
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpcycling of waste paper and cardboard to textiles†
Y.
Ma
a,
M.
Hummel
*a,
M.
Määttänen
b,
A.
Särkilahti
b,
A.
Harlin
b and
H.
Sixta
a
aDepartment of Forest Products Technology, Aalto University, P.O. Box 16300, FI-00076 Aalto, Finland. E-mail: michael.hummel@aalto.fi
bVTT Technical Research Centre of Finland, P.O. Box 1000, FI-02044 VTT, Finland
First published on 15th September 2015
Abstract
In continuation of previously reported results, the ionic liquid 1,5-diazabicyclo[4.3.0]non-5-ene-1-ium acetate was also found to be a powerful non-derivatizing solvent for cellulosic waste such as paper and cardboard. The ionic liquid could dissolve all the present bio-polymers (cellulose, hemicellulose, and lignin) in high concentrations, resulting in solutions with visco-elastic properties that were suitable for dry-jet wet fiber spinning. The cellulosic raw materials were refined gradually to identify the influence of residual components on the spinnability of the respective solution. Polymer degradation and losses in the spinning process could be avoided nearly entirely. With the exception of virtually unrefined cardboard, all the samples showed excellent spinnability, resulting in fibers with high tensile strength. Prototype textiles were produced to validate the quality of the fibers and demonstrate the possibility of using residual lignin in cardboard as a natural dye.
Introduction
The increasing disposable income of the growing middle class in emerging economies will trigger a significant rise in the global purchasing power. Amplified by an estimated population growth of 20% (1.4 billion) over the next two decades, we will not only face a hitherto unseen need for consumption goods, but also a new peak in waste production.1 Paper and paperboard represent almost 30% of the total generated municipal solid waste. Despite a high recycling rate of ca. 60% in the Northern countries, paper and cardboard waste still represent a substantial amount of total landfills, ca. 15% in the United States2 and roughly 18% in the European Union.3 At the same time the growing worldwide demand for end-use products is depleting natural resources at an alarming pace. The cultivation of natural products has started to lag behind their consumption. This also affects the global textile industry which showed an increase in production volume of 2.8% to 92.3 million tons in 2013.4 Although cellulosic fibers cover only a small share in the manmade fiber segment, their unique properties associated with moisture management, breathability and wearing comfort of the respective garment render cellulosic fibers indispensable for the apparel industry. The per-capita consumption of cellulosic products is predicted to rise from currently 3.7 to 5.4 kg in 2030. By contrast, the cultivation of cotton – the main source of cellulosic textile fibers – is no longer expandable and showed its first minus of 4.1% during the 2012/13 season. Man-made cellulosic fibers made from wood pulp have been proposed to compensate for this so called cellulose gap. Cellulose processing from pulp, however, requires special techniques to dissolve cellulose and regenerate it again as a shaped product. Several processes for the production of man-made cellulosic fibers have been developed5,6 but only two are currently of commercial relevance: In the viscose process cellulose is derivatized with CS2 to form the xanthate which is soluble in caustic soda and is then spun into a sulphuric acid coagulation bath where cellulose filaments are formed upon hydrolysis of the xanthate. Despite the implementation of substantial amounts of CS2 and hazardous byproducts this process is clearly dominating the man-made fiber market with 4.6 million tons in 2013.7 The alternative Lyocell process uses a solvent system (N-methylmorpholine N-oxide monohydrate) that allows for the direct dissolution of cellulose without derivatization.8 The term Lyocell, given in 1989 for solvent-spun fibers, owes its genesis to the Greek word lyein (meaning dissolve) from which lyo derives and to cell from cellulose. The respective cellulose solution is then spun into an aqueous coagulation bath. The abstinence of any additional hazardous chemicals renders this process the more environmentally benign option. However, stabilizers9,10 and high process temperatures in the Lyocell process add to the energy and material costs and cause higher emission to the environment.Both processes typically require so called dissolving pulp, i.e. highly purified cellulose from which lignin and hemicelluloses have been removed to a great extent. This limits their application when it comes to the use of less-refined materials such as cellulosic waste material.
