represent accumulated transcriptions.49 In the mreB case, to have nm = 9 mRNA copies per cell implies that the cell needs a transcription rate of 9 × δm, where δm is mreB mRNA degradation rate (m for mreB mRNA). As the degradation rate is the sum of active degradation by RNases and dilution by cell growth, the degradation rate δm = ln2/τhl-RNA + ln2/τcc-RNA where τhl-RNA is the mRNA half-life (3.15 min) and τcc-RNA is the cell doubling time (30 min). This indicates that the cell needs to produce ∼2.2 mRNA molecules per minute or ∼70 per doubling time. Protein number in steady state is Nm = [(βm × nm)/δM], where βm is mreB mRNA translation rate, nm is mreB mRNA copy number and δM is the MreB protein degradation rate (M for MreB protein); protein degradation rate δM = ln2/τhl-prot + ln2/τcc-prot. Since MreB protein is probably stable, the protein degradation rate is δM = ln2/τcc-prot. In the steady state, MreB protein is present at ∼10000 copies per cell and we measured that the number of mRNA copies is ∼9, so the translation rate is ∼25 protein copies per mRNA per minute. Since mreB mRNA mean life-time (1/k) is 4.5 min and translation rate is 25 copies per mRNA per minute, one mreB mRNA is translated on average 110 times before being degraded. This is in agreement with the estimated range of 1–150 translation events per mRNA in E. coli49 and with what should be expected for an efficiently produced protein such as MreB.
000 copies per cell).48 Nevertheless, MreB protein abundance is not necessarily correlated to the cellular concentration of its mRNA. If mreB mRNA is abundant as MreB, its translation and/or stability would unlikely be efficiently regulated by a sRNA of low abundance such as DsrA. Here we measured for the first time the mreB mRNAs steady-state level and showed that mreB mRNA is indeed present at a maximum of 9 copies per cell, a result in agreement with a possible regulation by DsrA. The copy number of mreB mRNA could appear too low for an abundant protein. Nevertheless, mRNA steady state copy number only reflects recent transcription events and can usually not be correlated with the protein concentration, which has a longer lifetime than the bacterial cell cycle and represent accumulated transcriptions.49 In the mreB case, to have nm = 9 mRNA copies per cell implies that the cell needs a transcription rate of 9 × δm, where δm is mreB mRNA degradation rate (m for mreB mRNA). As the degradation rate is the sum of active degradation by RNases and dilution by cell growth, the degradation rate δm = ln2/τhl-RNA + ln2/τcc-RNA where τhl-RNA is the mRNA half-life (3.15 min) and τcc-RNA is the cell doubling time (30 min). This indicates that the cell needs to produce ∼2.2 mRNA molecules per minute or ∼70 per doubling time. Protein number in steady state is Nm = [(βm × nm)/δM], where βm is mreB mRNA translation rate, nm is mreB mRNA copy number and δM is the MreB protein degradation rate (M for MreB protein); protein degradation rate δM = ln2/τhl-prot + ln2/τcc-prot. Since MreB protein is probably stable, the protein degradation rate is δM = ln2/τcc-prot. In the steady state, MreB protein is present at ∼10000 copies per cell and we measured that the number of mRNA copies is ∼9, so the translation rate is ∼25 protein copies per mRNA per minute. Since mreB mRNA mean life-time (1/k) is 4.5 min and translation rate is 25 copies per mRNA per minute, one mreB mRNA is translated on average 110 times before being degraded. This is in agreement with the estimated range of 1–150 translation events per mRNA in E. coli49 and with what should be expected for an efficiently produced protein such as MreB.
