
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive deconstruction of organosolv lignin catalyzed by zeolite supported nickel nanoparticles†
Stanislav
Kasakov
a,
Hui
Shi
b,
Donald M.
Camaioni
b,
Chen
Zhao‡
a,
Eszter
Baráth
*a,
Andreas
Jentys
a and
Johannes A.
Lercher
*ab
aTechnische Universität München, Department Chemie, Lichtenbergstraβe 4, Garching, D-85748, Germany. E-mail: Eszter.Barath@tum.de; Johannes.Lercher@ch.tum.de
bInstitute for Integrated Catalysis, Pacific Northwest National Laboratory, 902 Battelle Boulevard, Richland, WA 99352, USA
First published on 5th October 2015
Abstract
Mechanistic aspects of deconstruction and hydrodeoxygenation of organosolv lignin using zeolite (HZSM-5 and HBEA) and SiO2 supported Ni catalysts are reported. Lignin was deconstructed and converted to substituted alicyclic and aromatic hydrocarbons with 5 to 14 carbon atoms. Full conversion with total yield of 70 ± 5 wt% hydrocarbons was achieved at 593 K and 20 bar H2. The organosolv lignin used consists of seven to eight monolignol subunits and has an average molecular weight of ca. 1200 g mol−1. The monolignols were mainly guaiacyl, syringyl and phenylcoumaran, randomly interconnected through β-O-4, 4-O-5, β-1, 5-5′ and β–β ether bonds. In situ IR spectroscopy was used to follow the changes in lignin constituents during reaction. The reductive catalytic deconstruction of organosolv lignin starts with the hydrogenolysis of aryl alkyl ether bonds, followed by hydrogenation of the aromatic rings to cyclic alcohols. Oxygen is removed from the alcohols via dehydration on Brønsted acid sites to cyclic alkenes that are further hydrogenated.
1. Introduction
Lignin is one of the major components of lignocellulosic biomass, which constitutes also the most reduced carbon fraction.1–3 Conceptually, this minimizes the use of H2 to produce linear and cyclic alkanes. The methoxylated phenylpropane units of lignin form a three-dimensional structure incorporated in the so-called lignin-carbohydrate complex (LCC).4,5 Loosening the arrangement of LCC enables the separation of lignin and carbohydrates. One approach of isolating lignin from LCC is the so-called organosolv process, which is a chemically mild extraction through solvolysis, e.g., in ethanol/water mixtures at 473 K.6 This method leads to a lignin free of sulfur-containing compounds and other contaminants.7 Organosolv lignin is typically less condensed than the Kraft and soda lignins.8Conventionally, lignin is converted to liquid products in a two-stage process. The first stage includes rapid pyrolysis above 800 K or base-catalyzed deconstruction to yield bio-oil.9–12 The second stage involves upgrading of the thermally unstable bio-oils, which contain a variety of reactive oxygenates, into transportation fuels via hydrodeoxygenation (HDO). The HDO of various monomeric, bio-oil model compounds have been studied intensively.13–21 More recently, the cleavage of aryl ethers (i.e., dimeric phenolic compounds), as a key step in lignin depolymerization, has been a topic of increasing interest with focus on the effects of ring substituents, solvent and catalyst properties on the reactivities and selectivities during conversion of model ethers and real lignins.22–27 The complex structure of lignin has raised doubts concerning whether the direct hydrogenation of ring compounds as well as the hydrogenolysis is possible directly on the integral lignin polymer. To mitigate this perceived drawback, Rinaldi et al. explored RANEY® Ni catalyzed transfer hydrogenation of phenols, aldehydes and ketones as well as hydrogenolysis in aqueous iso-propanol solutions at autogenous pressures.28 Iso-propanol not only showed excellent hydrogen transfer properties, it simultaneously dissolved wood chips as the starting material.29 This strategy enables one-stage lignin isolation from whole cell wood to non-pyrolytic bio-oils, which are further hydrotreated. Non-polar solvents (e.g., methylcyclohexane) were found to be advantageous in terms of the higher selectivity to saturated products, compared to protic (e.g., alcohols) and polar aprotic (e.g., THF, dioxane) solvents, during the reductive deconstruction of organosolv lignin.30
Although partial catalytic deconstruction of lignin has been achieved with these and other attempts using solid catalysts,31–34 the heterogeneity and complexity of real lignins renders complete deconstruction and the mechanistic understanding on a molecular level challenging.35 To address this, physicochemical methods probing both the catalyst and the reaction mixture under working conditions have been used in the present contribution to link the previous model studies to a one-stage liquid phase process for hydro-deconstructing organosolv beech lignin (<600 K, 20 bar H2). In particular, attenuated total reflectance infrared spectroscopy (ATR-IR) is used to interrogate interaction between molecules and the surface of solid catalysts at operating conditions.
2. Experimental section
2.1. Chemicals
All chemicals were purchased from commercial suppliers and used without further purification: organosolv beech lignin (Fraunhofer Institute, Leuna), Ni(II) nitrate hexahydrate (Merck, 99%), n-heptane (Merck, 99%), n-hexadecane (Sigma-Aldrich, 99%), ethyl acetate (Fluka, 99.7%), tetrahydrofuran (Sigma-Aldrich, 99%), cyclohexane (Sigma-Aldrich, 99%), methylcyclohexane (Sigma-Aldrich, 99%), ethylcyclohexane (Sigma-Aldrich, 99%), n-propylcyclohexane (Sigma-Aldrich, 99%), dimethyl sulfoxide (DMSO, Fluka, 99.5%), ammonium hydroxide (Sigma-Aldrich, 28.0–30.0% aqueous solution), dimethyl glyoxime (Sigma-Aldrich, 99%), DMSO-d6 (Sigma-Aldrich, 99.9%), acetone (VWR, 95%), urea (Merck, 99.5%), HZSM-5 (H-form, Si/Al 45, Clariant AG), HBEA (H-form, Si/Al 75, Clariant AG), SiO2 (Aerosil 200, Evonik AG).2.2. Preparation of Ni-based catalysts
Nickel deposition on oxide supports was performed following the deposition precipitation (DP) procedure described in the literature.36 In a typical synthesis, a 250 ml aqueous solution of 0.14 M Ni(NO3)2 was prepared. 2 g of zeolite were suspended in 210 ml of that solution. The remaining 40 ml of Ni(II) solution was used to dissolve 10.2 g of urea, which was added dropwise to the zeolite suspension at 343 K. This mixture was heated further to 363 K to start the DP process. After 3 h, the suspension was cooled to ambient temperature, vacuum filtered, and the solid was washed three times with ultra-pure water. The recovered solid was dried at 383 K in air overnight. The dry solid was calcined at 673 K for 4 h in synthetic air with a heating rate of 1 K min−1 (flow rate: 100 ml min−1), and then reduced in pure hydrogen at 733 K for four hours with a heating rate of 1 K min−1 (hydrogen flow: 100 ml min−1). The synthesis of Ni/SiO2 catalyst followed the same procedure as described above, but with a DP time of 3.5 h.2.3. Catalyst characterization
2.4. Organosolv lignin characterization
The in situ ATR-IR study of organosolv lignin hydro-deconstruction was performed in a lab-built ATR-IR cell, with the idea of a double chamber cell design.39,40 The technical drawing and a detailed description of the ATR-IR cell are described in the ESI (technical drawing in Fig. S1 and photograph in Fig. S2†).
In a typical in situ ATR-IR experiment, a mixture of organosolv lignin and Ni/HBEA catalyst at a weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was mixed with isopropanol and sonicated for 5 min. The mixture was then drop-coated on the surface of ZnSe and the solvent evaporated at 353 K. The drop-coated ZnSe crystal was placed on the holder and the cell was closed, pressurized with H2 to 20 bar and heated to 523 K with a heating rate of 10 K min−1. At this condition, a background spectrum of chamber 1 (catalyst/lignin mixture) and chamber 2 (empty cell) was collected at a resolution of 4 cm−1 and 32 scans. After that, the reaction was initiated by pumping n-hexadecane through the cell at a rate of 0.05 ml min−1. The single beam spectra were collected every 30 min, with an acquisition time of 40 s. The time difference of spectra collection between chamber 1 and chamber 2 was 2 min. The baseline correction of each single beam spectrum was derived by using a blank ZnSe crystal as background spectrum. The difference resulted through subtraction between spectra collected from Chamber 1 (sample cell) and Chamber 2 (reference cell). During each in situ ATR-IR experiment, a sample of the liquid phase was taken at a contact time of 90 min and analyzed through GC-MS as described in section 2.5. The schematic drawing of the periphery for H2 and liquid phase supply, liquid phase sampling as well as the arrangement of the ATR-IR cell is shown in the ESI (Fig. S3†).