More than a decade ago, ionic liquids (ILs) have been suggested to be promising solvents for cellulose processing,11 stimulating worldwide intensive research activities in this field.12 Various cellulosic solutes, including structural analogs such as chitin13 and chitosan14 or complex polymer matrixes such as wheat straw15 or wood,16–18 were dissolved in different ILs and processed to produce fibers and films. However, nearly all reports on IL-based fiber spinning describe the use of imidazolium based ILs which show several intrinsic properties that can cause severe problems when advancing fiber spinning from a lab-scale syringe pump extruder to industrially applicable spinning equipment. Polar imidazolium-based ILs typically exhibit a limited thermal stability19,20 and are prone to degradation reactions of both the solvent itself and the cellulosic solutes.21–25
More importantly though, most ionic liquid–polymer solutions do not show the right visco-elastic properties required for a Lyocell-type fiber spinning.26 The stable extrusion of filaments alone is not sufficient for an industrially applicable spinning process. In the course of dry-jet wet spinning, the spun filaments first pass a small air gap before they enter the water bath where the solid cellulose fibers form upon instantaneous coagulation through rapid solvent exchange. In the air gap the filaments are stretched by adjusting the fiber take-up velocity to a multiple of the filament extrusion velocity. This so called draw exerts elongational stress on the polymer solutions and is the key feature of air-gap spinning. It causes the cellulose chains to align along the fiber axis. The highly oriented structure is preserved upon coagulation, resulting in filaments with high mechanical strength typical for Lyocell-type fibers. Filament draw is further detrimental to reduce the diameter of the final cellulosic fiber. Fibers in the range of 1–2 dtex (ca. 10–13 μm)‡ are needed to produce yarns for textile applications. The diameter of the spinneret orifices, however, can only be reduced to a certain extent as several breaching mechanisms in the air gap depend on the initial filament diameter.27,28 If the filament is too thin at its exit point it will break in the air gap and impede spinnability. Thus, the drawability of an initially thick filament is a key feature of the dry-jet wet spinning process to yield thin and strong fibers. The two archetypical ILs [bmim]Cl and [emim]OAc do not fully satisfy the criteria outlined above. [Bmim]Cl suffers from a high melting point, necessitating rather high process temperatures. Thus, stabilizers similar to in the case of NMMO are needed to avoid degradation of dissolved polymers.29,30 [Emim]OAc has been promoted as an excellent cellulose solvent due to its low viscosity. However, usually the reports do not address the problem that filaments extruded from [emim]OAc-cellulose solutions can only be stretched to a limited extent because they lack the required elasticity.26
We have recently introduced 1,5-diazabicyclo[4.3.0]non-5-ene-1-ium acetate ([DBNH]OAc) as a novel ionic liquid (IL) that shows excellent properties for the production of man-made cellulosic fibers (Fig. 1).26,31,32 [DBNH]OAc can dissolve a wide range of biopolymers including hemicelluloses and lignin at low temperatures and does not show side reactions as observed in imidazolium based ILs. More importantly though, even with a large share of secondary biopolymers such as lignin and hemicelluloses, the [DBNH]OAc solutions showed appropriate visco-elastic properties to withstand the high elongational stress caused by the draw in the air gap and, consequently, revealed excellent spinnability in a Lyocell-type dry-jet wet spinning process. In order to highlight the resemblance to Lyocell this IL-based process was termed Ioncell-F(iber).
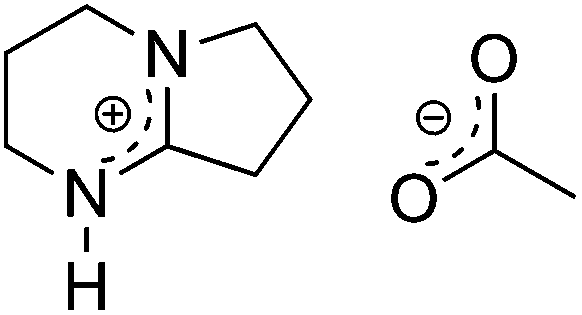 | ||
| Fig. 1 Chemical structure of 1,5-diazabicyclo[4.3.0]non-5-ene-1-ium acetate ([DBNH]OAc). | ||
Its broad tolerance towards various polymers allowed one to dissolve and process also less refined cellulosic materials. Instead of highly refined dissolving pulp, cellulosic waste material was used as feedstock for the production of textile fibers. Paper can typically undergo only a limited number of recycling cycles as the pulp fibers are shortened gradually. Extensively shortened fibers cannot be reused for paper production. If they are not destined for landfill they might find their final end-use as a filler material in composites or insulation materials. By contrast, the production of textile staple fibers is insensitive towards the length of the original paper pulp fiber and only depends on the cellulose's degree of polymerization which is typically still higher than that actually required. Thus, value-added products can be manufactured from low-grade waste materials. Earlier attempts to use NMMO for the production of textile fibers from cellulosic waste were confined to lignin free paper and resulted in fibers with very low mechanical strength (20 cN tex−1).33 No further reports on this topic appeared, presumably because of the complex side reactions that can occur at the rather high processing temperatures (110–130 °C) required for NMMO spinning. In particular lignin might undergo unexpected degradation reactions in the presence of the redox-active N-O moiety of NMMO, although cellulose–lignin blends have been spun successfully from NMMO solution.34 This problem is bypassed through the relative chemical inertness of [DBNH]OAc and moderate process temperatures (70–80 °C).
In the study at hand, recycled white fine paper and cardboard were used as raw materials to produce cellulosic textile fibers. The raw material was refined gradually to identify the minimum refining needed to turn waste material into feedstock for man-made cellulosic fibers.