The relative contribution of these transcriptional and post-transcriptional regulatory pathways remains to be determined. Intuitively, we might expect that sRNA regulation would allow the reduction of the delay to turn off mreB expression relative to the use of protein-based transcription control, although sRNA regulation can also introduce intrinsic noise.35,43,50,51 In addition, due to the high stability of DsrA in WT cells whose half-life was estimated to be 23 min at 25 °C in a previous work,38 this sRNA pathway is probably cost-effective for the bacterial cell, as the energy required to synthesize a sRNA is small compared with the energetic cost needed for the synthesis and translation of a mRNA. Note that steady state and half-life measurements of mreB mRNA could differ for a specific condition; for instance, at 16 °C, mreB mRNA half-life increases by a factor 10 in an hfq mutant compared to wild-type (Fig. 6), while steady state mRNA levels are slightly lower in the hfq mutant compared to those found in wild-type (3.5 copies per cell compared to 4.5 copies for WT, Fig. 5). This indeed reasserts that steady state measurements do not only result from the efficiency of RNA degradation but also from an intricate interplay with transcription. As we can see, the absence of Hfq greatly increases mreB mRNA stability whereas, as a pleiotropic regulator, it likely affects transcription independently of DsrA.52,53
Experimental
Chemicals, reagents, and oligodeoxyribonucleotides
All chemicals and reagents were purchased from Sigma-Aldrich or Thermo Fisher Scientific unless otherwise specified. Oligonucleotides were purchased from Eurogentec (Belgium).E. coli strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are shown in Table 1. The hfq::kan or hfq::cm alleles54,55 (when necessary for the compatibility with plasmids) were moved in appropriate genetic backgrounds by P1 transduction. For long-term growth under low temperature conditions, cells were grown at 16 °C and then induced by L-arabinose at the same temperature when indicated. For stationary phase conditions, cells were grown at 37 °C overnight. L-Arabinose concentrations used in the various experiments are indicated in the corresponding experiments.| Plasmid name | Description | Origin/marker | Source |
|---|---|---|---|
| pNM12 | Modified pBAD24,56 to generate a promoter-less DNA fragment, araC, PBAD, empty | ColE1/AmpR | 8 |
| pNM13 | pNM12, araC, PBAD, +DsrA insert | ColE1/AmpR | 8 |
| pBAD33-DsrA | Modified pBAD33,56araC, PBAD, +DsrA insert | p15A/CmR | This study |
| pBAD33-DsrAmut | Modified pBAD33DsrA, mutagenesis in dsrA sequence | p15A/CmR | This study |
| pZE12-DsrA | Modified pZE12-MCS,57 PLlacO-1 promoter, +DsrA insert | ColE1/AmpR | This study |
| pMec1 series | P1–2–3mreB, mreB-lacZ transcriptional and translational fusion | pSC101/KanR | 31 |
| pNM12mreB-lacZ | pNM12, araC, PBAD, mreB-lacZ translational fusion | ColE1/AmpR | This study |
| pNM12mreBmut-lacZ | Modified pNM12mreB-lacZ; mutagenesis in mreB 5′ sequence | ColE1/AmpR | This study |
Constructions of pNM13, a derivative of pBAD24 (ColE1, AmpR)56 mutated to generate a promoter-less dsrA DNA fragment under the control of the arabinose promoter PBAD, and the corresponding empty plasmid (pNM12), were previously described.8 pNM12 serves as a control for exposure to L-arabinose and antibiotic.