:1 was mixed with isopropanol and sonicated for 5 min. The mixture was then drop-coated on the surface of ZnSe and the solvent evaporated at 353 K. The drop-coated ZnSe crystal was placed on the holder and the cell was closed, pressurized with H2 to 20 bar and heated to 523 K with a heating rate of 10 K min−1. At this condition, a background spectrum of chamber 1 (catalyst/lignin mixture) and chamber 2 (empty cell) was collected at a resolution of 4 cm−1 and 32 scans. After that, the reaction was initiated by pumping n-hexadecane through the cell at a rate of 0.05 ml min−1. The single beam spectra were collected every 30 min, with an acquisition time of 40 s. The time difference of spectra collection between chamber 1 and chamber 2 was 2 min. The baseline correction of each single beam spectrum was derived by using a blank ZnSe crystal as background spectrum. The difference resulted through subtraction between spectra collected from Chamber 1 (sample cell) and Chamber 2 (reference cell). During each in situ ATR-IR experiment, a sample of the liquid phase was taken at a contact time of 90 min and analyzed through GC-MS as described in section 2.5. The schematic drawing of the periphery for H2 and liquid phase supply, liquid phase sampling as well as the arrangement of the ATR-IR cell is shown in the ESI (Fig. S3†).
2.5. Catalytic measurements
The catalytic reactions were carried out in a batch reactor (Parr Instruments, 300 ml). In a typical experiment, the reactor was loaded with 0.5 g Ni catalyst, 1 g organosolv lignin, 100 ml hexadecane and 1 ml of n-heptane as internal standard, sealed and purged with H2 three times. The reactor was pressurized with 20 bar of H2 at ambient temperature and heated up to either 523 K or 593 K while stirring at 700 rpm. The temperature was reached within 15 min, which was used as the zero reaction time. During the reaction, aliquots of 1.5 ml were sampled into GC vials. To end the reaction, stirring and heating was stopped, and the reactor was cooled down in air for the first 5 min, followed by quenching with an ice-water mixture to room temperature which lasted another 5 min.2.6. Analysis of gas, solution and solid phases of reaction mixture
The gas phase was collected into a gas bag after the reaction and subsequently analyzed with an Agilent Technologies 7890B GC equipped with a HP-1 column (30 m × 0.4 μm) and thermal conductivity and flame ionization detectors (TCD and FID). Based on the external calibration with methane/ethane/propane standard mixture (1 vol% for each) diluted in Argon from Westfalen, the integrated signals were quantified.Subsequently, a liquid phase aliquot of the organic mixture was collected. This sample was analyzed on an Agilent Technologies 7890B GC equipped with a HP-5 column GC-FID (quantification) (30 m × 0.25 μm) connected to Agilent Technologies 5977A MSD GC-MS (identification of products). The yield in wt% was calculated based on the internal standard n-heptane with the following equation:
 | (1) |
The 0.68 g is the absolute weight of the added 1 ml of n-heptane and 1 g is the starting weight of the lignin sample. The selectivity in C% was calculated based on the following equation:
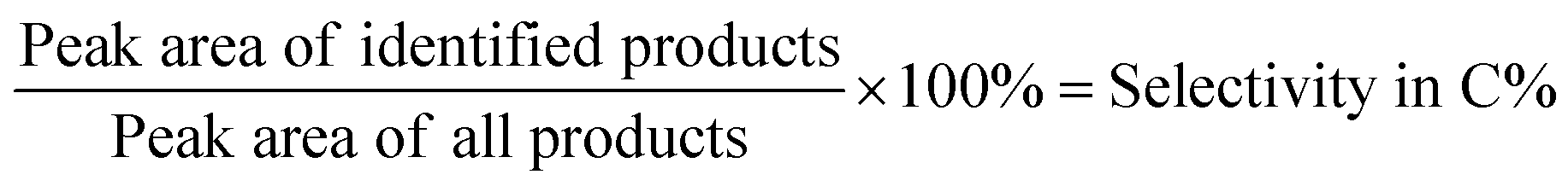 | (2) |
The fragmentation pattern in the MS has been compared using the NIST Mass Spectral Search Program for the NIST/EPA/NIH Mass Spectral Library Version 2.0 g, build Dec 4 2012 to match the identified products with probabilities higher than 73%. The MS ion intensities of the typical compounds, as well as their match and probabilities are summarized in the ESI (Fig. S16†). To further identify the liquid product fraction, naphthenes as standard materials were mixed at defined concentrations (mimicking the concentrations of products in a real experiment) in the non-polar n-hexadecane. This standard series was used to calibrate the FID response factors of major products, such as methyl-, ethyl-, and n-propylcyclohexane. This quantification method produced almost identical quantification results as the one using n-heptane as the internal standard. Additionally as a cross-check, a mixture of branched saturated alkanes (2 mg L−1) was identified with MS detector in order to evaluate the NIST database. The formed aqueous phase (mainly water containing traces of dissolved fractions, the exact concentrations of which were not determined) was not miscible with n-hexadecane, so it was readily separable and determined volumetrically. The solid residue was freeze-dried and weighed. The mass balance was 95 ± 5% in this work.
2.7. Recycle runs
Catalyst recycling runs have been performed under the same conditions as described in section 2.5. The catalyst was recovered by filtration, freeze-dried and reactivated in hydrogen flow at 733 K. A small fraction of the recovered catalyst (0.1 g) has been stored for further analysis (TEM, BET, Pyridine-IR), the remaining 0.4 g of the catalyst has been mixed with 0.1 g of a fresh fraction of Ni catalyst to perform the recycling runs.3. Results and discussion
3.1. Physicochemical properties of Ni catalysts
Physicochemical characteristics of the Ni catalysts used in this work are summarized in Table 1. The textural properties of the catalysts and supports are compiled in Table S2.† The Ni loadings are comparable at 21, 20, and 21 wt% for HZSM-5, HBEA and SiO2 supported catalysts, respectively. Catalysts with lower Ni loadings (10%, 15%) were less efficient for catalytic deconstruction of lignin, on a total Ni basis (i.e., normalized to the mass of Ni per gram of catalyst).| Catalyst | Nia (wt%) | Acidityb (μmol g−1) | Ni particle size | ||
|---|---|---|---|---|---|
| BAS | LAS | d Ni (nm) | Ni dispersiond (%) | ||
| a Determined by AAS. b Determined by IR spectra of adsorbed pyridine, BAS: Brønsted acid site, LAS: Lewis acid site. c Determined by TEM. d Determined by H2 chemisorption. | |||||
| Ni/HZSM-5 | 21 | 36 | 91 | 4.8 ± 1.2 | 13 |
| Ni/HBEA | 20 | 19 | 71 | 5.1 ± 1.1 | 13 |
| Ni/SiO2 | 21 | — | 39 | 4.5 ± 1.0 | 13 |
The concentrations of Brønsted (BAS) and Lewis acid sites (LAS) of the Ni catalysts as well as the parent support materials (Table S1†) were determined from the IR spectra of adsorbed pyridine after outgassing at 423 K. The acid site concentrations of the catalysts differed substantially from those in the supports before deposition of Ni. The BAS concentrations in Ni/HZSM-5 (36 μmol g−1) and Ni/HBEA (19 μmol g−1) were reduced to 10% of the concentrations in the parent zeolites (360 and 190 μmol g−1, respectively), caused by ion-exchange of Brønsted acid sites with Ni2+.41 In contrast, the LAS concentrations (91 and 45 μmol g−1 for Ni/HZSM-5 and Ni/HBEA, respectively) were slightly higher than in the parent zeolites (71 and 40 μmol g−1, respectively). The Ni/SiO2 catalyst showed 39 μmol g−1 LAS without detectable BAS.
The Ni particle sizes have been determined through transmission electron microscopy (TEM pictures shown in Fig. S5†). According to the statistical analyses, the three catalysts contain Ni particles with 5–10 nm average diameter and very similar size distributions. The Ni dispersion measured by H2 chemisorption was 13% for all three catalysts, in good agreement with TEM measurements.