Experimental
Materials
A4 copy paper sheets of 80 g m−2 grammage were used as fine paper. Cutting residues from a Finnish fluting board mill were used as a cardboard source. The fine paper contained about 73% of fully bleached chemical pulp and 27% of inorganic material analyzed as ash content. The cardboard was a mixture of mechanical, semi-chemical and chemical pulps having a high lignin content of over 10% and an ash content of about 5%. Beechwood organosolv lignin was received from Fraunhofer Institutes (Germany) and added to a pulp solution for the final demonstrator garment. Ecopulp® was purchased from AB Enzymes, Finland. H2SO4 (2 M) and NaOH solutions (1 N) were obtained from OY FF-Chemicals Ab, Finland; 50% NaOH solution from Algol Chemicals Oy, Finland; solid NaOH flakes and H2O2 (30% solution) from Merck-KGaA, Germany; Na2S from Tessenderlo Chemie, Switzerland. Cl2O solution was prepared as described earlier.35Raw material refining
The refining steps for the recycled materials are illustrated in Scheme 1.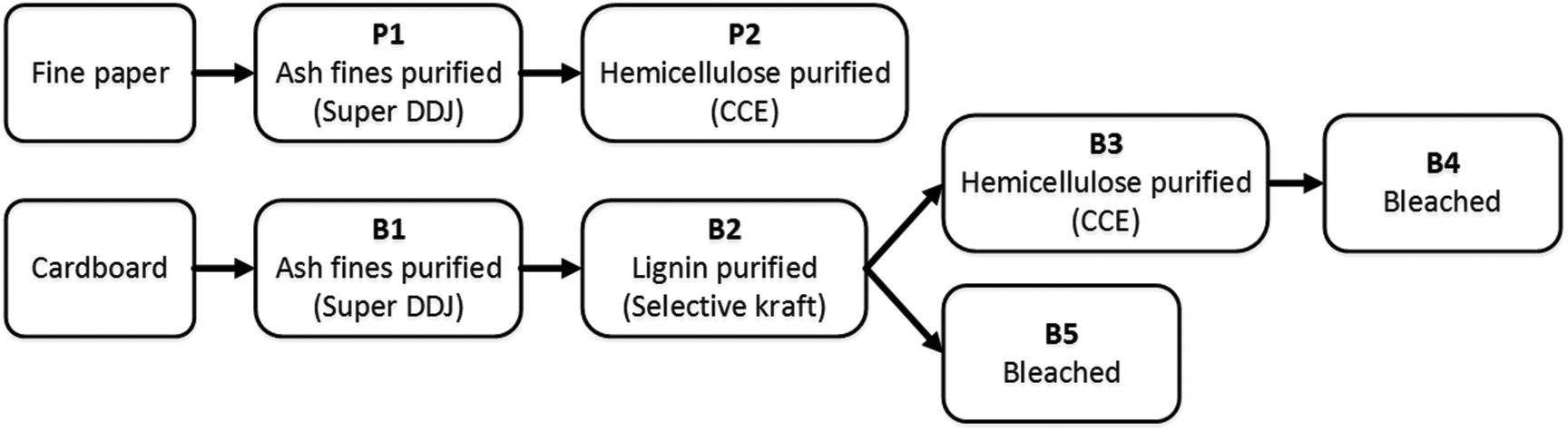 | ||
| Scheme 1 Purification procedure steps of fine paper and cardboard. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 liquor to wood ratio, 160 °C, 190 min). Subsequently, the pulp was washed, homogenized and characterized.
:1 liquor to wood ratio, 160 °C, 190 min). Subsequently, the pulp was washed, homogenized and characterized.
Pulp dissolution and filtration
Prior to dissolution of refined paper and cardboard, [DBNH]OAc was liquefied at 70 °C. The spinning dopes were prepared in a vertical kneader system as described previously.36 Air dried pulps were mixed with the IL and kneaded for 1.5 hours at 80 °C and 10 rpm under reduced pressure (50–200 mbar). The obtained solutions were filtered via a hydraulic press filter device (metal filter mesh with 5 μm absolute fineness, Gebr. Kufferath AG, Germany) with 2 MPa at 80 °C to remove undissolved substrate impurities which would deteriorate the spinnability. The dope was then shaped into the dimension of the spinning cylinder and solidified upon storage at room temperature.Rheological measurements
Shear rheology of all the solutions was measured with an Anton Paar MCR 300 rheometer with a parallel plate geometry (25 mm plate diameter, 1 mm gap size). The viscoelastic domain was determined by performing a dynamic strain sweep test and a strain of 0.5%, which fell well within the linear viscoelastic regime, was chosen for the frequency sweep measurements. Each sample was subjected to a dynamic frequency sweep between 60 and 90 °C over an angular velocity range of 0.1–100 s−1. Complex viscosity (|η*|) and storage (G′) and loss (G′′) moduli were recorded. Assuming that the Cox–Merz rule§ was valid,37,38 the zero shear viscosity was determined by fitting the complex viscosity data with the three-parameter Cross viscosity model.39Spinning, washing, and drying
Multi-filaments were spun on a customized laboratory piston spinning system (Fourné Polymertechnik, Germany) as described previously (Fig. 2).26 The solidified solution was placed in the cylinder in which the dope is heated to 70 °C to form a highly viscous, air bubble free spinning dope. The molten solution was then extruded through an 18-hole spinneret with a capillary diameter of 100 μm and a length to diameter ratio (L/D) of 0.2. After passing through a 1 cm air gap, the filaments were coagulated in a water bath (15 °C) in which the filaments were guided by Teflon rollers to the Godet couple. The extrusion velocity (ve) was set to 0.80 ml min−1 (5.66 m min−1) while the take-up velocity (vtu) of the Godet varied from 5 to 100 m min−1 depending on different samples. The draw ratio is defined as DR = vtu/ve. The fibres were washed in hot water (60 °C) and air dried.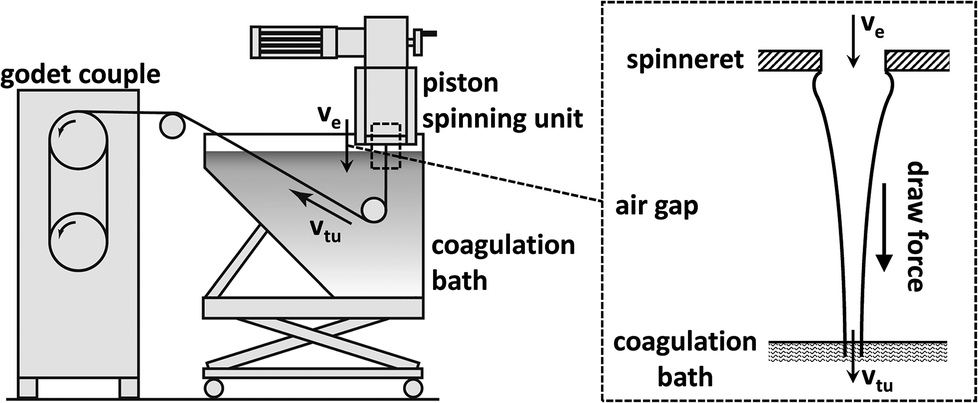 | ||
| Fig. 2 Left: schematic depiction of the customized laboratory piston spinning system (ve = extrusion velocity, vtu = take-up velocity); right: liquid filament in the air gap. After exiting the spinneret capillary the polymer solution shows dye swelling (exaggerated depiction) before the filament is thinning rapidly due to the draw force generated by the increased take-up velocity.26 | ||
Yarn spinning and knitting