For the construction of pZE12-DsrA (ColE1, AmpR), a dsrA PCR fragment was ligated into the pZE12 plasmid under the control of the constitutive PLlacO-1 promoter.57 When necessary, ampicillin resistance was replaced by kanamycin in order to allow co-transformation with other plasmids. For the construction of pBAD33-DsrA, a dsrA PCR fragment was inserted into pBAD33 (p15A, CmR) under the control of the PBAD promoter.56 Construction of the pMEC1 series (pSC101, KanR) was previously described.31 Note that mreB, mreC and mreD genes form an operon and that pMEC1 series allow the expression of the three proteins from the respective 3 promoters P1mreB, P2mreB and P3mreB for pMEC1, from P1mreB and P2mreB for pMEC1s and from P1mreB only for pMEC1ss (Fig. S1, ESI†).31 For the construction of pNM12mreB-lacZ, a PCR product carrying −43 to +495 nts of mreB relative to its Start codon was amplified from genomic DNA (MG1655) using the Phusion polymerase and primers carrying homologies to PBAD promoter and lacZ. lacZ was amplified from pMEC1. mreB and lacZ PCR fragments were assembled by PCR and inserted into the pBAD24 derivative pNM12.8
β-Galactosidase assays
Overnight cultures of cells-containing plasmids were diluted into fresh LB (supplemented with glucose and ampicillin, chloramphenicol or kanamycin as indicated), grown at the indicated temperature and harvested at OD600 = 0.3 for exponential phase analyses. For stationary phase analyses, overnight cultures were used. Expression of the lacZ fusions was induced by the addition L-arabinose. To remove glucose before induction, the culture was spun down and resuspended in LB containing glycerol (0.2%) and L-arabinose for 1 hour (concentration of L-arabinose is indicated in the legend of corresponding figures). β-Galactosidase activity was assayed using 0.1 ml culture as described in Miller.58 The β-galactosidase activities are the average value of at least three independent experiments.Construction of complementary mutations
As pNM13 and pNM12mreB-lacZ are incompatible plasmids, we used for this analysis the pNM12mreB-lacZ plasmid (ColE1, AmpR) and DsrA was expressed from pBAD33 (p15A, CmR). The following sequences have been used for WT and complementary mutations (the mutated region is underlined).DsrAWT: 5′ACACAUCAGAUUUCCUGGUGU![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/i_char_0047_0332.gif)
![[U with combining low line]](https://www.rsc.org/images/entities/i_char_0055_0332.gif) GCUUCUUGCUUAAGCAAGUUUCAUCCCGACCCCCUCAGGGUCGGGAUUU3′
GCUUCUUGCUUAAGCAAGUUUCAUCCCGACCCCCUCAGGGUCGGGAUUU3′
DsrAmut: 5′ACACAUCAGAUUUCCUGGUGUGCUUCUUGCUUAAGCAAGUUUCAUCCCGACCCCCUCAGGGUCGGGAUUU3′
mreB WT (AUG codon is indicated in bold):
5′…UUAGUAUGGGCAUGUUUUCCAAUGACUUGU… 3′
mreB mut : (Note that we conserve the in frame translation for mreBmut-lacZ sequence)
5′…UUAGUAUGGGCAUGUUUUCCAAUGACUUGU… 3′
Mutant RNAs annealing was confirmed with the RNAfold web server (Fig. S4, ESI†).37
Changes in DNA sequence of dsrA and mreB were introduced using PCR-based site-directed mutagenesis using the FastStart High Fidelity PCR System (Roche) with 1.25 pmol of appropriate primers listed in Table 3 and 50 ng of pBAD33-DsrA or pNM12mreB-lacZ vectors as template in 12.5 μl reaction volumes. PCR conditions were as follows: denaturation at 95 °C for 5 min; 18 cycles of 95 °C for 50 s, 50 °C for 50 s and 68 °C for 1 min per each 1 kb of template DNA + additional 1 min; final extension at 68 °C for 7 min. PCR products were subjected to DpnI digestion at 37 °C for 1 hour to eliminate the template DNA and then used for bacterial transformation of DJ480 competent cells. Resulting plasmids, pBAD33-DsrAmut and pNM12mreBmut-lacZ, are listed in Table 1.