3.2. Analysis of organosolv lignin
Fig. 1 compiles the main results obtained from different analytical approaches used to elucidate the structure of the starting organosolv lignin. The ATR-IR spectrum of the organosolv lignin (Fig. 1a) shows a series of bands characteristic of aromatic structures, i.e., ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations (1593 for symmetric and 1504 cm−1 for asymmetric mode), aryl ring breathing combined with Caromatic–O vibration (1326 cm−1) and CO stretching vibration (1270 cm−1) and aromatic skeletal vibrations combined with C–H in plane bending (1422, 1156 cm−1). Likewise, the oxy-functionalization is documented by bands of aromatic and aliphatic hydroxyl groups (3500 cm−1), alkyl aldehyde and/or ketone groups (1722 cm−1), aryl conjugated CO (the shoulder peak at 1680 cm−1), as well as methoxy groups (evidenced by a bending mode at 1369 cm−1). The linkages between the aromatic structures of the lignin have bands characteristic of aliphatic C–O–H and ether C–O–C groups.42–44
C stretching vibrations (1593 for symmetric and 1504 cm−1 for asymmetric mode), aryl ring breathing combined with Caromatic–O vibration (1326 cm−1) and CO stretching vibration (1270 cm−1) and aromatic skeletal vibrations combined with C–H in plane bending (1422, 1156 cm−1). Likewise, the oxy-functionalization is documented by bands of aromatic and aliphatic hydroxyl groups (3500 cm−1), alkyl aldehyde and/or ketone groups (1722 cm−1), aryl conjugated CO (the shoulder peak at 1680 cm−1), as well as methoxy groups (evidenced by a bending mode at 1369 cm−1). The linkages between the aromatic structures of the lignin have bands characteristic of aliphatic C–O–H and ether C–O–C groups.42–44
 | ||
| Fig. 1 Elucidating the structure of beech extracted organosolv lignin. (a) ATR-IR spectrum; (b) 2D HSQC NMR (DMSO-d6); (c) representation of monolignolic units (S, S′ and G) and its lignol linkages (β-5, β-O-4, β–β); (d) schematic lignin fragment showing the seven monolignols identified. | ||
The starting lignin material was further analyzed by liquid phase HSQC NMR in DMSO-d6 (Fig. 1b). The main monolignols are guaiacyl (G), syringyl (S) and carbonyl functionalized syringyl (S′), as indicated by the resonances in the aromatic region (100–130 ppm), characteristic of substituted benzene rings. Specifically, the signals correspond to the –CHCH– of guaiacyl (G5/6; G2), the syringyl and keto type syringyl –CHCH– linkages of the unsaturated ring.
In the aliphatic carbon region (60–90 ppm), peaks were detected for methoxy, CH– and HO–CH2– groups associated with the β-O-4 (β-aryl ether) (A), β–β (resinol) (C) and β-5 (phenylcoumaran) linkages. Signals corresponding to the tetrahydrofuran ring were also observed: –CH–O– (1α and 2α), OH–CH– (Aα), –CH–O– (Aβ syringyl and guaiacyl), and –CH2–OH (Aγ, 1γ) and –CH2–O– (2γ). The 13C signals detected at 53 ppm correspond to –CH–CH–O– (1β, 2β) carbons in the saturated furan ring. The –CH3 carbons of the methoxy group appeared at 55 ppm. The assignment was based on published attributions in the literature.45–47 From the 2D HSQC NMR analysis, we conclude that this organosolv lignin contains mainly guaiacyl and (CO conjugated) syringyl units (Fig. 1c), which are randomly connected through β-5, β-O-4 and β–β ether linkages.
According to the MALDI-TOF MS spectrum shown in Fig. S7,† the average molecular weight of the Beech extracted organosolv lignin was ca. 1200. Therefore, we conclude that this organosolv lignin is an oligomer comprised of approximately seven interconnected monolignols. GPC analysis (Fig. S8†) showed a number-averaged molecular weight of 1107 g mol−1 and a weight-averaged molecular weight of 2281 g mol−1, corresponding to a dispersity of 2.1. MALDI-TOF MS is comparable to the GPC result (1200 vs. 1107 g mol−1), where the molecular weight was slightly underestimated because of the difference of hydrodynamic volume of the external linear unbranched polystyrene and the branched or irregularly connected lignin sample. From the above analysis, we propose the structure shown in the Fig. 1d as a model, including all structural characteristics of the starting organosolv lignin. This structure has a chemical formula of C68H74O23 and a molecular weight of 1258 g mol−1, which is consistent with the elemental analysis (C70H80O24), GPC (1108 g mol−1) and MALDI-TOF MS measurements (1239 g mol−1). Note, however, that this proposed structure does not specifically take into account the abundance of each type of linkage, and is not meant to represent the only possible sequence, as lignin structures are intrinsically irregular.
3.3. Ni catalyzed lignin deconstruction and upgrading in the presence of H2 and hexadecane
The deconstruction and upgrading of organosolv lignin to cyclic saturated hydrocarbons were performed using Ni based catalysts. In preliminary tests, reaction conditions were optimized by varying reaction temperatures (473–543 K), pressure (10–50 bar H2) and the catalyst-to-lignin weight ratio of 0.2–0.5 (Table S3†). Results obtained at an optimized (among these preliminary tests) set of conditions of 523 K, 20 bar H2, and a catalyst/lignin weight ratio of 0.5 are summarized in Table 2. The products are divided into gas phase (methane, propane and methanol), liquid phase (C5–C14 naphthenes, H2O) and remaining solids (carefully excluding unconverted lignin). The unconverted organosolv lignin was separated from the solid mixture (including the catalyst) by dissolving it in tetrahydrofuran (THF), evaporating the THF fraction, and drying.| Catalyst | Hydrocarbons [wt%] | Waterc [wt%] | Solid residued [wt%] | Unconverted lignine [wt%] |
|---|---|---|---|---|
| a Reaction conditions: 1 g lignin, 0.5 g Ni catalyst, 100 mL (77 g) hexadecane as solvent, 20 bar H2 initially charged into reactor at RT, 6 h, 523 K, 700 RPM stirring. b ±: Standard deviations for multiple reaction tests. c Determined by volumetric analysis. d THF-insoluble solids. e THF-soluble solids. | ||||
| Ni/SiO2 | 27 ± 2 | 10 | 31 ± 5 | 25 |
| Ni/HZSM-5 | 34 ± 2 | 16 | 26 ± 5 | 18 |
| Ni/HBEA | 42 ± 4 | 18 | 22 ± 5 | 12 |
Converting organosolv lignin on Ni/SiO2 (1 g lignin, 0.5 g catalyst, 100 mL hexadecane, 20 bar H2, 523 K for 6 h) led to a total yield of 27 wt% for both gas and liquid phase products, mainly as hydrocarbons, while most of the oxygen was removed as water (10 wt%). On this catalyst, solid byproducts accounted for 31 (±5) wt% yields. After 6 h, 25 wt% of the lignin remained unreacted. Higher yields of gas and liquid phase hydrocarbons were achieved at the same conditions with Ni/HZSM-5 and Ni/HBEA (34 and 42 wt%, respectively) pointing to the importance of acids and the available space in the pores. The higher yields of hydrocarbons are consistent with the larger concentrations of water and the smaller concentrations of solid residues with the two zeolite-supported Ni catalysts than with Ni/SiO2.
As all three catalysts had similar concentrations of accessible Ni, the higher yields of hydrocarbons on Ni/HBEA and Ni/HZSM-5 are attributed to the presence of Brønsted acid sites, catalyzing the dehydration of the intermediately formed alcohols. The slightly higher yields in hydrocarbons are tentatively attributed to more stable Ni particles on the two zeolite supports.
A breakdown of the yields of gas and liquid phase products, as well as the selectivity within liquid phase products, is shown in Table 3. The distributions of liquid phase products on the three Ni catalysts are depicted schematically in Fig. 2. For Ni/SiO2 catalyzed reaction, 4 wt% of the lignin was converted to methane, propane and methanol. Naphthenes were produced at a yield of 23 wt%, the majority being cycloalkanes, primarily cyclohexane with methyl, ethyl and propyl side chains (80 C% selectivity). Cyclic saturated alcohols, namely, cyclohexanol and its methyl-, ethyl-, propyl derivatives, were also present (20 C%). A typical GC trace of the liquid reaction mixture catalyzed by Ni/SiO2 is shown in the ESI, see Fig. S4a.† Variations of the reaction time between 1 and 24 h (Fig. S10†) showed the selectivity to cyclic saturated alcohols to be the highest after 2 h reaction time. Based on our studies of model compounds, we conclude that the cyclic alcohols are formed via hydrogenolysis and subsequent hydrogenation of the lignol on Ni.25 The deoxygenation of cyclic saturated alcohols on Ni/SiO2 suggests that surface reactions of Ni2+ with SiO2 or small concentrations of NiO have generated Lewis acid sites, able to catalyze dehydration of alcohols. At this point we would not fully exclude, however, that some direct hydrogenolysis of cyclic alcohols occurs on Ni/SiO2.