The spun continuous filament was cut into staple fibers and then spun to yarns at the Swedish School of Textiles (University of Borås, Sweden) using a Mesdan lab mini spinning line comprising a Laboratory carding machine, a Stiro-Roving-Lab, and Ring-Lab Fibers. A two-ply yarn was produced and then knitted with a Stoll flat bed knitting machine. A detailed description of the yarn preparation and knitting was given earlier.40Molecular weight distribution
Prior to the analysis, the samples were cut into small pieces and – if needed – delignified in aqueous sodium chlorite solution according to Wise et al.41The intrinsic viscosity of the blends was determined in cupriethylenediamine (CED) according to the standard method SCAN-CM 15:99.
The molar mass distribution was characterized by gel permeation chromatography (GPC). The GPC-system consisted of a pre-column (PLgel Mixed-A, 7.5 × 50 mm), four analytical columns (4 × PLgel Mixed-A, 7.5 × 300 mm) and a RI-detector (Shodex RI-101). After a solvent exchange sequence, to remove the residual water and activate the sample in N,N-dimethylacetamide, the samples were dissolved in 90 g l−1 LiCl/DMAc at room temperature under constant slow speed magnetic stirring. In order to facilitate the dissolution in LiCl/DMAc, B1 pulp and fibre samples were additionally treated with ethylenediamine (EDA) following the protocol of Yamamoto et al. (2011).42 The dissolved cellulose samples were then diluted in pure DMAc to reach a concentration of 1 mg ml−1 in 9 g l−1 LiCl/DMAc. Filtration with a 0.2 μm syringe filter was carried out and a volume of 100 μl was separated at 25 °C at a flow rate of 0.750 ml min−1 with 9 g l−1 LiCl/DMAc as an eluent. Pullulan standards with molecular weights ranging from 343 Da to 708000 Da were selected for the calibration. A correction of the molar mass distribution obtained by direct-standard-calibration was performed with an algorithm calculating cellulose-equivalent molar masses of pullulan standards (MMcelluose = q* MMppullulan, with q = 12.19 and p = 0.78) as described earlier.43,44
Chemical composition
The chemical compositions (carbohydrate, total lignin) of the raw materials and fibers were analyzed according to NREL/TP-510-42618. The amount of carbohydrates was detected by high performance anion exchange chromatography with pulse amperometric detection (HPAEC-PAD) using a Dionex ICS-300 system. The cellulose and hemicellulose contents were calculated according to the amount of monosaccharides following the Janson formula.45Mechanical analysis of fibers
Linear density (dtex), tenacity (cN tex−1) and elongation to break (%) were determined in both conditioned (23 °C, 50% RH) and wet states using a Vibroskop–Vibrodyn system (Lenzing Instruments GmbH & Co KG, Austria). Ten fibres per sample were tested. The gauge length was 20 mm, pretension 5.9 ± 1.2 mN tex−1, and speed 20 mm min−1 according to DIN 53816. The Young's modulus of the spun fibres was calculated from the slope of the entire elastic region of the stress–strain curves with a Matlab script according to the ASTM standard D2256/D2256M.The total average orientation of both amorphous and crystalline cellulose in each fiber sample was determined using a polarized light microscope (Zeiss Axio Scope) equipped with a 5λ Berek compensator. The birefringence Δn of the specimen was obtained by dividing the retardation of the polarized light by the thickness of the fiber, which was calculated from the linear density (titer) using a cellulose density value of 1.5 g cm−3.46 The total orientation was then derived by dividing Δn by the maximum birefringence of cellulose of 0.062.47,48
Results and discussion
Chemical changes through raw material pre-treatment