| Gene name | Product size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| dsrA | 69 | CACATCAGATTTCCTGGTGTAACG | GGGGTCGGGATGAAACTTGC |
| mreB | 130 | ACGGTGTGGTTTACTCCTCTTC | ATTTCGTGCTTGATACGTTCTG |
| kan | 103 | GCGAGTGATTTTGATGACGA | CATGAGTGACGACTGAATCC |
| rrsB | 130 | TGTCGTCAGCTCGTGTTGTG | ATCCCCACCTTCCTCCAGTT |
| Gene | Primer sequence (changed sequence is underlined) |
|---|---|
| dsrA | Forward:CAGATTTCCTGGTGT![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif) GCTTCTTGCTTAAG GCTTCTTGCTTAAG |
| Reverse:CTTAAGCAAGAAGCACACCAGGAAATCTG |
|
| mreB | Forward:GGATTATCCCTTAGTATGGGCATGTTTTCCAATG |
| Reverse:CATTGGAAAACATGCCCATACTAAGGGATAATCC |
| WT (DJ480) | ΔdsrA | Δhfq | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 37 °C | 16 °C | S. Ph. | 37 °C | 16 °C | S. Ph. | 37 °C | 16 °C | S. Ph. | |
| MreB protein level (relative unit, %) | 100 ± 13 | 70 ± 11 | 50 ± 7 | 98 ± 9 | 97 ± 8 | 70 ± 8 | 95 ± 6 | 105 ± 7 | 105 ± 5 |
| Translational fusion (Miller unit) | 405 ± 33 | 425 ± 124 | 55 ± 25 | 380 ± 60 | 900 ± 160 | 95 ± 41 | 805 ± 90 | 800 ± 65 | 95 ± 41 |
| mreB mRNA (copy per cell) | 9 ± 1 | 4.5 ± 0.5 | 0.3 ± 0.12 | 8 ± 0.5 | 5.5 ± 0.8 | 0.5 ± 0.15 | 3.5 ± 0.5 | 3.5 ± 0.5 | ND |
| mreB mRNA half-life (min) | 3.15 ± 0.52 | 1.91 ± 0.35 | ND | 2.23 ± 0.52 | 6.41 ± 0.12 | ND | 1.21 ± 0.22 | 12 ± 1.23 | ND |
| Cell roundness (percentage/roundness) | 70%/0.36, 30%/0.48 | 64%/0.37, 36%/0.49 | 100%/0.59 | 87%/0.42, 13%/0.65 | 100%/0.45 | 100%/0.50 | ND | ND | ND |
RNA extraction, reverse transcription and RT-qPCR analysis
Total RNA preparation, reverse transcription and RT-qPCR analysis were performed as described previously.35 Briefly, 3 pmol of a fragment of the kan transcript fragment (103 nt, T7 in vitro transcription) was added to the bacterial lysate prior to RNA extraction to normalize quantitation. RNAs were then extracted by hot-phenol and ethanol precipitated from cell lysate made from 20 ml cultures grown to OD600 0.6. Samples were subjected to a RQ1 DNase treatment (RNase-free) for 1 hour at 37 °C followed by phenol/chloroform extraction. The cDNA synthesis was performed at 37 °C using the M-MuLV RT (Fermentas) and the indicated reverse primer (Eurogentec) and incubating at 37 °C for 60 min followed by 5 min at 85 °C to inactivate the reverse-transcriptase. Negative controls were incubated in the absence of reverse transcriptase.Quantification of transcript was performed by real time RT-qPCR using a LightCycler® 480 instrument (Roche) and a SYBR Green I based method as described previously.35 Four sequences were amplified: dsrA, mreB, kan and rrsB. rrsB housekeeping gene was used to normalize RNA quantity in each reaction and kan to calculate the efficiency of RNA extraction. The primers used for amplification are shown in Table 2.