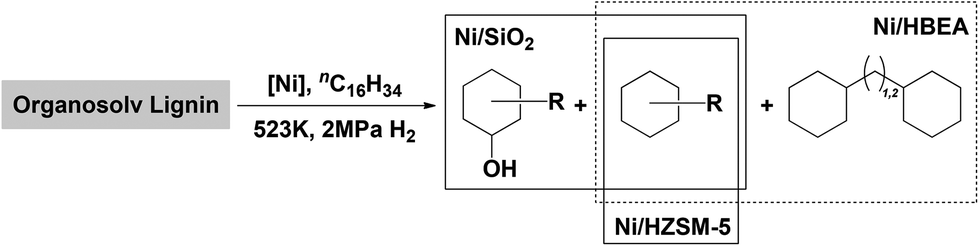 | ||
| Fig. 2 Schematic product distributions in the organosolv lignin deconstruction over Ni/SiO2, Ni/HZSM-5 and Ni/HBEA catalysts. | ||
| Catalysts | Gas phase productsc [wt%] | Liquid phase products [wt%] | Selectivity within liquid mix [wt%] | ||
|---|---|---|---|---|---|
| Monocyclic alcohols | Monocyclic alkanesd | Bicyclic alkanese | |||
| a Reaction conditions: 1 g lignin, 0.5 g Ni catalyst, 100 mL (77 g) hexadecane as solvent, 20 bar H2 initially charged into reactor at RT, 6 h, 523 K, 700 RPM stirring. b ±: Standard deviations for multiple reaction tests. c Methane, propane and methanol are the main components in the gas phase. d Monocyclic products and its isomers ranging from C5 to C9. e Bicyclic products greater than C9. | |||||
| Ni/SiO2 | 4.1 | 23 ± 2 | 20 | 80 | 0 |
| Ni/HZSM-5 | 5.5 | 28 ± 2 | 0 | 90 | 10 |
| Ni/HBEA | 6.8 | 35 ± 4 | 0 | 83 | 17 |
With Ni/HZSM-5, gas phase products were formed at a combined yield of 5.5 wt%. The product distribution in the liquid phase with Ni/HZSM-5 (Fig. S4b†) shows saturated cyclic hydrocarbons in the range of C5–C10 only, i.e., ranging from cyclopentanes (formed via acid catalyzed isomerization of cyclohexyl rings) to decalin. Performing the reaction for longer durations up to 24 h on Ni/HZSM-5 (Fig. S11†) resulted in naphthenes yields of 28 wt% after 6 h and 36 wt% after 24 h with mainly (>90%) monocyclic (isomerized) C5 to C9 and bicyclic C9 to C11 hydrocarbons (ca. 10%).
In comparison, Ni/HBEA yielded 7 wt% gas phase products and 35 wt% of liquid hydrocarbons, with carbon numbers ranging from C5 to C14 (Fig. S4c†). The higher selectivities towards bicyclic products (18%) contributed to the overall higher yield. Based on extensive studies about the alkylation during hydrodeoxygenation of phenol,48 we conclude that the medium pore of HZSM-5 hinders the alkylation of aromatic monomers by alcohols or olefins formed during hydrogenation or hydrodeoxygenation, leading to the low selectivity to bicyclic products with Ni/HZSM5. Note that some bicyclic products may originate from C–C bonds present in the starting lignin.
To show that the catalysts could be recycled, experiments were performed, in which the Ni catalyst was recovered and reused for total of three cycles. The yield of liquid phase naphthenes (Fig. S11†) decreased by approximately 5% with each reuse. Analysis of the TEM images of catalysts before and after reactions (Fig. S5†) showed that the average sizes of Ni particles were bigger in recovered catalysts. Therefore, the decline in activity with reuse is attributed to particle sintering.
Recent reports on lignin depolymerization on carbon-supported Ru,49 Ni31,32 and Pd/Zn catalysts34,50 in aqueous and/or alcoholic liquid phases have reported yields of aromatic monolignols ranging from 12 to 54 wt%, with little subsequent hydrogenation/deoxygenation even under high pressures of H2. In the present work, the catalytic lignin conversion at 523 K, in the presence of H2 and hexadecane solvent, produced saturated hydrocarbons with comparable liquid phase yields (23–35 wt%) on Ni based catalysts. This is in agreement with Rinaldi and Wang suggesting that Lewis basic solvents (H2O, methanol, THF, 1,4-dioxane) reduce the rate of catalytic hydrogenation on Ni catalysts, while the hydrogenolysis activity was not affected to the same extent during conversion of diphenyl ether or organosolv lignin.30 In the aprotic non-Lewis basic solvent, methylcyclohexane as an example, the main products derived upon organosolv lignin conversion on RANEY® Ni were saturated cyclic alcohols and hydrocarbons.
While the effects of reaction conditions, such as temperature, gas environment and its pressure, solvent and catalyst systems, have been widely explored for converting lignin, these studies9,24,26,32,34,51 have been addressing mechanistic aspects of the C–O bond cleavage of model compounds or on the comparison of ex situ analysis of lignins before and after catalyzed reactions. The kinetics of lignin deconstruction on a molecular level and the potential for a complete lignin conversion has not been explored.23,35 To follow these conversions, we use attenuated total reflection infrared (ATR-IR) spectroscopy for monitoring changes in the structure of lignin under the same reaction conditions used throughout this study.
3.4. ATR-IR spectroscopy during reductive deconstruction of lignin on Ni/HBEA
We decided to use the most active catalysts Ni/HBEA for following the changes of the lignin structure during its conversion under hydrogenation conditions using ATR-IR spectroscopy in situ during conversion (Fig. 1a). For this purpose a physical mixture of Ni/HBEA and organosolv lignin was deposited onto the ZnSe crystal (Fig. S1–S3†). n-Hexadecane saturated with 2 MPa H2 was pumped through the cell at 523 K to initiate the deconstruction of lignin; no spectroscopic changes occurred in the absence of this solvent. The surface of the ZnSe crystal was also probed with HR-SEM (Fig. 3) showing that the deposited (porous) catalyst/lignin layer (10–20 μm) was approximately an order of magnitude thicker than the penetration depth of the evanescent wave (1–2 μm). | ||
| Fig. 3 HR-SEM cross section of the lignin-catalyst physical mixture being deposited as a layer on the silicon crystal through drop coating. | ||
EDX element mapping in that film showed the distribution of Ni particles to be uniform across the coated film (Fig. S13†), thus, ensuring that the ATR-IR technique probes a region of the sample in which both lignin and Ni-catalyst are in contact. Fig. 4 shows a series of stacked background-corrected ATR-IR difference spectra of organosolv lignin reacting on Ni/HBEA catalyst as a function of contact time.