Two recycled materials were chosen as cellulose sources: white fine paper and cardboard. These two feedstock materials have undergone different refining procedures during their manufacture which was reflected in their initial composition summarized in Table 1. Cardboard consisted of a mix of different, low refined pulps and showed a lignin content of 16.6%. Both cellulose sources contained ca. 20% of hemicelluloses. In order to determine the influence of the solute composition on the spinnability and final fiber properties, the raw material was refined stepwise as depicted in Scheme 1. The samples were taken after each refining step, dissolved in [DBNH]OAc and spun into fibers. Both paper and cardboard contained a significant amount of inorganic material (determined as ash content) that had to be removed together with fines in a first purification step using a Super Dynamic Drainage Jar to produce pulp samples P1 and B1, respectively. Residual lignin in cardboard was largely removed via classical kraft cooking (B2). Cold caustic extraction led to a reduction in hemicellulose to yield paper and cardboard-derived samples P2 and B3, respectively. Final bleaching in the cardboard stream reduced the lignin content further and increased the brightness of the final fiber (B4 and B5). The intrinsic viscosity of all pulps was then adjusted via enzymatic treatment to a level of 400–500 ml g−1. It was found earlier that pulps within this particular viscosity range showed the highest spinnability upon dissolution in the IL which is in line with the requirements of the industrial NMMO-based Lyocell process. Both paper and cardboard contained a considerable amount of metal ions introduced primarily during pulping and product manufacturing (see ESI, Table S2†). In particular, cardboard showed high levels of calcium, silicon, magnesium, and aluminum. Metal ions would accumulate in the ionic liquid solvent medium and can catalyze side and degradation reactions. Since isolation of metal ions from an ionic liquid would be a considerable challenge, their introduction into the system should be avoided in the first place. They were thus removed in a final acid washing step before dissolution of the respective pulp.| Pulp | Fibera | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intrinsic viscosity | Cellulose | Hemi-cellulose | Lignin | Cellulose | Hemi-cellulose | Lignin | Titer | Dry tenacity | Wet tenacity | Dry modulus | Wet modulus | Max. draw | |
| [ml g−1] | % | % | % | % | % | % | [dtex] | [cN tex−1]b | [cN tex−1]b | [GPa] | [GPa] | ||
| a Fiber properties are from fibers spun at a draw ratio of 14.1 with the exception of B1. b Values in [MPa] in the ESI. | |||||||||||||
| Fine paper | 76.6 | 20.8 | 2.4 | ||||||||||
| P1 11.5% | 470 | 75.5 | 24.1 | 0.4 | 75.9 | 23.2 | 1.0 | 1.34 | 37.6 | 28.7 | 21.7 | 16.7 | 14.1 |
| P2 13% | 428 | 88.1 | 11.3 | 0.6 | 87.8 | 11.1 | 1.1 | 1.60 | 44.1 | 32.0 | 24.5 | 14.9 | 16.8 |
| cardboard | 57.6 | 20.8 | 16.6 | ||||||||||
| B1 13% | 447 | 63.8 | 20.9 | 15.3 | 67.4 | 16.5 | 16.1 | 5.81 | 13.2 | 10.0 | 7.4 | 4.6 | 3.5 |
| B2 11.5% | 500 | 80.2 | 16.6 | 2.9 | 80.8 | 16.4 | 1.1 | 1.31 | 44.5 | 36.9 | 23.4 | 19.9 | 15.9 |
| B3 13% | 470 | 88.3 | 9.8 | 1.9 | 88.8 | 9.7 | 1.4 | 1.54 | 48.9 | 43.9 | 27.7 | 22.1 | 17.8 |
| B4 13% | 460 | 89.1 | 10.0 | 0.9 | 89.4 | 9.9 | 0.8 | 1.40 | 47.4 | 42.6 | 23.6 | 19.9 | 15.9 |
| B5 13% | 436 | 82.4 | 16.7 | 0.9 | 82.4 | 16.4 | 1.2 | 1.66 | 45.8 | 37.4 | 24.1 | 19.2 | 19.8 |
Molar mass distribution of refined materials
Paper derived sample P1 showed a typical bimodal molar mass distribution (MMD) deriving from hemicelluloses at lower molar mass and cellulose at higher molar mass, respectively (Fig. 3, top and ESI, Table S3†). In accordance with the carbohydrate analysis, the hemicellulose peak for the P2 sample was notably decreased after the CCE step. | ||
| Fig. 3 Molar mass distribution of paper (top) and cardboard (bottom) pulps and fibers spun thereof. | ||
Fig. 3 (bottom) shows the MMD of the board samples. The bimodal MMD of the carbohydrate fraction is preserved in all pretreatment steps with the exception of B4 where the removal of the hemicelluloses via CCE is again reflected by the disappearance of the peak at low molar mass. The cellulose fraction, however, remains virtually undegraded (see also ESI, Table S3†). The MMD curve of B1 is clearly shifted to lower molar mass values. This, however, is an artefact deriving from the delignification and solvent exchange procedures needed to render B1 pulp and fiber samples soluble in LiCl/DMAc. Thereby, the carbohydrates undergo degradation. In untreated B1 samples cellulose is connected to lignin and cannot be dissolved properly for analysis. The effects of these supramolecular structures on the spinnability will be discussed in the next chapters.