Western Blot analysis
E. coli cell pellets were re-suspended in TE buffer containing 4% w/v SDS and 10% v/v β-mercaptoethanol and boiled 95 °C for 5 min. Equal amounts of total protein were loaded on 10% SDS-PAGE gels. After a transfer at 4 °C on a nitrocellulose membrane using a mini-protean transfer blot module (Bio-Rad), the blocking step was done in 5% Defatted Milk in PBS 1× containing 0.1% tween 20. Membranes were then successively incubated with a purified rabbit anti-MreB antibody,59 secondary anti-rabbit HRP conjugated antibody (GE Healthcare) and revealed with ECL Western Blot substrates (Pierce) using a G-box imager (SynGene). Intensities of bands were evaluated using the ImageJ software.60 For accurate measurement, the protein concentrations were chosen where linear relationship existed between the protein concentration and the immunostaining intensity. The assay was repeated for 3 independent extracts under each condition. ΔmreBCD strain was used as negative control.mRNA stability assay
E. coli strains were grown at 37 °C and 16 °C to the exponential phase. Stationary phase cells were grown at 37 °C. Rifampicin was added at 200 μg ml−1 to stop transcription and then samples were taken at several time points after rifampicin addition (time ranging between 0 and 60 min). Each sample was chilled on ice in the presence of 30 mM sodium azide and then total RNAs were extracted using High Pure RNA Isolation Kit (Roche) according to the manufacturer's instructions, which include DNase I treatment of the samples. The total concentration of RNA was measured. Finally, reverse transcription was performed using Thermo Scientific RevertAid H minus Reverse Transcriptase and specific primers, according to manufacturer's instructions. The relative quantification of the transcript was performed by real time RT-qPCR. Normalization was performed by using the amount of total RNA in each RT reaction (250 ng) and by the level of expression of a reference gene (rrsB) at time zero point (before addition of rifampicin). Relative mRNA level in each sample was normalized to time zero point for each strain. The half-lives of transcripts were estimated after curve fitting of the kinetics with an exponential decay model (A = A0 × exp(−k × t) + B).Cryo-transmission electron microscopy (cryo-TEM)
Cells grown at 16 °C or 37 °C were prepared for cryo-transmission electron microscopy as described previously.61 Z-loss images (25 eV slit) of each sample were recorded on a JEOL 2200FS cryo-transmission electron microscope operated at 200 kV by using a Gatan 2kx2k Ultascan ssCCD camera at average defocus and nominal magnification of −10 μm and 5000× respectively. Length and width of bacteria were measured using ImageJ software (http://rsb.info.nih.gov/ij/)62 to determine their roundness defined as the ratio between the minimal and the maximal axes of each cell. Populations in each condition were analyzed by using the Mixtool package,63 to examine multimodal distributions by Gaussian fitting determining the average size and their associated variances (σ2) in different experimental conditions.RNA secondary structure modeling
RNA secondary structure models were predicted by the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) with default options.37Conclusion
Taken together, our data suggest that at least two riboregulatory pathways are involved and coordinated for mreB expression: direct post-transcriptional regulation by DsrA, which occurs via the DsrA/mreB pathway, whereas indirect transcriptional regulation takes place via the DsrA/σS/BolA/H-NS pathway (Fig. 8). Although deletion of DsrA does not appear to impact the growth rate at low-temperature and is not detrimental under laboratory conditions, DsrA might nevertheless be important in natural environments where bacteria live under a combination of stresses that could make sRNA-mediated mreB regulation important for cell elongation and survival.Furthermore, taking into account the evolutionary conservation of DsrA in many bacteria,8 this regulatory mechanism might also apply to other bacterial species other than E. coli. Indeed morphological changes associated with mreB regulation during various stresses have been reported in Vibrio parahaemolyticus.64 Nevertheless, whether an sRNA mediates the regulation in this bacterium is not known.