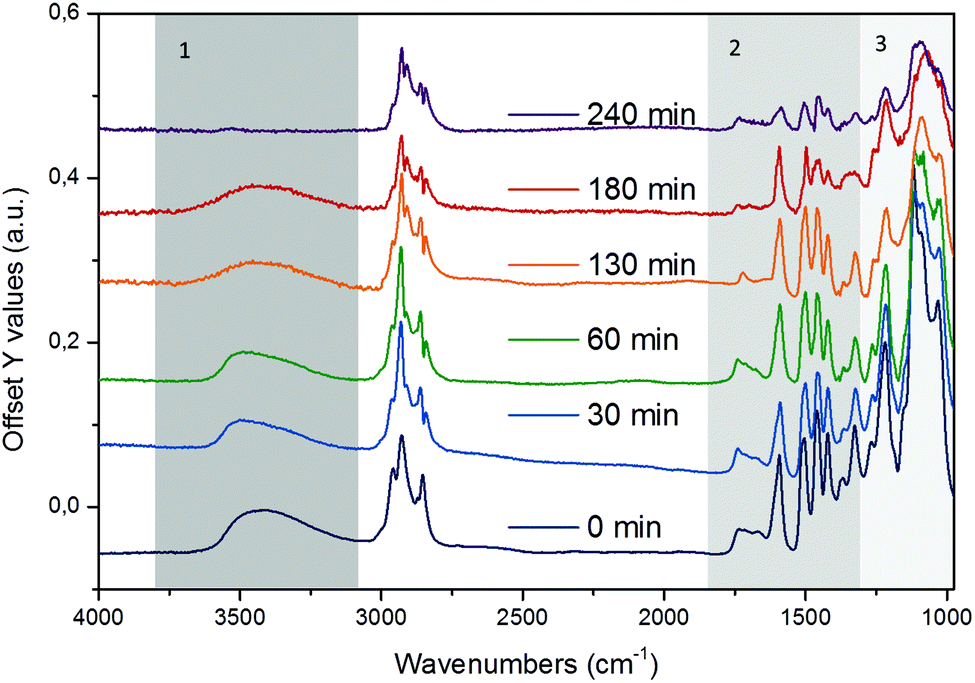 | ||
| Fig. 4 Time-resolved in situ ATR-IR difference spectra of organosolv lignin reacting over Ni/HBEA catalysts in n-hexadecane at 523 K and 20 bar H2. | ||
The time-resolved ATR-IR spectra clearly indicate a systematic change of the lignin structure with bands characteristic of individual functionalities in the lignin and intermediate products evolving at different reaction times. We have identified three characteristic regions, which are discussed in the sequence of decreasing wavenumbers, i.e., (i) O–H and C–H stretching, 3800–2500 cm−1, (ii) aryl CC, CO and C–OMe stretching, 1800–1400 cm−1, and (ii) lignol C–O–C linkages 1400–950 cm−1.
In the region of the O–H stretching vibration between 3640 and 3150 cm−1 (Fig. 5a), the broad band with a maximum at 3500 cm−1 is attributed to hydrogen bonded OH groups attached to alkyl or aryl groups. The bands between 2990 and 2816 cm−1 are assigned to the asymmetric and symmetric CH3 and CH2 stretching vibrations, which overlap with the signals of asymmetric and symmetric C–H stretching vibrations in CH2–O groups (Table S4†).44 The presence of n-hexadecane as solvent, however, makes the assignment and further deconvolution of the C–H stretching vibrations unfeasible at present. With increasing reaction time (Fig. 5b), the integrated peak areas of the O–H stretching vibration decrease quite rapidly (black trace), indicating gradual removal of the hydroxy groups.
 | ||
| Fig. 5 (a) ATR-IR spectra in the spectral region of 3800–2500 cm−1 (O–H and C–H stretching vibrations) for organosolv lignin reacting over Ni/HBEA in n-hexadecane at 523 K and 20 bar H2. (b) The evolution of integrated peak area at 3430 cm−1, representing the O–H stretching vibration, as a function of reaction time. | ||
While qualitative changes have been very reproducible, we caution against the quantitative interpretation of these observed changes, as during continuous reaction in the ATR cell the catalyst/lignin layer may change composition at the interface with the ZnSe element, i.e., more or less material may be present in the zone probed by ATR spectroscopy. Considering the sum of all C–H stretching bands on a semi-quantitative basis, it is concluded that the increase of the total band area within the first 60 min is associated with the partial hydrogenation of the aromatic rings (in products in the layer), while the decreasing intensity points to a depletion of organic matter close to the probed ZnSe surface.
The bands in the region between 1800 and 1400 cm−1 (Fig. 6) are attributed to CO stretching vibrations (1738 cm−1 and 1674 cm−1), aryl ring stretching vibrations (1591 cm−1, symmetric and 1503 cm−1 asymmetric), O–C–H (methoxy) bending vibrations (1457 cm−1), aromatic skeletal vibrations combined with C–H in-plane bending (1423 cm−1), and aryl ring breathing with CO stretch (1324 cm−1).43,44,52 These bands were deconvoluted to determine the changes of individual vibrational bands, which are plotted in Fig. 6b as a function of reaction time.
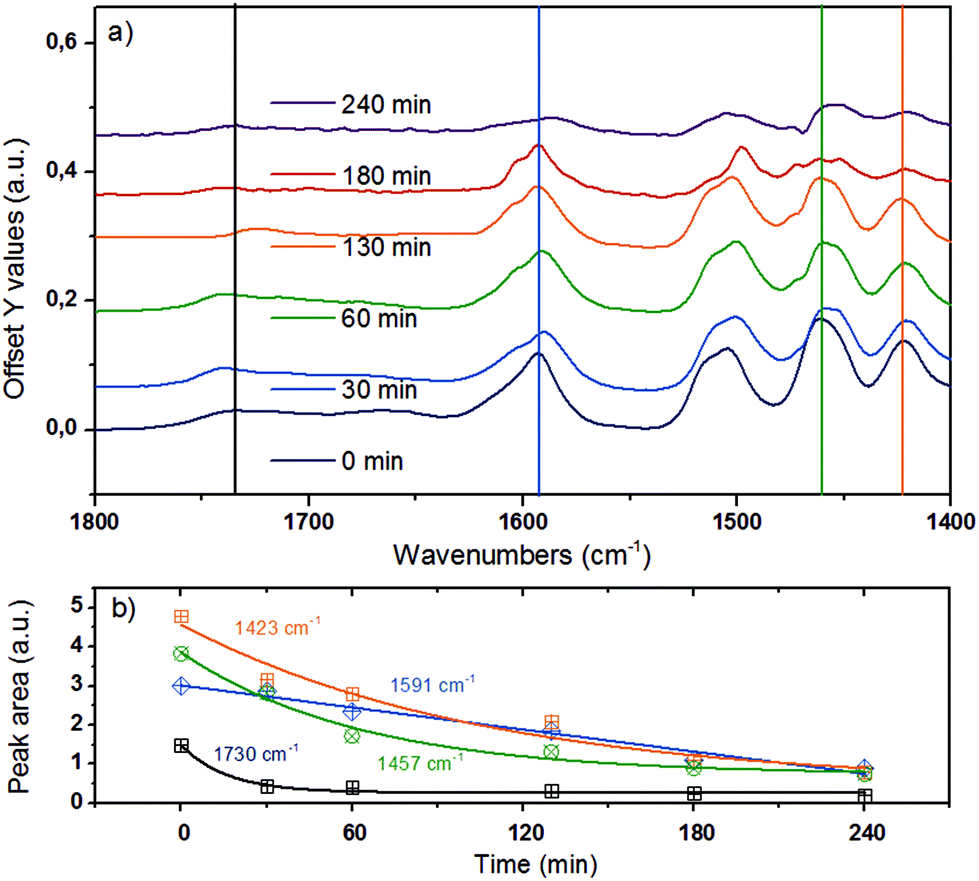 | ||
| Fig. 6 (a) ATR-IR spectra in the spectral region of 1800–1300 cm−1 for organosolv lignin reacting over Ni/HBEA in n-hexadecane at 523 K and 20 bar H2. (b) Qualitative trend for peak areas at 1730 cm−1 for CO stretching vibration, 1591 cm−1 for symmetric aryl ring stretch, 1457 cm−1 for CH3–O bend and 1423 cm−1 for aromatic skeletal vibration.43,44,52 | ||
By monitoring the temporal evolution of two vibrational modes, the symmetric aryl ring stretching at 1591 cm−1 and aromatic skeletal vibration 1423 cm−1, we follow systematically the loss of aromaticity as lignin is being deconstructed and the fragments are being hydrogenated. The band intensity of the carbonyl group associated with the syringyl-′ (S′) unit at 1740 cm−1 (ref. 43 and 44) also dropped significantly, pointing again to the reduction of the CO groups and/or the deconstruction of lignin. Note that the bending mode of C–H attached to oxygen is linked to the peak at 1457 cm−1, whose integrated area declines as a function of time.43,44 This suggests that lignin was demethoxylated also via hydrogenolysis.
Fig. 7a displays the third spectroscopic region, comprising vibrational modes in 1400–1000 cm−1. The abundant ether linkages connecting the monolignols lead to a group of pronounced bands. Due to the variety of ether linkages in lignin, it is impossible to resolve individual vibrational modes. To facilitate further analyses, we deconvoluted the broad IR peaks into components (denoted with vertical lines), assigning the IR bands at 1219 cm−1 to phenolic C–C–O asymmetric stretching and both of the bands at 1122 cm−1 and 1080 cm−1 to aliphatic C–C–O asymmetric stretching vibration.43,44,52 The time-resolved evolution of these peaks is presented in Fig. 7b.