Dissolution in [DBNH]OAc
Independent of the degree of refining and residual lignin and hemicelluloses all pulps dissolved readily in [DBNH]OAc. Typically, solutions of 13 wt% pulp in [DBNH]OAc were prepared following the protocol described earlier.26 The viscoelastic properties were assessed by means of oscillatory shear measurements. Frequency sweeps were recorded at different temperatures to capture the frequency dependent dynamic moduli and complex viscosity. Assuming the validity of the Cox–Merz rule, the zero-shear viscosity (η0) was calculated and the cross-over point (COP) of storage and loss modulus determined. It was found that stable fiber spinning required a η0 of around 30000 Pa and a COP located at ca. 1 s−1 and 3000–5000 Pa. This finding was crucial as it sets the upper and lower limits of solute concentration and allowed us to predict the temperature at which stable spinning was anticipated. For instance, the intrinsic viscosity of P1 and B2 was a bit higher and it proved beneficial for the spinnability to lower the solute concentration to 11.5 wt%. The rheological key values are summarized in ESI, Table S4.†
Fiber spinning
The obtained solutions were spun using a customized piston spinning unit (spinning temperatures are given in ESI, Table S4†). Good spinnability in the context of dry-jet wet spinning is reflected in a high possible draw-ratio (ratio of take-up velocity to extrusion velocity); that is, the filament can be stretched to a great extent to reduce the final diameter of the fiber and increase the total cellulose orientation in the resulting fibers.Fine paper could be spun already after removal of fines. The high hemicellulose content of more than 20% did not impair the spinnability and a draw of 14 was accessible. The resulting fibers showed already a high tensile strength of almost 38 cN tex−1 (600 MPa). Cold caustic extraction reduced the hemicellulose content to 11% and improved the spinnability even further. The reduction in hemicellulose content and, consequently, the relative gain in high molecular weight cellulose (ESI, Table S3†) are reflected in the higher mechanical properties of the P2 fibers. In the course of the spinning process the biopolymer composition changes only to a very minor extent. The hemicellulose content is reduced slightly (see Table 1) indicating that a small share is not coagulated but kept in the spin bath solution. It was shown earlier that low molecular weight hemicelluloses can be solubilized by IL–water mixtures.49,50 Due to the low processing temperatures and the high inertness of the ionic liquid solvent almost no cellulose degradation was observed. The molar mass distribution of the carbohydrates was preserved nearly entirely (Fig. 3 and ESI, Table S3†).
All board-derived pulps exhibited excellent spinnability in [DBNH]OAc with the exception of B1 which showed improper visco-elastic properties. The gel-like state of B1-solutions prevailed also at elevated temperatures and impaired the extrusion of filaments with the correct viscoelasticity to be drawn in the air gap. Despite being fully solubilized, residual native lignin intimately bound to cellulose forms macromolecular network structures in solution which cannot withstand the high uniaxial elongational stress exerted during the draw. Thus, only a moderate draw ratio of 3.5 was possible which resulted in relatively thick fibers with weak mechanical properties. Mild kraft cooking removed lignin to a great extent and reduced the hemicellulose content slightly (B2). The difference in spinnability was marked. High draw ratios and high tenacity fibers were accessible instantaneously. Further hemicellulose extraction (B3) and lignin reduction via bleaching (B4) improved the final fiber properties only slightly. However, the final bleaching step did reduce the brown hue of the fiber. Thus, the CCE step was skipped and the fibers were bleached directly after kraft cooking to obtain only slightly colored high-strength fibers (B5).
As observed for paper-based fibers, a very small share of hemicelluloses was not recovered in the spun fiber (Table 1). Small lignin losses were detected for B2 and B3 but not for the bleached fibers B4 and B5. Interestingly, lignin was fully recovered in the case of lignin-rich B1. This again indicates that the lignin is bound to the carbohydrates and the composite macromolecules are fully precipitated upon coming in contact with water.
Also in the case of cardboard, the molar mass distribution of the carbohydrates was preserved entirely during the spinning procedure (Fig. 3 and ESI, Table S3†).
Mechanical properties of spun fibers
The mechanical properties of the spun fibers depend on the orientation of the polymers, which is influenced by the composition of the pulp in two ways. The total orientation of the fiber is mainly determined by the cellulose chains having a considerably higher molar mass than lignin or hemicellulose. Thus, a large share of hemicellulose or lignin will inevitably weaken the fiber compared to pure cellulose fibers spun under the same conditions. The composition, however, also affects the visco-elastic properties of the spin solution which defines the maximum draw. Non-ideal elasticity will impede high draw ratios, resulting in fibers with low orientation. This is particularly visible in the case of B1 where only a draw ratio of 3.5 was possible. As the draw is increased the fibers develop both total orientation and strength (see ESI, Tables S5–S11†).32,51,52The mechanical properties of the fibers spun at different draw ratios are summarized for each sample in Tables S5–S11 in the ESI.† The tenacities (tensile strength) of all fibers at maximum draw in conditioned and wet states are illustrated in Fig. 4. With the exception of B1, fibers with tenacities of around 40 cN tex−1 (600 MPa) or higher could be spun from all pulps. Even at a hemicellulose and lignin content of 16% and 1%, respectively, B2 and B5 derived fibers showed tenacities of ca. 45 cN tex−1 (675 MPa). CCE treated B3 and B4 touched even 50 cN tex−1 (750 MPa). All waste material fibers showed only a minor drop in mechanical strength under wet conditions. This is a typical feature of Lyocell-type fibers.
 | ||
| Fig. 4 Conditioned (23 °C, 50% RH) and wet tenacities of spun fibers at maximum draw ratio (indicated on the top of each column pair). | ||
The mechanical properties of the spun fibers were compared to commercial man-made cellulose fibers and fibers spun with the same IL and conditions using a hardwood pre-hydrolysis kraft pulp (Fig. 5). The respective stress–strain curves can be found in the ESI (Fig. S1).† The Young's moduli were calculated from the entire elastic region. The waste-material based fibers were slightly inferior to those spun from dissolving pulp but exhibited nevertheless excellent properties, rendering them attractive also as reinforcing fibers for composite materials.