Funding
This work was supported by FP7 DIVINOCELL Health-F3-2009-223431, PHC Polonium No. 27701VG, University Paris Diderot, CNRS and CEA (VA), National Science Center Poland grant no. 2012/04/M/NZ1/00067 (GW) and by NIH grant R37 GM060632 (AT).Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.Author contribution
Author contribution: BC, EF, CM, MB, SC, GC & AK performed most experiments. ST & SM designed and performed experiments, analyzed the data and wrote parts of the manuscript. FB, AT, GW & VA designed experiments, analyzed the data and wrote the manuscript.Abbreviations
| Cryo-TEM | Cryo-transmission electron microscopy |
| EtBr | Ethidium bromide |
| nt | Nucleotide |
| PAGE | Polyacrylamide gel electrophoresis |
| PG | Peptidoglycan |
| rbs | Ribosome binding site |
| RT-qPCR | Reverse transcription quantitative PCR |
| sRNA | Small regulatory noncoding RNA |
Acknowledgements
We are grateful to N. Majdalani, S. Gottesman (NIH, Bethesda, MD) and M. Wachi (Tokyo Institute of Technology, Japan) for providing strains and plasmids. We are indebted to D. Joseleau-Petit (Université Paris Diderot, Paris, France) who contributed to this work at an early stage, to P. J. Choi (Harvard Medical School, Boston, MA) and R. Guantes (Universidad Autónoma de Madrid, Spain) for many fruitful discussions. We thank PICT-Ibisa for providing access to JEOL 2200FS cryo-TEM. We are finally indebted to L. Rothfield (University of Connecticut Health Center, USA), J. Voss (University of Cambridge, UK), N. Figueroa-Bossi and L. Bossi (CGM/CNRS, Gif sur Yvette, France) for their critical reading of the manuscript.References
- G. Storz, J. Vogel and K. M. Wassarman, Mol. Cell., 2011, 43, 880–891 CrossRef CAS PubMed.
- S. Gottesman and G. Storz, Cold Spring Harbor Perspect. Biol., 2011, 3, a003798 Search PubMed.
- D. Lalaouna, M. Simoneau-Roy, D. Lafontaine and E. Masse, Biochim. Biophys. Acta, 2013, 1829, 742–747 CrossRef CAS PubMed.
- R. G. Brennan and T. M. Link, Curr. Opin. Microbiol., 2007, 10, 125–133 CrossRef CAS PubMed.
- J. Vogel and B. F. Luisi, Nat. Rev. Microbiol., 2011, 9, 578–589 CrossRef CAS PubMed.
- D. Sledjeski and S. Gottesman, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2003–2007 CrossRef CAS.
- F. Repoila and S. Gottesman, J. Bacteriol., 2003, 185, 6609–6614 CrossRef CAS.
- N. Majdalani, C. Cunning, D. Sledjeski, T. Elliott and S. Gottesman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12462–12467 CrossRef CAS.
- R. A. Lease and S. A. Woodson, J. Mol. Biol., 2004, 344, 1211–1223 CrossRef CAS PubMed.
- W. Hwang, V. Arluison and S. Hohng, Nucleic Acids Res., 2011, 39, 5131–5139 CrossRef CAS PubMed.
- P. K. Brown, C. M. Dozois, C. A. Nickerson, A. Zuppardo, J. Terlonge and R. Curtiss, 3rd, Mol. Microbiol., 2001, 41, 349–363 CrossRef CAS.
- A. Olsen, A. Arnqvist, M. Hammar, S. Sukupolvi and S. Normark, Mol. Microbiol., 1993, 7, 523–536 CrossRef CAS.
- D. D. Sledjeski, A. Gupta and S. Gottesman, EMBO J., 1996, 15, 3993–4000 CAS.
- C. A. White-Ziegler, S. Um, N. M. Perez, A. L. Berns, A. J. Malhowski and S. Young, Microbiology, 2008, 154, 148–166 CrossRef CAS.
- R. A. Lease and M. Belfort, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9919–9924 CrossRef CAS PubMed.
- P. Mandin and M. Guillier, Curr. Opin. Microbiol., 2013, 16, 125–132 CrossRef CAS.
- F. Repoila, N. Majdalani and S. Gottesman, Mol. Microbiol., 2003, 48, 855–861 CrossRef CAS.