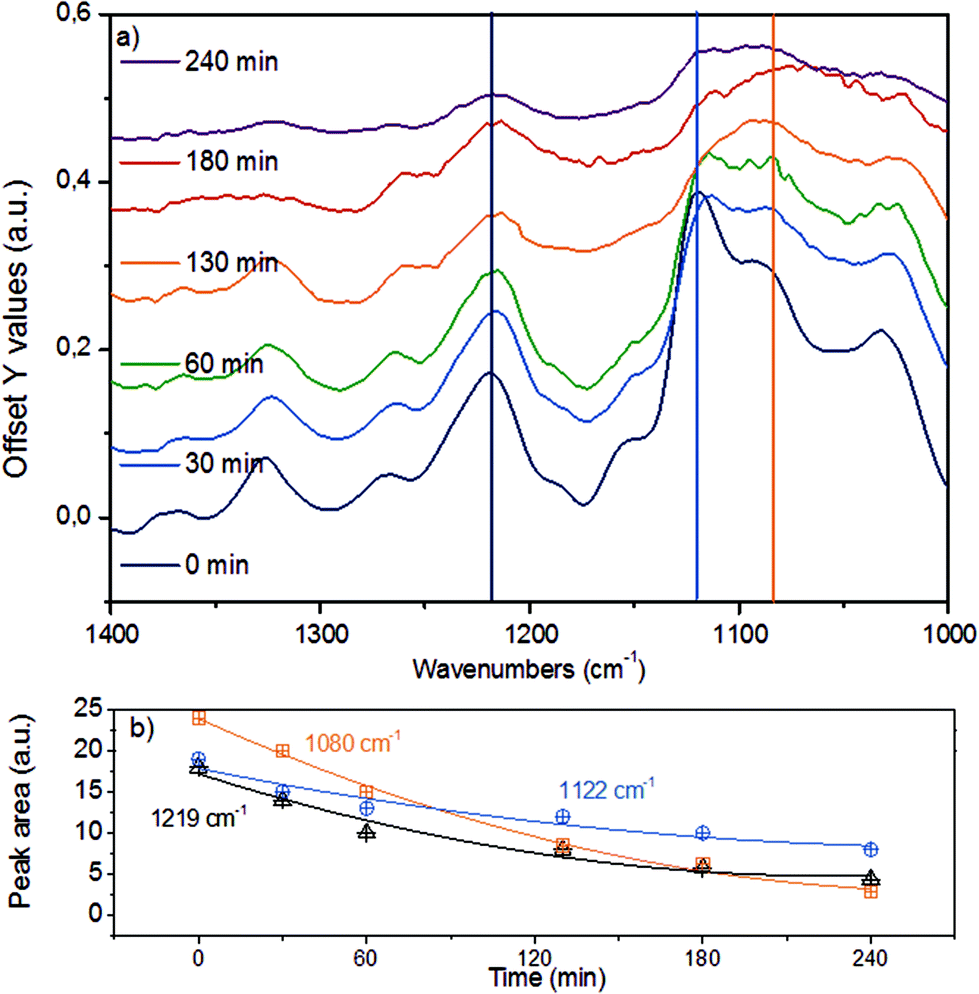 | ||
| Fig. 7 (a) ATR-IR spectra in the spectral region of 1400–1000 cm−1 for organosolv lignin reacting over Ni/HBEA in n-hexadecane at 523 K and 20 bar H2. (b) Deconvoluted and integrated peak areas at 1219 cm−1, 1122 cm−1 and 1080 cm−1 representing the aromatic C–C–O stretching vibration, the aliphatic secondary and primary C–C–O stretching vibrations, respectively. | ||
The monotonous decrease in intensity and the observation of primarily mononuclear products allow us to conclude that the changes observed are associated with C–O–C ether bond cleavage, i.e., that the cleavage of aryl alkyl ether bonds like in β-O-4 and α-O-4 linkages result in the parallel disappearance of IR bands at 1219 cm−1, 1122 cm−1 and 1080 cm−1. If we assume that the phenolic and aliphatic C–C–O stretching vibrations have comparable molar extinction coefficients, the higher initial disappearance rate of the 1080 cm−1 together with the 1122 cm−1 peaks compared to the 1219 cm−1 mode suggests that the ether bonds connecting lignols are preferably cleaved at the Caliphatic–O site, in line with observations for diphenyl ethers in aqueous and non-polar liquid phase.22,25
3.5. A proposed mechanism for Ni-catalyzed reductive deconstruction of lignin
The analyses presented above and the product analyses by GC-MS (Fig. S14†) allow us to propose a sequential mechanistic pathway for lignin deconstruction, reduction and hydrodeoxygenation. Taking as an example the β-O-4 linkage, the predominant ether bond linkage, we have drawn three possible reaction pathways in Scheme 1, including (a) Caromatic–O bond scission (b) Caliphatic–O bond scission and (c) aryl ring hydrogenation.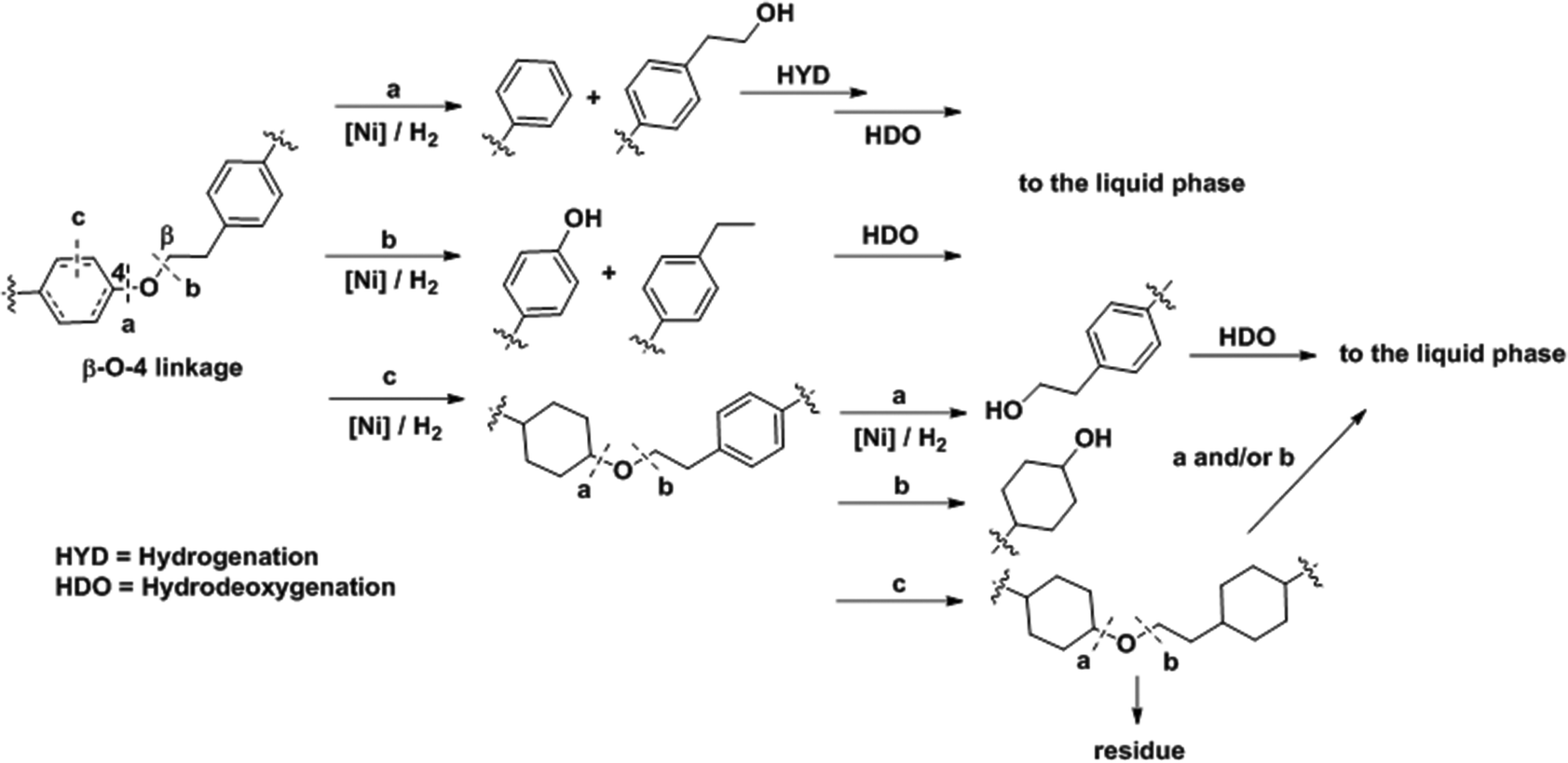 | ||
| Scheme 1 Proposed reaction pathways for Ni catalyzed C–O bond cleavage in a representative β-O-4 bond interconnecting monolignols. | ||
The direct C–O bond hydrogenolysis at the aromatic carbon through pathway a is a minor route, as evidenced by the less marked decrease in the phenolic C–O stretching band at 1219 cm−1 than the aliphatic C–O bands at 1122 and 1080 cm−1. This is consistent with the higher bond dissociation energies of Caromatic–O bonds than those of the Caliphatic–O bonds in aryl ethers, e.g., 322 vs. 289 kJ mol−1 for phenethyl phenyl ether.53 Hydrogenolysis of Caliphatic–O bonds (route b) and arene hydrogenation (route c) are major pathways. Dimer model compound studies on Ni catalysts in aqueous phase showed that the hydrogenolysis rate is one order of magnitude faster than arene ring hydrogenation.20,25 Our ATR IR results indicate that Caliphatic–O bond hydrogenolysis is faster than ring hydrogenation also in hexadecane. Once one of the ether bonded aryl rings is hydrogenated (route c), the rate of C–O bond cleavage becomes very slow.25 Thus, hydrogenation of the aromatic rings would lead to large ethers. Table 4 shows that increasing reaction temperatures selectively enhances dehydration, hydrogenolysis and hydrogenation, retarding on the other hand retrograde reactions. By this, it is possible to nearly quantitatively deconstruct lignin to substituted cycloalkanes and some light products. While increasing the temperature from 523 to 543 K led to marginal increases in hydrocarbon yield and lignin conversion, temperatures at 573 K and above helped overcome the bottlenecks for full lignin conversion.