 | ||
| Fig. 5 Mechanical properties of fibers spun from waste material. For comparison, the properties of fibers spun from dissolving pulp using the same ionic liquid and commercial fibers (Viscose, Modal, Tencel adopted from Adusumali et al.53) are included. | ||
Demonstrator garment production
The combination of kraft cooking and bleaching at various intensity levels allowed for the preparation of naturally dyed fibers covering a wide spectrum of brown hues. The spun filaments were cut into staple fibers and converted into yarns as described in detail earlier.26,40 Depending on the amount of lignin, the yarns exhibited different shades of brown. Fig. 6 shows photographs of small prototype fabrics knitted with different yarns to demonstrate the potential of lignin as a dye substituent. | ||
| Fig. 6 Yarns and final knitted garments made of fibers spun from bleached pulp (white), cardboard (beige), and pulp with additional lignin added (brown). | ||
Recycling of the ionic liquid and coagulation water
A virtually quantitative recovery of the ionic liquid solvent is mandatory from an economic and ecological perspective and is a prerequisite to advance laboratory demonstrations to commercially feasible concepts. Currently, water evaporation trials are conducted using a thin film short path evaporation system to reduce the thermal impact on the [DBNH]OAc–water mixture. At the same time the chemical stability and toxicity of neat and aqueous [DBNH]OAc are tested. Also, separation methods to remove oligosaccharides that remain dissolved in the coagulation bath are developed. Some but not all techniques established in the NMMO-based Lyocell process can be adopted. The research is ongoing and results will be reported elsewhere in due time.Conclusions
Capitalizing on the recently identified solubility power of the ionic liquid 1,5-diazabicyclo[4.3.0]non-5-ene-1-ium acetate it was possible to dissolve the cellulosic waste material to produce high strength fibers suitable for textile applications. Only mild refining of the raw material was needed once the lignin–carbohydrate complexes were destroyed. The adjustment of the cellulose DP was a prerequisite. The obtained solutions showed excellent spinnability at moderate temperatures resulting in cost-efficient fibers with high tensile strength and Young's modulus. During the spinning process, carbohydrates were preserved almost entirely, i.e. the molar mass distribution was not changed and only small amounts of hemicelluloses were not coagulated within the fibers but lost in the spinning bath instead. Bleaching was possible but not necessary as the resulting color could serve as a natural dye in the garment production. Evidently, the color is irrelevant when using the described fibers as a high strength reinforcement in non-transparent composite materials.In continuation of the results presented herein, research on the use of genuine newsprint paper waste as feedstock is ongoing. Newsprint typically consists of a large share of coarsely refined thermo-mechanical wood pulp which requires additional pulping procedures to unlock the cellulose in the matrix. Alternative methods to save chemicals and energy such as e-beam treatment are currently pursued.
Acknowledgements
This study was conducted within the Design driven World of Cellulose program funded by the Finnish Funding Agency for Innovation (TEKES).Notes and references
- F. M. Hämmerle, Lenzinger Ber., 2011, 89, 12–21 Search PubMed.
- EPA report: Municipal Solid Waste Generation, Recycling, and Disposal in the United States: Facts and Figures for 2012, 2014.
- EEA report: Managing municipal solid waste – a review of achievements in 32 European countries, 2013.
- The Fiber Year 2014 – World Survey on Textiles & Nonwovens, 2014.
- T. Röder, J. Moosbauer, G. Kliba, S. Schlader, G. Zuckerstätter and H. Sixta, Lenzinger Ber., 2009, 87, 98–105 Search PubMed.
- T. Liebert, in Cellulose Solvents: For Analysis, Shaping and Chemical Modification, ACS Symposium Series, ed. T. Liebert, T. J. Heinze and K. J. Edgar, American Chemical Society, 2010, vol. 1033, pp. 3–54 Search PubMed.
- N. Bywater, Lenzinger Ber., 2011, 89, 22–29 Search PubMed.
- H. P. Fink, P. Weigel, H. J. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473–1524 CrossRef CAS.
- F. A. Buijtenhuijs, M. Abbas and A. J. Witteveen, Papier, 1986, 40, 615–619 CAS.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968–971 RSC.
- B. Ma, A. Qin, X. Li and C. He, Carbohydr. Polym., 2013, 97, 300–305 CrossRef CAS PubMed.
- A. Lehmann, J. Bohrisch, R. Protz and H.-P. Fink, WO 2013/144082A1, 2013.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodriguez and R. D. Rogers, Green Chem., 2009, 11, 646–655 RSC.
- N. Sun, W. Li, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 1158–1161 RSC.
- A. Brandt, J. Grasvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- M. Kosmulski, J. Gustafsson and J. B. Rosenholm, Thermochim. Acta, 2004, 412, 47–53 CrossRef CAS.
- N. Meine, F. Benedito and R. Rinaldi, Green Chem., 2010, 12, 1711–1714 RSC.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- M. Hummel, C. Froschauer, G. Laus, T. Roder, H. Kopacka, L. K. J. Hauru, H. K. Weber, H. Sixta and H. Schottenberger, Green Chem., 2011, 13, 2507–2517 RSC.
- F. Wendler, L.-N. Todi and F. Meister, Thermochim. Acta, 2012, 528, 76–84 CrossRef CAS.