- P. Mandin and S. Gottesman, EMBO J., 2010, 29, 3094–3107 CrossRef CAS.
- K. M. Wassarman, F. Repoila, C. Rosenow, G. Storz and S. Gottesman, Genes Dev., 2001, 15, 1637–1651 CrossRef CAS.
- N. Zambrano, P. P. Guichard, Y. Bi, B. Cayrol, S. Marco and V. Arluison, Cell Cycle, 2009, 8, 2470–2472 CrossRef CAS.
- M. Vicente, A. I. Rico, R. Martinez-Arteaga and J. Mingorance, J. Bacteriol., 2006, 188, 19–27 CrossRef CAS.
- F. Tetart and J. P. Bouche, Mol. Microbiol., 1992, 6, 615–620 CrossRef CAS.
- A. Takada, M. Wachi and K. Nagai, Biochem. Biophys. Res. Commun., 1999, 266, 579–583 CrossRef CAS.
- J. W. Shaevitz and Z. Gitai, Cold Spring Harbor Perspect. Biol., 2010, 2, a000364 Search PubMed.
- M. J. Osborn and L. Rothfield, Curr. Opin. Microbiol., 2007, 10, 606–610 CrossRef CAS PubMed.
- C. L. White and J. W. Gober, Trends Microbiol., 2012, 20, 74–79 CrossRef CAS.
- J. Dominguez-Escobar, A. Chastanet, A. H. Crevenna, V. Fromion, R. Wedlich-Soldner and R. Carballido-Lopez, Science, 2011, 333, 225–228 CrossRef CAS PubMed.
- H. Strahl, F. Burmann and L. W. Hamoen, Nat. Commun., 2014, 5, 3442 Search PubMed.
- W. Margolin, Curr. Biol., 2009, 19, R812–R822 CrossRef CAS.
- M. Doi, M. Wachi, F. Ishino, S. Tomioka, M. Ito, Y. Sakagami, A. Suzuki and M. Matsuhashi, J. Bacteriol., 1988, 170, 4619–4624 CAS.
- M. Wachi, K. Osaka, T. Kohama, K. Sasaki, I. Ohtsu, N. Iwai, A. Takada and K. Nagai, Biosci., Biotechnol., Biochem., 2006, 70, 2712–2719 CrossRef CAS.
- R. Lange and R. Hengge-Aronis, J. Bacteriol., 1991, 173, 4474–4481 CAS.
- M. Aldea, C. Hernandez-Chico, A. G. de la Campa, S. R. Kushner and M. Vicente, J. Bacteriol., 1988, 170, 5169–5176 CAS.
- P. Freire, R. N. Moreira and C. M. Arraiano, J. Mol. Biol., 2009, 385, 1345–1351 CrossRef CAS.
- R. Guantes, B. Cayrol, F. Busi and V. Arluison, Mol. BioSyst., 2012, 8, 1707–1715 RSC.
- M. Wachi, M. Doi, S. Tamaki, W. Park, S. Nakajima-Iijima and M. Matsuhashi, J. Bacteriol., 1987, 169, 4935–4940 CAS.
- A. R. Gruber, R. Lorenz, S. H. Bernhart, R. Neubock and I. L. Hofacker, Nucleic Acids Res., 2008, W70–W74 CrossRef CAS.
- F. Repoila and S. Gottesman, J. Bacteriol., 2001, 183, 4012–4023 CrossRef CAS.
- C. A. McCullen, J. N. Benhammou, N. Majdalani and S. Gottesman, J. Bacteriol., 2010, 192, 5559–5571 CrossRef CAS.
- I. Moll, T. Afonyushkin, O. Vytvytska, V. R. Kaberdin and U. Blasi, RNA, 2003, 9, 1308–1314 CrossRef CAS.