| Temperature (K) | Hydrocarbons [wt%] | Selectivity [C%] | Waterc [wt%] | Solid residued [wt%] | Unconv. Lignine [wt%] |
|---|---|---|---|---|---|
| a Reaction conditions: 1 g lignin, 0.5 g Ni/HBEA catalyst, 100 mL (77 g) hexadecane as solvent, 20 bar H2 initially charged into reactor at RT, 6 h, temperature range from 493 to 593 K, 700 RPM stirring. b ±: Standard deviations for multiple reaction tests. c Determined by volumetric analysis. d THF-soluble solids. e THF-insoluble solids. | |||||
| 493 | 29 ± 2 | 40 | 10 | 31 ± 5 | 26 |
| 523 | 42 ± 4 | 57 | 18 | 22 ± 5 | 12 |
| 543 | 44 ± 4 | 60 | 18 | 23 ± 5 | 11 |
| 573 | 60 ± 5 | 82 | 19 | 11 ± 5 | 4 |
| 593 | 70 ± 5 | 96 | 21 | 4 ± 5 | 0.1 |
Combining these findings, the mechanism for catalytic reductive deconstruction of organosolv lignin on Ni catalysts is proposed (Scheme 2). Lignin is schematically represented by a pentameric lignol fragment, containing methoxylated phenylpropanoid moieties connected through β-O-4, α-O-4 and β–β ether bonds as prevailing in lignin1,29 and in the proposed structure (Fig. 1d). The first step is the hydrogenolysis of the ether bonds (pathway A). Under conditions used in this work, all types of ether linkages (β-O-4, α-O-4 and 4-O-5), while being intrinsically different in reactivities, were cleaved on Ni surfaces. In this step, phenolic intermediates are formed, which subsequently follow the hydrodeoxygenation pathways B, C and D on the Ni and zeolite catalyst components as described previously.16,19,21,54 As cyclic alcohols were also deoxygenated on Ni/SiO2 it is concluded that acid sites are formed with fractions of oxidized Ni (Ni-phyllosilicates),55 while we do not rigorously exclude Ni-catalyzed hydrogenolysis of C–O bonds under the experimental conditions. Especially at lower temperatures, the phenolic molecules recombine (pathway F) to form undesired higher molecular weight oligomers.25,29 We propose that retrograde reactions occur in competition with pathway B, especially at lower temperatures (T < 543 K), where the phenolic molecules recombine (pathway F) to form refractory compounds25,29 probably of high molecular weight since the solid material after reaction (Table 4) is insoluble in THF or acetone. The increased conversion of lignin at higher temperatures (≥573 K, Table 4) is a consequence of the rates of hydrogenation of phenolics (pathway B) increasing more with temperature than the retrograde reactions (pathway F). Thus, an almost complete conversion of lignin was achieved (96% at 593 K, Table 4). The modest effects of temperature on both hydrocarbon yields and lignin conversion, however, suggest that the mobility of lignin on the Ni surface, rather than chemical reactions such as hydrogenolysis of bonds (with barriers typically >100 kJ mol−1), limits the conversion of the solid lignin material. It should be noted in passing that in the presence of water hydrolysis may occur, the extent of which is dictated by the free energy barrier for this pathway and the availability of water at the surface active sites.25
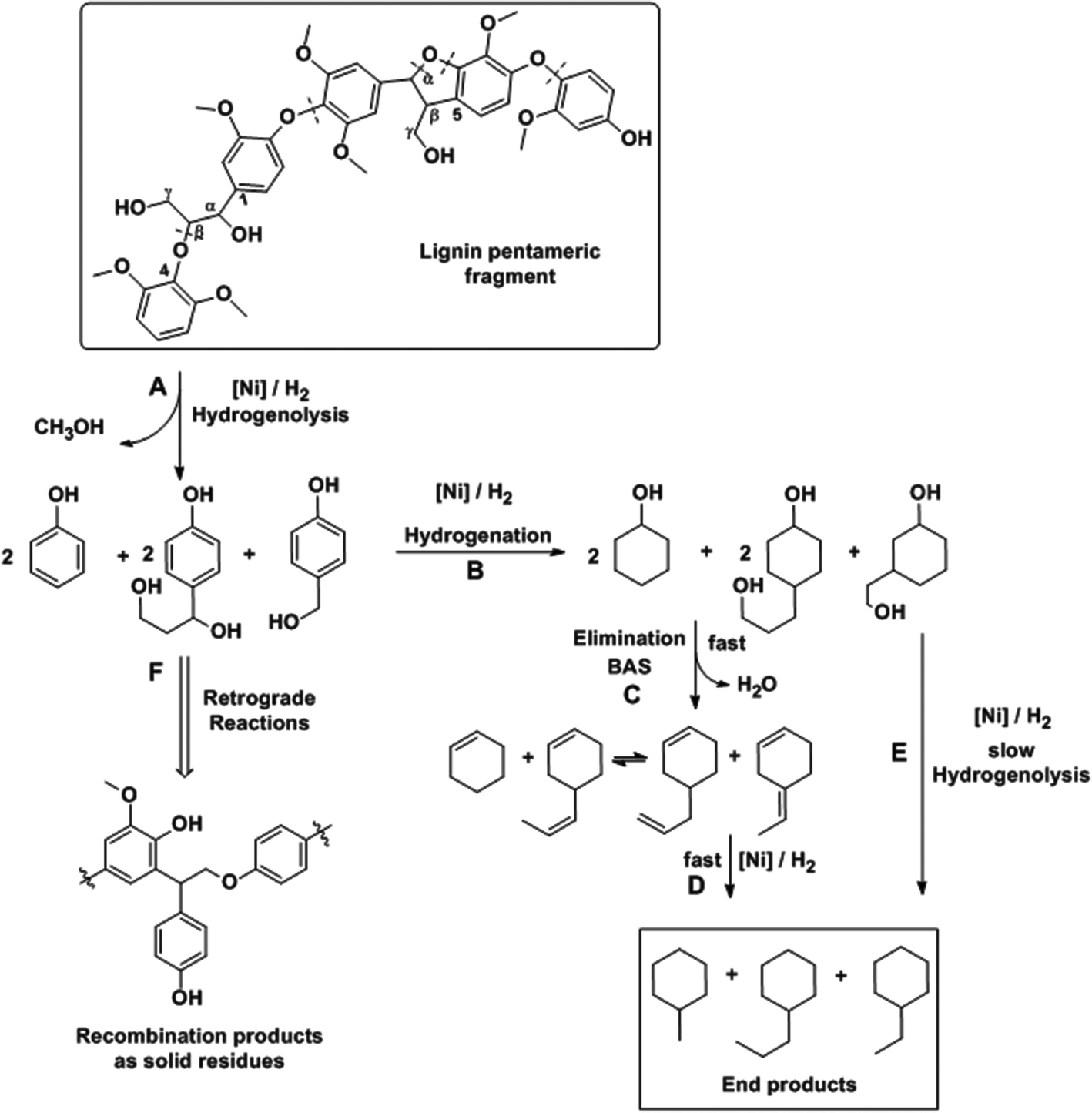 | ||
| Scheme 2 Proposed pathways for organosolv lignin deconstruction and hydrodeoxygenation on Ni based catalysts. | ||
4. Conclusions
We have shown that a one stage process for complete reductive deconstruction of organosolv lignin is possible using Ni catalysts supported on zeolites under relatively mild conditions (500–600 K, 20 bar H2) in a non-polar liquid phase such as n-hexadecane. The organosolv lignin used for the successful demonstration in the current study was a lignol oligomer with an average molecular weight of ca. 1200 Da and with 7–8 monolignols interconnected through a variety of aryl alkyl ether bonds. Full selective (to saturated hydrocarbons) conversion was achieved with Ni/H-BEA catalysts nearly avoiding retrograde reactions to high molecular weight compounds by increasing the temperature to 593 K. The HBEA zeolite catalyzed, however, some alkylation, leading to bicyclic products with up to 14 carbon atoms.The kinetics of the reduction and transformation of the aromatic structures and individual oxy-functional groups to reduced species were explored using ATR IR spectroscopy. It is shown that the reductive deconstruction of lignin proceeds via stepwise hydrogenolysis of the ether bonds followed by hydrogenation of the aromatic rings, and alkylation between partially reduced lignin monomers and substituted phenols. The results show that small lignin oligomers can be reductively deconstructed, opening a pathway to a full utilization of a wider range of lignin polymers.