- A. Michud, M. Hummel, S. Haward and H. Sixta, Carbohydr. Polym., 2015, 117, 355–363 CrossRef CAS PubMed.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. J. Hauru, A. Parviainen, A. T. King, I. Kilpeläinen and H. Sixta, Adv. Polym. Sci., 2015 DOI:10.1007/1012_2015_1307.
- A. Ziabicki and R. Takserman-Krozer, Kolloid Z. Z. Polym., 1964, 198, 60–65 Search PubMed.
- A. Ziabicki and R. Takserman-Krozer, Kolloid Z. Z. Polym., 1964, 199, 9–13 CAS.
- G. Laus, G. Bentivoglio, H. Schottenberger, V. Kahlenberg, H. Kopacka, H. Roeder, T. Roeder and H. Sixta, Lenzinger Ber., 2005, 84, 71–85 CAS.
- G. Bentivoglio, T. Roeder, M. Fasching, M. Buchberger, H. Schottenberger and H. Sixta, Lenzinger Ber., 2006, 86, 154–161 CAS.
- L. J. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471–4481 CrossRef CAS.
- H. Sixta, A. Michud, L. K. J. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpeläinen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 43–57 CAS.
- H. Firgo, D. Eichinger and M. Eibl, WO 96/07778A1, 1996.
- A. Lehmann, H. Ebeling and H. P. Fink, US 2014/0194603A1, 2014.
- T. Lehtimaa, S. Kuitunen, V. Tarvo and T. Vuorinen, Holzforschung, 2010, 64, 555–561 CrossRef CAS.
- L. K. J. Hauru, Y. Ma, M. Hummel, M. Alekhina, A. W. T. King, I. Kilpelaeinen, P. A. Penttilae, R. Serimaa and H. Sixta, RSC Adv., 2013, 3, 16365–16373 RSC.
- F. Lu, B. Cheng, J. Song and Y. Liang, J. Appl. Polym. Sci., 2012, 124, 3419–3425 CrossRef CAS.
- W. P. Cox and E. H. Merz, J. Polym. Sci., 1958, 28, 619–622 Search PubMed.
- R. J. Sammons, J. R. Collier, T. G. Rials and S. Petrovan, J. Appl. Polym. Sci., 2008, 110, 1175–1181 CrossRef CAS.
- A. Michud, M. Tanttu, S. Asaadi, Y. Ma, E. Netti, P. Kääriäinen, A. Persson, A. Berntsson, M. Hummel and H. Sixta, Text. Res. J., 2015 DOI:10.1177/0040517515591774.
- L. E. Wise, M. Murphy and A. A. D'Addieco, Pap. Trade J., 1946, 122, 35–42 CAS.
- M. Yamamoto, R. Kuramae, M. Yanagisawa, D. Ishii and A. Isogai, Biomacromolecules, 2011, 12, 3982–3988 CrossRef CAS PubMed.
- R. Berggren, F. Berthold, E. Sjöholm and M. Lindström, J. Appl. Polym. Sci., 2003, 88, 1170–1179 CrossRef CAS.
- M. Borrega, L. K. Tolonen, F. Bardot, L. Testova and H. Sixta, Bioresour. Technol., 2013, 135, 665–671 CrossRef CAS PubMed.
- J. Janson, Pap. Puu, 1970, 5, 323–329 Search PubMed.
- J. Maenner, D. Ivanoff, R. J. Morley and S. Jary, Lenzinger Ber., 2011, 89, 60–71 CAS.
- R.-B. Adusumalli, J. Keckes, K. Martinschitz, P. Boesecke, H. Weber, T. Roeder, H. Sixta and W. Gindl, Cellulose, 2009, 16, 765–772 CrossRef CAS.
- J. Lenz, J. Schurz and E. Wrentschur, Holzforschung, 1994, 48, 72–76 CrossRef CAS.
- C. Froschauer, M. Hummel, M. Iakovlev, A. Roselli, H. Schottenberger and H. Sixta, Biomacromolecules, 2013, 14, 1741–1750 CrossRef CAS PubMed.
- A. Roselli, M. Hummel, A. Monshizadeh, T. Maloney and H. Sixta, Cellulose, 2014, 21, 3655–3666 CrossRef CAS.
- M. G. Northolt, Lenzinger Ber., 1985, 59, 71–78 CAS.
- W. Gindl, M. Reifferscheid, R. B. Adusumalli, H. Weber, T. Roeder, H. Sixta and T. Schoeberl, Polymer, 2008, 49, 792–799 CrossRef CAS.
- R.-B. Adusumali, M. Reifferscheid, H. Weber, T. Roeder, H. Sixta and W. Gindl, Macromol. Symp., 2006, 244, 119–125 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01679g |
| ‡ dtex (decitex) is a unit for the linear density of filaments common in textile research. 1 dtex indicates a weight of 1 g at a length of 10000 m. The fiber diameter can be calculated assuming a circular cross section and a cellulose density of 1.5 g ml−1. |
| § The Cox–Merz rule is an empirical postulate stating that the complex and dynamic viscosities of a polymer solution or melt are identical at corresponding angular frequencies and shear rates, respectively. It is thus possible to calculate the zero-shear viscosity from the complex viscosity curve obtained in an oscillatory measurement. |
| This journal is © The Royal Society of Chemistry 2016 |