- H. Aiba, Curr. Opin. Microbiol., 2007, 10, 134–139 CrossRef CAS.
- A. Leroy, N. F. Vanzo, S. Sousa, M. Dreyfus and A. J. Carpousis, Mol. Microbiol., 2002, 45, 1231–1243 CrossRef CAS.
- R. Hussein and H. N. Lim, Nucleic Acids Res., 2012, 40, 7269–7279 CrossRef CAS PubMed.
- A. Battesti, N. Majdalani and S. Gottesman, Annu. Rev. Microbiol., 2011, 65, 189–213 CrossRef CAS PubMed.
- J. M. Santos, P. Freire, M. Vicente and C. M. Arraiano, Mol. Microbiol., 1999, 32, 789–798 CrossRef CAS.
- R. N. Moreira, C. Dressaire, S. Domingues and C. M. Arraiano, Biochem. Biophys. Res. Commun., 2011, 411, 50–55 CrossRef CAS PubMed.
- N. De Lay, D. J. Schu and S. Gottesman, J. Biol. Chem., 2013, 288, 7996–8003 CrossRef CAS PubMed.
- T. Kruse, J. Moller-Jensen, A. Lobner-Olesen and K. Gerdes, EMBO J., 2003, 22, 5283–5292 CrossRef CAS PubMed.
- Y. Taniguchi, P. J. Choi, G. W. Li, H. Chen, M. Babu, J. Hearn, A. Emili and X. S. Xie, Science, 2010, 329, 533–538 CrossRef CAS PubMed.
- P. Mehta, S. Goyal and N. S. Wingreen, Mol. Syst. Biol., 2008, 4, 221 CrossRef PubMed.
- Y. Shimoni, G. Friedlander, G. Hetzroni, G. Niv, S. Altuvia, O. Biham and H. Margalit, Mol. Syst. Biol., 2007, 3, 138 CrossRef PubMed.
- J. Le Derout, I. V. Boni, P. Regnier and E. Hajnsdorf, BMC Mol. Biol., 2010, 11, 17 CrossRef PubMed.
- F. Geinguenaud, V. Calandrini, J. Teixeira, C. Mayer, J. Liquier, C. Lavelle and V. Arluison, Phys. Chem. Chem. Phys., 2011, 13, 1222–1229 RSC.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2, 0008 CrossRef.
- H. C. Tsui, G. Feng and M. E. Winkler, J. Bacteriol., 1997, 179, 7476–7487 CAS.
- L. M. Guzman, D. Belin, M. J. Carson and J. Beckwith, J. Bacteriol., 1995, 177, 4121–4130 CAS.
- R. Lutz and H. Bujard, Nucleic Acids Res., 1997, 25, 1203–1210 CrossRef CAS.
- J. H. Miller, in A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, ed. C. S. Harbor, Cold Spring Harbor Laboratory Press, NY, edn, 1992 Search PubMed.
- Y. L. Shih, I. Kawagishi and L. Rothfield, Mol. Microbiol., 2005, 58, 917–928 CrossRef CAS.
- V. Girish and A. Vijayalakshmi, Indian J. Cancer, 2004, 41, 47 CAS.
- G. Pehau-Arnaudet, D. Joseleau-Petit and V. Arluison, Am. J. Mol. Cell. Biol., 2012, 1, 17–24 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- T. Benaglia, D. Chauveau, D. R. Hunter and D. Young, Journal of Statistical Software, 2009, 32 Search PubMed.
- S. W. Chiu, S. Y. Chen and H. C. Wong, Appl. Environ. Microbiol., 2008, 74, 7016–7022 CrossRef CAS.
- R. A. Lease and M. Belfort, Mol. Microbiol., 2000, 38, 667–672 CrossRef CAS.
- J. E. Cabrera and D. J. Jin, J. Bacteriol., 2001, 183, 6126–6134 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ib00102h |
| This journal is © The Royal Society of Chemistry 2015 |