Acknowledgements
The financial support from TUM-PNNL cooperation project “Development of new methods for in situ characterization in liquid phase reactions” (CN-177939) is highly appreciated. The work by S. K., H. S., and J. A. L. was partially supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), Division of Chemical Sciences, Geosciences & Biosciences. Pacific Northwest National Laboratory is a multi-program national laboratory operated for DOE by Battelle through Contract DE-AC05-76RL01830. Dr Sergei Vagin is acknowledged for conducting GPC measurements. Moreover, we want to thank Dr Jürgen Behr and Prof. Dr Rudi F. Vogel for conducting the MALDI-TOF MS analysis. Further, we want to acknowledge M.Sc. Moritz Schreiber for the help on the gas phase analysis. Dipl. Min. Katia Rodewald is acknowledged for HR-SEM EDX measurements. Lastly, we thank Stas Vaisman for the help on the graphical abstract.References
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. Holladay, J. Bozell, J. White and D. Johnson, DOE Report PNNL, 2007, 16983.
- T. Wiesenthal and A. Mourelatou, How much bioenergy can Europe produce without harming the environment?, 2006 Search PubMed.
- M. Lawoko, R. Berggren, F. Berthold, G. Henriksson and G. Gellerstedt, Holzforschung, 2004, 58, 603 CrossRef CAS.
- R. P. Overend and K. G. Johnson, in Enzymes in Biomass Conversion, American Chemical Society, 1991, ch. 21, vol. 460, pp. 270–287 Search PubMed.
- A. Johansson, O. Aaltonen and P. Ylinen, Biomass, 1987, 13, 45–65 CrossRef CAS.
- N.-E. E. Mansouri and J. Salvadó, Ind. Crops Prod., 2006, 24, 8–16 CrossRef.
- M. H. Hussin, A. A. Rahim, M. N. Mohamad Ibrahim and N. Brosse, Ind. Crops Prod., 2013, 49, 23–32 CrossRef CAS.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- B. A. V, Appl. Catal., A, 1994, 116, 5–47 CrossRef.
- C. J. Zhang Qi, W. Tiejun and Xu Ying, Energy Convers. Manage., 2007, 48, 87–92 CrossRef.
- A. A. Boateng, C. A. Mullen, N. Goldberg, K. B. Hicks, H.-J. G. Jung and J. F. S. Lamb, Ind. Eng. Chem. Res., 2008, 47, 4115–4122 CrossRef CAS.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- K. Li, R. Wang and J. Chen, Energy Fuels, 2011, 25, 854–863 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412–414 RSC.
- R. K. K. M. L. H. A. Gutierrez, R. Slioor and A. O. I. Krause, Catal. Today, 2009, 147, 239–246 CrossRef.
- E. Furimsky, Catal. Rev., 1983, 25, 421–458 CrossRef CAS.
- S. Kasakov, C. Zhao, E. Baráth, Z. A. Chase, J. L. Fulton, D. M. Camaioni, A. Vjunov, H. Shi and J. A. Lercher, Chem. – Eur. J., 2015, 21, 1567–1577 CrossRef CAS PubMed.
- C. Zhao, J. He, A. A. Lemonidou, X. Li and J. A. Lercher, J. Catal., 2011, 280, 8–16 CrossRef CAS.
- C. Zhao, S. Kasakov, J. He and J. A. Lercher, J. Catal., 2012, 296, 12–23 CrossRef CAS.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- J. He, L. Lu, C. Zhao, D. Mei and J. A. Lercher, J. Catal., 2014, 311, 41–51 CrossRef CAS.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed.
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 Search PubMed.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- I. Klein, B. Saha and M. M. Abu-Omar, Catal. Sci. Technol., 2015, 5, 3242–3245 Search PubMed.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 Search PubMed.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef CAS.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttamaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- R. Nares, J. Ramírez, A. Gutiérrez-Alejandre, C. Louis and T. Klimova, J. Phys. Chem. B, 2002, 106, 13287–13293 CrossRef CAS.
- A. Richel, C. Vanderghem, M. Simon, B. Wathelet and M. Paquot, Anal. Chem. Insights, 2012, 7, 79–89 CAS.
- D. S. Kosyakov, N. V. Ul'yanovskii, E. A. Sorokina and N. S. Gorbova, J. Anal. Chem., 2014, 69, 1344–1350 CrossRef CAS.
- F. Meemken, P. Müller, K. Hungerbühler and A. Baiker, Rev. Sci. Instrum., 2014, 85, 084101 CrossRef PubMed.
- J.-M. Andanson and A. Baiker, Chem. Soc. Rev., 2010, 39, 4571–4584 RSC.
- P. Burattin, M. Che and C. Louis, J. Phys. Chem. B, 1998, 102, 2722–2732 CrossRef CAS.
- O. Faix, Holzforschung, 1991, 45, 21 CrossRef CAS.
- S. Y. Lin and C. W. Dence, Methods in lignin chemistry, Springer, 1992 Search PubMed.
- B. C. Smith, Infrared spectral interpretation: a systematic approach, CRC press, 1998 Search PubMed.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 Search PubMed.
- S. Dabral, J. Mottweiler, T. Rinesch and C. Bolm, Green Chem., 2015 10.1039/C5GC00186B.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- C. Zhao, D. M. Camaioni and J. A. Lercher, J. Catal., 2012, 288, 92–103 CrossRef CAS.
- D. D. Laskar, M. P. Tucker, X. Chen, G. L. Helms and B. Yang, Green Chem., 2014, 16, 897–910 RSC.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- O. Derkacheva and D. Sukhov, Macromol. Symp., 2008, 265, 61–68 CrossRef CAS.
- Y.-R. Luo, Comprehensive handbook of chemical bond energies, CRC press, 2007 Search PubMed.
- W. Song, Y. Liu, E. Baráth, C. Zhao and J. A. Lercher, Green Chem., 2015, 17, 1204–1218 RSC.
- Z. A. Chase, S. Kasakov, H. Shi, A. Vjunov, J. L. Fulton, D. M. Camaioni, M. Balasubramanian, C. Zhao, Y. Wang and J. A. Lercher, Chem. – Eur. J., 2015 DOI:10.1002/chem.201502723.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc02160j |
| ‡ Current address: Department of Chemistry, East China Normal University, North Zhongshan Road 3663, 200062, Shanghai, China |
| This journal is © The Royal Society of Chemistry 2015 |